
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Hematol., 16 January 2025
Sec. Blood Cancer
Volume 3 - 2024 | https://doi.org/10.3389/frhem.2024.1501337
 Carolina Ottati1†‡Inés Gervaz1,2†‡Martín Yandian3,4‡
Carolina Ottati1†‡Inés Gervaz1,2†‡Martín Yandian3,4‡ Matilde Boada1,2Gabriela Vidal-Senmache5‡Percy Ortiz-Guerra5‡Ana I. Catalán1‡Patricia Kutscher6‡Diego Lopez6‡Lilian Diaz4‡
Matilde Boada1,2Gabriela Vidal-Senmache5‡Percy Ortiz-Guerra5‡Ana I. Catalán1‡Patricia Kutscher6‡Diego Lopez6‡Lilian Diaz4‡ Sofia Grille1,2*
Sofia Grille1,2*Introduction: VEXAS syndrome (Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic) is a recently identified disorder associated with somatic mutations in the UBA1 gene. Predominantly affecting adult males, it is characterized by a wide range of autoinflammatory symptoms and hematologic abnormalities.
Methods: We present three cases from Latin America, marking the first reported occurrences in this region, to illustrate the clinical variability and diagnostic challenges of VEXAS syndrome.
Results: Each patient exhibited unique clinical presentations, including refractory autoinflammatory symptoms, myelodysplastic syndrome, and bone marrow vacuolization. All cases were confirmed via genetic testing, revealing pathogenic UBA1 mutations alongside other genetic variants commonly linked with myeloid neoplasms.
Discussion: These findings underscore the importance of considering VEXAS syndrome in patients with unexplained inflammatory and hematologic symptoms. The coexistence of UBA1 mutations with other genetic variants suggests a potential overlap with clonal hematopoiesis, complicating the clinical picture. These cases contribute to the understanding of VEXAS syndrome and highlight the need for increased awareness and diagnostic testing in diverse populations to ensure early and accurate diagnosis.
VEXAS (Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic) syndrome is a recently described disorder characterized by a distinctive combination of autoinflammatory symptoms, hematologic abnormalities, and somatic mutations in the UBA1 gene (1). Since its identification, VEXAS syndrome has been recognized as a rare condition that primarily affects adult males due to its X-linked inheritance pattern.
UBA1 gene, located on the X chromosome, encodes the ubiquitin-activating enzyme 1 (E1 enzyme). The E1 enzyme plays a critical role in the ubiquitin-proteasome system (UPS), which is essential for cellular protein homeostasis. Ubiquitin, a small regulatory protein, is covalently attached to target proteins via a multi-step process involving E1 (activation), E2 (conjugation), and E3 (ligation) enzymes. This process governs key cellular functions, including protein degradation, signal transduction, and immune response regulation. Mutations in the UBA1 gene disrupt the enzymatic activity of E1, leading to impaired ubiquitination and subsequent dysregulation of protein turnover and cellular signaling pathways. In the context of VEXAS syndrome, defective ubiquitination is hypothesized to contribute to the accumulation of misfolded proteins and the activation of proinflammatory cascades. This pathogenic mechanism may explain the syndrome’s hallmark features, including systemic inflammation, cytopenias, and the presence of vacuoles in myeloid precursors (2).
The syndrome presents with a wide range of clinical manifestations, including fever, cytopenias, bone marrow vacuoles, and various systemic inflammatory responses (3). Despite its rarity, the clinical importance of VEXAS Syndrome lies in its potential for severe outcomes and diagnostic challenges. Early recognition and accurate diagnosis are crucial for appropriate management. To date, most cases have been reported in North America and Europe, with limited data available from other regions.
In this report, we present three cases of VEXAS Syndrome, the first documented in Latin America. These cases highlight the variability of clinical presentations and the diagnostic challenges in identifying this syndrome in diverse populations. Our findings contribute to the growing body of knowledge on VEXAS Syndrome and emphasize the need for increased awareness of this condition among clinicians in Latin America.
A 72-year-old male from Montevideo, Uruguay, first presented in 2018 with symptoms of uveitis, arthralgia, superficial venous thrombosis in the right saphenous vein, and deep venous thrombosis in the right subclavian vein. Extensive evaluations failed to identify an underlying neoplasm, leading to a provisional diagnosis of an unspecified autoimmune or autoinflammatory disease. The patient was treated sequentially with corticosteroids, mycophenolate mofetil, and hydroxychloroquine. Despite this therapeutic approach, the patient did not achieve clinical or laboratory remission, as evidenced by persistently elevated acute-phase reactants.
In 2020, the patient developed pancytopenia with macrocytosis. Bone marrow examination showed preserved cellularity with trilineage dysplasia but no excess blasts. Immunophenotyping revealed a predominance of granular elements with moderate dysgranulopoiesis, characterized by maturation arrest and disrupted pathways, without an increase in CD34+ events. Bone marrow biopsy confirmed hypercellularity with a predominance of the granulocytic lineage and dysgranulopoiesis, without blast proliferation. Cytogenetic and FISH analyses were normal. By 2021, the patient’s anemia and thrombocytopenia worsened, leading to transfusion dependence. A disease flare subsequently occurred, presenting with fever, arthralgia, oral ulcers, chondritis, perniosis, and erythema with periorbital edema (Figure 1). The patient was treated with methylprednisolone pulses and colchicine, followed by low-dose methotrexate and erythropoietin. Due to the worsening pancytopenia, methotrexate was discontinued, and treatment with tocilizumab was initiated. In 2022, the patient developed a tongue lesion, which was biopsied and revealed a granulomatous inflammatory process. Given the combination of multiple inflammatory manifestations and bone marrow dysplasia, VEXAS syndrome was suspected. Repeat bone marrow studies showed trilineage dysplasia with more than 10% vacuolated granulocytic precursors (Figure 2). Cytogenetic analysis was normal (46, XY). Molecular studies performed in bone marrow confirmed the pathogenic variants c.122T>C; p.Met41Thr in the UBA1 gene, establishing a diagnosis of VEXAS syndrome (Figure 3). Further analysis using a targeted next-generation sequencing (NGS) panel covering 63 myeloid genes revealed two additional variants: TET2 c.2746C>T (VAF 37.2%) and DNMT3A c.1483del (VAF 41.8%), supporting a concomitant diagnosis of myelodysplastic syndrome (MDS). The patient was initiated on hypomethylating therapy with subcutaneous 5-Azacitidine, which resulted in transfusion independence and a marked improvement in overall health and quality of life.
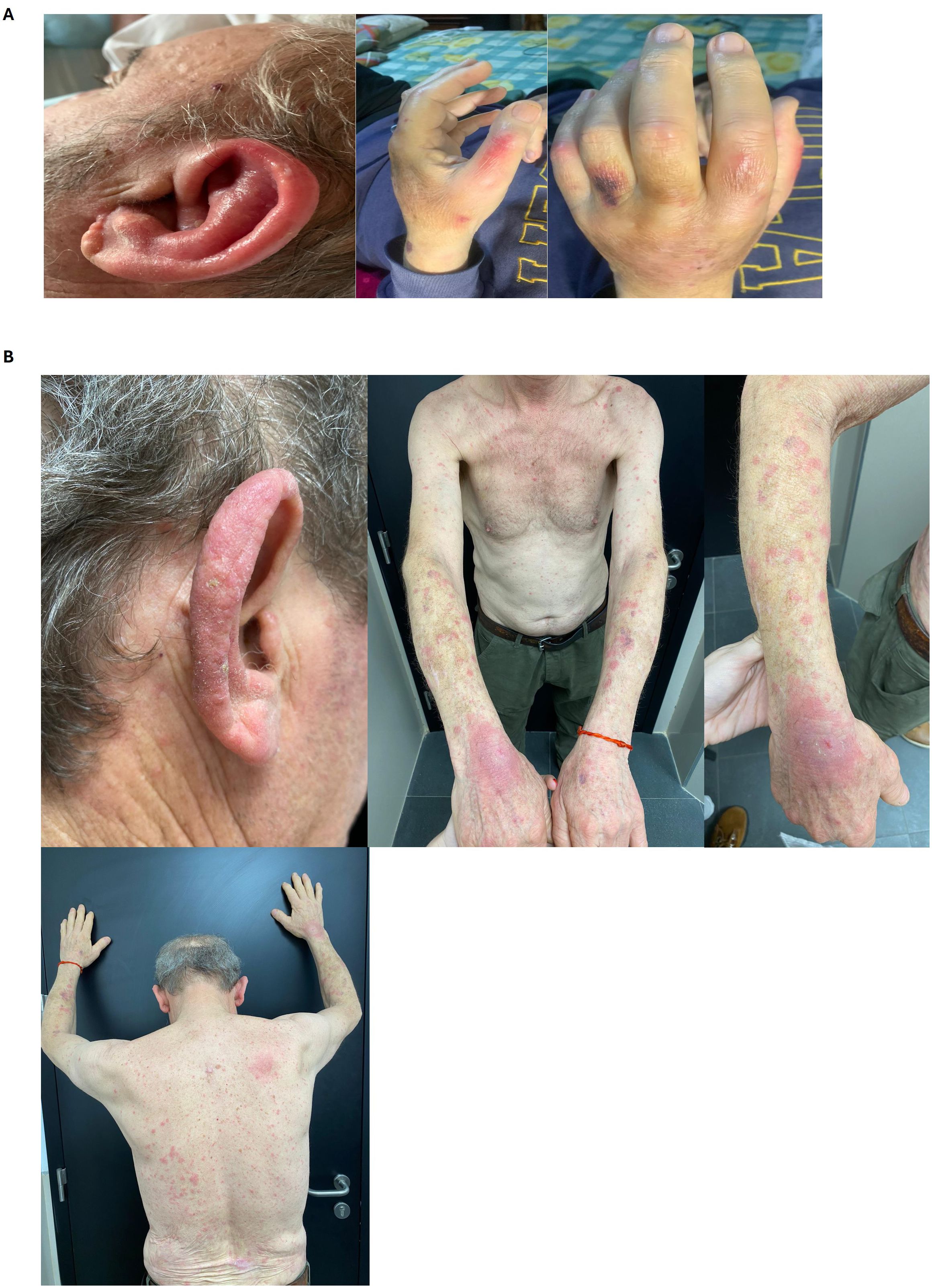
Figure 1. Cutaneous manifestations of VEXAS syndrome. (A) Case 1. Erythema and swelling of the auricle consistent with chondritis and perniosis in hands. (B) Case 2. Erythematous, edematous plaques without desquamation on the trunk and limbs. Erythema and edema of the helix and antihelix of both ears (chondritis).
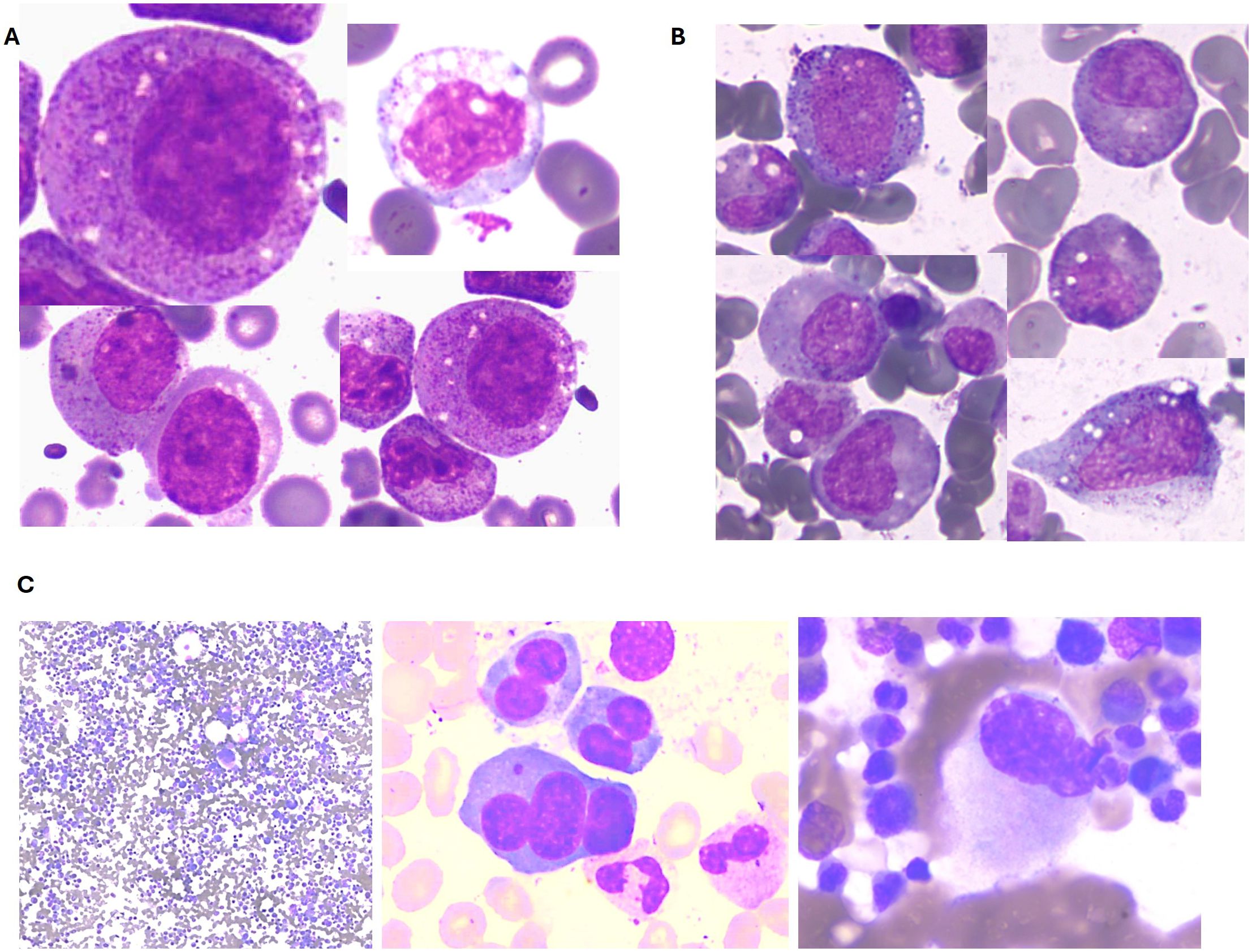
Figure 2. Illustrative bone marrow findings from VEXAS patients. (A) Case 1: Vacuoles in granulocytic precursors. (B) Case 2: Vacuoles in granulocytic precursors. (C) Case 3: Multilineage dysplasia.

Figure 3. Sanger sequencing results showing UBA1 variants in VEXAS patients. (A) Case 1, (B) Case 2 (C) Case 3.
A 62-year-old male from Montevideo, Uruguay, with a history of smoking and type 2 diabetes mellitus, had a squamous cell carcinoma on his lower back surgically resected in 2017. In August 2022, the patient presented with skin lesions, polychondritis and was diagnosed with Sweet syndrome based on biopsy results (Figure 1). He was started on treatment with steroids and colchicine. Multiple attempts to taper and discontinue corticosteroids led to relapses of his dermatologic condition. Additionally, the patient developed polyarthralgia and conjunctival erythema. In 2023, the patient developed macrocytic anemia, requiring transfusions since August 2023. A thorough evaluation of the anemia ruled out metabolite deficiencies and other common causes. Bone marrow examination revealed trilineage dysplasia, with a notable presence of vacuolated granulocytic precursors and 1% blasts (Figure 2). Immunophenotyping of the bone marrow showed 2.2% myeloid blasts, with an Ogata score of 3. Cytogenetic and FISH analyses were normal.
A targeted NGS panel covering 63 myeloid genes, performed on bone marrow samples, identified a likely pathogenic variant in DNMT3A c.1802G>A, with a VAF of 44.6%, supporting a diagnosis of MDS. Given the presentation of an elderly male with autoinflammatory symptoms and MDS with vacuoles in the granulocytic lineage, VEXAS syndrome was suspected. Sanger sequencing of the UBA1 gene confirmed the presence of the pathogenic variant c.122T>C; p.Met41Thr, establishing the diagnosis of VEXAS syndrome (Figure 3). The patient was started on azacitidine therapy and continued corticosteroid treatment. He is currently on his ninth cycle of azacitidine, showing improvement in cytopenias, although he continues to experience corticosteroid-dependent skin lesions that recur when steroids are tapered. Due to persistent symptoms, mycophenolate mofetil was introduced into his treatment regimen.
An 82-year-old male from Lima, Peru, with a history of atrial fibrillation treated with apixaban, was diagnosed with relapsing polychondritis in 2022 and received corticosteroid treatment. Later that year, he developed an edematous syndrome and began experiencing evening sweats, alongside the onset of macrocytic anemia. On examination, the patient appeared pale, with a palpable spleen extending 2 cm below the left costal margin. Painful lymphadenopathy, measuring 2 cm, was detected in both the right cervical and left inguinal regions. Abdominal ultrasound and computed tomography (CT) scans revealed hepatosplenomegaly without additional abnormalities. Laboratory findings from December 2023 indicated macrocytic anemia. Bone marrow studies demonstrated marked trilineage dysplasia without an excess of blasts and with a some vacuolated granulocytic precursors. Flow cytometry confirmed the absence of blast proliferation. The bone marrow biopsy was hypercellular for the patient’s age, with dysplastic changes predominantly affecting the megakaryocytic lineage and no evidence of increased blasts. Immunohistochemistry showed p53 positivity in 5-10% of cells, with fibrosis graded as I. Cytogenetic analysis revealed a normal karyotype (46, XY). Targeted NGS panel of peripheral blood identified a pathogenic variant in DNMT3A c.1628dup, with a VAF of 40.2%, consistent with a diagnosis of MDS. The patient was started on erythropoietin, but no response was observed, and transfusion requirements increased. Additionally, the patient developed respiratory infections and bronchial hyperreactivity, which were managed with steroid treatment. Subsequently, he exhibited signs of respiratory insufficiency and a right pleural effusion, which required drainage. A thoracic CT scan revealed a bilateral perihilar and basal alveolo-interstitial pattern along with a right pleural effusion. Given the presentation of an elderly male with autoinflammatory symptoms and MDS, VEXAS syndrome was considered. A DNA sample was sent to Uruguay for further analysis, where sequencing of the UBA1 gene confirmed the presence of the pathogenic variant c.121A>C; p.Met41Leu, establishing a diagnosis of VEXAS syndrome (Figure 3).
The patient was started on azacitidine therapy and is currently on his third cycle with clinical improvement.
The three cases presented in this report are significant as they represent the first documented occurrences of VEXAS Syndrome in Latin America. This contribution to the existing literature is crucial, given the emerging recognition of VEXAS Syndrome as a distinct clinical entity characterized by somatic mutations in the UBA1 gene. The syndrome predominantly affects adult males due to its X-linked inheritance and presents with a diverse range of symptoms that often overlap with those of other hematologic and autoinflammatory disorders (3). The findings from these cases provide valuable insights into the diagnostic complexities, genetic associations, and therapeutic challenges associated with VEXAS syndrome.
Due to its rarity and the diversity of its clinical manifestations, VEXAS syndrome presents a significant diagnostic challenge. The cases in this report underscore the variability in symptom presentation, which includes uveitis, chondritis, skin lesions, pulmonary symptoms, venous thrombosis, and macrocytic anemia. These symptoms initially led to misdiagnoses or delayed recognition of the condition. This variability highlights the importance of maintaining a high index of suspicion for VEXAS syndrome in patients who present with refractory autoinflammatory symptoms, unexplained cytopenias, and bone marrow vacuolization. The delayed diagnosis observed in these cases, despite comprehensive clinical evaluations, emphasizes the need for increased awareness of VEXAS syndrome among healthcare professionals and the integration of genetic testing into diagnostic protocols for patients with atypical presentations.
VEXAS syndrome is an X-linked, adult-onset disorder, primarily affecting males over the age of 50, as demonstrated in our case series. However, it is important to note that female patients with a UBA1 variant and monosomy X have also been reported (4–6). The mutation of the UBA1 gene in hematopoietic stem and progenitor cells (HSPCs) leads to clonal expansion of mutant clones in the bone marrow. This results in myeloid-skewed differentiation and abnormal activation of innate immune pathways, causing systemic autoinflammation. Patients with VEXAS syndrome not only exhibit autoinflammatory symptoms but are also frequently associated with hematologic disorders, such as myelodysplastic syndromes (MDS) or plasma cell dyscrasia (3).
The identification of somatic mutations in the UBA1 gene is a hallmark of VEXAS Syndrome. The most frequently reported mutations in the UBA1 gene affect the methionine at amino acid position 41. Currently, the pathogenic variants p.Met41Thr (c.122T>C), p.Met41Val (c.121A>G), and p.Met41Leu (c.121A>C) are the most commonly identified. Additionally, the likely pathogenic p.Ser56Phe (c.167C>T) and pathogenic p.Gly40_Lys43del (c.118–1G>C) variants have also been detected in patients without p.Met41 mutations but presenting a typical VEXAS phenotype (7, 8). In our cases, all patients had mutations affecting methionine at position 41, with two patients having p.Met41Thr (c.122T>C) and one patient having p.Met41Leu (c.121A>C). The p.Met41Val variant has been found to result in lower translation of UBA1b isoform compared to the p.Met41Leu or p.Met41Thr mutations (9).
It is also noteworthy that other somatic mutations may coexist with UBA1 variants, particularly in the DNMT3A (9.2–22% of cases) and TET2 (5–11% of cases) genes (5). The main mutation in DNMT3A is p.Arg882His (c.2645G>A) (6). In our case series, all patients had somatic mutations in DNMT3A, and one also had a mutation in TET2. A loss-of-function mutation in DNMT3A, which acts as a repressor of inflammation, may further contribute to the proinflammatory state observed in VEXAS syndrome. Deficiency of this protein has been linked to the activation of innate immune inflammatory signaling in myeloid cells. The coexistence of these mutations raises important questions about the pathophysiological relationship between VEXAS syndrome and other hematologic disorders, such as MDS. It is plausible that the presence of these additional mutations may influence the clinical course and severity of VEXAS syndrome, contributing to variability in patient outcomes. Understanding the interplay between these genetic alterations is crucial for refining diagnostic criteria and developing targeted therapies.
Clinically, VEXAS syndrome presents with a highly heterogeneous profile, as reflected in our case series, since all organs and tissues may be involved by inflammation. The manifestations of VEXAS syndrome can mimic various systemic rheumatologic disorders associated with MDS, including small vessel vasculitis, rheumatoid arthritis, seronegative spondyloarthritis, Sweet syndrome, relapsing polychondritis, polyarteritis nodosa, and even Behcet’s disease (1, 9). Common symptoms include fever (observed in approximately 65% of patients), weight loss (55% of patients), fatigue, night sweats, and muscle aches (7). Hematologic involvement manifests with MDS in more than half of the patients, with subtypes including MDS with ring sideroblasts or multilineage dysplasia. In our case series, all patients presented with the multilineage dysplasia subtype. A small percentage of patients with MDS also develop monoclonal gammopathy of unknown significance (MGUS) (7, 10). Obiorah et al. reported that macrocytic anemia was observed in all patients, and thrombocytopenia was identified in half of the patients. Absolute lymphopenia and monocytopenia were noted in 80% and 50% of patients, respectively, while neutropenia was less common, affecting 13% of patients (10). This aligns with our case series, where all patients presented with macrocytic anemia, and one also had thrombocytopenia and neutropenia. Bone marrow aspirates can reveal pathognomonic alterations, especially cytoplasmic vacuolization of erythroid and myeloid precursors in almost all patients. Overall, 15% of myeloid and erythroid cells present vacuoles, with an average of 5–7 vacuoles per cell. Some degree of dysplasia is found in megakaryocytes and myeloid and erythroid precursors in nearly all bone marrow aspirates; however, dysplasia exceeding 10% in a lineage was seen only in patients with MDS (11). Figure 1 shows the vacuolization observed in our patients. Bone marrow biopsy findings include characteristics resembling MDS. Similar to our cases, Obiorah et al. reported that bone marrow hypercellularity was found in 87.5% of patients, with cellularity ranging from 25 to 100%. Myeloid hyperplasia was described in more than half of the patients, with myeloid:erytroid ratios of 7:1 or greater, particularly in patients with concurrent MDS. The percentage of bone marrow blasts was less than 5% (11).
Skin involvement is the most frequently encountered manifestation and can be highly heterogeneous. Two of our patients exhibited skin involvement with erythema, and the other with Sweet syndrome. Skin manifestations can include Sweet syndrome, vasculitic features, erythematous papules, urticaria, periorbital edema, pathergy, Sweet syndrome-like nodules, and more (7, 12, 13). Chondritis occurs in 36–60% of patients and provides an important clinical clue to a diagnosis of VEXAS syndrome. This typically presents with erythema, swelling, and pain of the cartilaginous portion of the ear and nose (7, 12, 13).
In our cases, two patients exhibited ocular involvement, including uveitis, periorbital edema, and conjunctival edema. Ocular involvement occurs in up to 40.5% of cases, most commonly presenting as episcleritis (12.1%), uveitis (9.5%), scleritis (8.6%), orbital mass (3.4%), and blepharitis. Orbital and periorbital inflammation may also occur (14).
One patient in our series presented with pulmonary involvement, including bronchial hyperreactivity, respiratory insufficiency, and pleural effusion. Pulmonary involvement is very common (about half of the patients), with pulmonary infiltrates and pleural effusion being the most frequent manifestations (12).
Another patient experienced venous thrombosis. Episodes of venous and arterial thrombosis have been reported in approximately one-third of patients, with 60% occurring within the first two years of symptom onset (7, 15). The prevalence of venous thromboembolism (VTE) is reported to be between 10% and 56% of cases (15).
Therapeutic management of VEXAS syndrome remains a significant challenge due to its complex clinical course and the lack of established treatment guidelines. To date, treatments have included glucocorticoids, hypomethylating agents, conventional disease-modifying antirheumatic drugs, biological agents targeting IL-1 and IL-6, and Janus kinase (JAK) inhibitors. Allogenic hematopoietic stem cell transplantation (HSCT) and future gene-editing therapies are potential therapies worth investigating (1, 12, 16–20). The cases reported here demonstrate the use of a combination of corticosteroids, immunosuppressants, and hypomethylating agents in managing the diverse symptoms of VEXAS syndrome, as these represent the most accessible therapeutic options in Latin America. Glucocorticoids are commonly used as a first-line treatment due to their ability to rapidly control systemic inflammation and alleviate symptoms. However, their long-term use is limited by significant adverse effects, including infections, cardiovascular complications, and steroid dependence, as evidenced by recurrent disease flares upon tapering.Notably, the administration of azacitidine, a hypomethylating agent, appeared to provide clinical benefit in patients with concurrent MDS, leading to improved hematologic parameters and reduced transfusion dependency (21–23). Azacitidine not only improves hematologic parameters but also exhibits anti-inflammatory effects. While it has a favorable safety profile, its efficacy may vary among patients, and some may experience hematologic toxicities (2). However, the persistence of corticosteroid-dependent symptoms in some patients highlights the need for novel therapeutic approaches that address the underlying pathophysiology of VEXAS syndrome rather than merely managing symptoms. Further research into the efficacy of targeted therapies, such as JAK inhibitors and IL-1 and IL-6 antagonists, may offer new avenues for treating this complex syndrome (1, 16–20). Targeted biologics, such as IL-6 inhibitors (e.g., tocilizumab) and IL-1 inhibitors (e.g., anakinra and canakinumab), provide targeted suppression of cytokine-driven inflammation. IL-6 inhibitors have demonstrated clinical improvement and a reduction in steroid dependency, though their impact on hematologic abnormalities, such as cytopenias, remains limited. On the other hand, IL-1 inhibitors show varying responses, often limited by injection site reactions or incomplete symptom resolution (2). JAK inhibitors, including ruxolitinib, have emerged as a promising class of therapies by modulating inflammatory pathways. They offer symptom relief and reduce steroid dependency in many cases. However, their efficacy in addressing hematologic features is inconsistent, and there is a potential risk of infections and thrombotic events (2). Each treatment modality presents distinct advantages and limitations. Emerging treatments, such as JAK inhibitors and potential gene-editing approaches, represent exciting avenues for further exploration, although their long-term efficacy and safety profiles need to be established (2).
The cases described in this report also have significant implications for clinical practice and research in Latin America. The recognition of VEXAS syndrome in this region is particularly important, given the limited data available and the potential for underdiagnosis. There is a pressing need for educational initiatives to raise awareness among clinicians about the clinical features and genetic basis of VEXAS syndrome. Additionally, the establishment of diagnostic protocols that include genetic testing for UBA1 mutations in patients with unexplained autoinflammatory symptoms and hematologic abnormalities could facilitate earlier diagnosis and intervention, ultimately improving patient outcomes.
In conclusion, the first documented cases of VEXAS syndrome in Latin America contribute to the growing body of knowledge on this rare and complex disorder. These cases underscore the diagnostic challenges posed by VEXAS syndrome and the importance of considering it in the differential diagnosis of patients with refractory autoinflammatory symptoms and unexplained hematologic abnormalities. Continued efforts to improve diagnostic accuracy, enhance awareness, and develop effective therapeutic strategies will be crucial in advancing our understanding of VEXAS syndrome and improving outcomes for affected patients.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
CO: Conceptualization, Methodology, Writing – original draft. IG: Methodology, Writing – review & editing. MY: Conceptualization, Supervision, Writing – review & editing. MB: Conceptualization, Writing – review & editing. GV: Investigation, Writing – review & editing. PO: Methodology, Writing – review & editing. AC: Conceptualization, Methodology, Writing – review & editing. PK: Conceptualization, Writing – review & editing. DL: Conceptualization, Writing – review & editing. LD: Supervision, Writing – review & editing. SG: Conceptualization, Formal analysis, Methodology, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank the patient and his family for their participation in this study. OpenAI GPT-4.0 was used to improve the writing and clarity of certain parts of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. New Engl J Med. (2020) 383:2628–38. doi: 10.1056/nejmoa2026834
2. AlQatari S, Alqunais AA, Alali SM, Alharbi MA, Hasan M, Al Shubbar MD. VEXAS syndrome: A comprehensive review of current therapeutic strategies and emerging treatments. J Clin Med. (2024) 13. doi: 10.3390/jcm13226970
4. Barba T, Jamilloux Y, Durel CA, Bourbon E, Mestrallet F, Sujobert P, et al. VEXAS syndrome in a woman. Rheumatol (Oxford England). (2021) 60:E402–3. doi: 10.1093/RHEUMATOLOGY/KEAB392
5. Arlet J, Terrier B, Kosmider O. Mutant UBA1 and severe adult-onset autoinflammatory disease. New Engl J Med. (2021) 384:2163–5. doi: 10.1056/nejmc2102124
6. Tsuchida N, Kunishita Y, Uchiyama Y, Kirino Y, Enaka M, Yamaguchi Y, et al. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheumatic Dis. (2021) 80:1057–61. doi: 10.1136/ANNRHEUMDIS-2021-220089
7. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. (2022) 186:564–74. doi: 10.1111/BJD.20805
8. Shaukat F, Hart M, Burns T, Bansal P. UBA1 and DNMT3A mutations in VEXAS syndrome. A case report and literature review. Modern Rheumatol Case Rep. (2022) 6:134–9. doi: 10.1093/MRCR/RXAB021
9. Ferrada MA, Savic S, Cardona DO, Collins JC, Alessi H, Gutierrez-Rodrigues F, et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood. (2022) 140:1496–506. doi: 10.1182/BLOOD.2022016985
10. Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello AK, et al. Benign and Malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. (2021) 5:3203–15. doi: 10.1182/BLOODADVANCES.2021004976
11. Onuora S. Somatic mutations cause VEXAS syndrome. Nat Rev Rheumatol. (2020) 17:1–1. doi: 10.1038/s41584-020-00559-x
12. Vitale A, Caggiano V, Bimonte A, Caroni F, Tosi GM, Fabbiani A, et al. VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases. Internal Emergency Med. (2023) 18:711–22. doi: 10.1007/s11739-023-03193-z
13. Sterling D, Duncan ME, Philippidou M, Salisbury JR, Kulasekararaj AG, Basu TN. VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) for the dermatologist. J Am Acad Dermatol. (2023) 89:1209–14. doi: 10.1016/j.jaad.2022.01.042
14. Martín-Nares E, Vargas-Serafín C, Delgado-de la Mora J, Montante-Montes de Oca D, Grayson PC, Larios E, et al. Orbital and periorbital inflammation in VEXAS syndrome. Scandinavian J Rheumatol. (2022) 51:338–41. doi: 10.1080/03009742.2022.2045791/ASSET//CMS/ASSET/41BCCAD1-B813-40EE-9E52-2D3C7C860EFA/03009742.2022.2045791.FP.PNG
15. Groarke EM, Dulau-Florea AE, Kanthi Y. Thrombotic manifestations of VEXAS syndrome. Semin Hematol. (2021) 58:230–8. doi: 10.1053/J.SEMINHEMATOL.2021.10.006
16. Campochiaro C, Tomelleri A, Cavalli G, De Luca G, Grassini G, Cangi MG, et al. Successful use of cyclosporin A and interleukin-1 blocker combination therapy in VEXAS syndrome: a single-center case series. Arthritis Rheumatol. (2022) 74:1302–3. doi: 10.1002/ART.42101
17. Islam S, Cullen T, Sumpton D, Damodaran A, Heath D, Bosco A, et al. VEXAS syndrome: lessons learnt from an early Australian case series. Internal Med J. (2022) 52:658–62. doi: 10.1111/IMJ.15742
18. Goyal A, Narayanan D, Wong W, Laga AC, Connell NT, Ritter SY, et al. Tocilizumab for treatment of cutaneous and systemic manifestations of vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome without myelodysplastic syndrome. JAAD Case Rep. (2022) 23:15–9. doi: 10.1016/j.jdcr.2022.02.022
19. Kunishita Y, Kirino Y, Tsuchida N, Maeda A, Sato Y, Takase-Minegishi K, et al. Case report: tocilizumab treatment for VEXAS syndrome with relapsing polychondritis: A single-center, 1-year longitudinal observational study in Japan. Front Immunol. (2022) 13:901063/BIBTEX. doi: 10.3389/FIMMU.2022.901063/BIBTEX
20. Heiblig M, Ferrada MA, Koster MT, Barba T, Gerfaud-Valentin M, Mékinian A, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood. (2022) 140:927–31. doi: 10.1182/BLOOD.2022016642
21. Raaijmakers MHGP, Hermans M, Aalbers A, Rijken M, Dalm VASH, Van Daele P, et al. Azacytidine treatment for VEXAS syndrome. HemaSphere. (2021) 5:E661. doi: 10.1097/HS9.0000000000000661
22. Comont T, Heiblig M, Rivière E, Terriou L, Rossignol J, Bouscary D, et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. (2022) 196:969–74. doi: 10.1111/BJH.17893
Keywords: VEXAS syndrome, myelodysplastic syndrome, diagnosis, management, Latin America
Citation: Ottati C, Gervaz I, Yandian M, Boada M, Vidal-Senmache G, Ortiz-Guerra P, Catalán AI, Kutscher P, Lopez D, Diaz L and Grille S (2025) Case report: VEXAS syndrome: first documented cases in Latin America. Front. Hematol. 3:1501337. doi: 10.3389/frhem.2024.1501337
Received: 24 September 2024; Accepted: 23 December 2024;
Published: 16 January 2025.
Edited by:
Stefano Molica, Hull University Teaching Hospitals NHS Trust, United KingdomReviewed by:
Dorota Monika Rowczenio, University College London, United KingdomCopyright © 2025 Ottati, Gervaz, Yandian, Boada, Vidal-Senmache, Ortiz-Guerra, Catalán, Kutscher, Lopez, Diaz and Grille. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sofia Grille, c29maWFncmlsbGVAZ21haWwuY29t
†These authors have contributed equally to this work and share first authorship
†ORCID: Carolina Ottati, orcid.org/0000-0001-6625-5880
Inés Gervaz, orcid.org/0000-0002-0006-0498
Martin Yandian, orcid.org/0000-0001-9446-4778
Gabriela Vidal-Senmache, orcid.org/0000-0002-5812-8811
Percy Ortiz-Guerra, orcid.org/0000-0003-4641-6127
Ana I. Catalán, orcid.org/0009-0003-2458-7272
Patricia Kutscher, orcid.org/0000-0003-1883-4663
Diego Lopez, orcid.org/0000-0002-2459-1584
Lilian Diaz, orcid.org/0000-0002-1056-2379
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.