 Lars L. P. Hanssen
Lars L. P. Hanssen Deena Iskander
Deena Iskander
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Hematol., 26 February 2025
Sec. Red Cells, Iron and Erythropoiesis
Volume 4 - 2025 | https://doi.org/10.3389/frhem.2025.1540152
This article is part of the Research TopicUnraveling Molecular Pathways in Stress Erythropoiesis for Therapeutic AdvancementsView all 4 articles
Erythropoiesis is a critical homeostatic process responsible for the production of red blood cells, essential for oxygen delivery to tissues. This review provides a brief overview of erythropoiesis: the maturation of hematopoietic stem cells to circulating red blood cells. We examine the role of glucocorticoids (GCs) in modulating this process, highlighting how they influence erythroid progenitor proliferation and differentiation through various mechanisms, including transcriptional repression and non-genomic pathways. GCs have been shown to inhibit erythroid differentiation while promoting progenitor cell expansion, particularly under stress conditions such as anaemia or blood loss. These mechanisms are likely central to understanding the role of GC signalling in the treatment of haematological diseases such as Diamond-Blackfan anaemia syndrome and myelodysplastic syndromes, emphasising the need for further research into the genetic and epigenetic factors affecting individual responses to glucocorticoid therapy. By elucidating the intricate interplay between GCs and erythropoiesis, this work aims to provide insights into potential therapeutic strategies for managing disorders related to red blood cell underproduction.
Erythropoiesis is a multistep homeostatic process, tightly balanced with red cell destruction, that maintains the pool of circulating red blood cells at the level necessary to assure effective oxygen delivery to tissues. This process requires the steady-state production of approximately 120 million red blood cells per minute (1).
Definitive, adult erythropoiesis occurs in the bone marrow (BM) in three phases. In the first phase, multipotent haematopoietic stem cells (HSCs) and progenitors (MPP) differentiate sequentially via intermediate progenitors to committed progenitor cells originally defined on the basis of the colonies which they generate in semisolid assays as burst-forming unit-erythroid (BFU-E) and colony-forming unit-erythroid (CFU-E) (2). This latter part of this phase is characterised by rapid expansion of erythroid progenitors that lose their limited self-renewal capacity after the BFU-E stage (3, 4). The first phase of erythroid differentiation from HSCs is supported by the perivascular niche of the sinusoids in the trabecular bone, while the second phase occurs in the context of the erythroblastic island (EBI) in the medulla; cell-cell contacts between pro-erythroblasts positioned around a central macrophage promote cell survival and proliferation (5). From pro-erythroblast, differentiation progresses via morphologically distinct nucleated precursor stages: basophilic erythroblast, polychromatic erythroblast, and orthochromatic erythroblast. In the final phase, terminal maturation, the nucleus that has been condensed during the previous stages is extruded from the cell and phagocytosed by the EBI macrophage, after which the remaining reticulocyte undergoes further maturation and is finally released into peripheral blood (6). While the stages of erythroid differentiation are both morphologically and transcriptionally distinct, recent single-cell studies have highlighted the continuous nature of this process and revealed heterogeneity within stages (7, 8).
Endogenous glucocorticoids (GCs) are steroid hormones that regulate the stress response of physiological processes as varied as metabolism, development, mood and cognitive function, and the immune/inflammatory responses (9). The observations that deficiency of mineralocorticoids and GCs in Addison’s disease results in reduced red cell mass and normocytic anaemia (10, 11), while an excess of GCs in Cushing’s disease leads to erythrocytosis (12, 13), provided the first evidence that GC’s regulatory function extends to erythropoiesis. Furthermore, synthetic glucocorticoids are the first-line treatment of anaemia in Diamond-Blackfan anaemia syndrome (DBAS), a rare inherited bone marrow (BM) failure syndrome characterised by anaemia, usually from infancy (14). There is also evidence for the efficacy of low-dose steroids in treatment of anaemia in certain myeloproliferative disorders (15–17) and in the anaemia of chronic disease associated with autoimmune disease or infection (18).
In this review we will outline current knowledge of the role of GCs in erythropoiesis. We will briefly discuss the general mechanism of action of GCs across distinct cell types, examine the specific mechanisms by which GCs alter erythropoiesis, and explore how GC signalling has been exploited for the treatment of haematological disease.
GCs activate the glucocorticoid receptor (GR), a transcription factor (TF) and ligand-dependent nuclear receptor of the steroid family, encoded by the NR3C1 gene located on chromosome 5q31.3. Cytoplasmic GR is primarily associated with a chaperone complex that determines its ligand receptivity (19, 20). Upon binding to its GC ligand, GR dissociates from chaperones and undergoes translocation to the nucleus, where GR acts on chromatin either directly or via association with other TFs. Classically, GR forms a homodimer and activates target genes via binding to cis-regulatory regions, such as gene enhancers and promoters, containing a GC Response Element (GRE, Figure 1A). More recent studies have suggested that GR forms a tetramer or higher order associations upon association with chromatin (21, 22). In many cells, GR target genes include anti-inflammatory and pro-apoptotic genes, as well as mediators of cell metabolic changes. Alternatively, GRs also directly interact with TFs with roles in cell survival and inflammation (Activator Protein 1 (AP-1), Signal Transducer and Activator of Transcription family (STAT), Nuclear Factor-Kappa B (NF- κB)) to negatively regulate their transcriptional activity.
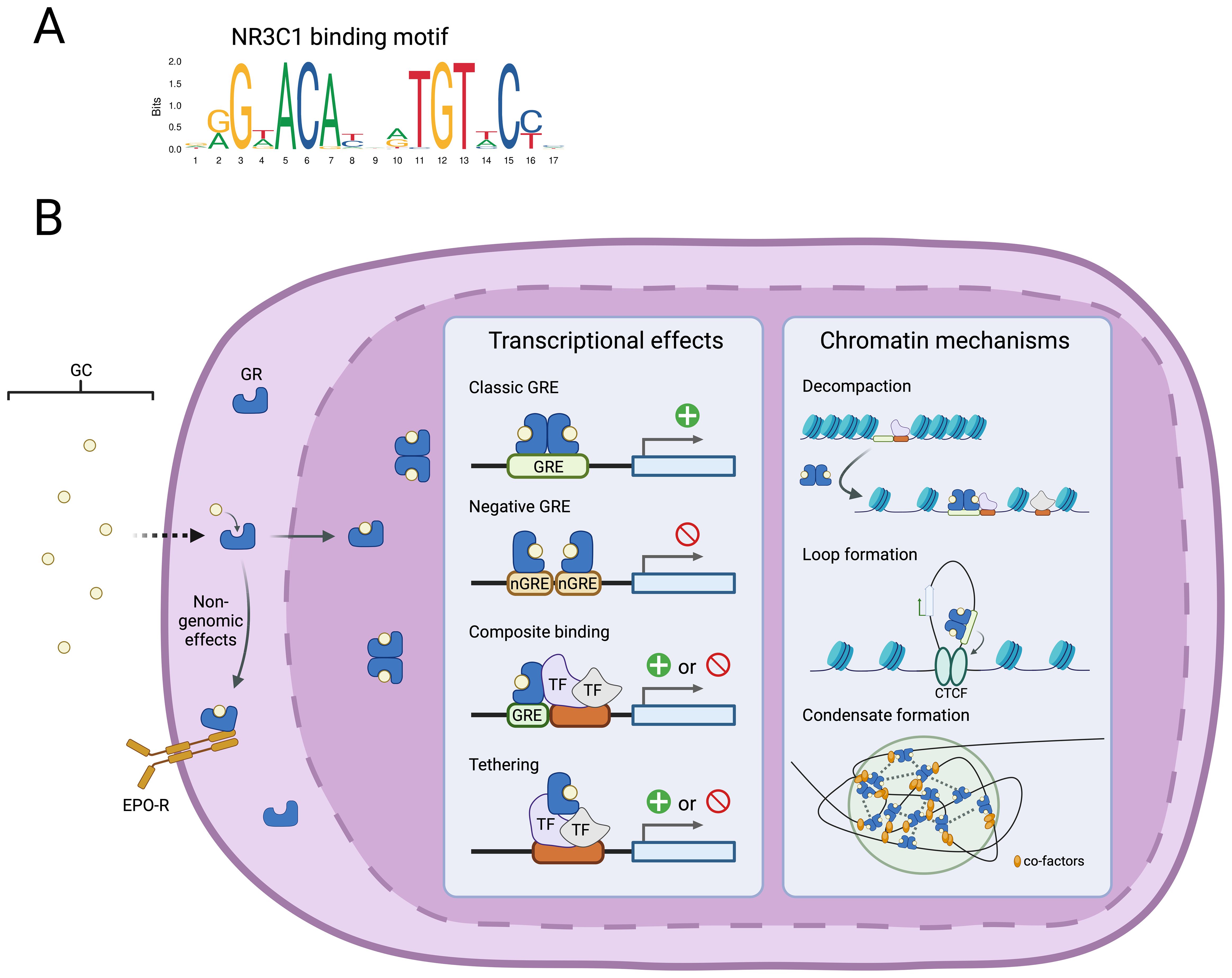
Figure 1. Cellular effects of GCs are mediated by GR chromatin association. (A) DNA binding motif for the glucocorticoid receptor identified in GREs (JASPAR database). (B) Schematic overview of GC association with the GR and the diverse downstream effects on regulation of gene transcription and the chromatin state.
However, the activity of GR is heavily dependent on tissue-type and cell state and there is little overlap in genome-wide GR binding between different cell types (23, 24). This heterogeneity may be mediated by crosstalk via the association of GR with tissue-specific coregulators (25) or heterodimer formation with other nuclear receptors (26) (Figure 1B). The observation that GR binds nearly exclusively to regions of open chromatin, has led to the suggestion that potentiation of chromatin accessibility by cell-type specific TFs is needed to allow GR association (23, 24). Indeed, abrogation of AP-1 binding reduced GR chromatin binding and attenuated the cellular GC response (27) The exact mechanisms by which GR binding affects target gene transcription are not fully defined, but include further chromatin decompaction (28), mediation of regulatory chromatin interactions (29), and nuclear condensate formation (22) (Figure 1B). Finally, there is increasing evidence that GC exert rapid non-genomic effects by altering cAMP signalling and membrane-association with other receptors (30, 31).
Considerable genetic variation of the NR3C1 gene exists in the human population (32), comprising single-nucleotide polymorphisms (SNP) that affect expression of different GR isoforms (33). These SNPs have been linked to human disease and are potential contributors to significant variation in response to synthetic GR agonist treatment (34, 35). GRα is the most widely expressed, transcriptionally active isoform that mediates most GC actions described above. The dominant-negative isoform, GRβ, is expressed in a cell-type specific manner and forms an inactive heterodimer with GRα that is retained in the nucleus, thus inhibiting its activation by cytoplasmic ligand. The SNP rs6198 (A3669G) in the exon 9 3’ UTR stabilises GRβ mRNA and increases its cellular abundance (36), suppressing GRa activity. This SNP has been linked to GC resistance in several autoimmune conditions (37–40). The frequency of the rs6198 polymorphism is increased in patients with the myeloproliferative disorder polycythaemia vera relative to the population, highlighting that genetic variation in the GR and in turn, downstream GC signalling, may affect the penetrance of these diseases (33, 41). An initial study of this variant in a small cohort of DBAS patients tentatively reported an increased rs6198 allele frequency in this patient group, raising the possibility that this variant could explain the variable response to GC treatment (42). However, a more robust subsequent study by the same group did not confirm this finding, and instead identified two SNPs (rs6196 and rs860457) that were enriched in DBAS patients presenting with anaemia before 4 months of age (43). While the biological significance of these variants is currently unknown, it was highlighted that the close proximity to cis-regulatory elements suggests a possible role in the regulation of GR expression in development. Larger cohort studies combined with functional studies are needed to confirm this hypothesis.
Red blood cell production is regulated by an internal gene regulatory circuit that includes the master megakaryocyte-erythroid TF GATA1, expressed early, as well as other erythroid-specific TFs, such as KLF1, NFE2, and TAL1/SCL (44) (Figure 2). Proteomic studies of erythroid TF networks revealed progressive positive-feedback regulatory links between these TFs during erythroid differentiation, as well as cross-antagonism with key TFs involved in cell fate decisions such as GATA1:PU.1 (myeloid lineage), GATA1:GATA2, and KLF1:FLI1 (megakaryocyte lineage) antagonism (45, 46).
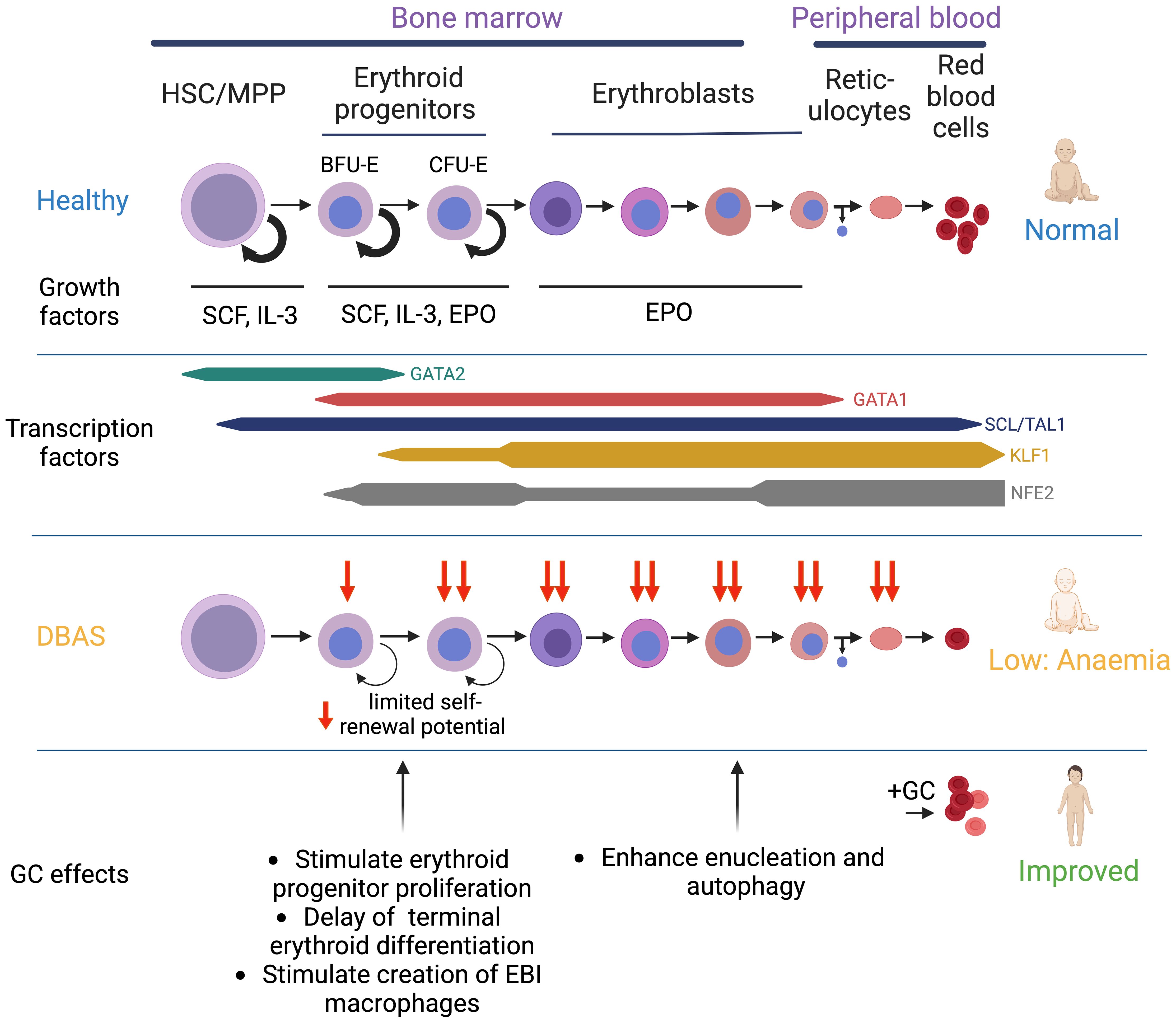
Figure 2. Overview of erythroid differentiation in health and DBAS patients. Schematic overview of erythroid differentiation, including selection of key cytokines and selected transcription factors. Additional transcription factors with an important role in erythropoiesis include LDB1, LMO2, MYB, and FOG1. Impaired erythropoiesis in DBAS and the mechanisms by which GCs restore erythroid output are shown.
To maintain adequate levels of circulating red blood cells in dynamic conditions, each phase of erythropoiesis is also sensitive to regulation by extrinsic factors (47). IL-3 enhances expansion of haematopoietic progenitors including BFU-Es in the early stages of erythropoiesis (48, 49). Stem Cell Factor (SCF)/KIT ligand promotes cell survival and proliferation of BFU-Es, CFU-Es, and proerythroblasts by binding to the KIT receptor (CD117) (50, 51). In the subsequent stages of maturation beginning with CFU-E progenitors, erythropoietin (EPO) signalling via the EPO receptor (EPO-R) is critical for both the proliferation and survival of differentiating erythroid cells before gradually losing EPO-R expression after entering terminal maturation (51–55) (Figure 2). These cytokines and growth factors stimulate several intracellular effector pathways including JAK/STAT, MAPK, and phosphatidylinositol 3-kinase (PI3K), that are known to promote cell growth and survival (56). Gain-of-function mutations in these pathways, such as constitutively activating JAK2 mutations, result in erythrocytosis and myeloproliferative disease (57–59). Similarly, a human EPO mutation resulting in altered EPO-R binding kinetics and reduced JAK2 signalling was described as a rare cause of DBAS (60, 61), whereas EPO gain-of-function mutations are associated with familial erythrocytosis (62).
Under conditions of erythroid stress, such as severe acute or chronic blood loss or hypoxia, red cell production is dramatically increased in response to stress, in a process termed stress erythropoiesis (63, 64). For example, a sustained increase in serum EPO levels, driven by tissue hypoxia, enhances the differentiation of erythroid progenitors and induces an erythroid lineage-bias in multipotent progenitor cells (65, 66). This physiological response has classically been considered part of stress erythropoiesis, but it has been proposed that this mechanism should instead be labelled enhanced steady-state erythropoiesis to distinguish it from a separate inflammation-driven pathway (53). Importantly, inflammation inhibits steady-state erythropoiesis (67–69), skews haematopoiesis toward myelopoiesis (70–72), and increases red cell turnover (73, 74), but simultaneously compensates for these processes by driving inflammation-driven stress erythropoiesis (63, 75). This involves two mechanisms: first, murine experiments have shown that BMP4 and Hedgehog signalling drive the recruitment of additional haematopoietic stem cell populations to the erythroid lineage (76, 77); second, amplification of lineage-committed erythroid progenitors is enhanced to increase red cell production (78–83).
Cell culture systems used to study erythropoiesis include primary CD34+ stem/progenitors, human umbilical cord blood derived erythroid progenitor (HUDEP) cells (84), and systems used to generate large numbers of red blood cells to develop alternatives to allogeneic red blood cell transfusion [termed Human Erythroid Massive Amplification (HEMA) cultures] (85–88). These systems exploit knowledge of the phases of erythroid differentiation in their design. EPO and SCF are used throughout in vitro differentiation, although EPO levels are increased in the latter phases. IL-3 and dexamethasone are used to promote erythroid expansion in early erythropoiesis, but are withdrawn in the last phase, when transferrin and insulin are added to promote terminal erythroid differentiation. Other cytokines and growth factors have been added with varying success in the context of human induced pluripotent stem cells (iPSCs) and murine erythroid differentiation, including pro-inflammatory cytokines such BMP4 and TGF-β signalling effectors that have been implicated in stress erythropoiesis in mice (63, 78, 89, 90).
Intrinsic gene regulatory circuits- composed of TFs that determine cell fate- and extrinsic signalling by growth factors and cytokines, including GCs, are intimately intertwined. GC signalling via the GR has an important role in regulating erythroid output both in steady-state and stress conditions. The cellular mechanisms by which GCs affect erythropoiesis are discussed below.
GC signalling via the GR receptor plays an important role in modulating the decision between self-renewal and differentiation in erythroid cells. Early observations in avian erythroid cells noted that in the absence of GR signalling, progenitors lose their ability to self-renew and enter terminal differentiation (82). Subsequent studies of mice defective for GR (79) have confirmed a cell-autonomous role for GCs in enhancing proliferation of erythroid progenitors (91), and shown that treatment of mice with synthetic GC dexamethasone simultaneously increased the proliferative capacity of BFU-E and CFU-E and inhibited the expression of key differentiation markers (3, 92). The slower rate of differentiation was proposed to allow more BFU-E cell divisions before self-renewal capacity was lost. This model fits with more recent single-cell studies that suggest the switch from late CFU-E to terminal differentiation governs the rate of erythropoiesis (8).
There is a debate in the field on the identity of the erythroid cells that are induced to self-replicate by GC. Data support target populations comprising BFU-E (3, 92, 93), CFU-E/pro-erythroblast (94), or a stress-specific erythroid progenitor cell. The latter was originally defined in mice as specific BMP4 and GC-dependent multi-potent progenitor and BFU-E that generated extramedullary erythropoiesis in the context of irradiation-induced anaemia (95). Subsequent work showed that murine and human bone marrow contain immunophenotypically distinct erythroid progenitors that express foetal haemoglobin during stress-induced differentiation (96). It is not yet clear whether ‘stress’ and steady state progenitor cells are mutually exclusive entities or a dynamic cell state. Further studies are also needed to resolve the impact of species, age and tissue (spleen and foetal liver versus bone marrow) on the cells targeted by GC in both steady state and stress erythropoiesis.
The transcriptional mechanisms by which GC delay erythroid differentiation, particularly in stress conditions, have been explored in numerous studies. Several direct transcriptional effects of GC via their GRE have been observed in erythroid progenitors. First, GATA1, the key TF driving erythroid differentiation in response to EPO-R signalling, is directly inhibited by GC activity via transcriptional repression in mouse erythroleukemia cells and human primary erythroblasts (31, 97), while expression of TFs driving erythroid proliferation such as LMO2 and C-MYB is induced (69, 75). Second, the ZFP36L2 gene, encoding an RNA-binding protein, is upregulated by GC during murine foetal erythropoiesis and has been shown to maintain a progenitor self-renewal state via downregulation of key erythroid differentiation genes by binding to the 3’ UTR ATTA motif of target transcripts (81). The observation that ZFP36L2 is downregulated in patient erythroid progenitors in DBAS, a condition in which expansion of erythroid progenitors is severely abrogated, provides further support for this (98), though the link between ribosomal haploinsufficiency in DBAS and downregulation of ZFP36L2 is unknown.
Direct non-transcriptional mechanisms of GC action have also been proposed. For example, a study of human proerythroblasts showed that GC inhibit erythroid maturation by interacting with EPO signalling and delaying STAT-5 phosphorylation through a rapid, membrane-associated pathway. When erythroid cells were stimulated with both dexamethasone and EPO, the GR associated with the EPO receptor and STAT-5. This association prevented the phosphorylation and subsequent nuclear translocation of STAT-5, thus inhibiting the transcriptional activation of the erythroid differentiation gene programme (31).
The delayed initiation of the terminal erythroid maturation gene programmes may at least in part be mediated by the ability of GCs to regulate cell cycle progression. Single-cell studies of erythropoiesis have revealed extensive changes to the cell cycle in BFU-E and CFU-E progenitors, including a gradual shortening of G1 and regulation of the S-phase at the transition from CFU-E to erythroid maturation (8, 99). Specifically, the cell fate switch from erythroid progenitor to terminal erythroid maturation was shown to be dependent on regulation of the S-phase by p57/KIP2, which mediates replication fork slowing (8, 100). This work highlighted that slowing of replication fork progression by p57/KIP2 in mouse erythroid progenitors prolongs the S-phase, which in turn promotes the CFU-E proliferation state over the switch to erythroid maturation. Deletion of p57/KIP2 resulted in a short S-phase and a failure to maintain a self-renewal state, resulting in deficient erythropoiesis in mouse embryos and reduced erythroid progenitor expansion in vitro (99, 101). GC were shown to modulate this process by inducing expression of p57/KIP2 to prevent S phase shortening and increase CFU-E proliferation, resulting in a larger CFU-E pool and erythropoietic rate. When the GC dexamethasone was withdrawn, p57/KIP2 expression was rapidly lost combined with induction of erythroid maturation transcriptional programmes (100). The expression of p57/KIP2 in CFU-E from DBAS patients was subsequently found to be essential for their GC responsiveness, highlighting the potential importance of this mechanism in humans (94).
Furthermore, the study of a genetic variant in an enhancer of the CCND3 gene identified in genome-wide association studies identified Cyclin D3 as an important regulator of the cell cycle during erythropoiesis. Genetic variation affecting CCND3 expression was linked to erythrocyte traits including both erythrocyte size and number (102). Although the effect of GC on Cyclin D3 has not been directly studied in erythropoiesis, Cyclin D3 expression is known to be sensitive to GC in T-cell lymphoma and lymphoblastic leukaemia where its downregulation and increased p27/KIP1 expression in response to dexamethasone results in increased apoptosis and growth suppression (103). Indeed, these mechanisms may play a role in erythropoiesis as cell cycle exit at the start of terminal erythroid maturation is also regulated by p27/KIP1 (104).
Cross-talk between GC signalling and the p53 response is well-established, but is complex and multivalent (105–107). P53 regulates the cell cycle via downstream activation of p21/CDKN1A (108). The effects of GC on p53 signalling in erythropoiesis have been best studied in the context of DBAS, in which ribosomal protein mutations lead to p53 activation in both patient cells and experimental model systems (98, 109–115). In an RPS19-deficient DBAS mouse model, GCs dampen the upregulation of cell cycle regulator p21/CDKN1A by antagonising the p53 response, stimulating maintenance of erythroid progenitors while delaying differentiation (93).
Cross-talk between GCs and other signalling pathways involved in erythroid differentiation and the stress response synergise to promote erythroid proliferation. GCs modulate gene expression in concert with cytokine receptors involved in erythroid differentiation such as EPO-R and SCF/KIT ligand to drive progenitor proliferation (116). Induction of CXCR4/CXCL12 by combined GC and EPO/SCF stimulation may be an important mediator of this effect by promoting BM retention of CD34+ progenitors and formation of erythroblastic islands (116–118).
Similar evidence exists for crosstalk between GC signalling and other signalling pathways involved in regulation of the stress response and metabolism. The simultaneous stimulation of the hypoxia-signalling transcription factor alpha (HIF1a) and dexamethasone treatment resulted in synergistic BFU-E expansion (3). Similarly, GC signalling and regulation of lipid metabolism may interact via bi-directional cross-talk in erythroblasts (4, 119). The lipid-sensing nuclear receptor PPAR-α co-occupies chromatin sites with GR when activated by agonists GW7647 and fenofibrate, resulting in a synergistic effect on BFU-E self-renewal (4).
Finally, there is increasing evidence that in addition to the direct effects on erythroid progenitors, GCs may stimulate erythroid differentiation indirectly by promoting the generation of macrophages with erythroid supporting potential in the context of the EBI. The surface protein CD169 is present on a subset of macrophages with an important nurturing role in murine erythropoiesis (120) and may be directly involved in EBI formation (121). In human HEMA cultures derived from CD34+ haematopoietic stem cells, dexamethasone promoted the generation of CD169+ macrophages that were observed to associate with proerythroblasts and stimulate cytokinesis to increase proerythroblast proliferation (122). Similar properties were described in culture of CD169+ macrophages that were derived from human monocytes by exposure to dexamethasone (118), highlighting the ability of GC to increase erythroid output by expanding the erythropoietic niche.
Before these cellular effects of GCs on erythropoiesis had been characterised, the ability of GCs to restore erythropoietic output in humans suffering from anaemia caused by certain types of red cell aplasia had been observed (123–125). Evidence for the efficacy of low-dose steroids in treatment of anaemia in myeloproliferative disorders (15–17) and for improved haemoglobin recovery after a haemolytic crisis in hereditary spherocytosis (126, 127) have also been suggested to be driven by enhancing erythropoiesis.
Although adrenal insufficiency causes a reduction in red cell mass and normocytic anaemia, the molecular mechanisms underlying this have not been studied directly (10, 11). The erythrocytosis observed in Cushing syndrome has only recently been exploited as a natural model for the study of erythropoiesis in chronic GC excess (13). CD34+ erythroid progenitors isolated from hypercortisolaemic Cushing patients were able to generate large numbers of immature erythroid cells in vitro, both in the presence and absence of further GC stimulation, suggesting that constitutive GR activation or epigenetic memory may be established upon chronic exposure. This phenotype disappeared in CD34+ cells isolated from patients in remission following pituitary adenoma removal, which instead showed resistance to GC stimulation and reduced erythroid cell numbers. The mechanisms underpinning this long-lasting desensitisation to GC stimuli has not yet been studied, but may be related to cytoplasmic retention of GRα (117).
GCs remain the only widely used drug class in the treatment of red cell failure in DBAS (14, 128), since it was first used over half a century ago (129). This congenital BM failure syndrome is rare (5-7 cases per million births) and is characterised by poor growth, congenital abnormalities, and severe anaemia, usually from infancy (14, 128, 130). The anaemia initially responds to high-dose GC treatment in 60-80% of patients, after which the dose is gradually tapered to minimise treatment-related morbidity. Unfortunately, the development of treatment-resistance is common and only 20-30% of patients respond to low-dose GCs long-term (131). GC treatment failure commits patients to a regular transfusion programme or allogeneic haematopoietic stem cell transplantation (HSCT), which remains the only curative treatment available for the BM-mediated effects of DBAS. Both treatments carry considerable treatment-related morbidity and mortality (130). While lentiviral vector-based gene therapies expressing RPS19 or GATA1 are currently being explored, these are not yet in use clinically (128).
DBAS is a ribosomopathy and more than 70% of DBAS cases are caused by heterozygous loss-of-function mutations in one of the ribosomal proteins (RP) of the large (RPL) or small (RPS) ribosomal subunits resulting in haploinsufficiency. Mutations most commonly affect the RPS19 gene (25% of patients) but have been described in over 20 RPS and RPL genes (132). Pathogenic variants in other genes (TSR2, GATA1, ADA2, EPO, and HEATR3) have been described to phenocopy DBAS in rare cases and in a quarter of patients, no pathogenic cause can be identified in genetic screens. While the pathophysiology of the erythroid lineage defect seen in DBAS has not been fully elucidated, multiple cellular aberrations arise due to reduced or selective translation of mRNAs (133) and nucleolar stress both caused by RP dysfunction (134). These include p53-mediated apoptosis, reduced levels of key erythroid TF GATA1, heme toxicity due imbalanced heme/globin production, and increased inflammation in the BM niche [for detailed review see (128, 132)].
Progress has been made in elucidating the mechanisms by which GCs influence these pathogenic processes. GC treatment did not ameliorate heme toxicity in RPL11-haploinsufficient mice (135). Instead of increasing GATA1 levels, GCs further inhibit GATA1 expression (31, 97), thus delaying erythroid differentiation and simultaneously stimulating expansion of progenitor cells via the stress erythropoiesis pathway (Figure 2, see above). This process may be further stimulated by TNFα and IFNγ-mediated inflammation in bone marrow niche that was observed in studies of DBAS patient bone marrow (98) and circulating red blood cells from DBAS patients (136). Moreover, GCs counter p53 activation in DBAS. The study of RPS19-mutant patient-derived CFU-Es and HUDEP2 erythroid cells showed reduced p53 signalling and apoptosis upon GC-treatment (101) and GC prevent p53 activation in a RPS19 mouse model (93).
Recent hypotheses propose that GCs may also support enucleation via c-Myc repression and modulate autophagy via mTor signalling in DBAS but direct evidence for these roles in erythropoiesis is awaited (137). Mytophagy, a form or autophagy specific to erythroid maturation, is an important regulatory component in erythropoiesis which facilitates the removal of mitochondria and other organelles during terminal maturation (138, 139). GATA1-mediated induction of mytophagy gene transcription requires silencing of mTor signalling (140), and increased mTor signalling results in macrocytic anaemia in mice (141). Interestingly, a screen for novel therapeutic approaches for DBAS identified the small-molecule SMER28 that was able to rescue erythroid differentiation of patient iPSCs by inducing mTor-independent mytophagy (142). The mTor signalling pathway is increased in DBAS, suggesting inhibition may be a therapeutic option to further enhance mytophagy (143). Paradoxically, a subset of DBAS patients respond to the mTor-inducing amino acid L-Leucine (144). This bifunctional response is explained by the dual, stage-dependent function of mTor in erythropoiesis, both stimulating proliferation of BFU-E and CFU-E proliferation in early erythropoiesis and inhibiting mytophagy in definitive erythroid maturation (140) (Figure 2). Inhibition of mTor signalling by GCs has been described in several non-haematopoietic cell types (145, 146), suggesting that GCs may contribute to erythropoiesis in DBAS by stimulating mytophagy while simultaneously restoring erythroid progenitor proliferation via stress erythropoiesis (137).
Resistance to GC treatment in DBAS is only partially understood (94), and may be driven by a wide range of mechanisms related to GR genetic variation (such as the A3669G polymorphism in GR) and transcriptional and post-transcriptional regulation of the GR (32, 147). Interestingly, foetal haemoglobin (HbF) expressing cord blood-derived CD34+ progenitors do not respond to GC stimulation, whereas peripheral blood-derived progenitors do (94). This was recently linked to the finding that expression of developmental globin-switching regulator BCL11A correlated to the ability of erythroid progenitors to proliferate upon GC treatment (148).
MDS is a heterogeneous clonal stem cell disorder characterised by rapid proliferation in the BM paired with dysplastic maturation and high cell turnover due to excessive apoptosis, explaining the peripheral cytopenias associated with the disease. The most common cytopenia is anaemia which is present in 80-90% of all patients (149). Cytopenic and dysplastic features are used to classify and risk-stratify MDS, based on the risk of transformation to AML present in this disorder (150). The only form to be classified predominantly by the presence of a cytogenetic abnormality is MDS with isolated del(5q), characterised clinically by a low risk of disease progression and a macrocytic anaemia with erythroid hypoplasia often resulting in transfusion-dependence (151, 152).
Interestingly, the hemizygous del(5q) deletion on the long arm of chromosome 5 contains the RPS14 gene. Analogous to DBAS, haploinsufficiency in del(5q) MDS drives ribosomal stress and activation of the p53 response in erythroid cells, predisposing cells to apoptosis (153). Unlike DBAS, GCs are not routinely used in treatment of del(5q) MDS, whereas lenalidomide restores transfusion-independence and improves survival (154). The effects of lenalidomide and GCs on erythropoiesis were compared in the in vitro erythroid differentiation of CD34+ stem cells. Whereas GCs stimulated erythroid progenitor expansion at the BFU-e stage, Lenalidomide stimulated CFU-e proliferation suggesting separate mechanisms of action. Both also stimulated erythropoiesis in RPS19 and RPS14 mutant CD34+ cells and showed additive effects when used in combination (155, 156). These in vitro findings were confirmed in del(5q) MDS patients who developed lenalidomide resistance. Transfusion-independence was restored in five out of eight patients when dexamethasone was added to lenalidomide treatment. This effect was suggested to be mediated by observed reduction of the p53 response, and could be replicated in vitro via specific suppression of p53 in patient-derived CD34+ cells by antisense oligonucleotide, Cenersen, which increased erythroid burst recovery (157).
GCs play a key regulatory role in erythropoiesis, particularly during erythropoietic stress such as chronic anaemia or acute blood loss. Mechanistic studies point to important roles in (1) inhibiting erythroid differentiation to prolong progenitor cell expansion in synergy with other signalling pathways and by (2) modulating cell cycle progression to enhance proliferation before entering terminal maturation. The full repertoire of GC effects, both transcriptionally and post translationally are yet to be determined.
Direct study of gene-regulatory networks in HSCs and erythroid progenitors following GC stimulation through single-cell genomics will provide further insight into the cell lineage decisions needed to drive effective erythropoiesis and how these are modulated by endogenous or exogenous GC. DBAS especially serves as a useful model for the role of GCs in stress erythropoiesis, having already highlighted its influence on p53 signalling and cell cycle regulation. Spatial approaches will help to establish whether GC also have an effect on the homing of erythroid cells within and from the BM.
Finally, the impact of genetic factors such as GR SNPs as well as epigenetic factors on the responsiveness of patients with a variety of haematological diseases to the effects of GC, are yet to be determined.
LH: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. DI: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. LH is supported by a UKRI LMS Chain-Florey Academic Clinical Fellowship and Imperial College NIHR Biomedical Research Centre grant. DI is supported by an NIHR Imperial College Post-Doctoral Fellowship and receives funding from the DBA foundation DBA UK and an ASH-EHA TRTH award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. (2014) 5:3. doi: 10.3389/fphys.2014.00003
2. Iskander D, Psaila B, Gerrard G, Chaidos A, En Foong H, Harrington Y, et al. Elucidation of the EP defect in Diamond-Blackfan anemia by characterization and prospective isolation of human EPs. Blood. (2015) 125:2553–7. doi: 10.1182/blood-2014-10-608042
3. Flygare J, Estrada VR, Shin C, Gupta S, Lodish H. Stress erythropoiesis: HIF-1 alpha synergizes with glucocorticoids to induce BFU-E progenitor self-renewal. Blood. (2010) 116:814–4. doi: 10.1182/blood.V116.21.814.814
4. Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, et al. PPAR-alpha and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature. (2015) 522:474–7. doi: 10.1038/nature14326
5. May A, Forrester LM. The erythroblastic island niche: modeling in health, stress, and disease. Exp Hematol. (2020) 91:10–21. doi: 10.1016/j.exphem.2020.09.185
6. Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature. (2005) 437:754–8. doi: 10.1038/nature03964
7. Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. (2017) 19:271–81. doi: 10.1038/ncb3493
8. Tusi BK, Wolock SL, Weinreb C, Hwang Y, Hidalgo D, Zilionis R, et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. (2018) 555:54–60. doi: 10.1038/nature25741
9. Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Front Immunol. (2019) 10:1545. doi: 10.3389/fimmu.2019.01545
10. Lichtman MA, Kaushansky K, Prchal JT, Levi MM, Burns LJ, Armitage JO. Anemia of endocrine disorders. In: Williams manual of hematology, 9e. New York, NY: McGraw-Hill Education (2017).
11. Ihara T, Tsujikawa T, Hodohara K, Inoue H, Fujiyama Y, Bamba T, et al. Severe anemia in a patient with isolated adrenocorticotropin deficiency. Intern Med. (1996) 35:898–901. doi: 10.2169/internalmedicine.35.898
12. Gursoy A, Dogruk Unal A, Ayturk S, Karakus S, Izol AN, Bascil Tutuncu N, et al. Polycythemia as the first manifestation of Cushing’s disease. J Endocrinol Invest. (2006) 29:742–4. doi: 10.1007/BF03344186
13. Varricchio L, Geer EB, Martelli F, Mazzarini M, Funnell A, Bieker JJ, et al. Patients with hypercortisolemic cushing disease possess a distinct class of hematopoietic progenitor cells leading to erythrocytosis. Haematol. (2022) 108:1053–67. doi: 10.3324/haematol.2021.280542
14. Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. (2010) 116:3715–23. doi: 10.1182/blood-2010-02-251090
15. Hernández-Boluda J-C, Martínez-Trillos A, García-Gutiérrez V, Ferrer-Marín F, Xicoy B, Alvarez-Larrán A, et al. Long-term results of prednisone treatment for the anemia of myelofibrosis. Leukemia Lymphoma. (2016) 57:120–4. doi: 10.3109/10428194.2015.1046866
16. Mesa RA, Yao X, Cripe LD, Li CY, Litzow M, Paietta E, et al. Lenalidomide and prednisone for myelofibrosis: Eastern cooperative oncology group (ECOG) phase 2 trial E4903. Blood. (2010) 116:4436–8. doi: 10.1182/blood-2010-05-287417
17. Mesa RA, Steensma DP, Pardanani A, Li C-Y, Elliott M, Kaufmann SH, et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood. (2003) 101:2534–41. doi: 10.1182/blood-2002-09-2928
18. Khalil M, He Z, Sangal A, Agheli A, Arora A, Dumlao T, et al. High dose glucocorticoids potentiate the reponse to erythropoietin. Blood. (2006) 108:3775–5. doi: 10.1182/blood.V108.11.3775.3775
19. Wang RY-R, Noddings CM, Kirschke E, Myasnikov AG, Johnson JL, Agard DA. Structure of Hsp90–Hsp70–Hop–GR reveals the Hsp90 client-loading mechanism. Nature. (2022) 601:460–4. doi: 10.1038/s41586-021-04252-1
20. Noddings CM, Johnson JL, Agard DA. Cryo-EM reveals how Hsp90 and FKBP immunophilins co-regulate the glucocorticoid receptor. Nat Struct Mol Biol. (2023) 30:1867–77. doi: 10.1038/s41594-023-01128-y
21. Johnson TA, Paakinaho V, Kim S, Hager GL, Presman DM. Genome-wide binding potential and regulatory activity of the glucocorticoid receptor’s monomeric and dimeric forms. Nat Commun. (2021) 12:1987. doi: 10.1038/s41467-021-22234-9
22. Stortz M, Presman DM, Bruno L, Annibale P, Dansey MV, Burton G, et al. Mapping the dynamics of the glucocorticoid receptor within the nuclear landscape. Sci Rep. (2017) 7:6219. doi: 10.1038/s41598-017-06676-0
23. Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. (2013) 32:1568–83. doi: 10.1038/emboj.2013.106
24. John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. (2011) 43:264–8. doi: 10.1038/ng.759
25. Fadel L, Dacic M, Fonda V, Sokolsky BA, Quagliarini F, Rogatsky I, et al. Modulating glucocorticoid receptor actions in physiology and pathology: Insights from coregulators. Pharmacol Ther. (2023) 251:108531. doi: 10.1016/j.pharmthera.2023.108531
26. De Bosscher K, Desmet SJ, Clarisse D, Estébanez-Perpiña E, Brunsveld L. Nuclear receptor crosstalk — defining the mechanisms for therapeutic innovation. Nat Rev Endocrinol. (2020) 16:363–77. doi: 10.1038/s41574-020-0349-5
27. Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. (2011) 43:145–55. doi: 10.1016/j.molcel.2011.06.016
28. Jubb AW, Boyle S, Hume DA, Bickmore WA. Glucocorticoid receptor binding induces rapid and prolonged large-scale chromatin decompaction at multiple target loci. Cell Rep. (2017) 21:3022–31. doi: 10.1016/j.celrep.2017.11.053
29. Jing D, Huang Y, Liu X, Sia KCS, Zhang JC, Tai X, et al. Lymphocyte-specific chromatin accessibility pre-determines glucocorticoid resistance in acute lymphoblastic leukemia. Cancer Cell. (2018) 34:906–921.e8. doi: 10.1016/j.ccell.2018.11.002
30. Nuñez FJ, Johnstone TB, Corpuz ML, Kazarian AG, Mohajer NN, Tliba O, et al. Glucocorticoids rapidly activate cAMP production via G αs to initiate non-genomic signaling that contributes to one-third of their canonical genomic effects. FASEB J. (2020) 34:2882–95. doi: 10.1096/fj.201902521R
31. Stellacci E, Di Noia A, Di Baldassarre A, Migliaccio G, Battistini A, Migliaccio AR. Interaction between the glucocorticoid and erythropoietin receptors in human erythroid cells. Exp Hematol. (2009) 37:559–72. doi: 10.1016/j.exphem.2009.02.005
32. Leventhal SM, Lim D, Green TL, Cantrell AE, Cho K, Greenhalgh DG. Uncovering a multitude of human glucocorticoid receptor variants: an expansive survey of a single gene. BMC Genet. (2019) 20:16. doi: 10.1186/s12863-019-0718-z
33. Varricchio L, Migliaccio AR. The role of glucocorticoid receptor (GR) polymorphisms in human erythropoiesis. Am J Blood Res. (2014) 4:53–72.
34. Zhou J, Cidlowski J. The human glucocorticoid receptor: One gene, multiple proteins and diverse responses. Steroids. (2005) 70:407–17. doi: 10.1016/j.steroids.2005.02.006
35. Hu W, Jiang C, Kim M, Yang W, Zhu K, Guan D, et al. Individual-specific functional epigenomics reveals genetic determinants of adverse metabolic effects of glucocorticoids. Cell Metab. (2021) 33:1592–1609.e7. doi: 10.1016/j.cmet.2021.06.004
36. Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol. (2003) 23:4319–30. doi: 10.1128/MCB.23.12.4319-4330.2003
37. Herrera C, Marcos M, Carbonell C, Mirón-Canelo JA, Espinosa G, Cervera R, et al. Association between allelic variants of the human glucocorticoid receptor gene and autoimmune diseases: A systematic review and meta-analysis. Autoimmun Rev. (2018) 17:449–56. doi: 10.1016/j.autrev.2017.11.034
38. Pac M, Krata N, Moszczuk B, Wyczałkowska-Tomasik A, Kaleta B, Foroncewicz B, et al. NR3C1 glucocorticoid receptor gene polymorphisms are associated with membranous and IgA nephropathies. Cells. (2021) 10:3186. doi: 10.3390/cells10113186
39. Nascimento M, Teixeira ES, Dal’ Bó IF, Peres KC, Rabi LT, Cury AN, et al. NR3C1 rs6198 variant may be involved in the relationship of graves’ disease with stressful events. Biomedicines. (2023) 11:1155. doi: 10.3390/biomedicines11041155
40. Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. (2001) 28:2383–8.
41. Varricchio L, Masselli E, Alfani E, Battistini A, Migliaccio G, Vannucchi AM, et al. The dominant negative β isoform of the glucocorticoid receptor is uniquely expressed in erythroid cells expanded from polycythemia vera patients. Blood. (2011) 118:425–36. doi: 10.1182/blood-2010-07-296921
42. Varricchio L, Godbold J, Scott SA, Whitsett C, Da Costa L, Pospisilova D, et al. Increased frequency of the glucocorticoid receptor A3669G (rs6198) polymorphism in patients with Diamond-Blackfan anemia. Blood. (2011) 118:473–4. doi: 10.1182/blood-2011-03-342139
43. Lonetti A, Indio V, Dianzani I, Ramenghi U, Da Costa L, Pospíšilová D, et al. The glucocorticoid receptor polymorphism landscape in patients with diamond blackfan anemia reveals an association between two clinically relevant single nucleotide polymorphisms and time to diagnosis. Front Physiol. (2021) 12:745032. doi: 10.3389/fphys.2021.745032
44. Anguita E, Hughes J, Heyworth C, Blobel GA, Wood WG, Higgs DR. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. (2004) 23:2841–52. doi: 10.1038/sj.emboj.7600274
45. Gillespie MA, Palii CG, Sanchez-Taltavull D, Shannon P, Longabaugh WJR, Downes DJ, et al. Absolute quantification of transcription factors reveals principles of gene regulation in erythropoiesis. Mol Cell. (2020) 78:960–974.e11. doi: 10.1016/j.molcel.2020.03.031
46. Palii CG, Cheng Q, Gillespie MA, Shannon P, Mazurczyk M, Napolitani G, et al. Single-cell proteomics reveal that quantitative changes in co-expressed lineage-specific transcription factors determine cell fate. Cell Stem Cell. (2019) 24:812–820.e5. doi: 10.1016/j.stem.2019.02.006
47. Caulier AL, Sankaran VG. Molecular and cellular mechanisms that regulate human erythropoiesis. Blood. (2022) 139:2450–9. doi: 10.1182/blood.2021011044
48. Migliaccio G, Migliaccio A, Adamson J. In vitro differentiation of human granulocyte/macrophage and erythroid progenitors: comparative analysis of the influence of recombinant human erythropoietin, g-CSF, GM-CSF, and IL-3 in serum-supplemented and serum- deprived cultures. Blood. (1988) 72:248–56. doi: 10.1182/blood.V72.1.248.248
49. Goodman JW, Hall EA, Miller KL, Shinpock SG. Interleukin 3 promotes erythroid burst formation in ‘serum-free’ cultures without detectable erythropoietin. Proc Natl Acad Sci USA. (1985) 82:3291–5. doi: 10.1073/pnas.82.10.3291
50. Sui X, Krantz SB, Zhao ZJ. Stem cell factor and erythropoietin inhibit apoptosis of human erythroid progenitor cells through different signalling pathways. Br J Haematol. (2000) 110:63–70. doi: 10.1046/j.1365-2141.2000.02145.x
51. Spivak J, Pham T, Isaacs M, Hankins W. Erythropoietin is both a mitogen and a survival factor. Blood. (1991) 77:1228–33. doi: 10.1182/blood.V77.6.1228.1228
52. Watowich SS. The erythropoietin receptor: Molecular structure and hematopoietic signaling pathways. J Invest Med. (2011) 59:1067–72. doi: 10.2310/JIM.0b013e31820fb28c
53. Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. (2011) 118:6258–68. doi: 10.1182/blood-2011-07-356006
54. Hidalgo D, Bejder J, Pop R, Gellatly K, Hwang Y, Maxwell Scalf S, et al. EpoR stimulates rapid cycling and larger red cells during mouse and human erythropoiesis. Nat Commun. (2021) 12:7334. doi: 10.1038/s41467-021-27562-4
55. Koury MJ, Bondurant MC. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science. (1990) 248:378–81. doi: 10.1126/science.2326648
56. Zhang Y, Wang L, Dey S, Alnaeeli M, Suresh S, Rogers H, et al. Erythropoietin action in stress response, tissue maintenance and metabolism. IJMS. (2014) 15:10296–333. doi: 10.3390/ijms150610296
57. James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. (2005) 434:1144–8. doi: 10.1038/nature03546
58. Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-Function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. (2005) 352:1779–90. doi: 10.1056/NEJMoa051113
59. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. (2005) 365:1054–61. doi: 10.1016/S0140-6736(05)71142-9
60. Kim AR, Ulirsch JC, Wilmes S, Unal E, Moraga I, Karakukcu M, et al. Discovery of the first pathogenic human EPO mutation provides mechanistic insight into cytokine signaling. Blood. (2016) 128:331–1. doi: 10.1182/blood.V128.22.331.331
61. Kim AR, Ulirsch JC, Wilmes S, Unal E, Moraga I, Karakukcu M, et al. Functional selectivity in cytokine signaling revealed through a pathogenic EPO mutation. Cell. (2017) 168:1053–1064.e15. doi: 10.1016/j.cell.2017.02.026
62. Zmajkovic J, Lundberg P, Nienhold R, Torgersen ML, Sundan A, Waage A, et al. A gain-of-Function mutation in EPO in familial erythrocytosis. N Engl J Med. (2018) 378:924–30. doi: 10.1056/NEJMoa1709064
63. Paulson RF, Ruan B, Hao S, Chen Y. Stress erythropoiesis is a key inflammatory response. Cells. (2020) 9:634. doi: 10.3390/cells9030634
64. Paulson RF, Hariharan S, Little JA. Stress erythropoiesis: definitions and models for its study. Exp Hematol. (2020) 89:43–54.e2. doi: 10.1016/j.exphem.2020.07.011
65. Grover A, Mancini E, Moore S, Mead AJ, Atkinson D, Rasmussen KD, et al. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med. (2014) 211:181–8. doi: 10.1084/jem.20131189
66. Singh RP, Grinenko T, Ramasz B, Franke K, Lesche M, Dahl A, et al. Hematopoietic stem cells but not multipotent progenitors drive erythropoiesis during chronic erythroid stress in EPO transgenic mice. Stem Cell Rep. (2018) 10:1908–19. doi: 10.1016/j.stemcr.2018.04.012
67. Xiao W, Koizumi K, Nishio M, Endo T, Osawa M, Fujimoto K, et al. Tumor necrosis factor-alpha inhibits generation of glycophorin a+ cells by CD34+ cells. Exp Hematol. (2002) 30:1238–47. doi: 10.1016/s0301-472x(02)00930-x
68. Tsopra OA, Ziros PG, Lagadinou ED, Symeonidis A, Kouraklis-Symeonidis A, Thanopoulou E, et al. Disease-related anemia in chronic lymphocytic leukemia is not due to intrinsic defects of erythroid precursors: A possible pathogenetic role for tumor necrosis factor-alpha. Acta Haematol. (2009) 121:187–95. doi: 10.1159/000220331
69. Rusten L, Jacobsen S. Tumor necrosis factor (TNF)-alpha directly inhibits human erythropoiesis in vitro: role of p55 and p75 TNF receptors. Blood. (1995) 85:989–96. doi: 10.1182/blood.V85.4.989.bloodjournal854989
70. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. (2017) 130:1693–8. doi: 10.1182/blood-2017-06-780882
71. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. (2016) 18:607–18. doi: 10.1038/ncb3346
72. Oduro KA, Liu F, Tan Q, Kim C-K, Lubman O, Fremont D, et al. Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood. (2012) 120:2203–13. doi: 10.1182/blood-2011-11-391342
73. Libregts SF, Gutiérrez L, De Bruin AM, Wensveen FM, Papadopoulos P, Van Ijcken W, et al. Chronic IFN-γ production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood. (2011) 118:2578–88. doi: 10.1182/blood-2010-10-315218
74. Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, et al. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science. (2019) 363:eaao5213. doi: 10.1126/science.aao5213
75. Ruan B, Paulson RF. Metabolic regulation of stress erythropoiesis, outstanding questions, and possible paradigms. Front Physiol. (2023) 13:1063294. doi: 10.3389/fphys.2022.1063294
76. Perry JM, Harandi OF, Porayette P, Hegde S, Kannan AK, Paulson RF. Maintenance of the BMP4-dependent stress erythropoiesis pathway in the murine spleen requires hedgehog signaling. Blood. (2009) 113:911–8. doi: 10.1182/blood-2008-03-147892
77. Harandi OF, Hedge S, Wu D-C, Mckeone D, Paulson RF. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J Clin Invest. (2010) 120:4507–19. doi: 10.1172/JCI41291
78. Hao S, Xiang J, Wu D-C, Fraser JW, Ruan B, Cai J, et al. Gdf15 regulates murine stress erythroid progenitor proliferation and the development of the stress erythropoiesis niche. Blood Adv. (2019) 3:2205–17. doi: 10.1182/bloodadvances.2019000375
79. Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, et al. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. (1999) 13:2996–3002. doi: 10.1101/gad.13.22.2996
80. Ganguli G, Back J, Sengupta S, Wasylyk B. The p53 tumour suppressor inhibits glucocorticoid-induced proliferation of erythroid progenitors. EMBO Rep. (2002) 3:569–74. doi: 10.1093/embo-reports/kvf114
81. Zhang L, Prak L, Rayon-Estrada V, Thiru P, Flygare J, Lim B, et al. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. (2013) 499:92–6. doi: 10.1038/nature12215
82. Wessely O. The glucocorticoid receptor is a key regulator of the decision between self-renewal and differentiation in erythroid progenitors. EMBO J. (1997) 16:267–80. doi: 10.1093/emboj/16.2.267
83. von Lindern M, Zauner W, Mellitzer G, Steinlein P, Fritsch G, Huber K, et al. The glucocorticoid receptor cooperates with the erythropoietin receptor and c-kit to enhance and sustain proliferation of erythroid progenitors in vitro. Blood. (1999) 94:550–9. doi: 10.1182/blood.V94.2.550
84. Kurita R, Suda N, Sudo K, Miharada K, Hiroyama T, Miyoshi H, et al. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PloS One. (2013) 8:e59890. doi: 10.1371/journal.pone.0059890
85. Anstee DJ. Production of erythroid cells from human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPS). Transfusion Clinique Biologique. (2010) 17:104–9. doi: 10.1016/j.tracli.2010.05.005
86. Migliaccio AR, Varricchio L. Concise review: Advanced cell culture models for diamond blackfan anemia and other erythroid disorders. Stem Cells. (2018) 36:172–9. doi: 10.1002/stem.2735
87. Migliaccio G, Sanchez M, Masiello F, Tirelli V, Varricchio L, Whitsett C, et al. Humanized culture medium for clinical expansion of human erythroblasts. Cell Transplant. (2010) 19:453–69. doi: 10.3727/096368909X485049
88. Claessen M, Varga E, Heshusius S, Heideveld E, Hansen M, Visser A, et al. Large scale culture and differentiation of erythroblasts from PBMC and iPSC. Blood. (2018) 132:2319–9. doi: 10.1182/blood-2018-99-118882
89. Ebrahimi M, Forouzesh M, Raoufi S, Ramazii M, Ghaedrahmati F, Farzaneh M. Differentiation of human induced pluripotent stem cells into erythroid cells. Stem Cell Res Ther. (2020) 11:483. doi: 10.1186/s13287-020-01998-9
90. Sivalingam J, Lam AT-L, Chen HY, Yang BX, Chen AK-L, Reuveny S, et al. Superior red blood cell generation from human pluripotent stem cells through a novel microcarrier-based embryoid body platform. Tissue Eng Part C: Methods. (2016) 22:765–80. doi: 10.1089/ten.tec.2015.0579
91. Dolznig H, Grebien F, Deiner EM, Stangl K, Kolbus A, Habermann B, et al. Erythroid progenitor renewal versus differentiation: genetic evidence for cell autonomous, essential functions of EpoR, Stat5 and the GR. Oncogene. (2006) 25:2890–900. doi: 10.1038/sj.onc.1209308
92. Li H, Natarajan A, Ezike J, Barrasa MI, Le Y, Feder ZA, et al. Rate of progression through a continuum of transit-amplifying progenitor cell states regulates blood cell production. Dev Cell. (2019) 49:118–129.e7. doi: 10.1016/j.devcel.2019.01.026
93. Sjogren SE, Siva K, Soneji S, George AJ, Winkler M, Jaako P, et al. Glucocorticoids improve erythroid progenitor maintenance and dampen Trp53 response in a mouse model of Diamond-Blackfan anaemia. Br J Haematol. (2015) 171:517–29. doi: 10.1111/bjh.13632
94. Ashley RJ, Yan H, Wang N, Hale J, Dulmovits BM, Papoin J, et al. Steroid resistance in diamond blackfan anemia associates with p57Kip2 dysregulation in erythroid progenitors. J Clin Invest. (2020) 130:2097–110. doi: 10.1172/JCI132284
95. Singbrant S, Mattebo A, Sigvardsson M, Strid T, Flygare J. Prospective isolation of radiation induced erythroid stress progenitors reveals unique transcriptomic and epigenetic signatures enabling increased erythroid output. Haematol. (2020) 105:2561–71. doi: 10.3324/haematol.2019.234542
96. Xiang J, Wu D-C, Chen Y, Paulson RF. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood. (2015) 125:1803–12. doi: 10.1182/blood-2014-07-591453
97. Chang TJ, Scher BM, Waxman S, Scher W. Inhibition of mouse GATA-1 function by the glucocorticoid receptor: possible mechanism of steroid inhibition of erythroleukemia cell differentiation. Mol Endocrinol. (1993) 7:528–42. doi: 10.1210/mend.7.4.8502237
98. Iskander D, Wang G, Heuston EF, Christodoulidou C, Psaila B, Ponnusamy K, et al. Single-cell profiling of human bone marrow progenitors reveals mechanisms of failing erythropoiesis in Diamond-Blackfan anemia. Sci Trans Med. (2021) 13:eabf0113. doi: 10.1126/scitranslmed.abf0113
99. Socolovsky M. The role of specialized cell cycles during erythroid lineage development: insights from single-cell RNA sequencing. Int J Hematol. (2022) 116:163–73. doi: 10.1007/s12185-022-03406-9
100. Hwang Y, Futran M, Hidalgo D, Pop R, Iyer DR, Scully R, et al. Global increase in replication fork speed during a p57KIP2-regulated erythroid cell fate switch. Sci Adv. (2017) 3:e1700298. doi: 10.1126/sciadv.1700298
101. Wang B, Wang C, Wan Y, Gao J, Ma Y, Zhang Y, et al. Decoding the pathogenesis of diamond–blackfan anemia using single-cell RNA-seq. Cell Discovery. (2022) 8:41. doi: 10.1038/s41421-022-00389-z
102. Sankaran VG, Ludwig LS, Sicinska E, Xu J, Bauer DE, Eng JC, et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. (2012) 26:2075–87. doi: 10.1101/gad.197020.112
103. Kullmann MK, Grubbauer C, Goetsch K, Jäkel H, Podmirseg SR, Trockenbacher A, et al. The p27–Skp2 axis mediates glucocorticoid-induced cell cycle arrest in t-lymphoma cells. Cell Cycle. (2013) 12:2625–35. doi: 10.4161/cc.25622
104. Hsieh FF, Barnett LA, Green WF, Freedman K, Matushansky I, Skoultchi AI, et al. Cell cycle exit during terminal erythroid differentiation is associated with accumulation of p27Kip1 and inactivation of cdk2 kinase. Blood. (2000) 96:2746–54. doi: 10.1182/blood.V96.8.2746
105. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. (2019) 20:199–210. doi: 10.1038/s41580-019-0110-x
106. Sengupta S, Wasylyk B. Physiological and pathological consequences of the interactions of the p53 tumor suppressor with the glucocorticoid, androgen, and estrogen receptors. Ann New York Acad Sci. (2004) 1024:54–71. doi: 10.1196/annals.1321.005
107. Li H, Qian W, Weng X, Wu Z, Li H, Zhuang Q, et al. Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PloS One. (2012) 7:e37030. doi: 10.1371/journal.pone.0037030
108. Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. (2022) 29:946–60. doi: 10.1038/s41418-022-00988-z
109. Bhoopalan SV, Yen JS, Mayuranathan T, Mayberry KD, Yao Y, Lillo Osuna MA, et al. An RPS19-edited model for Diamond-Blackfan anemia reveals TP53-dependent impairment of hematopoietic stem cell activity. JCI Insight. (2023) 8:e161810. doi: 10.1172/jci.insight.161810
110. Devlin EE, DaCosta L, Mohandas N, Elliott G, Bodine DM. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood. (2010) 116:2826–35. doi: 10.1182/blood-2010-03-275776
111. Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. (2011) 117:2567–76. doi: 10.1182/blood-2010-07-295238
112. Chakraborty A, Uechi T, Higa S, Torihara H, Kenmochi N. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PloS One. (2009) 4:e4152. doi: 10.1371/journal.pone.0004152
113. Moniz H, Gastou M, Leblanc T, Hurtaud C, Crétien A, Lécluse Y, et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. (2012) 3:e356–6. doi: 10.1038/cddis.2012.88
114. McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. (2008) 40:963–70. doi: 10.1038/ng.188
115. Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. (2008) 112:5228–37. doi: 10.1182/blood-2008-01-132290
116. Kolbus A, Blázquez-Domingo M, Carotta S, Bakker W, Luedemann S, Von Lindern M, et al. Cooperative signaling between cytokine receptors and the glucocorticoid receptor in the expansion of erythroid progenitors: molecular analysis by expression profiling. Blood. (2003) 102:3136–46. doi: 10.1182/blood-2003-03-0923
117. Strouboulis J, El Hoss S. The effects of chronic glucorticoid stimulation on erythropoiesis in cushing syndrome. Haematol. (2022) 108:947–8. doi: 10.3324/haematol.2022.281355
118. Heideveld E, Hampton-O’Neil LA, Cross SJ, Van Alphen FPJ, Van Den Biggelaar M, Toye AM, et al. Glucocorticoids induce differentiation of monocytes towards macrophages that share functional and phenotypical aspects with erythroblastic island macrophages. Haematologica. (2018) 103:395–405. doi: 10.3324/haematol.2017.179341
119. Zingariello M, Bardelli C, Sancillo L, Ciaffoni F, Genova ML, Girelli G, et al. Dexamethasone predisposes human erythroblasts toward impaired lipid metabolism and renders their ex vivo expansion highly dependent on plasma lipoproteins. Front Physiol. (2019) 10:281. doi: 10.3389/fphys.2019.00281
120. Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med. (2013) 19:429–36. doi: 10.1038/nm.3057
121. Bai J, Fan F, Gao C, Li S, Li W, Wei T, et al. CD169-CD43 interaction is involved in erythroblastic island formation and erythroid differentiation. Haematol. (2023) 108:2205–17. doi: 10.3324/haematol.2022.282192
122. Falchi M, Varricchio L, Martelli F, Masiello F, Federici G, Zingariello M, et al. Dexamethasone targeted directly to macrophages induces macrophage niches that promote erythroid expansion. Haematologica. (2015) 100:178–87. doi: 10.3324/haematol.2014.114405
123. Calvert RJ, Robson T. Cortisone therapy in erythrogenesis imperfecta. Arch Dis Childhood. (1956) 31:177–81. doi: 10.1136/adc.31.157.177
124. Finkel HE, Kimber RJ, Dameshek W. Corticosteroid-responsive acquired pure red cell aplasia in adults. Am J Med. (1967) 43:771–6. doi: 10.1016/0002-9343(67)90119-2
125. Bagby GJ, Goodnight S, Mooney W, Richert-Boe K. Prednisone-responsive aplastic anemia: a mechanism of glucocorticoid action. Blood. (1979) 54:322–33. doi: 10.1182/blood.V54.2.322.322
126. Duru F, Gürgey A. Effect of corticosteroids in hereditary spherocytosis. Pediatr Int. (1994) 36:666–8. doi: 10.1111/j.1442-200X.1994.tb03266.x
127. Ballin A, Waisbourd-Zinman O, Saab H, Yacobovich J, Zoldan M, Barzilai-Birenbaum S, et al. Steroid therapy may be effective in augmenting hemoglobin levels during hemolytic crises in children with hereditary spherocytosis. Pediatr Blood Cancer. (2011) 57:303–5. doi: 10.1002/pbc.22844
128. Liu Y, Karlsson S. Perspectives of current understanding and therapeutics of Diamond-Blackfan anemia. Leukemia. (2024) 38:1–9. doi: 10.1038/s41375-023-02082-w
129. Allen DM. Congenital (Erythroid) hypoplastic anemia: Cortisone treated. Am J Dis Child. (1961) 102:416. doi: 10.1001/archpedi.1961.02080010418021
130. Iskander D, Roy NBA, Payne E, Drasar E, Hennessy K, Harrington Y, et al. Diamond-Blackfan anemia in adults: In pursuit of a common approach for a rare disease. Blood Rev. (2023) 61:101097. doi: 10.1016/j.blre.2023.101097
131. Wlodarski MW, Vlachos A, Farrar JE, Da Costa LM, Kattamis A, Dianzani I, et al. Diagnosis, treatment, and surveillance of Diamond-Blackfan anaemia syndrome: international consensus statement. Lancet Haematology. (2024) 11:e368–82. doi: 10.1016/S2352-3026(24)00063-2
132. Iskander D, Karadimitris A, Roberts I. Harnessing single-cell technologies in the search for new therapies for diamond–blackfan anemia syndrome. Exp Hematol. (2024) 135:104235. doi: 10.1016/j.exphem.2024.104235
133. Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell. (2018) 173:90–103.e19. doi: 10.1016/j.cell.2018.02.036
134. Ellis SR. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim Biophys Acta (BBA) - Mol Basis Dis. (2014) 1842:765–8. doi: 10.1016/j.bbadis.2013.12.013
135. Doty RT, Yan X, Meng C, Lausted C, Tian Q, Abkowitz JL. Single-cell analysis of erythropoiesis in Rpl11 haploinsufficient mice reveals insight into the pathogenesis of diamond–blackfan anemia. Exp Hematol. (2021) 97:66–78.e6. doi: 10.1016/j.exphem.2021.02.010
136. Pesciotta EN, Lam H-S, Kossenkov A, Ge J, Showe LC, Mason PJ, et al. In-depth, label-free analysis of the erythrocyte cytoplasmic proteome in diamond blackfan anemia identifies a unique inflammatory signature. PloS One. (2015) 10:e0140036. doi: 10.1371/journal.pone.0140036
137. Macečková Z, Kubíčková A, De Sanctis JB, Hajdúch M. Effect of glucocorticosteroids in diamond-blackfan anaemia: Maybe not as elusive as it seems. Int J Mol Sci. (2022) 23:1886. doi: 10.3390/ijms23031886
138. Zhang J, Wu K, Xiao X, Liao J, Hu Q, Chen H, et al. Autophagy as a regulatory component of erythropoiesis. IJMS. (2015) 16:4083–94. doi: 10.3390/ijms16024083
139. Grosso R, Fader CM, Colombo MI. Autophagy: A necessary event during erythropoiesis. Blood Rev. (2017) 31:300–5. doi: 10.1016/j.blre.2017.04.001
140. Liu Q, Luo L, Ren C, Zou M, Yang S, Cai B, et al. The opposing roles of the mTOR signaling pathway in different phases of human umbilical cord blood-derived CD34+ cell erythropoiesis. Stem Cells. (2020) 38:1492–505. doi: 10.1002/stem.3268
141. Knight ZA, Schmidt SF, Birsoy K, Tan K, Friedman JM. A critical role for mTORC1 in erythropoiesis and anemia. eLife. (2014) 3:e01913. doi: 10.7554/eLife.01913
142. Doulatov S, Vo LT, Macari ER, Wahlster L, Kinney MA, Taylor AM, et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med. (2017) 9:eaah5645. doi: 10.1126/scitranslmed.aah5645
143. Heijnen HF, Van Wijk R, Pereboom TC, Goos YJ, Seinen CW, Van Oirschot BA, et al. Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway. PloS Genet. (2014) 10:e1004371. doi: 10.1371/journal.pgen.1004371
144. Payne EM, Virgilio M, Narla A, Sun H, Levine M, Paw BH, et al. L-leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood. (2012) 120:2214–24. doi: 10.1182/blood-2011-10-382986
145. Fu L, Wu W, Sun X, Zhang P. Glucocorticoids enhanced osteoclast autophagy through the PI3K/Akt/mTOR signaling pathway. Calcif Tissue Int. (2020) 107:60–71. doi: 10.1007/s00223-020-00687-2
146. Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. (2011) 13:170–82. doi: 10.1016/j.cmet.2011.01.001
147. Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. (2008) 134:394–401. doi: 10.1378/chest.08-0440
148. Mazzarini M, Cherone J, Nguyen T, Martelli F, Varricchio L, Funnell APW, et al. The glucocorticoid receptor elicited proliferative response in human erythropoiesis is BCL11A-dependent. Stem Cells. (2024) 42:1006–22. doi: 10.1093/stmcls/sxae049
149. Wouters HJCM, Conrads-Frank A, Koinig KA, Smith A, Yu G, De Witte T, et al. The anemia-independent impact of myelodysplastic syndromes on health-related quality of life. Ann Hematol. (2021) 100:2921–32. doi: 10.1007/s00277-021-04654-1
150. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Trans Internal Med. (2017) 5:139–43. doi: 10.1515/jtim-2017-0002
151. Venugopal S, Mascarenhas J, Steensma DP. Loss of 5q in myeloid malignancies – A gain in understanding of biological and clinical consequences. Blood Rev. (2021) 46:100735. doi: 10.1016/j.blre.2020.100735
152. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. (2012) 120:2454–65. doi: 10.1182/blood-2012-03-420489
153. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. (2008) 451:335–9. doi: 10.1038/nature06494
154. Reagan JL, Ollila T, Dubielecka PM, Olszewski AJ. Survival of patients with Del(5q) syndrome in the lenalidomide era. Blood. (2016) 128:1994–4. doi: 10.1182/blood.V128.22.1994.1994
155. Narla A, Dutt S, McAuley JR, Al-Shahrour F, Hurst S, McConkey M, et al. Dexamethasone and lenalidomide have distinct functional effects on erythropoiesis. Blood. (2011) 118:2296–304. doi: 10.1182/blood-2010-11-318543
156. Ellis S. R. DBA. del(5q): a reciprocal relationship. Blood. (2011) 118:2032–3. doi: 10.1182/blood-2011-05-352575
Keywords: glucocorticoids, Diamond-Blackfan anaemia syndrome, erythropoiesis, nuclear receptors, haematopoiesis
Citation: Hanssen LLP and Iskander D (2025) The role of glucocorticoids in erythropoiesis. Front. Hematol. 4:1540152. doi: 10.3389/frhem.2025.1540152
Received: 05 December 2024; Accepted: 03 February 2025;
Published: 26 February 2025.
Edited by:
John Strouboulis, King’s College London, United KingdomReviewed by:
Anna Rita Migliaccio, Campus Bio-Medico University, ItalyCopyright © 2025 Hanssen and Iskander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deena Iskander, ZC5pc2thbmRlckBpbXBlcmlhbC5hYy51aw==; Lars L. P. Hanssen, bC5oYW5zc2VuQGltcGVyaWFsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.