 Alok Kumar
Alok Kumar Sudip Bhattacharya
Sudip Bhattacharya- Department of Community and Family Medicine, All India Institute of Medical Sciences, Deoghar (AIIMS Deoghar), Deoghar, India
Sickle cell disease (SCD) is a genetic disorder caused by mutations in the β-globin chain of hemoglobin, leading to abnormal red blood cells, severe pain, anemia, organ damage, and increased infection risk. Inherited in an autosomal recessive manner, it mainly affects regions with high malaria incidence, like sub-Saharan Africa, the Middle East, and the Indian subcontinent. Management includes blood transfusions, hydroxyurea, folic acid, iron chelators, and hematopoietic stem cell transplantation (HSCT), the only curative option but limited by donor compatibility. Comprehensive healthcare management (CHCM) emphasizes patient education, nutrition, prophylactic antibiotics, and early intervention to reduce morbidity and improve quality of life. SCD presents a significant global health burden, particularly in regions with limited healthcare access, contributing substantially to child mortality. In sub-Saharan Africa, India, and the Middle East, SCD is prevalent, with approximately 300,000 infants born annually with the condition. In the United States, about 100,000 individuals, predominantly African Americans, are affected. National initiatives, such as Nigeria’s National Sickle Cell Disease Control Program and India’s National Sickle Cell Anaemia Elimination Mission, aim to improve outcomes through early screening, public education, and enhanced healthcare access. Global efforts by the WHO, GSCDN, and SCDC focus on strategic policies, advocacy, and international collaboration to improve care and reduce mortality. Challenges in implementing SCD prevention programs include the need for extensive genetic screening, robust healthcare infrastructure, and overcoming cultural stigmas. Research funding disparities between the Global North and South further hinder advancements. Future research avenues include gene editing technologies, improving prenatal diagnosis, enhancing newborn screening, understanding genetic modifiers, developing new pharmacological agents, and optimizing stem cell transplants. Collaborative efforts among researchers, healthcare providers, policymakers, and patient advocacy groups are essential to translate research into practical applications, ensuring accessible, effective, and equitable advancements in SCD prevention and treatment.
Introduction
Sickle cell disease (SCD) is an inherited disorder caused by mutations in the β-globin chain of hemoglobin, leading to hemoglobin S (HbS). This abnormal hemoglobin makes red blood cells stiff and sickle-shaped, blocking blood flow and causing severe pain, anemia, organ damage, and infections. SCD is inherited in an autosomal recessive pattern, with the homozygous state (HbSS) known as sickle cell anemia. Symptoms include acute and chronic pain, organ dysfunction, and increased infection risk, with a median life expectancy of 43 years (1). Management includes blood transfusions, hydroxyurea, folic acid, iron chelators, and hematopoietic stem cell transplantation (HSCT), the only curative treatment but limited by donor availability and compatibility (2). Comprehensive healthcare management (CHCM) is crucial for SCD patients which involves patient and parent education, proper nutrition and hydration, prophylactic antibiotics and antimalarials, folic acid supplementation, specific vaccines, continuous medical follow-up, and early complication management to reduce morbidity and improve quality of life. SCD is prevalent in sub-Saharan Africa, the Middle East, and the Indian subcontinent (3). In India, the sickle cell gene is common among marginalized tribal and economically disadvantaged populations, with carrier frequencies from 1 to 35% (4). The clinical presentation is milder than in Africa but varies widely, with common vaso-occlusive crisis. In Orissa, India, high alpha thalassemia and HbF levels, splenomegaly, painful events, febrile illness, and anemia are prevalent. In Maharashtra and Gujarat, non-tribal populations have higher rates of painful crisis, hospitalizations, and infections compared to tribal groups. Vaccinations, adequate fluids, and avoiding extreme temperatures have reduced crisis episodes (5–7).
Global and national burden
Sickle cell disease (SCD) presents a significant global health burden, with millions affected worldwide, particularly in sub-Saharan Africa, India, and the Middle East. The disease accounts for a substantial portion of child mortality in these regions due to limited access to adequate healthcare. According to Grosse et al., approximately 300,000 infants are born annually with SCD globally, with the highest incidence in Africa where it contributes to 5-16% of under-five mortality (8). The burden of SCD is also profound, particularly in countries with diverse populations such as the United States, where it predominantly affects African Americans. Hassell reports that SCD affects about 100,000 Americans, with significant healthcare costs and challenges in disease management (9). India ranks second in the world for the number of predicted SCD births, with an estimated 42,016 newborns (interquartile range: 35,347-50,919) born with sickle cell anemia in 2010 (10).
In Nigeria, India, and the Democratic Republic of the Congo, up to 2% of the population has SCD, with sickle cell trait prevalence of 10% to 30% (10, 11). These countries host 90% of the world’s SCD population, with 150,000 infants born with SCD annually in Nigeria (12).
In India, the sickle cell gene is prevalent among tribal populations in the Nilgiri Hills, Bihar, and Odisha, and socioeconomically disadvantaged groups. A pilot screening programs for SCD among newborn in Gujarat, Maharashtra, and Chhattisgarh revealed HbAS prevalence rates between 2% and 40%. The highest frequency of the βs allele, reaching up to 10%, is observed in Central India, spanning from South-Eastern Gujarat to South-Western Odisha (6, 13, 14). It is observed that the disease severity varies, with some tribal groups like those in Valsad experiencing milder symptoms due to high alpha thalassemia rates (15). Differences in severity are also influenced by assessment methods, with hospitalized cases showing more severe disease compared to outpatient reports (16–18). In Sindh Province, 40% of SCD patients had SS disease, with 16% having alpha thalassemia, compared to higher rates in Odisha and Valsad (18, 19).
Global and national initiatives
The WHO’s guidance frameworks for SCD in Africa and globally aim to enhance care through strategic policies and advocacy. The SICKLE technical package integrates interventions, education, and community empowerment (20). These frameworks emphasize early diagnosis through newborn screening, public education, and improved access to healthcare services. According to McGann et al., early diagnosis and intervention have resulted in decreased mortality rates among children with SCD in several African countries (21). The WHO’s efforts have also led to the establishment of national programs that enhance the availability of comprehensive care, including vaccination, prophylactic antibiotics, and pain management strategies. As Tshilolo et al. provides an overview of practices employed in neonatal screening and clinical care programmes for sickle cell disease in sub-Saharan African countries. The development of these programmes is pivotal to improving the health care of those affected by hemoglobin disorders. However, such programmes require major economic and organizational resources, which must take into account and balanced against other local health priorities (22). Additionally, the WHO’s advocacy for research and data collection has improved the understanding of SCD, facilitating more effective public health strategies. Overall, the WHO’s guidance frameworks have played a crucial role in mitigating the impact of SCD in Africa through coordinated efforts in early detection, comprehensive care, and public health policies. The Global Sickle Cell Disease Network (GSCDN) and Sickle Cell Disease Coalition (SCDC) focus on improving care and collaboration globally. The GSCDN, by establishing a global platform, has facilitated the sharing of best practices and resources, leading to improved standards of care across various regions. As reported by Telfer et al. in East London, UK documented the survival rate for children with HbSS at 16 years was 99.0% (95% CI, 93.2-99.9%), with a pneumococcal sepsis rate of 0.3 episodes per 100 patient-years (95% CI, 0.1-0.8). The risk of overt stroke was 4.3% (95% CI, 1.5-11.4%), which could be reduced further with early transcranial Doppler screening and transfusions for high-risk children (23). Similarly, the SCDC has been instrumental in uniting diverse stakeholders, including healthcare providers, researchers, and patient advocacy groups, to address the multifaceted challenges of SCD. Together, the GSCDN and SCDC have played a pivotal role in elevating the global response to SCD, ensuring that patients receive better care and support through enhanced collaboration and advocacy.
Nigeria’s National Sickle Cell Disease Control Program has made substantial strides in improving the management and outcomes of sickle cell disease (SCD). This program focuses on comprehensive strategies, including early screening, public education, and enhanced healthcare access. A study found that the program’s newborn screening initiatives have significantly improved early diagnosis and treatment, leading to better clinical outcomes and reduced mortality rates among children with SCD in Nigeria (24). Furthermore, the integration of community-based awareness campaigns has increased public knowledge about SCD, promoting timely medical interventions. According to a study, the program’s emphasis on training healthcare providers and improving treatment protocols has led to more effective management of SCD complications, enhancing patient quality of life (25). Overall, Nigeria’s National Sickle Cell Disease Control Program represents a critical model for addressing SCD through a multifaceted approach that combines screening, education, and improved care.
For the past eight years, the Sickle Cell Trust (Jamaica) has successfully run an efficient and cost-effective newborn screening program for sickle cell disease, using umbilical cord samples with only 0.05% maternal contamination. The program identified 130 rare hemoglobin variants: 15 alpha chain (all HbG Philadelphia), 57 beta chain (10 different variants), 23 gamma chain variants, with 30 still under analysis, and 5 cases lost to follow-up (26).
In the US, the Sickle Cell Treatment Act of 2003 supports Medicaid reimbursement for treatments and services, including genetic counseling and educational campaigns (27).
The National Sickle Cell Anaemia Elimination Mission, launched by the Prime Minister of India on July 1, 2023, in Shahdol, Madhya Pradesh, integrates with existing National Health Mission mechanisms to optimize resources and avoid duplication of efforts (28). The National Sickle Cell Elimination Mission is an ambitious healthcare initiative aimed at eradicating sickle cell disease through comprehensive screening, early diagnosis, and advanced treatment protocols. The components of screening programme are mentioned in Figure 1.
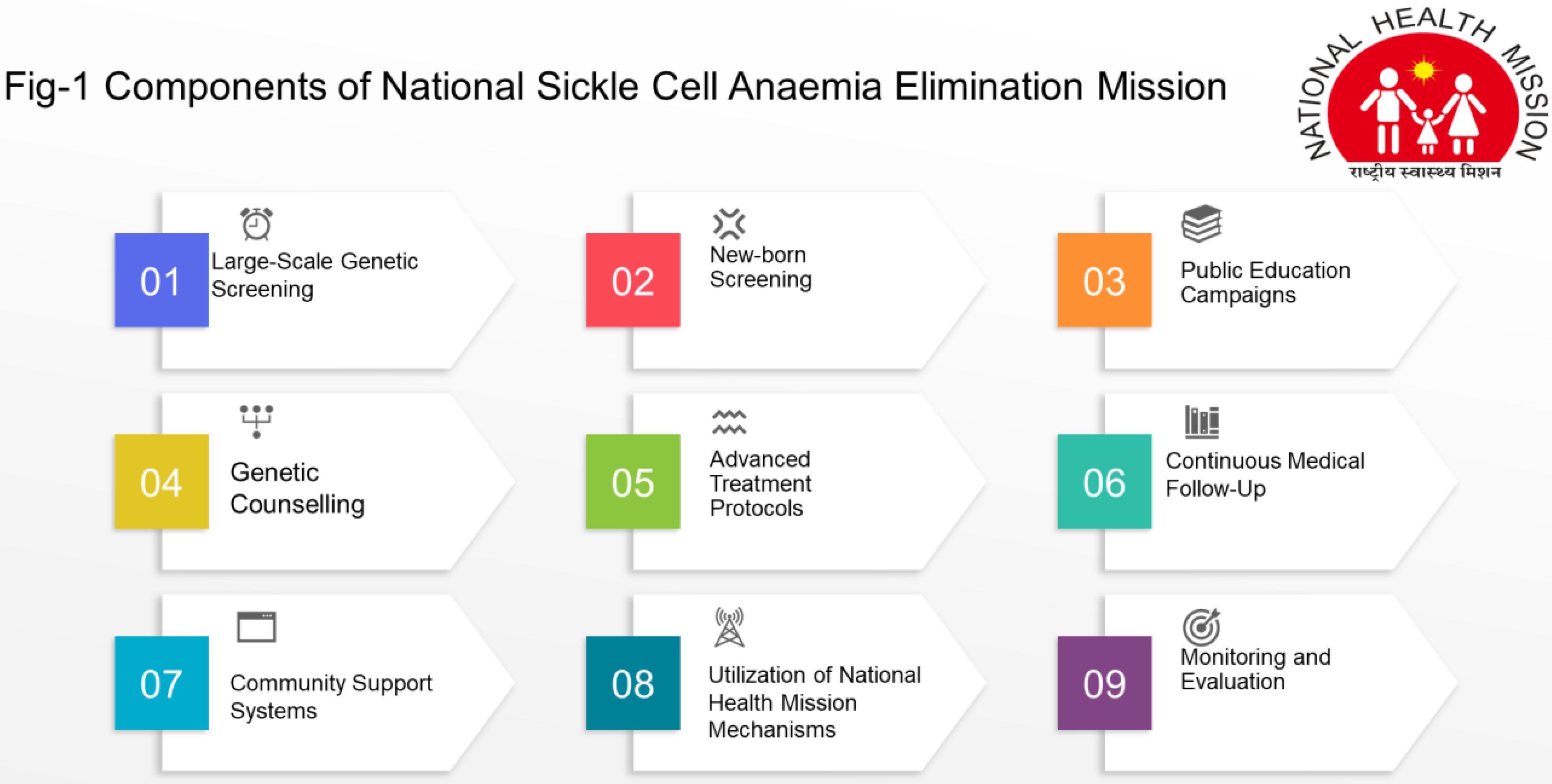
Figure 1. Components of national sickle cell anaemia ellimination mission.
These components collectively aim to reduce the burden of sickle cell disease and improve the quality of life for affected individuals in India. Although in Gujarat, the Sickle Cell Anaemia Control Program, initiated in 2006 and expanded state-wide, focuses on early diagnosis, treatment, and counseling. By March 2011, it screened one million three hundred ninety-six thousand nine hundred four tribal people, identified ten thousand six hundred seventy-three SCD patients, and provided necessary care (29). The on-going national mission aims to screen approximately seventy million people from fiscal year 2023-24 to 2025-26, achieving significant milestones such as screening over thirty-three million seven hundred thousand individuals, confirming nine hundred seventy-five thousand individuals as sickle cell trait carriers, diagnosing one hundred forty thousand with SCD, and verifying the status of over three hundred fifty-nine thousand individuals (30).
Challenges
Globally and nationally, implementing a sickle cell disease (SCD) prevention program presents a complex array of challenges, encompassing medical, social, economic, and infrastructural aspects (31). One of the primary obstacles is the need for widespread genetic screening and counseling, which necessitates substantial financial investment and robust healthcare infrastructure—resources often lacking in regions where SCD is most prevalent, such as sub-Saharan Africa and parts of India. Ensuring accurate and accessible genetic testing requires sophisticated laboratory facilities and trained personnel, which can be scarce (32). Moreover, cultural and social stigmas associated with genetic testing and SCD can significantly hinder community participation and acceptance of these programs (33). In many societies, there is a lack of awareness and understanding of the disease, compounded by misinformation and traditional beliefs, which can lead to resistance against preventive measures. Effective education and awareness campaigns are critical but challenging to implement, especially in areas with varying literacy levels and limited access to reliable information (34). Furthermore, once individuals are diagnosed, establishing a comprehensive care system is essential for managing the disease, yet this is often complicated by a shortage of healthcare professionals skilled in treating SCD and a lack of access to essential medications and treatments (31). The need for ongoing patient monitoring and the provision of specialized treatments further strain already overburdened healthcare systems. Additionally, socioeconomic factors play a significant role; families affected by SCD often face financial hardships due to medical expenses and the inability to work regularly, which can exacerbate the burden of the disease (35). Coordination among healthcare providers, government agencies, and non-profit organizations is crucial to address these multifaceted challenges effectively. Such collaboration must focus on integrating genetic services with existing healthcare frameworks, ensuring equitable access to care, and fostering community engagement to overcome cultural barriers (36). Without a comprehensive, multidisciplinary approach, the successful implementation of SCD prevention programs remains a daunting challenge at global as well as national level.
Apart from this, inequality in research funding for sickle cell disease (SCD) starkly contrasts between the Global North and Global South. Despite the majority of SCD sufferers residing in sub-Saharan Africa and parts of Asia, most research funding and resources are concentrated in high-income countries of the Global North. For instance, in the United States, the National Institutes of Health (NIH) allocates significant funds for SCD research, while African countries, where SCD prevalence is highest, struggle with limited financial resources for similar research endeavors (31). This disparity in funding creates a vicious cycle where resource-limited settings remain under-researched and underfunded, hindering the development of effective treatments and interventions tailored to the needs of those most affected.
Access to research funding in the Global South is impeded by several factors, including inadequate infrastructure, limited access to international research grants, and insufficient local funding mechanisms (37). Researchers in these regions often face significant challenges in securing financial support for their projects, resulting in a reliance on collaborations with institutions in the Global North. However, these partnerships are frequently imbalanced, with the Global North entities controlling the majority of the resources and decision-making power (38). This dynamic can marginalize the voices and contributions of local researchers and sufferers, who possess valuable insights into the disease and potential treatment strategies.
Involving sufferers from the Global South in SCD research is crucial for generating more relevant and context-specific knowledge. Patients and researchers in these regions can provide unique perspectives on the disease’s progression, response to treatments, and cultural factors influencing care (39). Enhancing their participation can lead to more effective and culturally appropriate interventions. Additionally, investing in research capacity building in resource-limited settings can empower local scientists, foster innovation, and ultimately improve health outcomes for SCD patients in these regions (40). Addressing the funding disparities and promoting equitable research collaboration are essential steps toward reducing the global burden of sickle cell disease.
Future research avenues
Future research avenues for sickle cell disease (SCD) prevention are multifaceted and highly promising. A key area of focus is the advancement of gene editing technologies, such as CRISPR-Cas9, which have the potential to correct the genetic mutation responsible for SCD at its source, offering a permanent cure. Research into safer and more efficient delivery methods for these gene therapies is critical to ensure their accessibility and effectiveness. Additionally, enhancing prenatal and preimplantation genetic diagnosis (PGD) techniques can prevent the transmission of SCD, allowing couples with the genetic trait to have healthy offspring through assisted reproductive technologies. Improving newborn screening programs is another vital research avenue. By identifying infants with SCD at birth, early interventions can be implemented to prevent complications and improve quality of life. This requires the development of more sensitive and cost-effective screening tools, especially for low-resource settings. Furthermore, understanding the genetic and environmental factors that influence the severity and manifestation of SCD can lead to personalized medicine approaches. This involves studying the genetic modifiers that may mitigate or exacerbate the disease, potentially leading to targeted therapies that can be tailored to individual patients. Investigating new pharmacological agents that can prevent the sickling of red blood cells or protect organs from damage is also a crucial research direction. Advances in biotechnology could lead to the discovery of novel drugs that are more effective and have fewer side effects. Moreover, exploring the role of stem cell transplants and improving the safety and success rates of these procedures can provide another curative option for patients with SCD.
The sickle cell cohorts in their studies frequently encounter major obstacles in obtaining advanced treatments like gene editing and new drugs, largely due to factors such as their geographic location, socioeconomic status, and the healthcare infrastructure available to them. In regions where sickle cell disease is most prevalent, particularly in sub-Saharan Africa, there is a critical shortage of specialized medical facilities and trained personnel, which hampers the availability of these cutting-edge treatments. To improve access, a global shift is necessary that includes increased investment in healthcare infrastructure, enhanced training for local healthcare providers, and the development of affordable treatment options. Moreover, fostering international collaborations and funding research initiatives can help bridge the gap, ensuring that these life-saving therapies reach those who need them most and reducing health disparities on a global scale. In addition to this, it is essential to translate these research findings into practical, real-world applications. This includes not only scientific and medical research but also social science research to address the cultural, ethical, and economic barriers to the adoption of new prevention and treatment strategies. By integrating these diverse perspectives, future research can ensure that advancements in SCD prevention are accessible, effective, and equitable, ultimately improving outcomes for individuals affected by this challenging condition.

Key messages on the global and national burden of sickle cell disease.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
AK: Writing – original draft, Writing – review & editing. SB: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Payne AB, Mehal JM, Chapman C, Haberling DL, Richardson LC, Bean CJ, et al. Trends in sickle cell disease-related mortality in the United States, 1979 to 2017. Ann Emerg Med. (2020) 76:S28–36. doi: 10.1016/j.annemergmed.2020.08.009
2. Krishnamurti L, Abel S, Maiers M, Flesch S. Availability of unrelated donors for hematopoietic stem cell transplantation for hemoglobinopathies. Bone Marrow Transplant. (2003) 31:547–50. doi: 10.1038/sj.bmt.1703887
3. Williams TN. Sickle cell disease in sub-saharan africa. Hematol Oncol Clin North Am. (2016) 30:343–58. doi: 10.1016/j.hoc.2015.11.005
4. Bhatia HM, Rao VR. Genetic atlas of the Indian tribes. Indian Council of Medical Research: Institute of Immunohaematology (1986). 449 p.
5. Colah RB, Mukherjee MB, Martin S, Ghosh K. Sickle cell disease in tribal populations in India. Indian J Med Res. (2015) 141:509.
6. Kaur M, Das GP, Verma IC. Sickle cell trait & disease among tribal communities in Orissa, Madhya Pradesh & Kerala. Indian J Med Res. (1997) 105:111–6.
7. Kate SL, Lingojwar DP, Kate SL, Lingojwar DP. Epidemiology of sickle cell disorder in the state of Maharashtra. Int J Hum Genet. (2002) 2:161–7. doi: 10.1080/09723757.2002.11885800
8. Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. (2011) 41:S398–405. doi: 10.1016/j.amepre.2011.09.013
9. Hassell KL. Population estimates of sickle cell disease in the U. S. Am J Prev Med. (2010) 38:S512–521. doi: 10.1016/j.amepre.2009.12.022
11. Kadima BT, Gini Ehungu JL, Ngiyulu RM, Ekulu PM, Aloni MN. High rate of sickle cell anaemia in Sub-Saharan Africa underlines the need to screen all children with severe anaemia for the disease. Acta Paediatr Oslo Nor 1992. (2015) 104:1269–73. doi: 10.1111/apa.13040
12. Oluwole EO, Adeyemo TA, Osanyin GE, Odukoya OO, Kanki PJ, Afolabi BB. Feasibility and acceptability of early infant screening for sickle cell disease in Lagos, Nigeria—A pilot study. PloS One. (2020) 15:e0242861. doi: 10.1371/journal.pone.0242861
13. Patra PK, Chauhan VS, Khodiar PK, Dalla AR, Serjeant GR. Screening for the sickle cell gene in Chhattisgarh state, India: an approach to a major public health problem. J Community Genet. (2011) 2:147–51. doi: 10.1007/s12687-011-0050-4
14. Rao VR. Genetics and epidemiology of sickle cell anemia in India. Indian J Med Sci. (1988) 42:218–22.
15. Mukherjee MB, Lu CY, Ducrocq R, Gangakhedkar RR, Colah RB, Kadam MD, et al. Effect of alpha-thalassemia on sickle-cell anemia linked to the Arab-Indian haplotype in India. Am J Hematol. (1997) 55:104–9. doi: 10.1002/(ISSN)1096-8652
16. Jain D, Italia K, Sarathi V, Ghoshand K, Colah R. Sickle cell anemia from central India: a retrospective analysis. Indian Pediatr. (2012) 49:911–3. doi: 10.1007/s13312-012-0217-z
17. Jain D, Bagul AS, Shah M, Sarathi V. Morbidity pattern in hospitalized under five children with sickle cell disease. Indian J Med Res. (2013) 138:317–21.
18. Kar BC, Satapathy RK, Kulozik AE, Kulozik M, Sirr S, Serjeant BE, et al. Sickle cell disease in Orissa State, India. Lancet Lond Engl. (1986) 2:1198–201. doi: 10.1016/S0140-6736(86)92205-1
19. Jain D, Warthe V, Dayama P, Sarate D, Colah R, Mehta P, et al. Sickle cell disease in central India: A potentially severe syndrome. Indian J Pediatr. (2016) 83:1071–6. doi: 10.1007/s12098-016-2081-7
20. WHO | Regional Office for Africa [Internet]. WHO sickle package of interventions for sickle cell disease management (2024). Available at: https://www.afro.who.int/publications/who-sickle-package-interventions-sickle-cell-disease-management (accessed August 9, 2024)
21. McGann PT, Hernandez AG, Ware RE. Sickle cell anemia in sub-Saharan Africa: advancing the clinical paradigm through partnerships and research. Blood. (2017) 129:155–61. doi: 10.1182/blood-2016-09-702324
22. Tshilolo L, Kafando E, Sawadogo M, Cotton F, Vertongen F, Ferster A, et al. Neonatal screening and clinical care programmes for sickle cell disorders in sub-Saharan Africa: lessons from pilot studies. Public Health. (2008) 122:933–41. doi: 10.1016/j.puhe.2007.12.005
23. Telfer P, Coen P, Chakravorty S, Wilkey O, Evans J, Newell H, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. (2007) 92:905–12. doi: 10.3324/haematol.10937
24. Nnodu OE, Sopekan A, Nnebe-Agumadu U, Ohiaeri C, Adeniran A, Shedul G, et al. Implementing newborn screening for sickle cell disease as part of immunisation programmes in Nigeria: a feasibility study. Lancet Haematol. (2020) 7:e534–40. doi: 10.1016/S2352-3026(20)30143-5
25. Adewoyin AS. Management of sickle cell disease: A review for physician education in Nigeria (Sub-saharan africa). Anemia. (2015) 2015:791498. doi: 10.1155/2015/791498
26. Serjeant GR, Serjeant BE, Mason KP, Gardner R, Warren L, Gibson F, et al. Newborn screening for sickle cell disease in Jamaica: logistics and experience with umbilical cord samples. J Community Genet. (2017) 8:17–22. doi: 10.1007/s12687-016-0283-3
27. Asekun-Olarinmoye E, Aishat U, Faramade I, Olarewaju S, Asekun-Olarinmoye T. NATIONAL POLICIES AND PROGRAMMES ON SICKLE CELL DISEASE. (2020), 307–37.
28. Prime Minister launches National Sickle Cell Anaemia Elimination Mission from Shahdol, Madhya Pradesh [Internet] (2024). Available at: https://pib.gov.in/pib.gov.in/Pressreleaseshare.aspx?PRID=1936735 (accessed August 9, 2024)
29. Saxena D, Yasobant S, Golechha M. Situational analysis of sickle cell disease in gujarat, India. Indian J Community Med Off Publ Indian Assoc Prev Soc Med. (2017) 42:218–21. doi: 10.4103/ijcm.IJCM_284_16
30. World Sickle Cell Disease Awareness Day Celebrated Across India [Internet] (2024). Available at: https://www.pib.gov.in/www.pib.gov.in/Pressreleaseshare.aspx?PRID=2027018 (accessed August 9, 2024)
31. National Academies of Sciences E, Division H and M, Practice B on PH and PH, Action C on ASCDASP and B for, Martinez RM, Osei-Anto HA, et al. Delivering high-quality sickle cell disease care with a prepared workforce. In: Addressing sickle cell disease: A strategic plan and blueprint for action. Washington (DC): National Academies Press (US) (2020). Available at: https://www.ncbi.nlm.nih.gov/books/NBK566459/.
32. Dusic EJ, Theoryn T, Wang C, Swisher EM, Bowen DJ. Barriers, interventions, and recommendations: Improving the genetic testing landscape. Front Digit Health. (2022) 4:961128. doi: 10.3389/fdgth.2022.961128
33. Zhong A, Darren B, Loiseau B, He LQB, Chang T, Hill J, et al. Ethical, social, and cultural issues related to clinical genetic testing and counseling in low- and middle-income countries: a systematic review. Genet Med. (2021) 23:2270–80. doi: 10.1038/s41436-018-0090-9
34. Egesa WI, Nakalema G, Waibi WM, Turyasiima M, Amuje E, Kiconco G, et al. Sickle cell disease in children and adolescents: A review of the historical, clinical, and public health perspective of sub-saharan africa and beyond. Int J Pediatr. (2022) 2022:3885979. doi: 10.1155/2022/3885979
35. Khan H, Krull M, Hankins JS, Wang WC, Porter JS. Sickle cell disease and social determinants of health – A scoping review. Pediatr Blood Cancer. (2023) 70:e30089. doi: 10.1002/pbc.30089
36. Hegemann L, Narasimhan V, Marfo K, Kuma-Aboagye P, Ofori-Acquah S, Odame I. Bridging the access gap for comprehensive sickle cell disease management across sub-saharan africa: learnings for other global health interventions? Ann Glob Health. (2023) 89:76. doi: 10.5334/aogh.4132
37. Aygun B, Odame I. A global perspective on sickle cell disease. Pediatr Blood Cancer. (2012) 59:386–90. doi: 10.1002/pbc.24175
38. Lee L, Smith-Whitley K, Banks S, Puckrein G. Reducing health care disparities in sickle cell disease: A review. Public Health Rep Wash DC 1974. (2019) 134:599–607. doi: 10.1177/0033354919881438
39. Lee LH, Whisenton LH, Benger J, Lanzkron S. A community-centered approach to sickle cell disease and clinical trial participation: an evaluation of perceptions, facilitators, and barriers. Blood Adv. (2021) 5:5323–31. doi: 10.1182/bloodadvances.2020003434
Keywords: sickle cell disease, health inequity, public health, National Sickle Cell Anaemia Elimination Mission, sickle cell anemia
Citation: Kumar A and Bhattacharya S (2024) Sickle cell disease: a comparative perspective on global and national initiatives. Front. Hematol. 3:1457158. doi: 10.3389/frhem.2024.1457158
Received: 12 July 2024; Accepted: 05 August 2024;
Published: 27 August 2024.
Edited by:
John Strouboulis, King’s College London, United KingdomReviewed by:
Hafsat Ahmad, Ahmadu Bello University, NigeriaArne De Kreuk, King’s College Hospital NHS Foundation Trust, United Kingdom
Copyright © 2024 Kumar and Bhattacharya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alok Kumar, YWxva2FpaW1zQGdtYWlsLmNvbQ==; Sudip Bhattacharya, ZHJzdWRpcDgxQGdtYWlsLmNvbQ==