 Nabih Maslah
Nabih Maslah Stephane Giraudier1,2†
Stephane Giraudier1,2† Bruno Cassinat
Bruno Cassinat- 1Service de Biologie Cellulaire, Assistance Publique Hôpitaux de Paris (APHP), Hôpital Saint-Louis, Paris, France
- 2Université Paris Cité, Institut National de la Santé et de la Recherche Médicale - Unite Mixte de Recherche (INSERM UMR)-S1131, Institut de Recherche Saint-Louis (IRSL), Hôpital Saint-Louis, Paris, France
- 3Hôpital Saint-Louis, Assistance Publique des Hôpitaux de Paris (APHP), Centre d’Investigations Cliniques (CIC 1427), Paris, France
Myeloproliferative Neoplasms comprise a heterogeneous group of diseases characterized over the past two decades by the acquisition of somatic mutations in hematopoietic stem cells, leading to a pre-leukemic state. The natural history of the disease is marked by the sequential acquisition of genetic events that play an essential role in the phenotype, evolution and response to treatment of the disease. Indeed, disease progression is as heterogeneous as the variety of genetic abnormalities found in individual patients, some of whom lead to disease evolution with a pejorative prognosis, while others persist in a benign manner. In order to better explore these questions, it is important to better understand: (1) the genetic structure of the tumor through dynamic reconstruction of clonal architecture (2) the factors favoring the development of certain clones and their expansion, some of which are governed by Darwinian laws. These parameters could help explain the heterogeneity between MPN patients with similar genetic profiles, and prevent the emergence of clones identified as aggressive by the use of innovative therapeutic strategies targeting new pathways to prevent early transformations in poor prognosis MPN subtypes.
Introduction
BCR-ABL1-negative myeloproliferative neoplasms (MPN) include essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (MF) (1, 2). They represent chronic hematological malignancies whose major risk is the occurrence of thrombosis, but they can also evolve into secondary myelofibrosis or acute leukemia in 10 to 30% of patients (3). There is a considerable heterogeneity in the disease evolution among patients, which is not clearly understood and cannot be accurately predicted. In the current state of knowledge, molecular abnormalities appear to be one of the main parameters associated with this heterogeneity. MPN are characterized by the acquisition in the hematopoietic stem cells of mutations that activate not only the JAK2/STAT5 pathway but also STAT3 and, either in parallel or consecutively, PI3K-AKT and MEK-ERK pathways. These mutations, considered as priming the phenotype and called “driver mutations”, affect the JAK2, MPL or CALR genes (4–9). Over the course of the disease, some patients may acquire one or multiple additional mutations that can be part of the MPN clone or not. Therefore early stages of MPN are usually tumors with a simple molecular profile that may acquire a much more complex clonal architecture over time. An important question is to understand how these tumors evolve at the genetic level which was reviewed elsewhere (10). The aim of this review is to better describe how the tumor develops a complex molecular profile and to highlight the main events and environmental factors that reshape the genetic structure of the MPN tumor cells over time.
An oligoclonal disease
Even before the discovery of the JAK2V617F mutation (4), it has been acknowledged that MPN are clonal (11, 12). An important question has long been to determine the timing of the acquisition of the mutations that trigger the disease. Indeed, several studies have shown that it is possible to detect mutations associated with myeloid hemopathies in the blood of healthy subjects, defining the notion of clonal hematopoiesis of undetermined potential (CHIP), among which the JAK2V617F mutation is quite frequently found (13, 14). Moreover, several studies have reported the possible detection of JAK2V617F mutation several years before the clinical development of an MPN (15, 16), suggesting that this mutation does not give an intense proliferative advantage as previously thought, and that it may take decades for the disease to develop. While one study reported that the JAK2V617F mutation could already be present in cord blood (17), the probable acquisition of this mutation during childhood or the prenatal period was suggested in several MPN patients (18, 19). In these studies, the latency period between the acquisition of JAK2V617F mutation and the diagnosis of MPN was several decades. It therefore appears that clones harboring MPN driver mutations can arise and likely take many years to expand to a clinically detectable level, possibly depending on the influence of intrinsic and extrinsic factors.
In addition to driver mutations, mutations considered “additional” can also be detected, targeting other genes involved in various cellular processes, all involved in the regulation of gene expression: intracellular signalling pathways, epigenetics (DNA methylation, post-transcriptional histone modifications), transcription factors, RNA splicing. Thus, from the early 2010s it appeared that MPN were less likely monoclonal diseases with accumulation of mutations in a single clone, but more likely oligoclonal with the coexistence of several molecularly distinct clones (20, 21). However, it was rapidly shown that the clonal origin of mutations is complex, as 2 different patterns can be distinguished: on the one hand, patients who first acquire a mutation in a driver gene and then additional mutations, and on the other hand, patients who acquire driver mutations within cells that have already acquired a mutation in genes such as TET2 or DNMT3A.
The presence of additional mutations has been shown to have a significant impact on patients’ prognosis. Indeed, the accumulation of several mutations in itself represents a poor prognostic factor in MPN patients, the number of mutations at diagnosis being associated with the risk of transformation (3, 22). Moreover, certain mutations are particularly associated with a poor prognosis. A group of high-molecular-risk mutations affecting the ASXL1, EZH2, SRSF2 and IDH1/2 genes has been defined (23), to which have been added mutations in the TP53 (24), NRAS/KRAS (25, 26) and NFE2 (27) genes. There are now several prognostic scores for MPN that have incorporated molecular data. Recently the presence of some mutations was shown to have an impact on treatment sensitivity. Indeed, the risk of developing resistance to hydroxyurea seems higher in patients with mutations in TP53 (28) or genes regulating RNA splicing (29), and mutations in the RAS pathway are more frequently associated with resistance to ruxolitinib (25, 26). Confirmation of the deleterious role of the co-occurrence of JAK2V617F mutations with some of these mutations has been provided by mouse models. For example, the transduction of JAK2V617F in the bone marrow cells of TP53 knockout mice increased the occurrence of leukemia in recipient mice (30).
Similarly, while deletion of the EZH2 gene does not induce a marked phenotype in mice, crossing EZH2 knockout mice with JAK2V617F transgenic mice showed an accelerated onset of myelofibrosis and a very marked shortening of the lifespan of the mice (31). Same observations were described with the inactivation of ASXL1 and IDH2 mutations (32, 33). Conversely, combined with SRSF2 mutants, JAK2 mutated MPN do not accelerate the evolution of the disease whereas patients seem to evolve (23, 34) highlighting the limits of those mouse models. The use of human xenotranplantation models would probably help resolving those discrepancies even though still technologically challenging. Overall, these observations demonstrate that certain additional mutations expressed in the JAK2V617F clone modify cell fate and can accelerate the transformation of MPN, confirming that clonal evolution is an important milestone linked to clinical evolution.
Clonal evolution in MPN: an example of multistep process in cancer evolution
Although it was quickly accepted that ET, PV and MF can clinically evolve from one entity to another, the concept that MPN could evolve at the genetic level took 50 years to establish. Indeed, studies of X chromosome inactivation patterns on female samples identified the clonal nature of the disease before the first molecular markers and additional mutations were discovered (12, 35). After the description of the first acquired mutations in MPN, one of the questions that quickly arose was how these mutations are acquired and distributed within the tumor. It is now well established that the driver mutation occurs in HSCs, giving rise to the MPN phenotype, while additional mutations acquired within the so-called driver clone contribute to the evolution of the disease, as shown by mouse models with JAK2V617F an additional mutations (31, 33, 34, 36). A linear pattern of mutation acquisition in the same cells is quite rare and a branching pattern of mutations is most often observed in MPN. Altogether, this leads to the frequent presence of various co-existing clones within the same tumor (10, 18, 19, 37–39). A novel and possibly more accurate vision of MPN could be envisaged to evaluate the prognosis of patients no longer based on the numbers and types of mutations but on the numbers and types of clones present at one time point in the tumor (Figure 1). Furthermore, the clonal architecture of MPN is a dynamic process, with clones appearing or disappearing and clones that can increase or decrease in length during disease evolution. Therefore, patients’ prognosis may rely on the clonal evolution of the disease and longitudinal study of clonal architecture could be of high clinical relevance.
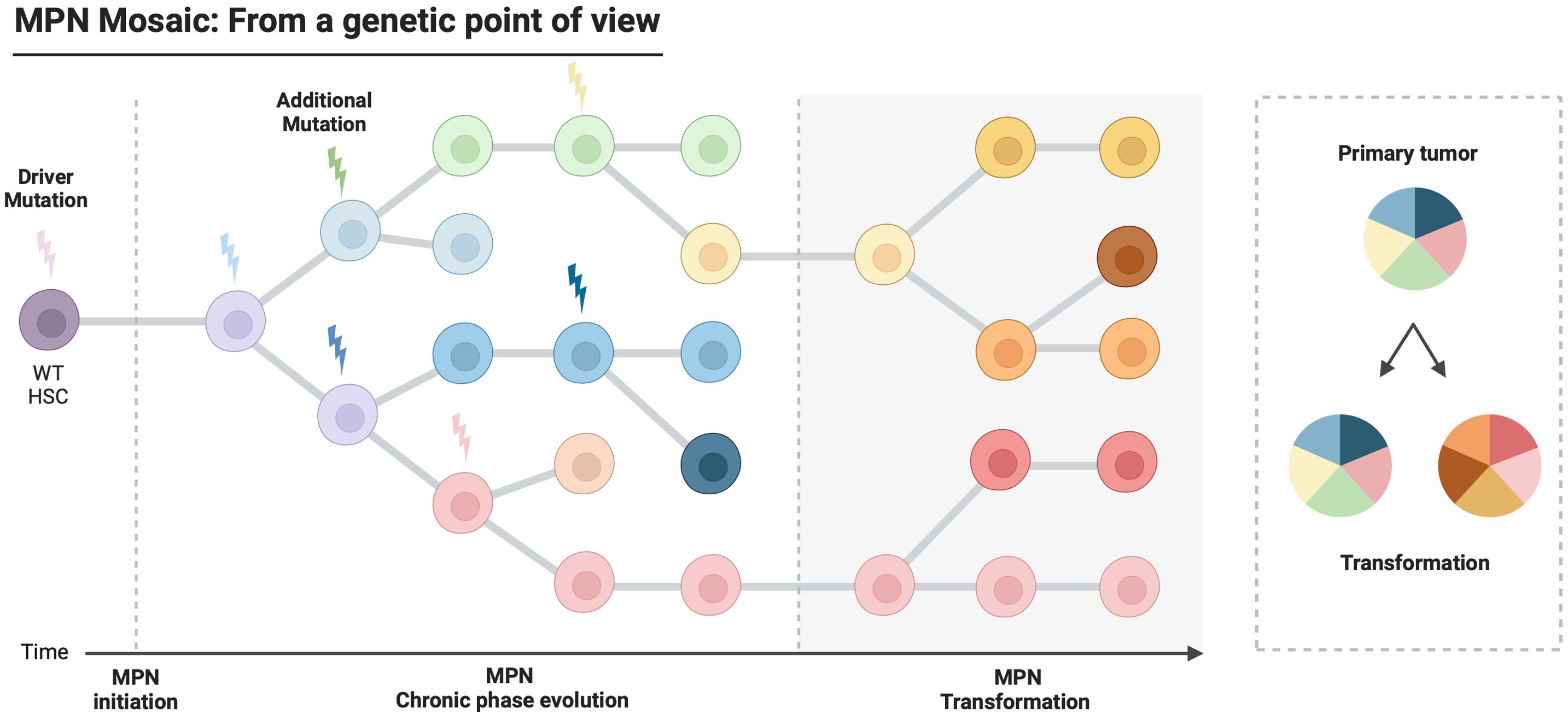
Figure 1. Model of leukemogenesis and clonal evolution in Myeloproliferative Neoplasms. Myeloproliferative Neoplasms are characterized by the acquisition of driver mutations in a hematopoietic stem cell. During disease progression, additional genetic events are acquired sequentially in cells carrying the driver mutation, leading to an oligoclonal disease. This intratumoral heterogeneity contributes to very different outcomes according to the patients.
Environmental factors modifying clonal evolution
From a Darwinian point of view, one of the major questions posed today, and because we know that multiple clones co-exist within the tumor, is to identify and better understand how environmental factors promote or, on the contrary, inhibit the expansion of a clone. The fitness of clones is the result of their own proliferation rate, their characteristics in terms of metabolism requirements and their ability to resist under a given selection pressure (40, 41).
Inflammation
Inflammation and driver mutations in MPN
Chronic inflammation is a hallmark of MPN which has been well described (42) and MF development is associated with the most severe level of inflammation (43). Among various mechanisms of inflammation, the most widely described in MPN is due to cytokine dysregulation. Indeed, a large amount of literature has been written on this topic, and many cytokines have been implicated. TGF-β was one of the first cytokine identified as hypersecreted in HSPCs from MF patients (44, 45). Its role in the development of myelofibrosis is well established in several models, including murine models overexpressing the thrombopoietin which rapidly develop myelofibrosis (46). The mechanism through which the TGF-β secreted by megakaryocytes in the bone marrow niche participates in myelofibrosis development was recently shown (47). More recently, the number of inflammatory cytokines identified as upregulated in MPN has been steadily increasing. For instance, IL-8 is overexpressed in MPN, via the transcription factor NFE2 which is frequently mutated in these diseases (48) and lipocalin-2 plays a pro-inflammatory but also a direct pro-fibrotic role in MPN (49). Interestingly, the JAK2V617F mutated cells were shown to be hypersensitive to TNFa (50), one of the most important inflammatory cytokines. Also, the IL-6 pathway is hyperactivated in CALR-mutated MPN, which is an important inflammatory pathway associated to myelofibrosis development (51–53). In addition, several inflammatory pathways are interdependent and may contribute to deregulation of key pathogenic processes involved in MPN. For example the IL-4/IL-13 axis that activates the TGF-β pathway has been shown to be deregulated in MPN, explaining in part the deregulation of this pathway (54). Very recently, overproduction of IL-1β by the JAK2V617F clone was implicated in bone marrow fibrosis in mouse models but was also shown to be a major factor favoring the clonal expansion of JAK2V617F-mutated cells (55, 56) providing a link between inflammation, fibrosis and clonal selection.
All these data given, it is still rather unclear whether inflammation is a cause or a consequence of MPN development (57). From a genetic point of view, although various models have been proposed, the question remains as to whether the presence of the mutation induces an inflammatory phenotype, or whether pre-existing inflammation is responsible at least in part for the clonal evolution (58). In the era of molecular characterization of tumors, a number of new data allow us to better answer the question. Several studies in various in vivo models of MPN showed strong evidence that constitutive activation of the JAK2/STAT5 pathway due to the acquisition of a driver mutation induces a major inflammatory phenotype, which is highly implicated in the disease (59–61).
Recently, studies combining at the single cell level the genotyping of specific genes with the analysis of transcriptome (60, 62) as well as chromatin accessibility showed that JAK2V617F mutated HSPCs exhibited specific proinflammatory signatures with accessibility to DNA motifs binding NF-kB or JUN/FOS factors. These approaches also allowed to highlight epigenetic modifications specific to mutated erythroid or megakaryocytic progenitors, demonstrating that the consequences of the mutations are different according to the differentiation stage and cellular context (63).
Finally, inhibitors of the JAK-STAT pathway such as ruxolitinib are widely used to treat MPN with important efficacy on symptom burden (64, 65). The main explanation for such efficacy seems to be linked to their potent anti-inflammatory effect (57, 64, 66). Some other anti-inflammatory therapies have also demonstrated activity against the mutated cells such as anti-IL1b therapies (52) and Interferon alpha has been reported to be effective at least in part through its anti-inflammatory property (56, 67).
Inflammation and additional (non-driver) mutations in MPN
The identification of circulating somatic mutations in the absence of clinical or biological evidence for any hematological disorder defines CHIP (ref 2014), which has been associated with an inflammatory phenotype. TET2 is one of the most frequently mutated genes in both MPN and CHIP. The TET2 protein represses IL-6 induction indirectly through the recruitment of histone deacetylases on its promoter (68). This is in line with the inflammatory phenotype observed during TET2 KO (69). The presence of TET2 mutations in persons with CHIP has been associated with an excess of various cardiovascular events (70) and a mechanism involving the inflammasome has been proposed (71). Similarly in pathological situations, TET2 mutations are strongly associated with inflammation in MPN and MDS (72, 73). DNMT3A-mutated HSPCs, on the other hand, have been shown to be selected in an inflammatory context (74). Single-cell studies capturing DNMT3A R882H mutations and coupling it to chromatin accessibility showed an inflammatory phenotype of mutated HSPCs compared to their wild-type counterpart (75), a finding in agreement with the anti-inflammatory properties of this gene (76) and explaining the described association between inflammatory diseases and DNMT3A clonal hematopoiesis (77). Finally, in MPN, JAK2/DNMT3A double mutants have been shown to cooperate to induce myelofibrosis via aberrant inflammation and self-renewal of HSPCs (78). Similarly, increased expression of IL-8 found in MPN (43) has been shown to be caused by overexpression of NFE2 (48), itself regulated by the histone demethylase JMJD1C (79). It would thus be interesting to study whether in an NFE2 mutated context, which has been shown to give a poor prognosis in MPN (27), IL-8 expression is increased and involved in clonal specific progression. Finally, after the acquisition of TP53 mutations clonal dominance of mutated cells is dependent on an inflammatory environment which participates to the transformation toward acute leukemia (60). This field of research is important since specific causes of mutation-independent inflammation could become useful new targets in MPN therapy for a subset of patients.
Altogether, because the genetic evolution of MPN appears to be partly shaped by inflammation, one can consider that MPN is a human inflammation model for cancer development (58) (Figure 2).
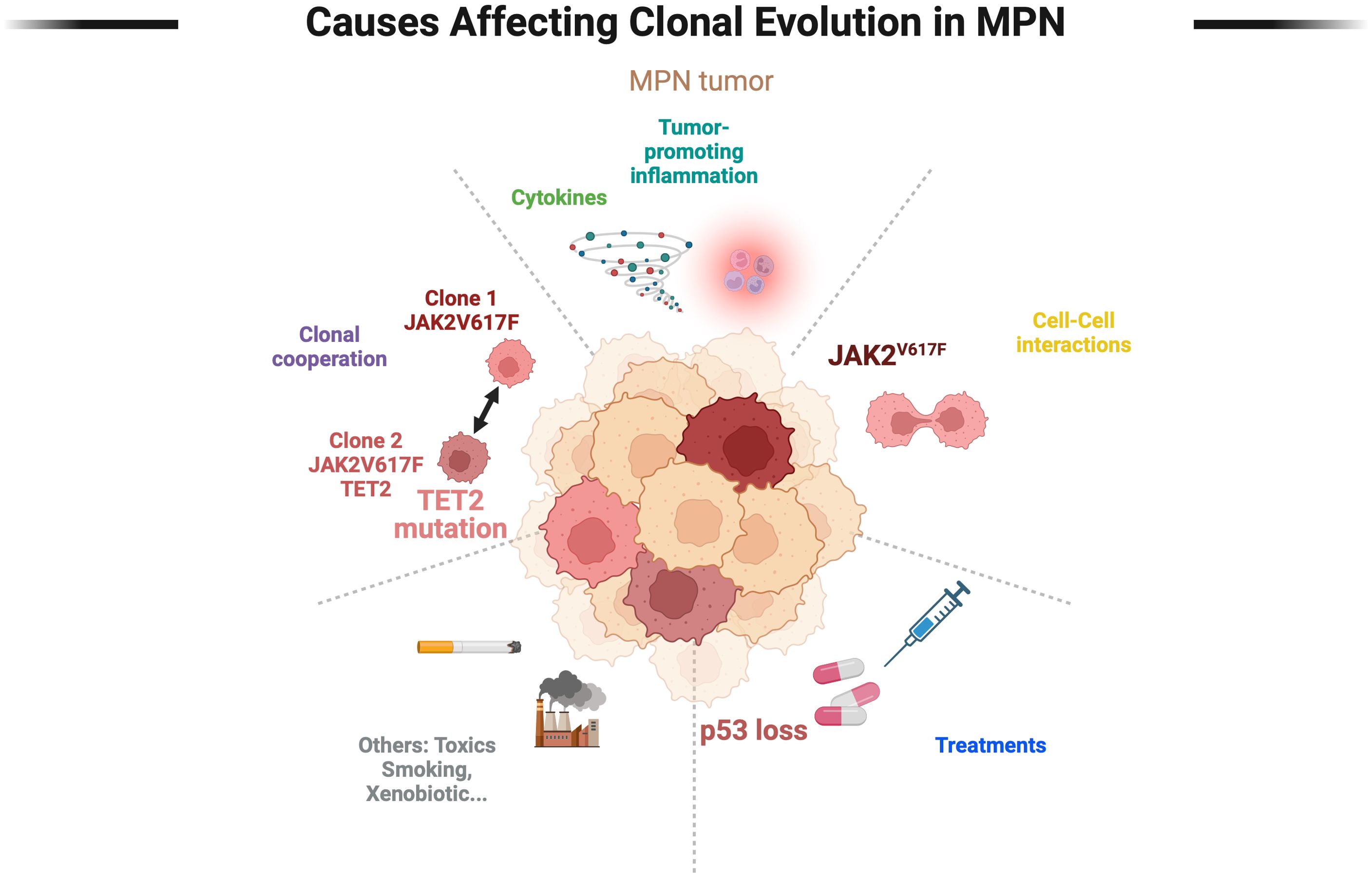
Figure 2. Main clonal selection factors in Myeloproliferative Neoplasms. The selection pressures exerted by the tumor are multiple and still poorly described and characterized. A distinction is made between extrinsic selection factors (toxins, treatments) and intrinsic selection factors (clonal cooperation, cell-cell interactions, microenvironment), the latter being poorly studied. The genetic evolution of tumors over time combines multiple factors, the real modeling of which remains a challenge.
Treatments
MPN are chronic diseases that, in a large proportion of patients, require cytoreductive therapies on the long term. However, the correlation of a given treatment with the risk of transformation in MPN patients is difficult to establish and still controversial. It has been strongly suggested that 32Phosphorus or alkylating agents such as pipobroman are associated to a higher rate of MDS/AML transformation and can promote the expansion of a subpopulation of more aggressive clones. Still, few studies have investigated the clonal composition of tumors developed after treatment with these agents (80, 81). The most frequently used cytoreductive treatment in MPN is hydroxyurea (HU), which has long been suspected to promote blastic transformation and also the selection of TP53-mutated cells in MPN (28, 29, 81). However, no definitive demonstration of a major role for HU in TP53-mutant clonal selection has emerged until now. To the contrary, the increase of TP53-mutations allelic frequencies under treatment with MDM2 inhibitors was recently reported in a phase II clinical trial in MF patients (82). An important element reinforcing the idea of environmental selection pressure by drugs was the regression of these clones at treatment discontinuation. We were able to confirm in vitro the potential for MDM2 inhibitors to promote the expansion of TP53-mutated clones. Furthermore, using single cell DNAseq analysis we observed that the selection pressure occurred in TP53 and JAK2V617F doubly mutant cells (37).
Another demonstration that treatments used in MPN could change the clonal composition of the tumor came with the discovery that Interferon alpha (IFNa) was able to specifically target JAK2V617F or CALR mutated clones (83–86). However, identification of resistance to IFNa in some patients with several mutations suggested that the presence of additional mutations could interfere with this process (87). Indeed, the role of DNMT3A mutations in the clonal resistance to IFNa has been recently shown (88, 89), although the mechanism remains only partially explained (90). In an MPN setting, Usart and al elegantly demonstrated very recently that loss of Dnmt3a increased self-renewal of of Jak2v617f mutant HSCs and mitigates their attrition by IFNa (91).
Intraclonal cooperation and competition
Since the discovery that MPN are oligoclonal diseases, the question has arisen of the distribution of mutations within the tumor, thus defining subclones. Depending on the study, between one and more than ten different clones can now be identified, giving an idea of the complexity of clonal hierarchy in MPN (37–39, 92). These clones probably interact with each other and may influence tumor evolution over time. Therefore it may be envisioned that the interactions between these clones may be synergistic defining positive clonal cooperation, or on the contrary these interactions may prove to be deleterious between clones, demonstrating a rather competitive character (93). Even though most interactions among subclones are neutral, an ecological interaction between clones has been reported in solid tumor models with detectable interactions that should be positive or negative, resulting in a phenotypic switch (94). Such cooperative or antagonistic interactions, due for instance to metabolite exchanges, could apply to MPN where multiple clones of various sizes coexist. In particular, the presence of a CHIP clone aside from the MPN clone could, directly or indirectly, regulate tumor development. The impact of such process on clinical evolution has been suggested (39) but the mechanisms explaining these effects remain to be studied.
Understanding the extent of cooperation and communication among MPN clones will be an important step in the future. Imaging technologies such as mass cytometry applied to bone marrow or spleen of patients with MPN could provide an accurate phenotypic characterization of diverse genotypic cell types. This has given greater granularity to early observations of clonal cooperation, and relationships between cells can now be described in detail (95–97). Multi-omic studies associated to the genotype of each clone within the same sample that can be mapped spatially to the tumor will help to further define functional examples of tumor heterogeneity.
Clonal cell-microenvironment interactions and communication
The differential interaction of clonal cells harboring different mutations with other cells in the microenvironment has been clearly demonstrated, but few studies showed a direct effect of these exchanges on clonal selection. In glioma models for example, it was shown that wild-type IDH tumors cause greater neuronal cell interaction and signaling than mutated IDH tumors (98). In lymphoid malignancies, it has been shown recently that TET2 and IDH2 double mutant cells in T follicular helper cells were able to alter cross-talk with germinal center B cells that promotes B cell clonal expansion while decreasing Fas-FasL interaction and reducing B cell apoptosis (99). Likewise, in myeloid malignancies, Asxl1-/- and nrasG12D/+ leukemic cells induced Host-derived wild-type T cells overexpressed programmed cell death protein 1 (PD-1) and T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) receptors, leading to a predominant exhausted T-cell phenotype (100). In hematopoietic models of CHIP, Partial transplantation with Dnmt3aR878H/+ bone marrow altered function of neutrophils and Tregs exacerbates CHIP-driven inflammation (101). Same model caused MSCs to upregulate the senescence markers SA-β-gal, BCL-2, BCL-xL, Cdkn1a (p21) and Cdkn2a (p16), ex vivo and in vivo. This effect was cell contact-independent and can be replicated by IL-6 or TNFα, which are produced by Dnmt3aR878H/+ HSPCs (102). In the medullary microenvironment of MPN, interaction between neuronal cells and death of mesenchymal stromal cells has been demonstrated, promoting the expansion of JAK2V617F clones (103, 104). Although soluble factors are involved in this process (IL-1b, CXCL12 and adrenergic agonists), direct interaction also seems to play a role. It would be interesting to study the impact of direct cellular interactions between different clones of the same tumor and stromal cells, even if demonstration of indirect interactions via cytokines have already been described (105). Another evidence of cell interaction mechanism leading to evolution of myeloid disease and involving interaction of clonal cells with the microenvironment was provided by transplantation of human MF HSPCs into immunocompromised mice experiments, leading to murine AML. This study showed that the secretome-driven myeloproliferation of murine cells can occur independently of both the type of mutation and the mutational burden of transplanted HSPC patient cells (106).
Clonal evolution: a path towards transformation?
Monitoring clonal evolution is obviously of clinical interest, as the risk of transformation has been correlated with genetic changes in the tumor. The genetic basis of transformation has been well described previously (107–110). As discussed above, it has been shown that the accumulation of mutations is a clear unfavorable prognostic factor in MPN, and that the number of HMR mutations at diagnosis correlates with the risk of transformation whatever the MPN subtype, as well as high-risk mutations such as TP53 (111, 112). However, to date, it is not exactly known why some patients with the same molecular lesions will experience leukemic progression while others will not. If we take the example of TP53 mutations, we can find contradictory evidence in the literature. Firstly, the risk of transformation in JAK2V617F-TP53 double mutation models is still debated as some mouse models do not progress to secondary AML while in patients this risk of progression is clearly present (30, 60, 113, 114). The second observation is that many patients with TP53 mutations can be followed for a long time without progressing to transformation, even without specific treatment. Finally, we know that a non-negligible proportion of transformations arise from a driver mutation-negative clone, pointing to the importance of the molecular composition of the tumor and the clones present throughout the tumor process (107) (Figure 1). Although it is well established that a second event is required to accelerate the transformation in a TP53-mutated context, we still do not know precisely how this event occurs in a TP53 heterozygous MPN clone. New single cell technologies combining genotype and transcriptomics or mutliomics on longitundinal samples of MPN throughout clinical evolution will help us get through these questions.
Conclusion
The complexity of myeloproliferative neoplasms (MPNs) reflects the intricate interplay between genetic mutations, clonal evolution, and microenvironmental factors. This review has highlighted how MPNs are driven by a sequential accumulation of mutations that not only initiate but also fuel disease progression and transformation. The dynamic nature of clonal evolution, where subclones with distinct genetic profiles compete and cooperate, adds further complexity to MPN pathogenesis. In addition, the microenvironment plays a crucial role in shaping clonal behavior, influencing both the expansion of malignant clones and their interaction with supporting cells. Lastly, acute or chronic environmental context including inflammation and selection pressure by the therapies used during the chronic course of the illness may further contribute to disease progression.
As we continue to unravel the molecular mechanisms underlying MPNs, it becomes evident that understanding clonal evolution in its full context is essential for improving therapeutic strategies. Future research, leveraging advanced technologies like single-cell sequencing and integrated multi-omics, will be key to uncovering new insights into MPN initiation, progression, and therapeutic resistance. By integrating these discoveries into clinical practice, we can better predict disease trajectories and optimize treatment approaches, ultimately aiming for more personalized and effective care for MPN patients.
Author contributions
NM: Conceptualization, Formal analysis, Validation, Writing – original draft, Writing – review & editing. SG: Validation, Writing – original draft, Writing – review & editing. BC: Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. JK: Conceptualization, Project administration, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We thank the Institut du Cancer INCa (Prev-Bio 2021, Project R21166HH) and la Ligue contre le Cancer (Research mobility 2023) for their support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka H-M, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
2. Thiele J, Kvasnicka HM, Orazi A, Gianelli U, Gangat N, Vannucchi AM, et al. The international consensus classification of myeloid neoplasms and acute leukemias: myeloproliferative neoplasms. Am J Hematol. (2023) 98:166–795. doi: 10.1002/ajh.26751
3. Tefferi A, Alkhateeb H, Gangat N. Blast phase myeloproliferative neoplasm: contemporary review and 2024 treatment algorithm. Blood Cancer J. (2023) 13:1–115. doi: 10.1038/s41408-023-00878-8
4. James C, Ugo V, Couédic J-P Le, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. (2005) 434:1144–48. doi: 10.1038/nature03546
5. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet (London England). (2005) 365:1054–61. doi: 10.1016/S0140-6736(05)71142-9
6. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. (2005) 7:387–97. doi: 10.1016/j.ccr.2005.03.023
7. Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. (2005) 352:1779–905. doi: 10.1056/NEJMoa051113
8. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PloS Med. (2006) 3:e270. doi: 10.1371/journal.pmed.0030270
9. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. (2013) 369:2391–405. doi: 10.1056/NEJMoa1312542
10. Maslah N, Benajiba L, Giraudier S, Kiladjian J-J, Cassinat B. Clonal architecture evolution in myeloproliferative neoplasms: from a driver mutation to a complex heterogeneous mutational and phenotypic landscape. Leukemia. (2023) 37:957–635. doi: 10.1038/s41375-023-01886-0
11. Michiels JJ, Kutti J, Stark P, Bazzan M, Gugliotta L, Marchioli R, et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic metaplasia and myelofibrosis. Netherlands J Med. (1999) 54:46–62. doi: 10.1016/s0300-2977(98)00143-0
12. Kiladjian J-J, Elkassar N, Cassinat B, Hetet G, Giraudier S, Balitrand N, et al. Essential thrombocythemias without V617F JAK2 mutation are clonal hematopoietic stem cell disorders. Leukemia. (2006) 20:1181–83. doi: 10.1038/sj.leu.2404214
13. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
14. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
15. Nielsen C, Bojesen SE, Nordestgaard BG, Kofoed KF, Birgens HS. JAK2V617F somatic mutation in the general population: myeloproliferative neoplasm development and progression rate. Haematologica. (2014) 99:1448–555. doi: 10.3324/haematol.2014.107631
16. McKerrell T, Park N, Chi J, Collord G, Moreno T, Ponstingl H, et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv. (2017) 1:968–71. doi: 10.1182/bloodadvances.2017007047
17. Hirsch P, Mamez AC, Belhocine R, Lapusan S, Tang R, Suner L, et al. Clonal history of a cord blood donor cell leukemia with prenatal somatic JAK2 V617F mutation. Leukemia. (2016) 30:1756–59. doi: 10.1038/leu.2016.31
18. Van Egeren D, Escabi J, Nguyen M, Liu S, Reilly CR, Patel S, et al. Reconstructing the lineage histories and differentiation trajectories of individual cancer cells in myeloproliferative neoplasms. Cell Stem Cell. (2021) 28:514–523.e9. doi: 10.1016/j.stem.2021.02.001
19. Williams N, Lee J, Mitchell E, Moore L, Baxter EJ, Hewinson J, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature. (2022) 602:162–68. doi: 10.1038/s41586-021-04312-6
20. Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. (2010) 70:447–52. doi: 10.1158/0008-5472.CAN-09-3783
21. Schaub FX, Looser R, Li S, Hao-Shen H, Lehmann T, Tichelli A, et al. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood. (2010) 115:2003–75. doi: 10.1182/blood-2009-09-245381
22. Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. (2014) 28:1804–10. doi: 10.1038/leu.2014.76
23. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. (2013) 27:1861–69. doi: 10.1038/leu.2013.119
24. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. (2014) 123:2220–28. doi: 10.1182/blood-2013-11-537167
25. Coltro G, Rotunno G, Mannelli L, Mannarelli C, Fiaccabrino S, Romagnoli S, et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. (2020) 4:3677–87. doi: 10.1182/bloodadvances.2020002175
26. O’Sullivan JM, Taylor J, Gerds A, Buckley S, Harrison CN, Oh S, et al. RAS-pathway mutations are common in patients with ruxolitinib refractory/intolerant myelofibrosis: molecular analysis of the PAC203 cohort. Leukemia. (2023) 37:2497–501. doi: 10.1038/s41375-023-02027-3
27. Marcault C, Zhao LP, Maslah N, Verger E, Oliveira RD De, Soret-Dulphy J, et al. Impact of NFE2 mutations on AML transformation and overall survival in patients with myeloproliferative neoplasms (MPN). Blood. (2021). doi: 10.1182/blood.2020010402
28. Kubesova B, Pavlova S, Malcikova J, Kabathova J, Radova L, Tom N, et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia. (2018) 32:450–61. doi: 10.1038/leu.2017.230
29. Alvarez-Larrán A, Díaz-González A, Such E, Mora E, Andrade-Campos M, García-Hernández C, et al. Genomic characterization of patients with polycythemia vera developing resistance to hydroxyurea. Leukemia. (2021) 35:623–27. doi: 10.1038/s41375-020-0849-2
30. Rampal R, Ahn J, Abdel-Wahab O, Nahas M, Wang K, Lipson D, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci USA. (2014) 111:E5401–5410. doi: 10.1073/pnas.1407792111
31. Sashida G, Wang C, Tomioka T, Oshima M, Aoyama K, Kanai A, et al. The loss of ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J Exp Med. (2016) 213:1459–775. doi: 10.1084/jem.20151121
32. McKenney AS, Lau AN, Somasundara AVH, Spitzer B, Intlekofer AM, Ahn J, et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J Clin Invest. (2018) 128:789–804. doi: 10.1172/JCI94516
33. Shi Z, Liu J, Zhao Y, Yang L, Cai Y, Zhang P, et al. ASXL1 mutations accelerate bone marrow fibrosis via EGR1-TNFA axis-mediated neoplastic fibrocyte generation in myeloproliferative neoplasms. Haematologica. (2022) 108:1359–73. doi: 10.3324/haematol.2021.280320
34. Willekens C, Laplane L, Dagher T, Benlabiod C, Papadopoulos N, Lacout C, et al. SRSF2-P95H decreases JAK/STAT signaling in hematopoietic cells and delays myelofibrosis development in mice. Leukemia. (2023) 37:1287–97. doi: 10.1038/s41375-023-01878-0
35. Tsukamoto N, Morita K, Maehara T, Okamoto K, Sakai H, Karasawa M, et al. Clonality in chronic myeloproliferative disorders defined by X-chromosome linked probes: demonstration of heterogeneity in lineage involvement. Br J Haematol. (1994) 86:253–58. doi: 10.1111/j.1365-2141.1994.tb04723.x
36. Guo Y, Zhou Y, Yamatomo S, Yang H, Zhang P, Chen S, et al. ASXL1 alteration cooperates with JAK2V617F to accelerate myelofibrosis. Leukemia. (2019) 33:1287–91. doi: 10.1038/s41375-018-0347-y
37. Maslah N, Verger E, Giraudier S, Chea M, Hoffman R, Mascarenhas J, et al. Single-cell analysis reveals selection of TP53-mutated clones after MDM2 inhibition. Blood Adv. (2022). doi: 10.1182/bloodadvances.2021005867
38. Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid Malignancies. Nature. (2020) 587:477–82. doi: 10.1038/s41586-020-2864-x
39. Luque Paz D, Bader MS, Nienhold R, Rai S, Fonseca TA, Stetka J, et al. Impact of clonal architecture on clinical course and prognosis in patients with myeloproliferative neoplasms. HemaSphere. (2023) 7:e8855. doi: 10.1097/HS9.0000000000000885
40. Greaves M, Maley CC. Clonal evolution in cancer. Nature. (2012) 481:306–13. doi: 10.1038/nature10762
41. Anderson K, Lutz C, Delft FWv, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. (2011) 469:356–61. doi: 10.1038/nature09650
42. Barbui T, Carobbio A, Finazzi G, Vannucchi AM, Barosi G, Antonioli E, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactive protein and pentraxin 3. Haematologica. (2011) 96:315–18. doi: 10.3324/haematol.2010.031070
43. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J Clin Oncol: Off J Am Soc Clin Oncol. (2011) 29:1356–635. doi: 10.1200/JCO.2010.32.9490
44. Martyré M-C, Magdelenat H, Bryckaert M-C, Laine-Bidron C, Calvo F. Increased intraplatelet levels of platelet-derived growth factor and transforming growth factor-β in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. (1991) 77:80–865. doi: 10.1111/j.1365-2141.1991.tb07952.x
45. Le Bousse-Kerdiles MC, Chevillard S, Charpentier A, Romquin N, Clay D, Smadja-Joffe F, et al. Differential expression of transforming growth factor-beta, basic fibroblast growth factor, and their receptors in CD34+ Hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood. (1996) 88:4534–46. doi: 10.1182/blood.V88.12.4534.bloodjournal88124534
46. Chagraoui H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F. Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in mice. Blood. (2002) 100:3495–35035. doi: 10.1182/blood-2002-04-1133
47. Yao J-C, Oetjen KA, Wang T, Xu H, Abou-Ezzi G, Krambs JR, et al. TGF-β Signaling in myeloproliferative neoplasms contributes to myelofibrosis without disrupting the hematopoietic niche. J Clin Invest. (2022) 132(11):e154092. doi: 10.1172/JCI154092
48. Wehrle J, Seeger TS, Schwemmers S, Pfeifer D, Bulashevska A, Pahl HL. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative neoplasms. Haematologica. (2013) 98:1073–805. doi: 10.3324/haematol.2012.071183
49. Lu M, Xia L, Liu Y-C, Hochman T, Bizzari L, Aruch D, et al. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood. (2015) 126:972–825. doi: 10.1182/blood-2014-12-618595
50. Fleischman AG, Aichberger KJ, Luty SB, Bumm TG, Petersen CL, Doratotaj S, et al. TNFα Facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. (2011) 118:6392–98. doi: 10.1182/blood-2011-04-348144
51. Panteli KE, Hatzimichael EC, Bouranta PK, Katsaraki A, Seferiadis K, Stebbing J, et al. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br J Haematol. (2005) 130:709–155. doi: 10.1111/j.1365-2141.2005.05674.x
52. Fisher DAC, Miner CA, Engle EK, Hu H, Collins TB, Zhou A, et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia. (2019) 33:1978–955. doi: 10.1038/s41375-019-0379-y
53. Balliu M, Calabresi L, Bartalucci N, Romagnoli S, Maggi L, Manfredini R, et al. Activated IL-6 Signaling Contributes to the Pathogenesis of, and Is a Novel Therapeutic Target for, CALR-Mutated MPNs. Blood Adv. (2021) 5:2184–955. doi: 10.1182/bloodadvances.2020003291
54. Melo-Cardenas J, Bezavada L, Crawford JC, Gurbuxani S, Cotton A, Kang G, et al. IL-13/IL-4 signaling contributes to fibrotic progression of the myeloproliferative neoplasms. Blood. (2022) 140:2805–17. doi: 10.1182/blood.2022017326
55. Rahman MF-U, Yang Y, Le BT, Dutta A, Posyniak J, Faughnan P, et al. Interleukin-1 contributes to clonal expansion and progression of bone marrow fibrosis in JAK2V617F-induced myeloproliferative neoplasm. Nat Commun. (2022) 13:53475. doi: 10.1038/s41467-022-32928-3
56. Rai S, Zhang Y, Grockowiak E, Kimmerlin Q, Hansen N, Stoll CB, et al. IL-1β Promotes MPN disease initiation by favoring early clonal expansion of JAK2-mutant hematopoietic stem cells. Blood Adv. (2024) 8:1234–49. doi: 10.1182/bloodadvances.2023011338
57. Hasselbalch HC, Bjørn ME. MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediators Inflamm. (2015) 2015:102476. doi: 10.1155/2015/102476
58. Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. (2012) 119:3219–25. doi: 10.1182/blood-2011-11-394775
59. Falanga A, Marchetti M, Vignoli A, Balducci D, Russo L, Guerini V, et al. V617F JAK-2 mutation in patients with essential thrombocythemia: relation to platelet, granulocyte, and plasma hemostatic and inflammatory molecules. Exp Hematol. (2007) 35:702–115. doi: 10.1016/j.exphem.2007.01.053
60. Rodriguez-Meira A, Norfo R, Wen S, Chédeville AL, Rahman H, O’Sullivan J, et al. Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat Genet. (2023) 55:1531–41. doi: 10.1038/s41588-023-01480-1
61. Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK-STAT pathway activation in Malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. (2015) 5:316–31. doi: 10.1158/2159-8290.CD-14-0736
62. Nam AS, Kim K-T, Chaligne R, Izzo F, Ang C, Taylor J, et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature. (2019) 571:355–60. doi: 10.1038/s41586-019-1367-0
63. Myers RM, Izzo F, Kottapalli S, Prieto T, Dunbar A, Bowman RL, et al. Integrated single-cell genotyping and chromatin accessibility charts JAK2V617F human hematopoietic differentiation. bioRxiv. (2022). doi: 10.1101/2022.05.11.491515
64. Harrison C, Kiladjian J-J, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. (2012) 366:787–98. doi: 10.1056/NEJMoa1110556
65. Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. (2015) 372:426–35. doi: 10.1056/NEJMoa1409002
66. Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian J-J, et al. Myeloproliferative neoplasms and inflammation: whether to target the Malignant clone or the inflammatory process or both. Leukemia. (2016) 30:1018–24. doi: 10.1038/leu.2016.12
67. Skov V, Riley C, Thomassen M, Kjær L, Larsen TS, Bjerrum OW, et al. Interferon-alfa2 treatment of patients with polycythemia vera and related neoplasms influences deregulated inflammation and immune genes in polycythemia vera and allied neoplasms. Blood. (2018) 132:5490. doi: 10.1182/blood-2018-99-118690
68. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting hdac2 to specifically repress IL-6. Nature. (2015) 525:389–93. doi: 10.1038/nature15252
69. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–849.e5. doi: 10.1016/j.stem.2018.10.013
70. Jaiswal S, Pradeep N, Silver Alexander J, Gibson Christopher J, Bick Alexander G, Eugenia S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
71. Lin AE, Bapat AC, Xiao L, Niroula A, Ye J, Wong WJ, et al. Clonal hematopoiesis of indeterminate potential with loss of tet2 enhances risk for atrial fibrillation through nlrp3 inflammasome activation. Circulation. (2024). doi: 10.1161/CIRCULATIONAHA.123.065597
72. Elessa D, Zhao L-P, de Oliveira RD, Maslah N, Soret-Dulphy J, Verger E, et al. Clinical features and genomic landscape of myeloproliferative neoplasm (MPN) patients with autoimmune and inflammatory diseases (AID). Leukemia. (2023) 37:1741–44. doi: 10.1038/s41375-023-01967-0
73. Zhao L-P, Boy M, Azoulay C, Clappier E, Sébert M, Amable L, et al. Genomic landscape of MDS/CMML associated with systemic inflammatory and autoimmune disease. Leukemia. (2021) 35:2720–24. doi: 10.1038/s41375-021-01152-1
74. Zioni N, Bercovich AA, Chapal-Ilani N, Bacharach T, Rappoport N, Solomon A, et al. Inflammatory signals from fatty bone marrow support DNMT3A driven clonal hematopoiesis. Nat Commun. (2023) 14:2070. doi: 10.1038/s41467-023-36906-1
75. Nam AS, Dusaj N, Izzo F, Murali R, Myers RM, Mouhieddine T, et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. bioRxiv. (2022). doi: 10.1101/2022.01.14.476225
76. Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, et al. Dnmt3a restrains mast cell inflammatory responses. Proc Natl Acad Sci. (2017) 114:E1490–995. doi: 10.1073/pnas.1616420114
77. Zhang CRC, Nix D, Gregory M, Ciorba MA, Ostrander EL, Newberry RD, et al. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Exp Hematol. (2019) 80:36–41.e3. doi: 10.1016/j.exphem.2019.11.008
78. Jacquelin S, Straube J, Cooper L, Vu T, Song A, Bywater M, et al. Jak2V617F and dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood. (2018) 132:2707–21. doi: 10.1182/blood-2018-04-846220
79. Peeken JC, Jutzi JS, Wehrle J, Koellerer C, Staehle HF, Becker H, et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood. (2018) 131:2065–73. doi: 10.1182/blood-2017-10-810622
80. Najean Y, Rain JD. Treatment of polycythemia vera: the use of hydroxyurea and pipobroman in 292 patients under the age of 65 years. Blood. (1997) 90:3370–77. doi: 10.1182/blood.V90.9.3370
81. Kiladjian J-J, Chevret S, Dosquet C, Chomienne C, Rain J-D. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncology: Off J Am Soc Clin Oncol. (2011) 29:3907–135. doi: 10.1200/JCO.2011.36.0792
82. Marcellino BK, Farnoud N, Cassinat B, Lu M, Verger E, McGovern E, et al. Transient expansion of TP53 mutated clones in polycythemia vera patients treated with idasanutlin. Blood Adv. (2020) 4:5735–44. doi: 10.1182/bloodadvances.2020002379
83. Kiladjian J-J, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. (2008) 112:3065–725. doi: 10.1182/blood-2008-03-143537
84. Verger E, Cassinat B, Chauveau A, Dosquet C, Giraudier S, Schlageter M, et al. Clinical and molecular response to interferon-α Therapy in essential thrombocythemia patients with CALR mutations. Blood. (2015) 126:2585–91. doi: 10.1182/blood-2015-07-659060
85. Dagher T, Maslah N, Edmond V, Cassinat B, Vainchenker W, Giraudier S, et al. JAK2V617F myeloproliferative neoplasm eradication by a novel interferon/arsenic therapy involves PML. J Exp Med. (2021) 218:e20201268. doi: 10.1084/jem.20201268
86. Kiladjian J-J, Giraudier S, Cassinat B. Interferon-alpha for the therapy of myeloproliferative neoplasms: targeting the Malignant clone. Leukemia. (2016) 30:776–81. doi: 10.1038/leu.2015.326
87. Kiladjian J-J, Massé A, Cassinat B, Mokrani H, Teyssandier I, le Couédic J-P, et al. Clonal analysis of erythroid progenitors suggests that pegylated interferon alpha-2a treatment targets JAK2V617F clones without affecting TET2 mutant cells. Leukemia. (2010) 24:1519–23. doi: 10.1038/leu.2010.120
88. Knudsen TA, Skov V, Stevenson KE, Werner L, Duke W, Laurore C, et al. Genomic profiling of a randomized trial of interferon-α versus hydroxyurea in MPN reveals mutation-specific responses. Blood Adv. (2021). doi: 10.1182/bloodadvances.2021004856
89. Zhao L-P, Maslah N, De Oliveira RD, Verger E, Soret-Dulphy J, Marcault C, et al. Chronic exposure to cytoreductive treatment shapes clonal evolution in myeloproliferative neoplasms. Blood. (2021) 138:3620. doi: 10.1182/blood-2021-152622
90. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives dnmt3a-loss-of-function clonal hematopoiesis via IFNγ Signaling. Cell Stem Cell. (2021) 28:1428–14425.e6. doi: 10.1016/j.stem.2021.03.002
91. Usart M, Stetka J, Paz DL, Hansen N, Kimmerlin Q, Fonseca TA, et al. Loss of dnmt3a increases self-renewal and resistance to pegIFN-α in JAK2-V617F–positive myeloproliferative neoplasms. Blood. (2024) 143:2490–503. doi: 10.1182/blood.2023020270
92. Maslah N, Roux B, Kaci N, Verger E, De Oliveira RD, Pasquer H, et al. JAK inhibition mediates clonal selection of RAS pathway mutations in myeloproliferative neoplasms. Blood. (2022) 140:795–96. doi: 10.1182/blood-2022-167719
93. Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in wnt-driven mammary cancers. Nature. (2014) 508:113–17. doi: 10.1038/nature13187
94. Hershey BJ, Barozzi S, Orsenigo F, Pompei S, Iannelli F, Kamrad S, et al. Clonal cooperation through soluble metabolite exchange facilitates metastatic outgrowth by modulating allee effect. Sci Adv. (2023) 9:eadh4184. doi: 10.1126/sciadv.adh4184
95. Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. (2019) 177:1330–1345.e18. doi: 10.1016/j.cell.2019.03.005
96. Ali HR, Jackson HW, Zanotelli VRT, Danenberg E, Fischer JR, Bardwell H, et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat Cancer. (2020) 1:163–75. doi: 10.1038/s43018-020-0026-6
97. Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature. (2020) 578:615–20. doi: 10.1038/s41586-019-1876-x
98. Varn FS, Johnson KC, Martinek J, Huse JT, Nasrallah MP, Wesseling P, et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell. (2022) 185:2184–2199.e16. doi: 10.1016/j.cell.2022.04.038
99. Julie L, Lemonnier F, Meydan C, Foox J, Ghamrasni SE, Mboumba DL, et al. IDH2 and TET2 Mutations Synergize to Modulate T Follicular Helper Cell Functional Interaction with the AITL Microenvironment. Cancer Cell. (2013) 41(2):323–39.e10.
100. You X, Liu F, Binder M, Vedder A, Lasho T, Wen Z, et al. Asxl1 Loss Cooperates with Oncogenic Nras in Mice to Reprogram the Immune Microenvironment and Drive Leukemic Transformation. Blood. (2022) 139(7). doi: 10.1182/blood.2021012519
101. Wang H, Divaris K, Pan B, Li X, Lim J-H, Saha G, et al. Clonal hematopoiesis driven by mutated DNMT3A promotes inflammatory bone loss. Cell. (2024) 187:3690–3711.e19. doi: 10.1016/j.cell.2024.05.003
102. Mistry JJ, Young KA, Díaz PAC, Maestre IF, Levine RL, Trowbridge JJ. Mesenchymal stromal cell senescence induced by dnmt3a-mutant hematopoietic cells is a targetable mechanism driving clonal hematopoiesis and initiation of hematologic Malignancy. bioRxiv. (2024). doi: 10.1101/2024.03.28.587254
103. Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. (2014) 512:78–81. doi: 10.1038/nature13383
104. Grockowiak E, Korn C, Rak J, Lysenko V, Hallou A, Panvini FM, et al. Different niches for stem cells carrying the same oncogenic driver affect pathogenesis and therapy response in myeloproliferative neoplasms. Nat Cancer. (2023) 4:1193–209. doi: 10.1038/s43018-023-00607-x
105. Ganesan S, Awan-Toor S, Guidez F, Maslah N, Aoun C, Gou P, et al. Comprehensive analysis of mesenchymal cells from myeloproliferative neoplasm (MPN) patients reveals the role of HOXB7 in myelofibrosis induction. Blood. (2022) 140:3852–54. doi: 10.1182/blood-2022-157490
106. Triviai I, Ziegler M, Bergholz U, Oler AJ, Stübig T, Prassolov V, et al. Endogenous retrovirus induces leukemia in a xenograft mouse model for primary myelofibrosis. Proc Natl Acad Sci USA. (2014) 111:8595–86005. doi: 10.1073/pnas.1401215111
107. Beer PA, Delhommeau F, LeCouédic J-P, Dawson MA, Chen E, Bareford D, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. (2010) 115:2891–900. doi: 10.1182/blood-2009-08-236596
108. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. (2017) 129:667–795. doi: 10.1182/blood-2016-10-695940
109. Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. (2020) 136:61–705. doi: 10.1182/blood.2019000943
110. Luque Paz D, Kralovics R, Skoda RC. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood. (2023) 141:1909–215. doi: 10.1182/blood.2022017578
111. Tefferi A, Guglielmelli P, Lasho TL, Coltro G, Finke CM, Loscocco GG, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. (2020) 189:291–302. doi: 10.1111/bjh.16380
112. Luque Paz D, Jouanneau-Courville R, Riou J, Ianotto J-C, Boyer F, Chauveau A, et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: genomic profiles predict time to transformation. Blood Adv. (2020) 4:4887–97. doi: 10.1182/bloodadvances.2020002271
113. Gou P, Liu D, Ganesan S, Lauret E, Maslah N, Parietti V, et al. Genomic and functional impact of trp53 inactivation in JAK2V617F myeloproliferative neoplasms. Blood Cancer J. (2024) 14:1. doi: 10.1038/s41408-023-00969-6
Keywords: myeloproliferative neoplasms, additional mutation, clonal evolution, inflammation, selection
Citation: Maslah N, Giraudier S, Cassinat B and Kiladjian J-J (2024) Clonal evolution: a landmark of MPN evolution. Front. Hematol. 3:1473920. doi: 10.3389/frhem.2024.1473920
Received: 31 July 2024; Accepted: 09 September 2024;
Published: 07 October 2024.
Edited by:
Hans Carl Hasselbalch, Zealand University Hospital, DenmarkReviewed by:
Ioanna Triviai, Max Planck Institute for Molecular Genetics, GermanyAngela Fleischman, University of California, Irvine, United States
Copyright © 2024 Maslah, Giraudier, Cassinat and Kiladjian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Jacques Kiladjian, amVhbi1qYWNxdWVzLmtpbGFkamlhbkBhcGhwLmZy; Nabih Maslah, bmFiaWgubWFzbGFoQGFwaHAuZnI=
†ORCID: Nabih Maslah, orcid.org/0000-0002-5553-462X
Stephane Giraudier, orcid.org/0000-0002-1817-2236
Bruno Cassinat, orcid.org/0000-0002-6514-3905
Jean-Jacques Kiladjian, orcid.org/0000-0002-8121-438X