 Lushen Li
Lushen Li Pankaj K. Mandal*
Pankaj K. Mandal*- Tumor Vaccines and Biotechnology Branch, Division of Cell Therapy 2, Office of Cellular Therapy and Human Tissue, Office of Therapeutic Products, Center for Biologics Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD, United States
β-hemoglobinopathies, including sickle cell disease (SCD) and β-thalassemia, are prevalent monogenic disorders causing abnormal hemoglobin structure or production that affect millions globally. Current available therapies for SCD and β-thalassemia are primarily symptomatic treatments and allogeneic hematopoietic stem cell transplant (HSCT). Allo-HSCT is the only curative treatment, which has limitations. Gene therapy using genetically modified hematopoietic stem cells (HSCs) holds promise to be an effective curative therapy. Recently approved ex vivo genetically modified HSC-based therapeutics (CASGEVY, LYFGENIA, ZYNTEGLO) have shown remarkable and durable therapeutic benefits for SCD and β-Thalassemia. In this review article, we discuss the current genetic approaches and innovative strategies to ensure safe and effective gene therapy for SCD and β-thalassemia and summarize findings from completed and ongoing clinical trials. We also discuss prospects and challenges of in vivo gene editing with CRISPR/Cas technology for SCD and beta-thalassemia that may simplify manufacturing and treatment process. In vivo gene therapy may minimize the risks associated with ex vivo gene therapy and may overcome multiple barriers associated with complex gene therapy products for wider patient access, especially in developing regions of the world where these diseases are highly prevalent.
Introduction of sickle cell disease and β-thalassemia
Hemoglobinopathies, including sickle cell disease (SCD) and β-thalassemia, are common monogenic disorders affecting millions of people worldwide. The World Health Organization estimates that over 5% of the global population carries hemoglobinopathies, with nearly 300,000 – 400,000 children born annually with severe forms of these diseases (1, 2). Hemoglobinopathies are caused by perturbed hemoglobin (Hb) biosynthesis, affecting the structure, function or production of Hb which is the oxygen-carrying protein in red blood cells (RBCs). The normal adult Hb molecule, known as Hemoglobin A (HbA), is composed of two α-globin chains and two β-globin chains (3). Hemoglobinopathies are broadly classified into two subgroups: a) Hemoglobinopathies with Hb structural aberrations that include SCD, Hemoglobin C (HbC), Hemoglobin D (HbD), Hemoglobin E (HbE), and Hemoglobin Constant Spring disease (HbCS), and b) Hemoglobinopathies with abnormal Hb production include thalassemia syndrome (α− and β−thalassemia). SCD is caused by a point mutation in β-globin (HBB) gene that substitutes Valine for Glutamic acid at codon 6 (βG6V). G6V mutation results in production of sickle hemoglobin (HbS) which forms long polymers under poor oxygenation. HbS polymerization distorts the shape of RBC with a characteristic sickle shape (4). SCD syndrome is characterized by hemolysis, vaso-occlusion crises (VOC), and infections causing recurring episodes of acute pain, tissue damage, strokes, and potentially resulting in multi-organ damage or death in most severe cases (5). β-thalassemias are caused by mutations in HBB gene resulting in absence (beta-zero (β0) Thalassemia) or quantitative reduction (beta-plus (β+) thalassemia) in β-globin production (6). With a reduction in β-globin, the formation of functional Hb complex is impaired and normal development of RBC is affected, leading to anemia and associated health complications. β-thalassemia is caused by more than 200 mutations in the HBB gene and are classified in to major, intermedia, and minor forms depending on clinical severities with symptoms like severe anemia, bone deformities, and organ damage (7).
Until recently, treatment options for SCD and β-thalassemia were primarily limited to palliative symptomatic managements and allogeneic hematopoietic stem cell transplant (HSCT). However, challenges like unavailability of compatible donors and immunological risks associated with allogeneic HSCT necessitate more effective solutions. As the root cause of SCD and β-thalassemia lies in mutations in the HBB gene, employing genetic approaches targeting the mutant HBB gene present the most promising and effective approach. In 2023, the U.S. Food and Drug Administration (FDA) granted approval to two groundbreaking therapies for SCD patients: CASGEVY (exagamglogene autotemcel) and LYFGENIA (lovotibeglogene autotemcel). The approval of two SCD gene therapies represents a significant advancement, showcasing the role of innovative technologies in transforming medical treatment.
In the early embryonic development ϵ-globin is predominantly expressed in the yolk sac erythroblasts. During fetal development, fetal hemoglobin (HbF), containing γ-globin (α2γ2) instead of β-globin, is predominantly expressed. γ-globin is produced by two closely related genes, HBG1 and HBG2, which are silenced after birth. Postnatally, γ-globin is gradually replaced by β-globin (8). This switching of globin gene expression from ϵ -globin to γ-globin and then to β-globin, is governed through precise chromatin modifications, selective hypomethylation of globin gene promoter (9, 10) and expression of stage-specific transcription factors (11, 12).
The asymptomatic adult SCD patients have abnormally high levels of HbF, a condition known as hereditary persistence of fetal hemoglobin (HPFH) which disrupts the switching of HBB gene (8). With HPFH mutations, the elevated HbF in RBCs reduces the polymerization of sickle hemoglobin, resulting in minimal hemolysis and vaso-occlusive events in affected patients. Similarly, co-inheritance of genetic variants elevating HbF production has the strongest disease modifying effect in β-thalassemia with milder phenotype (13–15). Elevated γ-globin can pair with excess of α-chain, resulting in a functional HbF, thereby ameliorating the severity of β-thalassemia. Therefore, several HPFH related mutations have been identified as targets for gene-editing therapies to cure hemoglobinopathies, including BCL11A, the γ-globin transcriptional repressor, the HBS1L-MYB intergenic region, and the δ- and β-globin gene locus (16–18). Gene editing strategies for β-thalassemia and SCD aim to increase HbF levels by 30% to prevent α-chain precipitation and HbS polymerization, respectively. Achieving this threshold can significantly impact treatment efficacy (19).
Gene therapy for blood disorders entails isolating the patient’s hematopoietic stem cells (HSCs), using vectors to introduce healthy genes ex vivo, and then reintroducing these modified cells into the bloodstream to produce healthy RBCs in the bone marrow (20). Ex vivo gene editing strategies encompass globin gene addition, β-globin gene correction, and HbF upregulation. For effective treatment, it’s crucial to accurately target the genetic mutations, efficiently deliver gene-editing tools to HSCs, and minimize off-target effects to ensure the creation of healthy RBCs. The in vivo approach aiming to directly administrate genome editing reagents emerged as a promising methodology. In this review, we summarize the current state of gene therapy for β-hemoglobinopathies by highlighting recent advancements in pre-clinical research, clinical trials, and provide perspectives on the challenges in achieving desired clinical outcomes.
Genetic approaches for SCD and β-thalassemia
Genetic approaches for beta-globinopathies can broadly be classified in to three categories: a) β-globin gene addition to restore HBB function, b) genetic correction of causal mutation, and c) HbF upregulation (Figure 1). Each of these approaches are described in detail in the following sections.
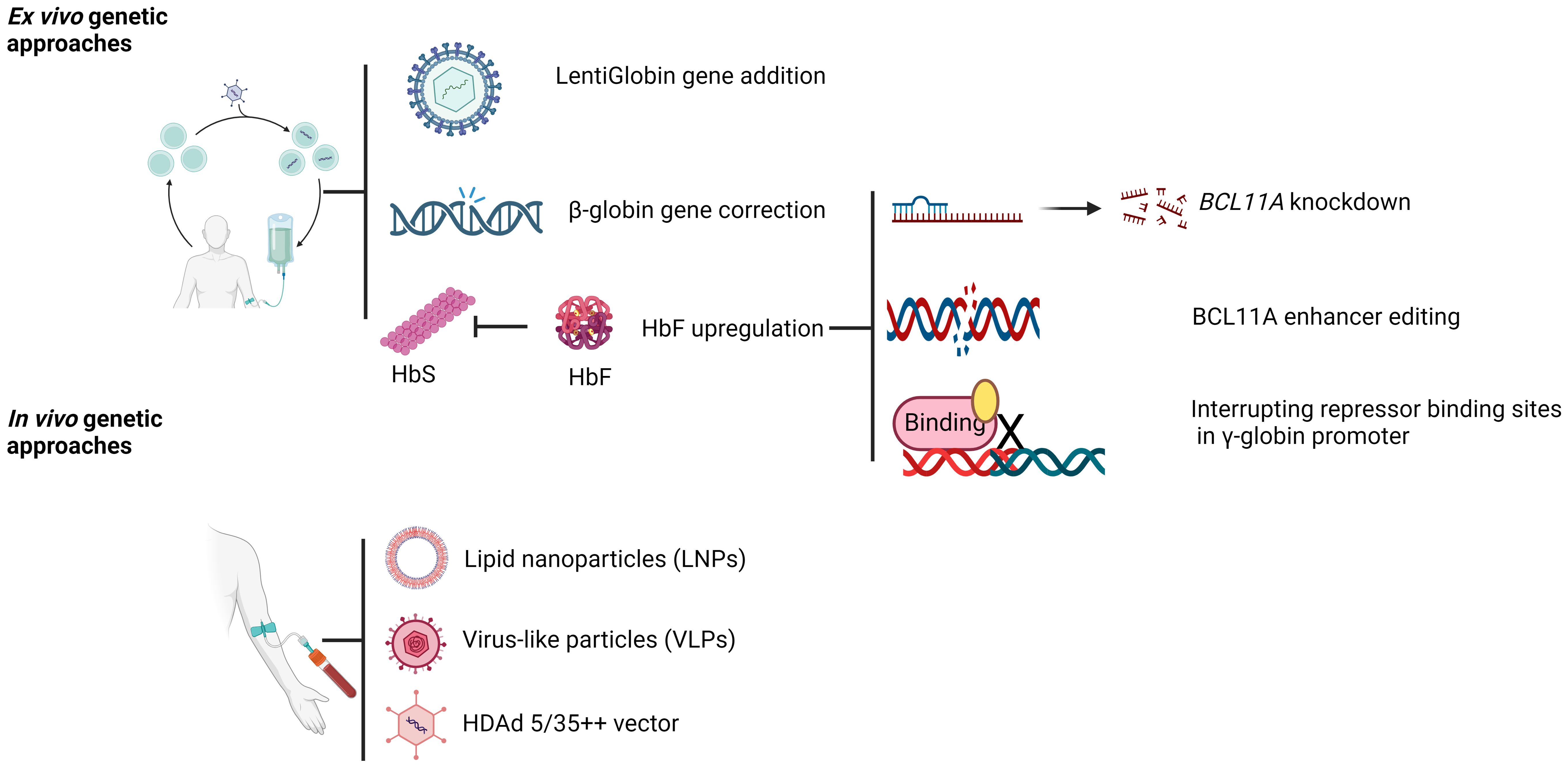
Figure 1. Gene therapy approaches for SCD and β-thalassemia. Created with BioRender.com.
β-globin gene addition
In this section, we discuss gene therapy approaches with functional β-globin gene addition. Earlier attempts to develop gene therapy approaches for SCD and β-Thalassemia co-evolved with the development of gamma-retroviral vector with the possibility of stable delivery of functional β-globin gene to hematopoietic stem and progenitor cells (HSPCs). In early 80s, Williams et al. demonstrated for the first time that genetic material can be stably transferred into murine HSPCs using a retroviral vector (21). Shortly thereafter stable production of human β-globin in mice reconstituted with human HSPCs ex vivo modified with retroviral vector encoding β-globin gene was demonstrated (22). However, in earlier studies, β-globin expression level was very low, position dependent, and dysregulated. Extensive characterization of β-globin gene locus led to the identification of distal regulatory elements located about 6 to 22 kb upstream of the ϵ-globin gene called the locus control region (LCR) (23–25), which is required for developmental stage regulated, erythroid-restricted, high level expression of globin gene. Initial attempts to incorporate LCR sub-fragments resulted in low viral titer, low β-globin expression and unstable vector. Through DNase hypersensitivity assay core regulatory regions (HS2, HS3, and HS4) within LCR were identified. Incorporation of core regulatory regions in retroviral vector generated high titer vector with elevated β-globin expression in MEL cells; however, the position-independent effect was lost due to shortening of the LCR, resulting in clonal variations in β-globin expression (26). With the development of HIV-based lentiviral vector (LV) (27), it became feasible to incorporate necessary elements of the complex β-globin gene (LCR HS elements, introns) to achieve erythroid-specific, high levels of β-globin expression. LV was very useful to achieve relatively high level of transduction in quiescent long-term hematopoietic stem cells as it can infect both dividing and non-dividing cells. The utility of such vector for gene therapy for β-globinopathies was demonstrated by successful use of the TNS9 β-globin LV in treating β-thalassemias intermedia and Cooley’s anemia in mice (28). The TNS9 incorporates essential elements of LCR (a 3.2 kb region containing HS2, HS3, and HS4), together with an intron 2 internal deletion and the β-globin gene under endogenous β-globin promoter. The TNS9 LV demonstrated high transduction efficiency into murine HSPCs. It allowed long-term, erythroid-specific expression of β-globin resulting in on an average increase of 4-6 g/dL of Hb per vector copy at steady state in peripheral blood, and rescued β-thalassemia (intermedia and major) in murine models. The TNS9 β-globin LV (TNS9.3.55) was evaluated in a Phase 1 study in adults with β-thalassemia (NCT01639690). Four patients were treated with autologous CD34+ HSPCs ex vivo modified with TNS9.3.55 globin vector. 6-8-year follow-up of these patients indicate that gene marking within hematopoietic system was very stable but low, and were able to reduce transfusion requirements in two patients (29). However, transfusion independence was not achieved due to low transduction (VCN 0.15). The TNS9.3.55 vector or its variant has been studied in three different Phase 1/2 clinical studies. The LentiGlobin BB305 vector encodes anti-sickling βA-T87Q-globin and contains both a β-globin promoter and the LCR for erythroid lineage-specific expression of βA-T87Q-globin (30). LentiGlobin BB305 was evaluated in patients with transfusion dependent β-thalassemia (TDT) for safety and efficacy in two Phase 1/2 studies HGB-204 (NCT01745120) and HGB-205 (NCT02151526) (31) (Table 1). Treatment with autologous CD34+ cells transduced with the BB305 vector reduced or eliminated the need for RBC transfusions in 22 patients with severe β-thalassemia.
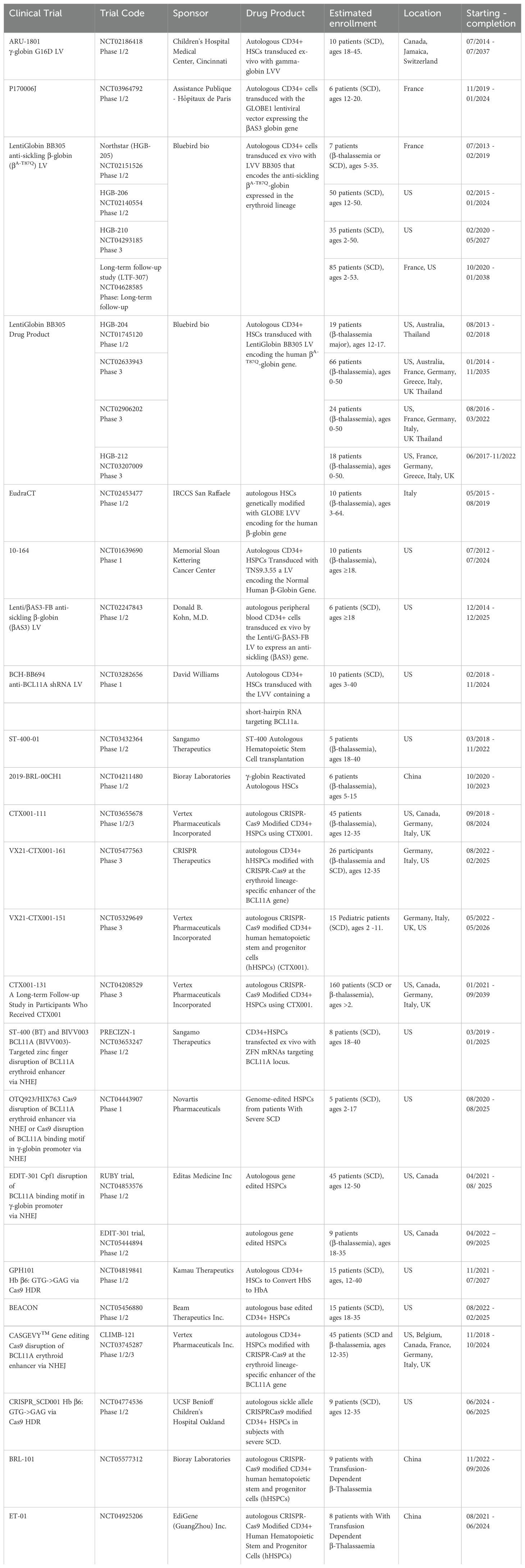
Table 1. Gene therapy clinical trials for β-hemoglobinopathies.
In another Phase 1/2 trial (TIGET-BTHAL, NCT02453477), a modified vector lacking HS4 LCR element (LV GLOBE) and encoding anti-sickling globin β-A-AS3 (described under SCD) was used to treat three adults and six children with β0 or severe β+ mutations (32). Three out of four pediatric patients discontinued RBC transfusion while transfusion frequency and volume requirement were reduced in the adults. Data from these clinical studies suggest that younger patients have better outcome from Lenti-globin gene therapy and a minimum transduction threshold of CD34+ HSPCs (VCN >0.5) is required to achieve a therapeutically meaningful outcome. Further optimization of this approach may provide a long-lasting therapeutic benefit for β-thalassemia.
Following favorable outcomes from Phase 1/2 study, two Phase 3 trials HGB-207 (Northstar-2, NCT02906202, non-β0/β0 genotypes) and HGB-212 (Northstar-3, NCT03207009, β0/β0, β0/β+ IVS-I-110 or β+ IVS-I-110/β+ IVS-I-110 genotype) were initiated to evaluate efficacy and safety of LentiGlobin BB305 in patients with TDT. In Northstar-2 study, 20 out of 22 patients had durable transfusion independence with a median HbAT87Q level of 8.7 g/dL (range, 5.2 to 10.6) at 12-month post infusion and an average hemoglobin level of 11.7 g/dL (range, 9.5 to 12.8) during transfusion independence (33). Northstar-3 study evaluated the efficacy and safety of LentiGlobin BB305 in 18 patients, 88.9% of patients achieved transfusion independence at 24 months post-transplant with a mean HbAT87Q level of 10.8 g/dL during transfusion independence (data source: clinicaltrials.gov). Based on the successful outcome from Phase 3 efficacy study, the European Commission granted BB305 (Proper name: betibeglogene autotemcel or beti-cel) conditional marketing authorization marketed as ZYNTEGLO™ gene therapy in 2019 for patients 12 years and older with TDT who do not have a β0/β0 genotype. ZYNTEGLO received the US FDA approval in 2022 for the treatment of adult and pediatric patients with β-thalassemia who require regular RBC transfusions. ZYNTEGLO is the first cell-based gene therapy marketed in the United States for the treatment of TDT. Preclinical and clinical studies have shown effectiveness of Lenti-globin gene addition approach for β-thalassemia. In patients with non- β0/β0 genotype transfusion independence was achieved in 12 out of 13 patients treated with Lenti-Globin. However, in patients with β0/β0 genotype or two copies of the IVS1-110 mutation (n=9), only three patients achieved transfusion independence while transfusion requirements were reduced for others, with a 73% reduction in the median annualized transfusion volume (31). This indicates that overall output of therapeutic β-globin together with balanced ratio of α/β-globin is critical to achieve transfusion independence in patients with β0/β0 genotype. To improve the efficacy of the Lenti-globin BB305 vector for the most severe β0/β0 genotype patients, a multiplex LV, LVβ-shα2 was developed (34) that allowed the coordinated expression of βA-T87Q-globin and selective reduction in α2-globin mRNA expression without affecting α1-globin mRNA levels. LVβ-shα2 is 1.7-fold more potent than BB305, potentially offering greater therapeutic efficacy and reliability for severe β-thalassemia irrespective of genotype (34).
Unlike β-thalassemia, ectopic expression of correct β-globin gene is not sufficient to prevent sickling of RBCs. Any therapeutic strategy will also require reduction or inhibition of HbS polymerization to prevent sickling of RBCs and resulting vaso-occlusion. G6V mutation is exposed on the surface of β-globin and can interact with α-globin even in the presence of normal β-globin forming a HbS hybrid tetramer (α2 βSβA). Several crucial observations led to the development of anti-sickling β-globin variants. Initial observation by Janet Watson showed that relatively few sickle cells were found in the circulation of newborn children with SCD (35) due to high level of HbF and its lack of sickling property. In fact, higher HbF level is positively correlated with increased life expectancy in SCD patients (36). HbF is the major genetic modulator of SCD clinical manifestations. Another line of evidence on the benefit of elevated HbF in SCD was provided by patients with compound heterozygosity for HbS and gene deletion HPFH with no symptoms of SCD or hemolytic anemia despite a very high level of HbS (37). In HPFH deletion, uniform distribution of HbF is observed among all erythrocytes and a level of 30% HbF is capable of preventing HbS polymerization (38). HPFH is a benign asymptomatic condition with continued HbF synthesis into adulthood. HbF is more potent inhibitor of HbS polymerization and very effective in preventing sickling of RBCs compared to HbA (39). The entrance of α2γβS hybrid tetramer in to HbS polymer fibers occurs at a much lower extent than α2β2S homotetramer or α2 βSβA hetrotetramer (40, 41) due to occurrence of glutamine (Q) at position 87 in γ-globin (instead of Threonine (T) in β-globin) that does not favor critical intermolecular contact with β6V. This led to the development of β-globin with T87Q substitution with anti-sickling property. LV with T87Q substituted β-globin was able to correct SCD in two transgenic mouse models (42). Long-term expression of anti-sickling protein was detected in 99% of circulating RBCs with up to 52% accumulation of anti-sickling βA-T87Q-globin of total hemoglobin. Similar to mouse models, stable expression of anti-sickling βA-T87Q-globin was achieved in erythroid progeny derived from Lenti-globin T87Q vector transduced CD34+ cells from cord blood (43), healthy donors and adult SCD patients (44), suggesting that LV expressing β-globin with T87Q substitution can be used to treat SCD in human patients. Biochemical and structural characterization studies of HbS and other globin variants indicated that the residues 22, 80, and 87 of β chain play critical roles in stabilization of the deoxy-HbS polymer through intermolecular interaction (45). Such insight led to the development of another anti-sickling globin, harboring three mutations (βAS3 with amino acid substitutions at position 16 (G16D), 22 (E22A), and 87 (T87Q) with superior anti-sickling properties than HbF (46). E22A and T87Q disrupt axial and lateral contacts between HbS tetramers, preventing HbS polymerization, whereas G16D substitution increases the binding affinity for α-globin. Oxygen binding affinity of HbAS3 is comparable with HbF. When tested in a mouse model of SCD, Lenti-βAS3 transduced HSCs were able to correct SCD-associated pathology (47).
Both iterations of anti-sickling globin have been tested for safety and efficacy in clinical trials. The Lenti-Globin BB305 vector was studied in patients with TDT and SCD for its safety and efficacy in clinical trials HGB-205 (NCT02151526). Initially one SCD patient was treated with Lenti-Globin BB305 under this trial. BB305 modified cells remained persistent 15 months post-infusion with approximately 50% of anti-sickling βA-T87Q-globin of total hemoglobin that resulted in no recurrence of sickle crises and correction of SCD hallmarks (48). In HGB-205 trial, 2 out of 3 SCD patients treated with BB305 transduced CD34+ cells achieved long-term clinical remission. In another patient, the need for transfusions was reduced (49).
A separate Phase 1/2 study (HGB-206, NCT02140554) evaluating safety and efficacy of Lenti-Globin BB305 (also known as bb1111) in approximately 50 adult and adolescent patients with severe SCD was conducted. Treatment process and outcome of this trial evolved over the study period. In the initial group (Group A), 7 patients were treated and modest level of HbAT87Q level (≥0.46 g/dL) due to lower VCN (≥0.08 c/dg) was achieved 6 months post-infusion, which were insufficient for substantial clinical benefits. Through improved cell collection and manufacturing changes, higher VCN was achieved in next group (Group B) of patients (n=2) that resulted in higher VCN (≥0.53 c/dg and ≥2.14 c/dg), higher HbAT87Q (≥2.69 g/dL and ≥6.40 g/dL), decreased hemolysis, and better clinical outcome (50). Notably, cases of acute myeloid leukemia (AML) (51) and myelodysplastic syndrome (MDS) (52) were observed in two patients treated with HGB-206, which were unlikely related to insertional oncogenesis. Though insertional mutagenesis was not the cause of AML or MDS cases observed with HGB-206, a recent published study observed an increased frequency of potential driver mutations associated with clonal hematopoiesis (such as DNMT3A and EZH2) in SCD patients (53). This suggests that pre-existing driver mutations of clonal hematopoiesis improve the fitness of HSCs post-gene therapy in SCD patients. Treatment of patients under Group C was initiated for pivotal evaluation of Lenti-Globin BB305 with changes in cell collection process (HSPC collection by plerixafor mobilization and apheresis) and increased cell dose. 36 patients were treated in Group C with modified cell collection and manufacturing process. While this study was ongoing, a Phase 3 study (HBG-210, NCT04293185) that also included pediatric patients, 12 to <18 years of age, was started to collect efficacy data. As of February 2023, 11 patients were treated in HGB-210 study. Combined data of 47 patients treated with BB305 showed median total Hb increased from 8.7 g/dL to 11.8 g/dL of which 40% or more was contributed by HbAT87Q. Complete resolution of vaso-occulusive events were observed in more than 90% of evaluable patients (n=33) (54). Furthermore, correlative analysis of drug product quality attributes and the clinical outcome suggests that post-infusion transduction efficiency is positively correlated with the reduction in hemolysis. DP VCN in the myeloid lineage was the most predictive attribute of PB VCN. Post-infusion transduction efficiency in peripheral blood is a strong predicator of biological efficacy and successful treatment outcome with Lenti-Globin (55). Clinical data from HGB-206 and HGB-210 demonstrated that a single infusion of Lenti-Globin BB305-transduced autologous CD34+ cells sustains HbAT87Q production and significantly reduces vaso-occlusive crises (54). Favorable outcomes from these studies led to the US FDA approval for the marketing of Lenti-Globin BB305 (under proper name: lovotibeglogene autotemcel or Lovo-cel, Trade name: LYFGENIA) for the treatment of patients 12 years of age or older with SCD and a history of vaso-occlusive events. Notably, LYFGENIA comes with a safety warning on the package insert as hematologic malignancy has occurred in two patients treated with LYFGENIA. The ongoing long-term follow-up study, LTF-307 (NCT04628585), will provide data on the long-term efficacy and safety of Lenti-Globin BB305 gene therapy for SCD.
The original Lenti-βAS3 vector contained LCR hypersensitive sites HS2, HS3, and HS4, produced poor viral titer, and showed low transduction efficiency (47). An optimized LV encoding β-A-AS3 developed by removing HS4 sequence (GLOBE LV) allowed production of high titer virus, efficient transduction of Plerixafor-mobilized HSPCs, and showed efficient correction of SCD phenotype in SCD patient cells ex vivo (56). Lenti-vector GLOBE1 encoding anti-sickling globin β-A-AS3 is evaluated for safety and efficacy in a Phase 1/2 study (DREPAGLOBE, NCT03964792). Data from 18 months follow-up of three patients showed variable efficacy. Two out of three patients clinically benefited from the treatment were no longer required transfusions, with HbAS3 levels correlated with their VCN. However, the 3rd patient did not show any benefit due to poor gene marking and rapid decline of genetically modified cells (57). Additional studies are required to assess the safety and efficacy of Lenti-vector GLOBE1.
γ-Globin is better than β-globin in preventing HbS polymerization and sickling of RBCs (58). LV expressing γ-Globin is also explored as therapeutic option for SCD in various pre-clinical models (59–61). The γ-Globin expression vector sGbG encodes γ-Globin under β-globin regulatory control elements. Study with sGbG vector showed that a stable, long-term upregulation of HbF can be achieved post-nataly and SCD-associated pathologies can be ameliorated with an HbF level of over 10% (60). Using the same vector, long-term, stable expression of HbF expression (12 – 15%) was achieved in non-human primates (59). This vector is currently being evaluated in a Phase 1/2 study (NCT02186418) for its safety and efficacy. Papanikolaou and colleague have developed an LCR-less, γ-Globin encoding LV GGHI incorporating -117 HPFH point mutation and the HPFH-2 enhancer and showed increased production of γ-globin by 32.9% (62). GGHI vector was further optimized utilizing 3D enhancer element of the naturally occurring HPFH-1 deletion together with the β-globin gene 3’ UTR (GGHI-mB-3D), which showed high and stable expression of HbF at low multiplicity of infection (63). Optimized GGHI-mB-3D vector showed significant increase in HbF level with concomitant decrease in HbS in SCD patient derived cells. Notably, GGHI and GGHI-mB-3D vectors are devoid of LCR present in Lenti-Globin vectors. LCR containing Lenti-Globin vectors are prone to genetic rearrangements and have lower viral titer (64, 65). LV-vector encoding γ-Globin may offer better safety and higher efficacy. However, safety and efficacy of such vectors need to be evaluated in clinical studies. Additional information on β-globin gene addition can be found in previously published reviews (66–69).
β-globin gene correction
Converting the SCD mutant codon GTG (encoding Valine) to the normal GAG (encoding Glutamic acid) in the β-globin gene is seen as an ideal approach for gene correction. Gene editing utilizes specific endonucleases, cellular repair mechanisms, and delivery methods for precise DNA changes (16). Site-specific DNA double-strand breaks (DSBs) created by engineered nucleases like ZFNs, TALENs, and CRISPR/Cas9 are repaired by cellular DNA repair machinery (such as nonhomologous end joining (NHEJ), homologous recombination (HR), homology-directed repair (HDR), mis-match repair (MMR) (70). NHEJ is the major repair pathway active throughout cell cycle (71). The erroneous NHEJ repair outcomes resulting from the incorporation of small insertion and deletion at the nuclease-induced DSB sites has been exploited for efficient gene disruption and has led to development of novel therapeutics (72). HDR, active in the late S or G2 phase, enables precise DNA repair or mutation correction using a homologous template (73). HBB gene correction in patient-derived HSCs followed by transplantation of HBB-corrected cells has the potential to provide long-term cure for SCD. Hoban et al. utilized ZFNs for correction of sickle mutation in the β-globin gene (74). Co-delivery of ZFN targeting β-globin gene together with homologous donor template (delivered as oligonucleotide or integrase-defective LV with wild-type sequence resulted in HDR-mediated correction of sickle mutation. Despite achieving high levels of gene correction in 10-20% of cells in vitro, the persistence of gene corrected human cells 16 weeks post transplantation was very low, suggesting that engagement of HDR pathway in long-term HSCs is infrequent (74), given HSCs are largely quiescent (75).
The CRISPR/Cas9 technology as an innovative genome editing tool not only simplified genome editing process but also proven to be highly effective across biological systems (76–80). Highly efficient ablation of genes in human HSCs with minimal off-target mutagenesis provided the framework for therapeutic genome editing in human HSCs and other primary cells (81). Delivery of Cas9 ribonucleoprotein (RNP) together with Adeno-associated virus (AAV) donor improved targeted gene knock-in efficiency by up to 12-fold (82). Of note, AAV is highly recombinogenic and exhibit a propensity of integration at the DSB sites in the genome, thus is a suitable HDR donor template carrier for targeted insertion (83). In addition, incorporation of chemical modifications in synthetic gRNA enhanced genome editing efficiency in human primary T cells and CD34+ HSPCs (84). Utilizing Cas9 protein complexed with chemically modified synthetic gRNA as RNP and AAV vector delivery of HDR template, Dever and colleague achieved efficient HDR at HBB locus with an average correction of 50% E6V mutation in SCD patient-derived CD34+ HSPCs (85). Significant reduction in cytotoxicity and off-target cleavage activity with the use of the RNP system was observed. Further optimization of this approach led to the successful correction of the HBB gene in SCD patient-derived HSCs. Stable production of HbA from the corrected HSCs was observed in a humanized globin-cluster SCD mouse model (85). Notably, robust HbA levels were achieved even with low chimerism levels, indicating effective gene correction and functional recovery of the HSCs (86). Another study employed a ribonucleoprotein (RNP) complex containing Cas9 protein and unmodified sgRNA, along with a single-stranded DNA oligonucleotide donor (ssODN), to correct the SCD mutation (87). The edited HSPCs showed a decrease in sickle hemoglobin RNA and protein levels, and an increase in wild-type hemoglobin upon differentiation into erythroblasts. When edited HSPCs were transplanted into immunocompromised mice, the corrected sequence at the SCD locus was retained for four months, showcasing the durability of the correction (87). Further optimization of this approach together with the use of a high-fidelity Cas9 variant led to efficient correction of >20% of HBB alleles (~30% cells with at least one corrected allele) in the long-term repopulating HSCs with minimal off-target effects (88). In addition, an enrichment of corrected β-globin allele was observed in erythroid lineage upon in vivo differentiation, suggesting selective survival advantage to erythroblasts carrying one or more corrected alleles. These studies demonstrated correcting SCD-causing mutation (E6V) in the HBB gene in patient-derived HSPCs using CRISPR/Cas9 could be useful in clinical settings.
Park et al. utilized a high-fidelity SpyCas9 variant alongside a single-stranded oligonucleotide template for correcting the sickle cell mutation in SCD patient-derived HSPCs from peripheral blood or bone marrow with 24.5 ± 7.6% efficiency without selection. Significant reduction in sickle cells was observed together with increased levels of normal hemoglobin (HbA) (25.3 ± 13.9%) (89). Using SpyFi Cas9 significantly reduced off-target effects including chromosomal rearrangement but retained on-target specificity. Gene corrected SCD HSPCs were able to engraft and persist for long-term in transplanted mice. The rate of HDR remained unchanged compared to the pre-transplantation levels for peripheral blood derived CD34+ HSPCs from SCD patients. However, a reduction in gene corrected cells was observed in mice transplanted with gene corrected HSPCs obtained from bone marrow of SCD patients. In plerixafor-mobilized CD34+ HSPCs from both healthy donor and SCD patients, up to 60% allelic correction at HBB locus was achieved in a pre-clinical feasibility study at a clinical-scale manufacturing (90). In transplanted NSG mice, multilineage engraftment was observed with long-term persistence of 20% gene correction cells. Comprehensive long-term studies demonstrated no drug product relevant adverse effects, indicating the safety, effectiveness, and consistency of this approach (90). High levels of HDR are relatively challenging to achieve compared to gene deletion in primary cells. However, recently published studies indicate that a therapeutically meaningful HBB gene correction is achievable through process optimization (88, 90). HBB gene correction approach is currently being evaluated in ongoing Phase 1/2 studies for safety and efficacy (NCT04819841; RESTORE trial, and NCT04774536) for the treatment of SCD. Clinical studies with HBB gene corrected HSPCs will provide data on safety, efficacy, and durability, of this approach. Though majority of recently developed HBB gene correction approaches utilized CRISPR/Cas9, a GMP-compatible TALEN-mediated gene editing method using viral or non-viral donor template was shown to correct HBB gene, resulting in over 50% of normal globin expression in RBCs without inducing β-thalassemic phenotype (91).
Gene correction approach described above requires a donor template (either AAV, or ssODN) to execute gene repair. From therapeutic development point of view, it requires manufacturing and controls of an additional critical component - the donor template. In addition, there are safety issues with unintentional integration of donor template within genome other than intended β-globin site. Gene editing approaches independent of a donor template is highly desirable for therapeutic gene correction. Such approaches may have better safety and efficacy profile. To this end, novel gene editing tools were created by tethering various enzymatic domains to the mutated Cas9 variants which retain DNA binding activity but are devoid of DSB nuclease activity (nickase or enzymatically dead Cas9) (92). Base editors (BEs) contain active domain from a nucleotide deaminase enzyme (Cytidine or Adenine deaminases) fused with Cas9 nickase. Cytosine BEs (CBEs) contains catalytic domain from cytidine deaminases (APOBEC1) and Uracil glycosylase inhibitor (UGI) that execute direct conversion of C to U thereby imparting C to T (or G to A) substitution at the targeted site (93). Adenine BEs (ABEs) contain adenosine deaminase domain from tRNA-specific deaminase TadA engineered through directed evolution that convert A to G at the target site (94). Both ABEs and CBEs generate nucleotide substitution and have comparable genome editing efficiency to wild-type Cas9. However, BEs produce significantly lower DSB, genomic deletion/translocation, p53 activation and p53-dependent response, and inflammation pathways (95) compared to wild-type Cas9 (96, 97) and may have lower genotoxicities.
Using a custom ABE (ABE8e-NRCH), Newby and colleague converted the SCD allele (HBBS) into a non-pathogenic Makassar β-globin (HBBG) with high efficiency in HSPCs from SCD patients (98). 80% conversion of HBBS to HBBG was observed ex vivo. When transplanted into the mice, 68% of converted allele persisted long term and led to production of non-sickling RBSs. Compared to Lenti-globin approach, greater reduction in βS was observed in erythroid cells. In contrast to Cas9, BE did not activate p53-dependent DNA damage response in HSPCs. In a humanized sickle cell mouse model, transplantation of BE-treated HSCs showed 80% of total globin contributed by Makassar β-globin. Persistence of converted alleles in secondary transplanted mice suggests efficient BE-mediated editing in long term repopulating HSCs. Using the structural information of Cas9 and TadA, Chu and colleague rationally designed a set of inlaid base editors (IBEs) with deaminase domain imbedded within Cas9 (99). Some of these IBEs exhibited shifted editing windows, greater editing efficiency with reduced DNA and RNA off-target editing activity and efficiently converted the pathogenic SCD mutation to a non-pathogenic, naturally occurring Makassar (HBBG) variant. Given the precise editing without creating DSB, BEs/IBEs will likely lead to development of safe and effective autologous treatment for SCD.
By fusing an engineered reverse transcriptase (RT) domain to nickase Cas9 Liu and co-workers developed Prime Editors (PEs) (100). Together with prime editing guide RNA (pegRNA), which specifies target site specificity and contains desired complimentary sequence for genetic modification, PE can introduce targeted nucleotide insertions, deletions, and substitutions (transitions or transversions) independent of a donor repair template and without introducing DSB (101). BEs can only make six of the 12 possible point mutation and can introduce nucleotide substitutions within a target window. Same nucleotide occurring within target window are also modified by BE, introducing undesirable bystander editing within target window. BE activity is limited by presence of a protospacer adjacent motif (PAM) sequence at a specific distance range (typically 15 ± 2 nucleotide). By overcoming multiple limitations associated with other gene editing tools, PEs increases the scope of genome editing and can correct majority of known disease-causing genetic variants. Using an engineered pegRNA (epegRNA)( (102) and an improved PE (103), Everette et al. corrected the sickle cell allele (HBBS) to wild-type (HBBA) in HSPCs from SCD patients with more than 20% correction frequency (104). When the PE-edited HSPCs were transplanted into mice, they successfully engrafted and produced normal, functional RBCs, effectively alleviating the symptoms of SCD in the animal model. PE HSPCs retained their differentiation potential, with 30-45% HbA in erythroid cells (104). Reticulocytes from mice transplanted with PE-treated HSPCs showed a significant reduction in sickling compared to untreated controls (37% vs 63%). In vivo prime editing of HSC in a sickle cell mouse model was recently reported (discussed below) (105).
Due to the genetic heterogeneity, developing β-globin gene correction approaches for β-thalassemia is comparatively challenging and will require specific guide RNAs and donor DNA templates tailored to unique mutation (6). Aberrant splicing of HBB observed with several of the most common HBB mutant alleles causes loss of β-globin expression. IVS1-110G>A (HBB:c.93-21G>A, rs35004220) is one of the most prevalent and severe β-thalassemia causing mutations (106) that generates a de novo splice acceptor site in HBB intron-1. Aberrantly spliced HBB mRNA resulting from 110G>A mutation contains pre-mature stop codon that leads to truncated β-globin expression (107). IVS2-654C>T (HBB:c.316-197C>T, rs34451549) is commonly occurring HBB mutation in East Asia that creates a de novo splice donor site in HBB intron-2. Disrupting these aberrant splice sites or targeted degradation of aberrant mRNA may restore normal splicing of HBB gene and β-globin expression respectively. shRNA-targeted degradation of aberrant spliced mRNA with 110G>A restored β-globin levels in a novel cell model and primary HSPC (108). Several genome-editing based approaches have demonstrated that disruption of aberrant splice site resulting from 110G>A mutation restores normal splicing and β-globin expression in β-thalassemia patient-derived CD34+ HSPCs with very high efficiency (109–111). A universal approach of knocking-in a HBB cDNA in exon 1 of HBB gene was tested to correct different HBB gene mutations in two TDT patients-derived iPSC cell lines (6). HBB protein production was restored in the erythroid cells derived from corrected iPSCs. Though this study provided an universal approach for HBB gene correction, utility of this approach for the treatment of SCD and β-thalassemia is yet to be proven. As the severity of β-thalassemia often correlates with the number of inherited α-globin genes (HBA1 and HBA2) and imbalance of α- and β-globin, deletion of the α-globin (HBA1 or HBA2) gene together with targeted insertion of a β-globin transgene was shown to restore α/β globin imbalance in thalassemic HSPCs-derived erythroblasts (112, 113). This knock-out, knock-in approach could be developed as a novel therapeutic to restore globin homeostasis in β-thalassemia patients.
HbF upregulation
Elevated HbF level is positively correlated with milder SCD manifestation and a level of 30% HbF is capable of preventing HbS polymerization (38). HbF-containing RBCs are called F-cells which survive much longer than non-F cells (114). HbF level of >8.6% was associated with improved survival of SCD patients (36). Therefore, reactivation of the γ-globin genes (HBG1/2) is the key to increasing the production of HbF, which may alleviate the symptoms of severe β-globin disorders. The asymptomatic adult SCD patients have abnormally high levels of HbF, a condition known as hereditary persistence of fetal hemoglobin (HPFH), which disrupts the switching of globin gene (8). Several HPFH mutations were identified that elevated HbF in RBCs resulting in reduced polymerization of sickle hemoglobin, minimal hemolysis and vaso-occlusive events in affected patients. Elevated level of HbF also ameliorates β-thalassemia severity. Genome-wide association studies (GWAS) identified three quantitative trait loci (QTLs) (BCL11A at 2p15, HBS1L-MYB intergenic region at 6q23 and XmnI-Gγ at 11p16) containing single-nucleotide polymorphisms (SNPs) that resulted in elevated level of HbF (115–120). Single-nucleotide variants (SNVs) and small deletions were identified in the proximal promoter of the HBG1 and HBG2 gene that resulted in upregulation of HbF levels (121). Mechanistic studies with these variants, provided insights on globin gene regulation (11, 122–129) that could be exploited to develop therapeutic strategies for SCD and β-thalassemia by targeting the repressors of γ-globin gene or their binding sites in the promoter region of γ-globin gene. BCL11A is a principal repressor of γ-globin gene activity in adult RBCs and is responsible for more than half of the postnatal suppression of γ-globin gene expression (128, 130, 131). Targeting γ-globin gene repressors, specifically BCL11A and ZBTB7A/LRF, to enhance HbF production emerged as a promising therapeutic approach (18, 128, 130, 131). The BCL11A gene is a preferred target because it specifically influences γ-globin expression without impacting other essential genes (132). Strategies for upregulating HbF include knocking down the BCL11A gene, disrupting its enhancer, or interfering with BCL11A binding sites on the γ-globin promoter (described below).
Knocking down BCL11A gene
In primary adult erythroid cells, BCL11A binds to various sites in β-globin gene cluster. Suppressing BCL11A boosts γ-globin production, leading to a rise in HbF (131). However, Bcl11a is also essential for postnatal development, normal lymphopoiesis, and may act as a T-cell tumor suppressor (133, 134). BCL11A deletion is associated with persistent HbF and neurodevelopmental disorders (122). Ubiquitous knock-down of BCL11A may adversely affects the long-term engraftment of edited cells (135). Erythroid lineage-specific silencing of BCL11A circumvented engraftment impairment observed with ubiquitous knockdown and led to 90% reduction in BCL11A in erythroid lineage (135) and increased γ-chain expression with concomitant elevation in HbF levels. Transplantation of HSCs transduced with LV-LCR-shRNAmiR-BCL11A into Berkeley SCD mouse models demonstrated a significant alleviation of disease symptoms (135). A refined version of this vector (called as BCH-BB694) showed 3- to 5-fold induction of HbF without affecting growth, differentiation, and engraftment potential of genetically modified CD34+ HSPCs in a pre-clinical study (136). In the ongoing clinical study (NCT03282656), as of October 2020, 6 patients were treated with BCH-BB694 with a median follow-up for 18 months. All treated patients showed robust and stable HbF induction with 9.0 to 18.6 pg HbF per F-cell, resulting in reduced or absent SCD clinical manifestations (137). This study demonstrated a favorable risk-benefit profile of BCH-BB694 in patients with SCD. However, an increased frequency of driver mutations (mutations in DNMT3A and EZH2) of clonal hematopoiesis was observed at 3.5 to 5 years of follow-up in these patients (53). Following gene therapy, an increase in total number of mutations per HSPC was observed in four of the six patients compared to the baseline mutation burden. Although, there was no unique clonal expansions larger than 1% observed post-gene theapy, findings from this study suggest that gene therapy related procedure allows selection of HSCs with pre-existing mutations in genes associated with clonal hematopoiesis (such as DNMT3A and EZH2) which may “imporve” fitness of HSC post-gene therapy in SCD patients. Therefore, a long-term follow-up post-gene therapy is required to monitor clonal evolution and expansion of mutated HSCs. A recent study introduced a novel LV that co-expresses two short hairpin microRNAs (shmiRs) targeting BCL11A and ZNF410. This dual-targeting strategy yielded up to a 70% reduction in targeted protein levels in erythroid cells and a synergistic increase in HbF levels by 10% compared to targeting BCL11A alone (138). When applied to erythroid cells from SCD patients, this approach markedly reduced the in vitro sickling phenotype, presenting a promising dual-target strategy for the treatment of β-hemoglobinopathies (138). These studies underscore the therapeutic potential of precise, lineage-specific genetic modifications which may offer a more refined approach to treating hemoglobinopathies.
Disrupting BCL11A enhancer
Common genetic variations associated with HbF level are located in the intron 2 of BCL11A with an erythroid-specific enhancer signature. Three erythroid-specific DNase Hypersensitive (DHS) sites named as DHS +62, +58, and +55 (based on their location) were identified in primary erythroid cell, which were not detected in other cell types (123). Furthermore, these sites were occupied by erythroid-specific transcription factors GATA1 and TAL1. Deletion of orthologous enhancer elements in mouse leukemia MEL cell line resulted in reduced expression of BCL11A and de-repression of γ-globin. Further interrogation of these sites through in situ saturating mutagenesis (124) using pooled sgRNA library led to the identification of +58 site as the most critical regulator of BCL11A. CRISPR-mediated deletion of +58 site in human CD34+ HSPCs led to the downregulation of BCL11A, upregulation of γ-globin, and increase in HbF in erythroid progeny derived from edited HSPCs. This suggests that disruption of critical sequence within +58 site within BCL11A enhancer with a single gRNA could be used to elevate HbF level as a therapeutic approach for β-globinopathies without impacting non-erythroid functions of BCL11A (such as lymphopoiesis) (133). Cas9:sgRNA ribonucleoprotein (RNP)-mediated disruption of GATA1 binding site at the +58 BCL11A erythroid-specific enhancer resulted in BCL11A downregulation and induction of HbF in erythroid cells differentiated from edited SCD and β-Thalassemia patient-derived HSPCs (139). Sickling of erythroid cells, differentiated from edited SCD-patient derived HSPCs, was prevented and globin chain homeostasis was restored in erythroid cells derived from edited β-Thalassemia patient-derived HSPCs. BCL11A enhancer edited HSCs persisted for more than 101 weeks and biallelic BCL11A enhancer editing resulted in robust HbF induction in rhesus monkey without any toxicity (140). Similar results were obtained with ZFN-mediated bi-allelic disruption of GATAA motif within erythroid-specific enhancer in human bone marrow derived CD34+ HSPCs (141). GATAA motif disruption resulted in upregulation of HbF without impairing HSPC functions or erythroid enucleation. However, biallelic disruption of BCL11A through exon 2 targeting led to erythroid enucleation impairment and poorer engraftment of edited HSPCs in mice (141). Another study reported erythroid-specific BCL11A enhancer editing in β-Thalassemia major patient-derived HSPCs using ZFN, showing BCL11A enhancer deletion resulted in increased γ-globin expression (132).
Encouraging results from per-clinical studies described above led to the translation of BCL11A enhancer editing approaches into the clinic. ZFN-mediated disruption of the GATA-binding region within erythroid-specific BCL11A enhancer is currently studied in a Phase 1/2 clinical trial, addressing safety and efficacy of this approach for TDT (ST-400, Thales trial, NCT03432364) (142). ST-400 is an ex vivo, ZFN-edited autologous HSC product for the treatment of TDT. At the time of interim reporting, 5 patients treated with ST-400 showed hematopoietic reconstitution. However, HbF level were not sustained (declined by 64% from peak level) to achieve transfusion independence. This decline in HbF level is presumably due to low levels of edited long-term HSCs in the ST-400. An analogous drug product SAR445136 (also known as BIVV003) is also being developed for SCD (NCT03653247, PRECIZN-1) (143, 144). Four patients treated with BIVV003 showed no SCD-related complications, with increased total hemoglobin and HbF along with improved clinical scores (143).
CRISPR/Cas9-mediated BCL11A enhancer deletion approach for SCD and β-Thalassemia (139) were evaluated for safety and efficacy under two clinical studies (CTX-001, NCT03655678; CLIMB THAL-111 and NCT03745287; CLIMB SCD-121). Initially, two patients — one with TDT and the other with SCD were treated with CRISPR-Cas9 edited autologous CD34+ HSPCs (CTX-001) (145). High levels of BCL11A enhancer editing were achieved in bone marrow and blood of both the patients with increased HbF. Transfusion independence and elimination of vaso-occlusive episodes were achieved in TDT and SCD patient respectively. Data from the pivotal CLIMB SCD-121 trial showed that a single dose of CTX-001 (Proper Name: Exagamglogene autotemcel or exa-cel) in first 31 patients aged 12 to 35 years with sever SCD led to upregulation of HbF >20% and all the treated patients were VOC-free (146). A phase 3 study was conducted and a total of 44 patients received exa-cel under CLIMB SCD-121 trial. Of the 30 patients with sufficient follwo-up, 29 were free from VOC for at least 12 consecutive months and all 30 patients were free from hospitalizations for VOC for at least 12 consecutive months (147). This study met its primary endpoint and both key secondary endpoints with 97% of patients free from VOC for at least 12 months and 100% were free from hospitalization. Exa-cel (Proprietary name: CASGEVY™) received FDA approval on December 8, 2023 for the treatment of SCD in patients 12 years of age or older with recurrent VOCs. exa-cel was also studied for its efficacy and safety in TDT patients in a Phase 3 trial (148). A total of 52 TDT patients received single infusion of exa-cel. Transfusion independence was achieved in 91% of patients and red cell transfusions were stopped at a mean of 35.2 ± 18.5 days after the exa-cel infusion. CASGEVY™ was approved by the FDA on January 16, 2024, for the treatment of patients aged 12 years and older with TDT. A long-term follow-up study continues to monitor hemoglobin levels, safety, potential secondary cancers, vaso-occlusive events, and patient-reported outcomes (CLIMB-131, NCT04208529). Several ongoing studies are evaluating CRISPR/Cas9-mediated BCL11A enhancer editing in autologous CD34+ HSPCs for SCD and TDT (NCT04211480, NCT05577312, NCT04211480, NCT04205435, NCT04925206) (149, 150) with encouraging results. Furthermore, editing the BCL11A enhancer at both +58 and +55 sites leads to disruption of alleles with high efficiency and specificity, resulting in targeted downregulation of BCL11A in erythroid cells, and a significant increase in HbF production (151). Taken together, these studies underscore the efficacy and safety of CRISPR/Cas9-mediated targeting of BCL11A enhancer in treating β-hemoglobinopathies.
Newly developed CRISPR tools such as BEs and PEs were also used to edit BCL11A enhancer or BCL11A binding sites in HBG1/HBG2 promoter (described in the following section) with high efficiency. Utilizing ABE8e base editor, a recent study enhanced HbF production either by targeting the BCL11A enhancer or the HBG promoter in the CD34+ HSPCs from β-thalassemia patients (152). Additionally, ABE8e-SpRY, a modified version of ABE designed for extensive targeting range, was used to directly correct HbE and IVS II-654 mutations in CD34+ HSPCs from patients. Sustained therapeutic modifications were validated in self-renewing, repopulating human HSCs. These findings underscore the efficacy of ABE-mediated base editing in HSCs as a promising therapeutic approach for SCD and TDT (152).
Recent studies have identified additional strategies to modulate BCL11A gene function. The gene HIC2 is a key regulator influencing hemoglobin switching, it represses BCL11A transcription by binding to erythroid BCL11A enhancers, thereby reducing chromatin accessibility and GATA1 transcription factor binding, leading to decreased enhancer activity. Overexpressing HIC2 in SCD patient-derived HSPCs improved HBG1/2 mRNA levels and inhibited sickling (153).
Interrupting repressor binding sites in γ-globin promoter
HPHF is a benign condition with elevated HbF production into adulthood often caused by deletions or point mutations in the HBB gene or the promoter region of HBG genes. Targeting regions encompassing the δ- and β-globin genes in HSPCs from patients with SCD has significantly boosted HbF synthesis in adult erythroblasts, ameliorating the sickle cell phenotype (154). In SCD patients with HPFH, natural point mutations in the HBG promoter led to a shift from producing faulty adult β-globin to fetal γ-globin that reduces the severity of the disease. Recent studies identified more HPFH variants, including GATA1, KLF1, or TAL1, which serve as new binding sites to reactivate HbF for β-Hemoglobinopathies (155, 156). Reactivation of γ-globin gene through recreating HPFH mutations that disrupt repressor binding sites could be used therapeutically to mitigate β-hemoglobinopathies. In a study utilizing CRISPR-Cas9 technology, HPFH-like mutations were mimicked in the HBG promoters through the introduction of insertions and deletions (157). These modifications disrupted the repressor binding sites for BCL11A and LRF, with particular emphasis on the LRF-binding site in patient-derived HSPCs. This genetic editing enhanced γ-globin production and ameliorated the sickling phenotype in these cells. Notably, the HSPCs which were modified at the LRF-binding site exhibited effective gene editing and demonstrated efficient repopulation after being transplanted into a xenograft model (157).
Editing specific DNA sequences in the HBG1/HBG2 gene promoters increased HbF production in erythroid cells, maintaining cell multipotency and lineage differentiation for extended periods without detectable off-target effects (158, 159). EDIT-301 is a gene editing product under investigation for the treatment of severe SCD and TDT. It consists of patient-derived CD34+ HSPCs edited at the γ-globin gene promoters HBG1/2. RBCs derived from EDIT-301 cells demonstrate a sustained increase in HbF. These data support two clinical trials. The ongoing RUBY trial (NCT04853576) is evaluating the efficacy and safety of EDIT-301 in adult and adolescent participants with severe SCD. In this study, 4 patients received a single administration of EDIT-301 (160). Preliminary data demonstrated successful engraftment of edited cells, sustained increase in total Hb, HbF, and F cells. The ongoing EDIT-301 trial (NCT05444894) is to evaluate the safety, tolerability, and efficacy of EDIT-301 in adult participants with TDT.
OTQ923 is another gene therapy product comprised of CD34+ HSPCs that were edited using CRISPR-Cas9 targeting the HBG1/2 promoters. Preclinical experiments showed sustained on-target editing without off-target mutations and elevated HbF levels in edited CD34+ HSPCs. A clinical study (NCT04443907) involving severe SCD patients assessed its safety and efficacy. Three study participants receiving OTQ923 experienced successful engraftment and a significant induction of HbF, resulting in a reduction of disease manifestations (161).
The heme-regulated eIF2α kinase (HRI, also known as EIF2AK1), regulates hemoglobin levels in SCD patients. Reducing HRI levels can increase HbF, and in turn, decrease the sickling of erythroid cells in culture, suggesting that HRI could be a potential target for increasing HbF levels and reducing the complications of SCD (162). BCL11A reduction is primarily accounted for the HbF increase following HRI depletion (162).
New regulatory elements have recently been identified as targets for enhancing HbF. A study showed that editing the -123 and -124 positions in the HBG promoter of erythroblasts from human CD34+ HSPCs led to a notable increase in HbF levels, surpassing the results from disrupting the BCL11A site. The introduction of HPFH-like mutations enhances γ-globin production, facilitated by the new binding site for the KLF1 protein, offering a potent method to augment HbF in patients (163). Another study identified a core-regulatory region known as PRR-bE1, an 11-kb core-regulatory region spanning from the putative repressor region to β-globin exon-1. Editing the PRR-bE1 region effectively shifted globin expression from β-globin to γ-globin and reversed β-hemoglobinopathy phenotypes (164).
A recent published study compared Cas9 and ABE and reported that compared to Cas9-generated indels at erythroid enhancer, ABE was more potent, with –175A>G causing the strongest induction of HbF (19). Utilizing a non-integrating, helper-dependent human CD46 targeting adenoviral vector (HDAd5/35++), Li and colleague delivered CRISPR/Cas9 (HDAd-HBG-CRISPR) in to HSC of β-YAC mice to disrupt a BCL11A binding site in the HBG1/2 promoter that resulted in reactivation of γ-globin in red blood cells of adult mice (165). Further refining their approach, they developed BE vectors to target BCL11A binding site in the HBG1/2 promoter, mimicking HPFH mutations (166). The BE effectively reactivated γ-globin in human erythroid cells. In vivo experiments showed significant γ-globin+ erythrocyte levels and confirmed the targeted genetic modification’s stability and safety, highlighting the potential of this base editing approach for treating β-hemoglobinopathies (166). Most importantly, use of BE resulted in marked reduction in intergenic HBG1/2 deletion and detectable genotoxicity. A phase 1/2 trial (NCT05456880) is exploring the use of ABE to mimic HPFH variants in SCD patients. Furthermore, multiplex editing combining BCL11A enhancer deletion together with disruption of LRF or BCL11A binding sites within HBG promoter region (-115 and -197 sites) resulted in significantly higher levels of γ-globin expression in thalassemic CD34+ cells (167). Utilizing this dual editing approach, Psatha and colleague further showed successful editing in thalassemic CD34+ cells in vivo that led to induction of γ-globin and pancellular HbF expression in a thalessemic xenotransplantation model with their adenoviral vector platform (HD-Ad-dualCRISPR) (167). HPFH related mutations are clustered within -200 to – 115 nucleotides upstream of the HBG1/2 transcriptional start sites. Using CBEs and ABEs, the dissection the -200 region of the HBG1/2 promoter showed that creation of novel KLF1 activator binding sites in this region are the most potent inducer of γ-globin, resulting in higher levels of HbF compared to disruption of BCL11A binding site (163, 168).
Taken together, gene therapy for SCD and β-thalassemia have advanced notably, with strategies like Lenti-globin gene addition, β-globin gene correction, BCL11A manipulation, and base editing showing promising results, backed by clinical successes and regulatory approvals. Ongoing research is expected to improve the safety and efficacy of these gene therapy treatments.
In vivo gene editing in HSCs for β-globinopathies
Ex vivo gene therapy (GT) and gene editing (GE) approaches developed thus far are complex and expensive (169). Ex vivo manufacturing of HSPCs-based therapeutics requires CD34+ HSPCs mobilization, collection of mobilized HSPCs through apheresis, shipment of apheresis material to the manufacturing facility, ex vivo manipulation of mobilized CD34+ HSPCs, harvest and formulation of genetically modified cells, it’s cryopreservation and shipment to the clinical site for infusion. Ex vivo manufacturing of HSPC products requires variety of resources, such as advanced manufacturing facility, complex logistics, and close coordination between doctors, patients, and manufacturers. Ex vivo culture of HSPCs can also result in loss of stemness and engraftment potential, which may reduce therapeutic efficacy (170, 171). In addition, successful transplantation of ex vivo modified HSPC products in patients requires myeloablative treatment, which is a high-risk procedure. HSCT remains a high-risk procedure with significant treatment related complications such as bacterial and fungal infections, toxicities, graft failure, safety issues arising from manufacturing process (insertional mutagenesis of lentiviral vectors, off-target editing, and myeloblative conditioning, and can result in death (172, 173). In vivo GE and GT via intravenous or intraosseous administration of GT vectors/GE reagents simplify the manufacturing process, and overcomes challenges associated with ex vivo manufacturing of HSPC-based products. Systemic in vivo delivery bypasses the need for conditioning and extensive care, and minimizes the risks associated with ex vivo methods like myeloablation, genotoxic effects. Unlike ex vivo methods, in vivo GT/GE offers a more accessible option for regions heavily burdened by β-hemoglobinopathies with shortage of advanced medical facilities and resources. Recently published reports demonstrated that therapeutically meaningful in vivo gene editing can be achieved (105, 174, 175). A recent study used a non-integrating, helper-dependent adenoviral (HDAd) vector (HDAd5/35++) with high affinity toward CD46 (a receptor that is uniformly expressed on primitive HSCs), to deliver PEs in vivo in a SCD mouse model (CD46/Townes mice) (105). HDAd5/35++ vector also allows selection of gene edited cells in vivo using a mutant O6-methylguanine DNA methyltransferase (MGMTP140K) gene that confers resistance to O6-BG/BCNU (O6-Benzylguanine/Carmustine). To overcome the physical barriers formed by the bone marrow stroma, granulocyte-colony stimulating factor (G-CSF)/plerixafor (AMD3100) were used to mobilize HSCs from bone marrow into peripheral blood. With a single intravenous injection of PE-expressing HDAd5/35++ vector, ~40% of HBBS alleles were corrected in HSCs with minimal generation of insertion/deletion and undetectable off-target editing. With this approach 43% of HbS was replaced with HbA. Efficient induction of γ-globin was also achieved in non-human primates following in vivo transduction of CD34+ HSPCs with HDAd5/35++ vector expressing γ-globin (176). Following G-CSF/AMD3100-mobilization, infusion of HDAd5/35++ vector resulted in preferential transduction of CD34+ cells and MGMTP140K-based in vivo selection of transduced cells resulted in up to 90% γ-globin+ RBCs, though a decline in γ-globin+ RBCs following selection was observed.
Non-viral delivery systems such as lipid nanoparticles (LNPs), virus-like particles (VLPs), and exosomes are also being studied as alternative to viral vectors to improve the safety and effectiveness of these gene therapies (174, 177). Systemic deliver via LNPs and VLPs are two commonly used non-viral-based delivery systems for in vivo delivery of GT/GE cargo (178, 179). LNPs are central to enhancing the delivery of genetic drugs with improved targeting capability and minimized side effects (180). For example, the LNPs targeting BM has shown the ability to genetically alter HSPCs in vivo in a humanized mouse model (175), providing a proof-of-concept for SCD treatment. A recently published report described targeted delivery of GE components to HSCs using anti-CD117 antibody conjugated LNP (174). anti-CD117/LNP were able to delivery mRNA cargos (Cre recombinase, Luciferase) to HSCs in vivo in mice. Cre-mediated recombination was detected in all the lineages in peripheral blood and bone marrow in a dose-dependent manner, suggesting targeting of HSCs. Using the same anti-CD117/LNP delivery platform, ex vivo delivery of ABEs to SCD HSPCs resulted in highly efficient (88%) conversion of HBBS to HBBG-Makassar with up to 91.7% increase in HBBG protein and a nearly complete absence of sickled RBCs in differentiated erythroid cells. These findings demonstrated that in vivo targeting of HSCs is achievable and may eventually be translated into clinics for the treatment of SCD and other monogenic blood disorders with a simple single intravenous infusion of targeted genetic medicines.
Outstanding challenges and future perspectives
Recent advances in gene therapy have revolutionized the treatment of SCD and β-thalassemia by targeting the underlying causal mutations. One time infusion of genetically modified cells may provide long-term therapeutic benefits and possibly cure SCD and β-thalassemia. Despite tremendous progress, several challenges remain for broader application of such therapies for SCD and β-thalassemia. Long-term safety and efficacy of gene therapy or gene editing approaches are primary concerns. Insertional oncogenesis from random integration of LV or editing at unintended sites in the genome could be detrimental to patients receiving such therapy. Lowering the VCN in the drug product and minimizing off-target editing by using improved Cas9 nuclease or limiting the duration of Cas9 expression will improve the safety profile of GT/GE products for SCD and β-thalassemia. High-fidelity CRISPR-Cas9 systems like HiFi Cas9, SpyCas9, SpCas9-HF1, eSpCas9, and HypaCas9 exhibit improved specificity (181). To achieve long-term therapeutic benefits, genetically modified CD34+ HSPCs must engraft and persist in the patients receiving such therapies. Ex vivo culturing and manipulation (LV transduction, gene editing) may lead to differentiation, activation of innate immune system, or activation of p53-dependent DNA damage pathways, which can adversely affect the engraftment potential of ex vivo modified CD34+ HSPCs. Improved culture conditions, better quality reagents and LV, and utilization of nucleases that do not induce DNA DSB may improve the quality of ex vivo modified CD34+ HSPCs resulting in better engraftment and persistence of these cells for long-lasting efficacy. Unlike Cas9, BEs and PEs do not generate DSBs and CD34+ cells modified with BE/PE have better engraftment potential than CD34+ cells modified with Cas9. In addition, use of milder conditioning regimen or antibody-mediated conditioning may improve the engraftment of ex vivo modified cells for better outcome.
Given the complexity of HSC-based gene therapy product manufacturing processes, scaling up the manufacturing is often challenging. Innovations in bioprocessing technologies and automation can help to improve product quality and reduce lot-to-lot variability. Modular and adaptable manufacturing processes may streamline the scalability and provide better control of critical variables that affect product quality. More effort is required to increase the accessibility of these genetic therapies to the resource limited regions of the world with the highest disease burden (182). Emerging in vivo HSPCs editing or targeted gene delivery to HSPCs would simplify the manufacturing and treatment process for wider applications.
Conclusive remarks
More than 50 years of basic research in vector biology, globin gene regulation, hemoglobin polymerization, coupled with newer technology such as CRISPR/Cas, and many years of pre-clinical and clinical research on SCD and β-thalassemia culminated with the US FDA approval of three HSC-based gene therapy products (ZYNTEGLO, LYFGENIA, and CASGEVY) for the treatment of SCD and β-thalassemia. SCD is often referred as poster child for gene therapy for monogenic disorders and along the way it helped to develop the framework for gene therapy and gene editing based therapeutics. Multiple approaches utilizing safer and effective gene editing tools (such as BEs and PEs) are currently being evaluated in pre-clinical and clinical studies with hope to develop safe, effective, and durable therapeutics, which may eventually cure SCD and β-thalassemia with a single, one-time infusion of genetically modified HSPCs. As these newer approaches correct SCD causal mutation, independent of a donor repair template and without generating DSB, correcting HBB gene mutation with BEs and PEs could be safer and more effective compared to wild-type Cas9. Recently published studies indicated that in vivo HBB gene editing is achievable in pre-clinical SCD model. Clinical translation of this approach would overcome several limitations associated with ex vivo manufacturing and transplantation related complications. In vivo editing of HBB will also simplify the treatment procedure and will likely make such therapy widely available for geographical areas where these diseases are prevalent with limited access to advanced healthcare infrastructure (5). Lessons learned with developing gene therapy products for β-globinopathies will also accelerate the development of such therapies for other monogenic blood disorders.
Author contributions
LL: Writing – original draft, Writing – review & editing. PM: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank Drs. Bharat Joshi, Sandip De, Nirjal Bhattarai, and Carolyn Laurencot of the Center for Biologics Evaluation and Research, FDA, Silver Spring, MD for critical review of the manuscript and providing helpful comments. This work was supported by the Intramural Research Program of the Center for Biologics Evaluation and Research (CBER), U.S. Food and Drug Administration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
This review article reflects the views of the authors and should not be construed to represent FDA’s views or policies. Authors declare no conflict of interest.
References
1. Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. (2011) 41:S398–405. doi: 10.1016/j.amepre.2011.09.013
2. Thomson AM, McHugh TA, Oron AP, Teply C, Lonberg N, Tella VV, et al. Global, regional, and national prevalence and mortality burden of sickle cell disease 2000-2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. (2023) 10: e585–99. doi: 10.1016/S2352-3026(23)00118-7
3. Thomas C, Lumb AB. Physiology of haemoglobin. continuing education in anaesthesia. Critical Care & Pain, vol. 12. (2012) 12: 251–6. doi: 10.1093/bjaceaccp/mks025
4. Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Internal Med. (1910) 6:517–21. doi: 10.1001/archinte.1910.00050330050003
5. Piel FB, Rees DC, DeBaun MR, Nnodu O, Ranque B, Thompson AA, et al. Defining global strategies to improve outcomes in sickle cell disease: a Lancet Haematology Commission. Lancet Haematol. (2023) 10:e633–86. doi: 10.1016/S2352-3026(23)00096-0
6. Cai L, Bai H, Mahairaki V, Gao Y, He C, Wen Y, et al. A universal approach to correct various HBB gene mutations in human stem cells for gene therapy of beta-thalassemia and sickle cell disease. Stem Cells Trans Med. (2018) 7:87–97. doi: 10.1002/sctm.17-0066
7. Sanchez-Villalobos M, Blanquer M, Moraleda JM, Salido EJ, Perez-Oliva AB. New insights into pathophysiology of β-thalassemia. Front Med. (2022) 9:880752. doi: 10.3389/fmed.2022.880752
8. Steinberg MH. Fetal hemoglobin in sickle hemoglobinopathies: high HbF genotypes and phenotypes. J Clin Med. (2020) 9:3782. doi: 10.3390/jcm9113782
9. Mavilio F, Giampaolo A, Care A, Migliaccio G, Calandrini M, Russo G, et al. Molecular mechanisms of human hemoglobin switching: selective undermethylation and expression of globin genes in embryonic, fetal, and adult erythroblasts. Proc Natl Acad Sci. (1983) 80:6907–11. doi: 10.1073/pnas.80.22.6907
10. Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harbor Perspect Med. (2013) 3:a011643. doi: 10.1101/cshperspect.a011643
11. Shen Y, Verboon JM, Zhang Y, Liu N, Kim YJ, Marglous S, et al. A unified model of human hemoglobin switching through single-cell genome editing. Nat Commun. (2021) 12:4991. doi: 10.1038/s41467-021-25298-9
12. Wilber A, Nienhuis AW, Persons DA. Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood J Am Soc Hematol. (2011) 117:3945–53. doi: 10.1182/blood-2010-11-316893
13. Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S, et al. Variants in genetic modifiers of beta-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. (2011) 96:1712–4. doi: 10.3324/haematol.2011.046748
14. Danjou F, Anni F, Perseu L, Satta S, Dessi C, Lai ME, et al. Genetic modifiers of beta-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. (2012) 97:989–93. doi: 10.3324/haematol.2011.053504
15. Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. (2012) 379:373–83. doi: 10.1016/S0140-6736(11)60283-3
16. Germino-Watnick P, Hinds M, Le A, Chu R, Liu X, Uchida N. Hematopoietic stem cell gene-addition/editing therapy in sickle cell disease. Cells. (2022) 11:1843. doi: 10.3390/cells11111843
17. Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. (2018) 173:430–442. e417. doi: 10.1016/j.cell.2018.03.016
18. Topfer SK, Feng R, Huang P, Ly LC, Martyn GE, Blobel GA, et al. Disrupting the adult globin promoter alleviates promoter competition and reactivates fetal globin gene expression. Blood J Am Soc Hematol. (2022) 139:2107–18. doi: 10.1182/blood.2021014205
19. Mayuranathan T, Newby GA, Feng R, Yao Y, Mayberry KD, Lazzarotto CR, et al. Potent and uniform fetal hemoglobin induction via base editing. Nat Genet. (2023) 55:1210–20. doi: 10.1038/s41588-023-01434-7
20. Doudna JA. The promise and challenge of therapeutic genome editing. Nature. (2020) 578:229–36. doi: 10.1038/s41586-020-1978-5
21. Williams DA, Lemischka IR, Nathan DG, Mulligan RC. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature. (1984) 310:476–80. doi: 10.1038/310476a0
22. Dzierzak EA, Papayannopoulou T, Mulligan RC. Lineage-specific expression of a human β-globin gene in murine bone marrow transplant recipients reconstituted with retrovirus-transduced stem cells. Nature. (1988) 331:35–41. doi: 10.1038/331035a0
23. Forrester WC, Takegawa S, Papayannopoulou T, Stamatoyannopoulos G, Groudine M. Evidence for a locus activation region: the formation of developmentally stable hypersensitive sites in globin-expressing hybrids. Nucleic Acids Res. (1987) 15:10159–77. doi: 10.1093/nar/15.24.10159
24. Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell. (1987) 51:975–85. doi: 10.1016/0092-8674(87)90584-8
25. Tuan D, Solomon W, Li Q, London IM. The” beta-like-globin” gene domain in human erythroid cells. Proc Natl Acad Sci. (1985) 82:6384–8. doi: 10.1073/pnas.82.19.6384
26. Sadelain M, Wang C, Antoniou M, Grosveld F, Mulligan RC. Generation of a high-titer retroviral vector capable of expressing high levels of the human beta-globin gene. Proc Natl Acad Sci. (1995) 92:6728–32. doi: 10.1073/pnas.92.15.6728
27. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. (1996) 272:263–7. doi: 10.1126/science.272.5259.263
28. May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, et al. Therapeutic haemoglobin synthesis in β-thalassaemic mice expressing lentivirus-encoded human β-globin. Nature. (2000) 406:82–6. doi: 10.1038/35017565
29. Boulad F, Maggio A, Wang X, Moi P, Acuto S, Kogel F, et al. Lentiviral globin gene therapy with reduced-intensity conditioning in adults with β-thalassemia: a phase 1 trial. Nat Med. (2022) 28:63–70. doi: 10.1038/s41591-021-01554-9
30. Negre O, Eggimann A-V, Beuzard Y, Ribeil J-A, Bourget P, Borwornpinyo S, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the βA (T87Q)-globin gene. Hum Gene Ther. (2016) 27:148–65. doi: 10.1089/hum.2016.007
31. Thompson AA, Walters MC, Kwiatkowski J, Rasko JE, Ribeil J-A, Hongeng S, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. New Engl J Med. (2018) 378:1479–93. doi: 10.1056/NEJMoa1705342
32. Marktel S, Scaramuzza S, Cicalese MP, Giglio F, Galimberti S, Lidonnici MR, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med. (2019) 25:234–41. doi: 10.1038/s41591-018-0301-6
33. Locatelli F, Thompson AA, Kwiatkowski JL, Porter JB, Thrasher AJ, Hongeng S, et al. Betibeglogene autotemcel gene therapy for non–β0/β0 genotype β-thalassemia. New Engl J Med. (2022) 386:415–27. doi: 10.1056/NEJMoa2113206
34. Nualkaew T, Sii-Felice K, Giorgi M, McColl B, Gouzil J, Glaser A, et al. Coordinated β-globin expression and α2-globin reduction in a multiplex lentiviral gene therapy vector for β-thalassemia. Mol Ther. (2021) 29:2841–53. doi: 10.1016/j.ymthe.2021.04.037
35. Watson J, Stahman AW, Bilello FP. The significance of the paucity of sickle cells in newborn Negro infants. Obstet Gynecol Survey. (1948) 3:819–20. doi: 10.1097/00006254-194812000-00022
36. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease–life expectancy and risk factors for early death. New Engl J Med. (1994) 330:1639–44. doi: 10.1056/NEJM199406093302303
37. Steinberg MH, Rodgers G. Pharmacologic modulation of fetal hemoglobin. Medicine (2001). 80:328–344. doi: 10.1097/00005792-200109000-00007
38. Ngo DA, Aygun B, Akinsheye I, Hankins JS, Bhan I, Luo HY, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. (2012) 156:259–64. doi: 10.1111/j.1365-2141.2011.08916.x
39. Bookchin RM, Nagel RL. Ligand-induced conformational dependence of hemoglobin in sickling interactions. J Mol Biol. (1971) 60:263–70. doi: 10.1016/0022-2836(71)90292-0
40. Benesch RE, Edalji R, Benesch R, Kwong S. Solubilization of hemoglobin S by other hemoglobins. Proc Natl Acad Sci. (1980) 77:5130–4. doi: 10.1073/pnas.77.9.5130
41. Goldberg MA, Husson MA, Bunn HF. Participation of hemoglobins A and F in polymerization of sickle hemoglobin. (1977) 252:3414–3421. doi: 10.1016/S0021-9258(17)40407-8
42. Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. (2001) 294:2368–71. doi: 10.1126/science.1065806
43. Imren S, Karen W, Fabry M, Pawliuk R, Tang P, Reid D, et al. High level and persistent expression of an anti-sickling beta-globin gene after lentiviral-mediated transduction of human hematopoietic stem cells. In 5. 525 B ST, STE 1900, SAN DIEGO, CA 92101 …: ACADEMIC PRESS INC ELSEVIER SCIENCE (2003) p. S406–7.
44. Oh I-H, Fabry ME, Humphries RK, Pawliuk R, Leboulch P, Hoffman R, et al. Expression of an anti-sickling β-globin in human erythroblasts derived from retrovirally transduced primitive normal and sickle cell disease hematopoietic cells. Exp Hematol. (2004) 32:461–9. doi: 10.1016/j.exphem.2004.02.001
45. Nagel RL, Bookchin RM, Johnson J, Labie D, Wajcman H, Isaac-Sodeye WA, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci. (1979) 76:670–2. doi: 10.1073/pnas.76.2.670
46. Levasseur DN, Ryan TM, Reilly MP, McCune SL, Asakura T, Townes TM. A recombinant human hemoglobin with anti-sickling properties greater than fetal hemoglobin. J Biol Chem. (2004) 279:27518–24. doi: 10.1074/jbc.M402578200
47. Levasseur DN, Ryan TM, Pawlik KM, Townes TM. Correction of a mouse model of sickle cell disease: lentiviral/antisickling β-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood. (2003) 102:4312–9. doi: 10.1182/blood-2003-04-1251
48. Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene therapy in a patient with sickle cell disease. New Engl J Med. (2017) 376:848–55. doi: 10.1056/NEJMoa1609677
49. Magrin E, Semeraro M, Hebert N, Joseph L, Magnani A, Chalumeau A, et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: the HGB-205 trial. Nat Med. (2022) 28:81–8. doi: 10.1038/s41591-021-01650-w
50. Kanter J, Thompson AA, Pierciey FJ Jr., Hsieh M, Uchida N, Leboulch P, et al. Lovo-cel gene therapy for sickle cell disease: Treatment process evolution and outcomes in the initial groups of the HGB-206 study. Am J Hematol. (2023) 98:11–22. doi: 10.1002/ajh.26741
51. Goyal S, Tisdale J, Schmidt M, Kanter J, Jaroscak J, Whitney D, et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N Engl J Med. (2022) 386:138–47. doi: 10.1056/NEJMoa2109167
52. Hsieh MM, Bonner M, Pierciey FJ, Uchida N, Rottman J, Demopoulos L, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. (2020) 4:2058–63. doi: 10.1182/bloodadvances.2019001330
53. Spencer Chapman M, Cull AH, Ciuculescu MF, Esrick EB, Mitchell E, Jung H, et al. Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease. Nat Med. (2023) 29:3175–83. doi: 10.1038/s41591-023-02636-6
54. Kanter J, Thompson AA, Kwiatkowski JL, Parikh S, Mapara MY, Rifkin-Zenenberg S, et al. Efficacy and Safety in Patients (Pts) with Sickle Cell Disease (SCD) Who Have Received Lovotibeglogene Autotemcel (Lovo-cel) Gene Therapy: Up to 60 Months of Follow-up. Transplant Cell Ther. (2024) 30:S230–1. doi: 10.1016/j.jtct.2023.12.302
55. Kinney MA, Shestopalov I, Christiansen L, Jiang H, Foos M, Elliot H, et al. Predictors of Biologic Efficacy with Lovotibeglogene Autotemcel (Lovo-cel) Gene Therapy in Patients with Sickle Cell Disease. Transplant Cell Ther. (2024) 30:S231–2. doi: 10.1016/j.jtct.2023.12.303
56. Weber L, Poletti V, Magrin E, Antoniani C, Martin S, Bayard C, et al. An optimized lentiviral vector corrects efficiently the human sickle cell disease phenotype. Mol Therapy-Methods Clin Dev. (2018) 10:268–280. doi: 10.1016/j.omtm.2018.07.012
57. Magrin E, Magnani A, Semeraro M, Hebert N, Joseph L, Leblanc O, et al. Clinical results of the Drepaglobe trial for sickle cell disease patients. Blood. (2021) 138:1854. doi: 10.1182/blood-2021-152331
58. Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood J Am Soc Hematol. (2020) 136:2392–400. doi: 10.1182/blood.2020007645
59. Kiem H-P, Arumugam P, Christopher B, Adair JE, Beard BC, Fox C, et al. Safety Of a Gamma Globin Expressing Lentivirus Vector In a Non-Human Primate Model For Gene Therapy Of Sickle Cell Disease. Blood. (2013) 122:2896. doi: 10.1182/blood.V122.21.2896.2896
60. Perumbeti A, Higashimoto T, Urbinati F, Franco R, Meiselman HJ, Witte D, et al. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood J Am Soc Hematol. (2009) 114:1174–85. doi: 10.1182/blood-2009-01-201863
61. Pestina TI, Hargrove PW, Jay D, Gray JT, Boyd KM, Persons DA. Correction of murine sickle cell disease using γ-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Mol Ther. (2009) 17:245–52. doi: 10.1038/mt.2008.259
62. Papanikolaou E, Georgomanoli M, Stamateris E, Panetsos F, Karagiorga M, Tsaftaridis P, et al. The new self-inactivating lentiviral vector for thalassemia gene therapy combining two HPFH activating elements corrects human thalassemic hematopoietic stem cells. Hum Gene Ther. (2012) 23:15–31. doi: 10.1089/hum.2011.048
63. Drakopoulou E, Georgomanoli M, Lederer CW, Panetsos F, Kleanthous M, Voskaridou E, et al. The Optimized γ-Globin Lentiviral Vector GGHI-MB-3D Leads to Nearly Therapeutic HbF Levels In Vitro in CD34+ Cells from Sickle Cell Disease Patients. Viruses. (2022) 14:2716. doi: 10.3390/v14122716
64. Magrin E, Miccio A, Cavazzana M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood J Am Soc Hematol. (2019) 134:1203–13. doi: 10.1182/blood.2019000949
65. Poletti V, Mavilio F. Designing lentiviral vectors for gene therapy of genetic diseases. Viruses. (2021) 13:1526. doi: 10.3390/v13081526
66. Cavazzana M, Antoniani C, Miccio A. Gene therapy for β-hemoglobinopathies. Mol Ther. (2017) 25:1142–54. doi: 10.1016/j.ymthe.2017.03.024
67. Sadelain M, Lisowski L, Samakoglu S, Rivella S, May C, Riviere I. Progress Toward the Genetic Treatment of the β-Thalassemias. Ann New York Acad Sci. (2005) 1054:78–91. doi: 10.1196/annals.1345.010
68. Segura E, Tam K, Fernandez BC, MacKenzie TC, Hollis R, Kohn DB. [amp]]Alpha;-Globin Lentiviral Vectors for Hematopoietic Stem Cell Gene Therapy of α-Thalassemia. Blood. (2023) 142:7131. doi: 10.1182/blood-2023-190619
69. Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discovery. (2019) 18:139–58. doi: 10.1038/s41573-018-0003-2
70. Kim H, Kim J-S. A guide to genome engineering with programmable nucleases. Nat Rev Genet. (2014) 15:321–34. doi: 10.1038/nrg3686
71. Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. (2008) 7:2902–6. doi: 10.4161/cc.7.18.6679
72. Chavez M, Chen X, Finn PB, Qi LS. Advances in CRISPR therapeutics. Nat Rev Nephrol. (2023) 19:9–22. doi: 10.1038/s41581-022-00636-2
73. Bashir S, Dang T, Rossius J, Wolf J, Kühn R. Enhancement of CRISPR-Cas9 induced precise gene editing by targeting histone H2A-K15 ubiquitination. BMC Biotechnol. (2020) 20:1–16. doi: 10.1186/s12896-020-00650-x
74. Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood J Am Soc Hematol. (2015) 125:2597–604. doi: 10.1182/blood-2014-12-615948
75. Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. (2010) 7:174–85. doi: 10.1016/j.stem.2010.06.014
76. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. (2013) 339:819–23. doi: 10.1126/science.1231143
77. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. (2014) 157:1262–78. doi: 10.1016/j.cell.2014.05.010
78. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science. (2012) 337:816–21. doi: 10.1126/science.1225829
79. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. (2013) 339:823–6. doi: 10.1126/science.1232033
80. Wang JY, Doudna JA. CRISPR technology: A decade of genome editing is only the beginning. Science. (2023) 379:eadd8643. doi: 10.1126/science.add8643
81. Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. (2014) 15:643–52. doi: 10.1016/j.stem.2014.10.004
82. Gaj T, Staahl BT, Rodrigues GM, Limsirichai P, Ekman FK, Doudna JA, et al. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. (2017) 45:e98–8. doi: 10.1093/nar/gkx154
83. Hirata R, Chamberlain J, Dong R, Russell DW. Targeted transgene insertion into human chromosomes by adeno-associated virus vectors. Nat Biotechnol. (2002) 20:735–8. doi: 10.1038/nbt0702-735
84. Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. (2015) 33:985–9. doi: 10.1038/nbt.3290
85. Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. (2016) 539:384–9. doi: 10.1038/nature20134
86. Wilkinson AC, Dever DP, Baik R, Camarena J, Hsu I, Charlesworth CT, et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat Commun. (2021) 12:686. doi: 10.1038/s41467-021-20909-x
87. DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Trans Med. (2016) 8:360ra134–360ra134. doi: 10.1126/scitranslmed.aaf9336
88. Magis W, DeWitt MA, Wyman SK, Vu JT, Heo S-J, Shao SJ, et al. High-level correction of the sickle mutation is amplified in vivo during erythroid differentiation. Iscience. (2022) 25:104374. doi: 10.1016/j.isci.2022.104374
89. Park SH, Lee CM, Dever DP, Davis TH, Camarena J, Srifa W, et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. (2019) 47:7955–72. doi: 10.1093/nar/gkz475
90. Lattanzi A, Camarena J, Lahiri P, Segal H, Srifa W, Vakulskas CA, et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci Trans Med. (2021) 13:eabf2444. doi: 10.1126/scitranslmed.abf2444
91. Moiani A, Letort G, Lizot S, Chalumeau A, Foray C, Felix T, et al. Non-viral DNA delivery and TALEN editing correct the sickle cell mutation in hematopoietic stem cells. Nat Commun. (2024) 15:4965. doi: 10.1038/s41467-024-49353-3
92. Pacesa M, Pelea O, Jinek M. Past, present, and future of CRISPR genome editing technologies. Cell. (2024) 187:1076–100. doi: 10.1016/j.cell.2024.01.042
93. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533:420–4. doi: 10.1038/nature17946
94. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature. (2017) 551:464–71. doi: 10.1038/nature24644
95. Fiumara M, Ferrari S, Omer-Javed A, Beretta S, Albano L, Canarutto D, et al. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nat Biotechnol. (2023) 42:877–891. doi: 10.1038/s41587-024-02142-1
96. Blattner G, Cavazza A, Thrasher AJ, Turchiano G. Gene editing and genotoxicity: targeting the off-targets. Front Genome Editing. (2020) 2:613252. doi: 10.3389/fgeed.2020.613252
97. Hunt JMT, Samson CA, du Rand A, Sheppard HM. Unintended CRISPR-Cas9 editing outcomes: a review of the detection and prevalence of structural variants generated by gene-editing in human cells. Hum Genet. (2023) 142:705–20. doi: 10.1007/s00439-023-02561-1
98. Newby GA, Yen JS, Woodard KJ, Mayuranathan T, Lazzarotto CR, Li Y, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. (2021) 595:295–302. doi: 10.1038/s41586-021-03609-w
99. Chu SH, Packer M, Rees H, Lam D, Yu Y, Marshall J, et al. Rationally designed base editors for precise editing of the sickle cell disease mutation. CRISPR J. (2021) 4:169–77. doi: 10.1089/crispr.2020.0144
100. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
101. Chen PJ, Liu DR. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet. (2023) 24:161–77. doi: 10.1038/s41576-022-00541-1
102. Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, et al. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol. (2022) 40:402–10. doi: 10.1038/s41587-021-01039-7
103. Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen P-F, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. (2021) 184:5635–5652. e5629. doi: 10.1016/j.cell.2021.09.018
104. Everette KA, Newby GA, Levine RM, Mayberry K, Jang Y, Mayuranathan T, et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat Biomed Eng. (2023) 7:616–628. doi: 10.1038/s41551-023-01026-0
105. Li C, Georgakopoulou A, Newby GA, Chen PJ, Everette KA, Paschoudi K, et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood J Am Soc Hematol. (2023) 141:2085–99. doi: 10.1182/blood.2022018252
106. Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M. IthaGenes: an interactive database for haemoglobin variations and epidemiology. PloS One. (2014) 9:e103020. doi: 10.1371/journal.pone.0103020
107. Busslinger M, Moschonas N, Flavell RA. Beta + thalassemia: aberrant splicing results from a single point mutation in an intron. Cell. (1981) 27:289–98. doi: 10.1016/0092-8674(81)90412-8
108. Patsali P, Papasavva P, Stephanou C, Christou S, Sitarou M, Antoniou MN, et al. Short-hairpin RNA against aberrant HBB(IVSI-110(G>A)) mRNA restores beta-globin levels in a novel cell model and acts as mono- and combination therapy for beta-thalassemia in primary hematopoietic stem cells. Haematologica. (2018) 103:e419–23. doi: 10.3324/haematol.2018.189357
109. Naiisseh B, Papasavva PL, Papaioannou NY, Tomazou M, Koniali L, Felekis X, et al. Context base editing for splice correction of IVSI-110 beta-thalassemia. Mol Ther Nucleic Acids. (2024) 35:102183. doi: 10.1016/j.omtn.2024.102183
110. Patsali P, Turchiano G, Papasavva P, Romito M, Loucari CC, Stephanou C, et al. Correction of IVS I-110(G>A) beta-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica. (2019) 104:e497–501. doi: 10.3324/haematol.2018.215178
111. Xu S, Luk K, Yao Q, Shen AH, Zeng J, Wu Y, et al. Editing aberrant splice sites efficiently restores beta-globin expression in beta-thalassemia. Blood. (2019) 133:2255–62. doi: 10.1182/blood-2019-01-895094
112. Cromer MK, Camarena J, Martin RM, Lesch BJ, Vakulskas CA, Bode NM, et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat Med. (2021) 27:677–87. doi: 10.1038/s41591-021-01284-y
113. Pavani G, Fabiano A, Laurent M, Amor F, Cantelli E, Chalumeau A, et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. (2021) 5:1137–53. doi: 10.1182/bloodadvances.2020001996
114. Franco RS, Yasin Z, Palascak MB, Ciraolo P, Joiner CH, Rucknagel DL. The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood. (2006) 108:1073–6. doi: 10.1182/blood-2005-09-008318
115. Bhatnagar P, Purvis S, Barron-Casella E, DeBaun MR, Casella JF, Arking DE, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J Hum Genet. (2011) 56:316–23. doi: 10.1038/jhg.2011.12
116. Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. (2010) 42:1049–51. doi: 10.1038/ng.707
117. Lettre G, Sankaran VG, Bezerra MA, Araujo AS, Uda M, Sanna S, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U.S.A. (2008) 105:11869–74. doi: 10.1073/pnas.0804799105
118. Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. (2007) 39:1197–9. doi: 10.1038/ng2108
119. Solovieff N, Milton JN, Hartley SW, Sherva R, Sebastiani P, Dworkis DA, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5’ olfactory receptor gene cluster. Blood. (2010) 115:1815–22. doi: 10.1182/blood-2009-08-239517
120. Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U.S.A. (2008) 105:1620–5. doi: 10.1073/pnas.0711566105
121. Giardine B, Borg J, Viennas E, Pavlidis C, Moradkhani K, Joly P, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. (2014) 42:D1063–1069. doi: 10.1093/nar/gkt911
122. Basak A, Hancarova M, Ulirsch JC, Balci TB, Trkova M, Pelisek M, et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest. (2015) 125:2363–8. doi: 10.1172/JCI81163
123. Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. (2013) 342:253–7. doi: 10.1126/science.1242088
124. Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. (2015) 527:192–7. doi: 10.1038/nature15521
125. Liu N, Xu S, Yao Q, Zhu Q, Kai Y, Hsu JY, et al. Transcription factor competition at the gamma-globin promoters controls hemoglobin switching. Nat Genet. (2021) 53:511–20. doi: 10.1038/s41588-021-00798-y
126. Martyn GE, Wienert B, Kurita R, Nakamura Y, Quinlan KGR, Crossley M. A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood. (2019) 133:852–6. doi: 10.1182/blood-2018-07-863951
127. Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. (2016) 351:285–9. doi: 10.1126/science.aad3312
128. Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. (2008) 322:1839–42. doi: 10.1126/science.1165409
129. Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. (2011) 365:807–14. doi: 10.1056/NEJMoa1103070
130. Crossley M, Christakopoulos GE, Weiss MJ. Effective therapies for sickle cell disease: are we there yet? Trends Genet. (2022). doi: 10.1016/j.tig.2022.07.003
131. Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. (2011) 334:993–6. doi: 10.1126/science.1211053
132. Psatha N, Reik A, Phelps S, Zhou Y, Dalas D, Yannaki E, et al. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with β-thalassemia major. Mol Therapy-Methods Clin Dev. (2018) 10:313–26. doi: 10.1016/j.omtm.2018.08.003
133. Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, et al. Bcl11a is essential for normal lymphoid development. Nat Immunol. (2003) 4:525–32. doi: 10.1038/ni925
134. Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med. (2012) 209:2467–83. doi: 10.1084/jem.20121846
135. Brendel C, Guda S, Renella R, Bauer DE, Canver MC, Kim Y-J, et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest. (2016) 126:3868–78. doi: 10.1172/JCI87885
136. Brendel C, Negre O, Rothe M, Guda S, Parsons G, Harris C, et al. Preclinical evaluation of a novel lentiviral vector driving lineage-specific BCL11A knockdown for sickle cell gene therapy. Mol Therapy-Methods Clin Dev. (2020) 17:589–600. doi: 10.1016/j.omtm.2020.03.015
137. Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. New Engl J Med. (2021) 384:205–15. doi: 10.1056/NEJMoa2029392
138. Liu B, Brendel C, Vinjamur DS, Zhou Y, Harris C, McGuinness M, et al. Development of a double shmiR lentivirus effectively targeting both BCL11A and ZNF410 for enhanced induction of fetal hemoglobin to treat β-hemoglobinopathies. Mol Ther. (2022) 30:2693–708. doi: 10.1016/j.ymthe.2022.05.002
139. Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. (2019) 25:776–83. doi: 10.1038/s41591-019-0401-y
140. Demirci S, Zeng J, Wu Y, Uchida N, Shen AH, Pellin D, et al. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J Clin Invest. (2020) 130:6677–87. doi: 10.1172/JCI140189
141. Chang K-H, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol Therapy-Methods Clin Dev. (2017) 4:137–48. doi: 10.1016/j.omtm.2016.12.009
142. Walters MC, Smith AR, Schiller GJ, Esrick EB, Williams DA, Gogoleva T, et al. Updated results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood. (2021) 138:3974. doi: 10.1182/blood-2021-147907
143. Alavi A, Krishnamurti L, Abedi M, Galeon I, Reiner D, Smith SE, et al. Preliminary safety and efficacy results from Precizn-1: an ongoing Phase 1/2 study on Zinc Finger Nuclease-Modified Autologous CD34+ HSPCs for sickle cell disease (SCD). Blood. (2021) 138:2930. doi: 10.1182/blood-2021-151650
144. Moran K, Ling H, Lessard S, Vieira B, Hong V, Holmes MC, et al. Ex vivo gene-edited cell therapy for sickle cell disease: disruption of the BCL11A erythroid enhancer with zinc finger nucleases increases fetal hemoglobin in plerixafor mobilized human CD34+ cells. Blood. (2018) 132:2190. doi: 10.1182/blood-2018-99-116998
145. Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New Engl J Med. (2021) 384:252–60. doi: 10.1056/NEJMoa2031054
146. Frangoul H, Locatelli F, Bhatia M, Mapara MY, Molinari L, Sharma A, et al. Efficacy and safety of a single dose of exagamglogene autotemcel for severe sickle cell disease. Blood. (2022) 140:29–31. doi: 10.1182/blood-2022-162353
147. Frangoul H, Locatelli F, Sharma A, Bhatia M, Mapara M, Molinari L, et al. Exagamglogene autotemcel for severe sickle cell disease. New Engl J Med. (2024) 390:1649–1662. doi: 10.1056/NEJMoa2309676
148. Locatelli F, Lang P, Wall D, Meisel R, Corbacioglu S, Li AM, et al. Exagamglogene Autotemcel for Transfusion-Dependent beta-Thalassemia. N Engl J Med. (2024) 390:1663–76. doi: 10.1056/NEJMoa2309673
149. Fu B, Liao J, Chen S, Li W, Wang Q, Hu J, et al. CRISPR–Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia. Nat Med. (2022) 28:1573–80. doi: 10.1038/s41591-022-01906-z
150. Zheng B, Liu R, Zhang X, Fu B, Xu Y, Shi J, et al. Efficacy and safety of brl-101, crispr-cas9-mediated gene editing of the bcl11a enhancer in transfusion-dependent β-thalassemia. Blood. (2023) 142:4995–5. doi: 10.1182/blood-2023-186031
151. Zeng J, Demirci S, Nguyen MA, Lin LY, Maitland SA, Mintzer E, et al. Combined+ 58 and+ 55 BCL11A enhancer editing Yields exceptional efficiency, specificity and HbF induction in human and NHP preclinical models. Blood. (2021) 138:1852. doi: 10.1182/blood-2021-152634
152. Liao J, Chen S, Hsiao S, Jiang Y, Yang Y, Zhang Y, et al. Therapeutic adenine base editing of human hematopoietic stem cells. Nat Commun. (2023) 14:207. doi: 10.1038/s41467-022-35508-7
153. Huang P, Peslak SA, Ren R, Khandros E, Qin K, Keller CA, et al. HIC2 controls developmental hemoglobin switching by repressing BCL11A transcription. Nat Genet. (2022) 54:1417–26. doi: 10.1038/s41588-022-01152-6
154. Antoniani C, Meneghini V, Lattanzi A, Felix T, Romano O, Magrin E, et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood J Am Soc Hematol. (2018) 131:1960–73. doi: 10.1182/blood-2017-10-811505
155. Shariati L, Khanahmad H, Salehi M, Hejazi Z, Rahimmanesh I, Tabatabaiefar MA, et al. Genetic disruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J Gene Med. (2016) 18:294–301. doi: 10.1002/jgm.v18.10
156. Wienert B, Martyn GE, Funnell AP, Quinlan KG, Crossley M. Wake-up sleepy gene: reactivating fetal globin for β-hemoglobinopathies. Trends Genet. (2018) 34:927–40. doi: 10.1016/j.tig.2018.09.004
157. Weber L, Frati G, Felix T, Hardouin G, Casini A, Wollenschlaeger C, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. (2020) 6:eaay9392. doi: 10.1126/sciadv.aay9392
158. Métais J-Y, Doerfler PA, Mayuranathan T, Bauer DE, Fowler SC, Hsieh MM, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. (2019) 3:3379–92. doi: 10.1182/bloodadvances.2019000820
159. Traxler EA, Yao Y, Wang Y-D, Woodard KJ, Kurita R, Nakamura Y, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. (2016) 22:987–90. doi: 10.1038/nm.4170
160. Hanna R, Frangoul H, McKinney C, Pineiro L, Mapara M, Chang KH, et al. S264: EDIT-301 shows promising preliminary safety and efficacy results in the phase i/ii clinical trial (ruby) of patients with severe sickle cell disease using highly specific and efficient ASCAS12A enzyme. Hemasphere. (2023) 7:e05170e0. doi: 10.1097/01.HS9.0000967968.05170.e0
161. Sharma A, Boelens J-J, Cancio M, Hankins JS, Bhad P, Azizy M, et al. CRISPR-Cas9 editing of the HBG1 and HBG2 promoters to treat sickle cell disease. New Engl J Med. (2023) 389:820–32. doi: 10.1056/NEJMoa2215643
162. Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, et al. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science. (2018) 361:285–90. doi: 10.1126/science.aao0932
163. Ravi NS, Wienert B, Wyman SK, Bell HW, George A, Mahalingam G, et al. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. Elife. (2022) 11:e65421. doi: 10.7554/eLife.65421.sa2
164. Venkatesan V, Christopher AC, Rhiel M, Azhagiri MKK, Babu P, Walavalkar K, et al. Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of β-hemoglobinopathies. Mol Therapy-Nucleic Acids. (2023) 32:671–88. doi: 10.1016/j.omtn.2023.04.024
165. Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, et al. Reactivation of gamma-globin in adult beta-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood. (2018) 131:2915–28. doi: 10.1182/blood-2018-03-838540
166. Li C, Georgakopoulou A, Mishra A, Gil S, Hawkins RD, Yannaki E, et al. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood Adv. (2021) 5:1122–35. doi: 10.1182/bloodadvances.2020003702
167. Psatha N, Georgakopoulou A, Li C, Nandakumar V, Georgolopoulos G, Acosta R, et al. Enhanced HbF reactivation by multiplex mutagenesis of thalassemic CD34+ cells in vitro and in vivo. Blood. (2021) 138:1540–53. doi: 10.1182/blood.2020010020
168. Antoniou P, Hardouin G, Martinucci P, Frati G, Felix T, Chalumeau A, et al. Base-editing-mediated dissection of a gamma-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nat Commun. (2022) 13:6618. doi: 10.1038/s41467-022-34493-1
169. Kohn DB, Chen YY, Spencer MJ. Successes and challenges in clinical gene therapy. Gene Ther. (2023) 30:738–46. doi: 10.1038/s41434-023-00390-5
170. Bastani S, Staal FJ, Canté-Barrett K. The quest for the holy grail: overcoming challenges in expanding human hematopoietic stem cells for clinical use. Stem Cell Invest. (2023) 10:15. doi: 10.21037/sci-2023-016
171. Sakurai M, Ishitsuka K, Becker HJ, Yamazaki S. Ex vivo expansion of human hematopoietic stem cells and clinical applications. Cancer Sci. (2024) 115:698–705. doi: 10.1111/cas.v115.3
172. Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood J Am Soc Hematol. (2014) 124:344–53. doi: 10.1182/blood-2014-02-514778
173. Hulbert ML, Shenoy S. Hematopoietic stem cell transplantation for sickle cell disease: Progress and challenges. Pediatr Blood Cancer. (2018) 65:e27263. doi: 10.1002/pbc.27263
174. Breda L, Papp TE, Triebwasser MP, Yadegari A, Fedorky MT, Tanaka N, et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science. (2023) 381:436–43. doi: 10.1126/science.ade6967
175. Burns S. Short Talk: In Vivo Genome Editing of Hematopoietic Stem and Progenitor Cells using Non-viral Delivery of CRISPR/Cas9. Keystone eSymposium: Precision Engineering of the Genome, Epigenome and Transcriptome (2021). Available online at: https://virtual.keystonesymposia.org/ks/live/587/page/4632 (Accessed March 10, 2021).
176. Li C, Wang H, Gil S, Germond A, Fountain C, Baldessari A, et al. Safe and efficient in vivo hematopoietic stem cell transduction in nonhuman primates using HDAd5/35++ vectors. Mol Ther Methods Clin Dev. (2022) 24:127–41. doi: 10.1016/j.omtm.2021.12.003
177. Kohn DB. Gene therapy for blood diseases. Curr Opin Biotechnol. (2019) 60:39–45. doi: 10.1016/j.copbio.2018.11.016
178. An M, Raguram A, Du SW, Banskota S, Davis JR, Newby GA, et al. Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat Biotechnol. (2024), 1–12. doi: 10.1038/s41587-023-02078-y
179. Verma M, Ozer I, Xie W, Gallagher R, Teixeira A, Choy M. The landscape for lipid-nanoparticle-based genomic medicines. Nat Rev Drug Discovery. (2023) 22:349–50. doi: 10.1038/d41573-023-00002-2
180. Cullis PR, Hope MJ. Lipid nanoparticle systems for enabling gene therapies. Mol Ther. (2017) 25:1467–75. doi: 10.1016/j.ymthe.2017.03.013
181. Al-Saif AM. Gene therapy of hematological disorders: current challenges. Gene Ther. (2019) 26:296–307. doi: 10.1038/s41434-019-0093-4
Keywords: sickle cell disease, beta-thalassemia, gene therapy, CRISPR/Cas (clustered regularly interspaced short palindromic repeats), fetal hemoglobin (HbF), beta-globinopathies, hematopoietic stem cells (HSC)
Citation: Li L and Mandal PK (2024) Recent advancements in gene therapy for sickle cell disease and β-thalassemia. Front. Hematol. 3:1468952. doi: 10.3389/frhem.2024.1468952
Received: 22 July 2024; Accepted: 30 August 2024;
Published: 27 September 2024.
Edited by:
Savita Rangarajan, University of Southampton, United KingdomReviewed by:
Shuaiying Cui, Boston University, United StatesNikoleta Psatha, Aristotle University of Thessaloniki, Greece
Copyright © 2024 Li and Mandal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pankaj K. Mandal, UGFua2FqLk1hbmRhbEBmZGEuaGhzLmdvdg==