 Cristian C. Taborda
Cristian C. Taborda Amer M. Zeidan
Amer M. Zeidan Lourdes M. Mendez
Lourdes M. Mendez- Section of Hematology, Department of Internal Medicine, Yale Cancer Center and Smilow Cancer Hospital, Yale University School of Medicine, New Haven, CT, United States
Cancer-related somatic genetic alterations are detectable in the blood of individuals without hematologic malignancy, reflecting the outgrowth of a mutated stem/progenitor cell population, a phenomenon termed clonal hematopoiesis (CH). When accompanied by an unexplained cytopenia(s), CH is further refined to clonal cytopenia of undetermined significance (CCUS) whereas, the finding of a mutation/alteration in the setting of a normal complement of blood counts is called clonal hematopoiesis of indeterminate potential (CHIP). CHIP and CCUS are now recognized precursor conditions to myeloid neoplasms. Advances in the understanding of the epidemiology and clonal metrics associated with evolution to a myeloid malignancy has permitted the elaboration of risk stratification tools poised for use in the clinic and initial clinical investigations seeking to disrupt the natural history of high risk CHIP and CCUS. In this review, we focus on CCUS and the current understanding of its classification, risk stratification and potential therapeutic targets
1 Clonal hematopoiesis: an introduction
Cancer-related somatic genetic alterations are detectable in the blood of ostensibly healthy individuals. Such somatic genetic alterations include SNV, Indels, and also large chromosomal alterations or mosaic chromosomal alterations (1–5). Somatic genetic mosaicism within the hematopoietic system has been termed clonal hematopoiesis (CH). Several concurrent papers employed next-generation sequencing technology to define the frequency of mutations in cancer-related genes in large population cohorts revealing recurrently mutated myeloid driver genes, many increasing in frequency with age (6–9). The frequency of CH in individuals aged 70-80 was found to be approximately 10-20% (6, 7). In fact, subsequent studies have shown that in elderly individuals, oligoclonal hematopoiesis is pervasive, compared to polyclonal hematopoiesis in children and young adults, related to positive selection of recurrent genetic mutations in hematopoietic stem progenitor cells (10).
The commonplace occurrence of clonal mosaicism involving cancer-related genes raised the question of whether this was associated with an increased incidence of hematologic malignancy. Interestingly, even “silent” variants, that is, synonymous mutations in protein-coding regions or non-splice site mutations in noncoding regions – may have predictive value in the development of hematologic malignancies (11). Individuals with CH involving cancer-related mutations taken as a whole, had an 11- to 12-fold increased risk of hematologic malignancies related to clone size (6, 7). Clonal hematopoiesis has been seen in about 29.7% of patients with solid tumors and was associated with a two-fold increase in the development of therapy-related hematologic malignancy (11).
Yet, it was apparent that the overwhelming majority of individuals would not develop a blood cancer leading to the term, clonal hematopoiesis of indeterminate potential (CHIP). The impact of clonal mosaicism on phenotypes beyond cancer has since been the subject of a burgeoning literature. Early on an increased risk of cardiovascular and cerebrovascular disease (7) was appreciated. Notably, CH was associated with a 1.4-fold increased risk of all-cause mortality (6, 7). CH has now been associated with a wide variety of age-related phenotypes (12). CH may mechanistically tie together the increase in systemic inflammation observed in older persons, termed “inflammaging” and many age-related diseases (13, 14). Clonal cytopenia of undetermined significance (CCUS) was first reported in 2015 (15, 16) on the heels of the CHIP reports, but studies of CCUS have been comparatively scarce despite the imperative to characterize its epidemiology and pathogenesis based on early evidence of remarkably high rates of progression to myeloid neoplasms in subsets of CCUS (17).
2 Clonal cytopenias: subtypes within classification systems
With a body of literature substantiating the premalignant potential of CH, its various subtypes were codified in recent updates to the myeloid/leukemia classification systems by the WHO and ICC. CHIP is defined as CH bearing mutations in genes that are recurrently mutated in myeloid malignancies at a variant allele fraction (VAF) of >=2% in the absence of a hematologic malignancy or unexplained cytopenia (18). Cytopenia in the context of clonal hematopoiesis is defined as acquired, sustained, otherwise unexplained anemia (Hb <12 in females and <13 in males), neutropenia (<1.8 X 109/L), thrombocytopenia (<150 X 109/L) and these definitions are uniform for clonal cytopenias, MDS, MDS/MPN (19–21). Clonal cytopenias include clonal cytopenia of undetermined significance (CCUS), clonal monocytosis of undetermined significance (CMUS), clonal cytopenia and monocytosis of undetermined significance (CCMUS). CCUS is a form of CHIP associated with persistent cytopenia(s) (16). CHIP and CCUS are recognized as precursor conditions that can progress to become MDS and AML whereas CMUS and CCMUS are precursor conditions to CMML (20–22).
A note is made in both the 5th Edition of the WHO and ICC classification systems of the recently described VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) syndrome, an acquired, adult-onset multisystem inflammatory disease characterized by hematologic abnormalities (macrocytic anemia, vacuolated myeloid and erythroid progenitors) described in 2020. CH in VEXAS stems from HSPC bearing mutations in the E1 ubiquitin activating enzyme, UBA1 (23), which lies at the apex of the protein ubiquitination cascade, critical for cellular protein homeostasis. VEXAS is associated with a high incidence of MDS and plasma cell dyscrasias (23, 24). Thus, VEXAS lies on the spectrum of clonal disorders with premalignant potential (25) and has been put forth as a form of CCUS (26). Clonal myeloid dominance stems from UBA1-mutant HSPC (27, 28) which upregulate inflammatory pathways (23, 28) underscoring the intertwined origins of CH and inflammation.
The recognition of subtypes of CH as new nosological entities reflects the emphasis on classification based on defining genetic abnormalities (20, 21). Along the same lines, the list of MDS-defining genetic abnormalities in the context of cytopenia(s), irrespective of dysplasia has been updated to include MDS with SF3B1 mutation (20) and is added to del(5q), -7/del(7q), or a complex karyotype. MDS with bilallelic TP53 inactivation is an additional MDS genetic subtype (21). Like MDS, the myeloid precursor states are also qualified as therapy-related if following exposure to chemotherapy or radiation (20). Data from longitudinal studies of cytopenia cohorts are expected to inform further refinements in genetically defined nosologic entities.
3 CCUS: incidence and genetics
3.1 Incidence of CCUS
Amongst individuals with idiopathic cytopenia who do not meet the diagnostic criteria for MDS, the frequency of CCUS has been reported to be on the order of 30% (16, 29). Concordant with previous studies, in the largest cohort of patients with cytopenias to date, 400 of 1485 patients with unexplained cytopenias and a non-diagnostic bone marrow evaluation were found to have ≥1 mutation in a targeted myeloid panel covering 27 genes thus giving a 26.9% frequency of CCUS (30).
3.2 Genetic profile of CCUS
The genetic mutational spectrum of CCUS overlaps with that of CHIP and MDS. Mutations in one of three genes involved in epigenetic regulation account for two-thirds of recurrent mutations in cancer related genes found in healthy individuals without a hematologic phenotype. These genes are DNMT3A, TET2 and ASXL1 (DTA) (6, 7). In a prospective cohort of 311 individuals with idiopathic cytopenia(s), the 10 most frequent myeloid malignancy-related genetic mutations detected in the peripheral blood of individuals with CCUS were identified as TET2 (18.6%), ASXL1 (14.5%), SRSF2 (13.9%), SF3B1 (11.4%), DNMT3A (9.5%), RUNX1 (8.5%), TP53 (5.4%), JAK2 (5.1%), U2AF1 (4.7%), and EZH2 (3.8%) (17). A distinct prospective, unexplained cytopenia cohort of 1485 patients, likewise identified TET2 and SRSF2 from the bone marrow of individuals with CCUS, but the third most frequently mutated gene was DNMT3A (30). There is a high level of concordance with the established mutational landscape of MDS where the most frequent mutations are in TET2, SF3B1, ASXL1, SRSF2, DNMT3A, RUNX1, U2AF1, ZRSR2, STAG2, TP53 (31). Co-mutations with DTA and RUNX1, EZH2, CBL, BCOR, CUX1, TP53, or IDH1/IDH2 are reported to be highly specific for a myeloid neoplasm (MN) with myelodysplasia (MDS, MDS/MPN, AML with myelodysplasia-related changes) (17).
Comparatively less has been reported on mosaic structural variants or chromosomal abnormalities (mCA) associated with CCUS. Niroula and colleagues analyzed the frequency of autosomal mCA in nearly 400,000 individuals in the UK Biobank without a previous hematologic malignancy diagnosis, the majority of which had normal blood counts, and found that 0.38% had mCA associated with myeloid neoplasms, 0.84% had mCA associated with lymphoid neoplasms, 0.32% had mCA common to both myeloid and lymphoid neoplasms and 1.99% demonstrated other unclassified mCAs as detected by SNP array (32). Myeloid-mCA included loss of heterozygosity in JAK2, gain 1q, gain 9p, loss of heterozygosity in EP300 in individuals who went on to develop an MPN; deletion 20q, deletion 5q in individuals subsequently diagnosed with MDS (32, 33). Huber and colleagues studied a cohort of 222 CCUS cases and 698 MDS patients and observed that chromosomal alterations were found in 27% of patients with CCUS compared to 43% of patients with MDS with the most frequent alteration being -Y. Loss of chromosome Y was more frequent in CCUS, while loss of the long arm of chromosome 5 (-5q) and complex karyotypes were more frequent in MDS. Recurrent copy neutral loss of heterozygosity was observed in 9 CCUS patients, in 4q and 7q (in a subset involving TET2 and EZH2 respectively) (34). Composite genotypes with concurrent chromosomal alterations and somatic mutations are uncommon in CCUS but commonplace in MDS, in one cohort with a frequency of 14% and 37% respectively (34) concordant with a previous report of mCA synergizing with gene mutations in the risk of progression to leukemia (35).
Clonal expansion is the result of positive selection (36) that is context specific and occurs on the backdrop of germline genetics. Germline genetic predisposition loci can influence mutagenesis and clonality by compromising genome integrity/DNA repair pathways (TERT, CHEK2), increasing HSC self-renewal (TET2), selecting for specific mutations in the same gene (ATM, MPL, TP53) (37–39). Both germline predisposition loci and CH spectrum vary with ancestry (37, 40). Studies of germline predisposition variants and CCUS are rare. In an unselected cytopenia cohort found to have a hypoplastic marrow, targeted germline and somatic sequencing revealed that 27 of 402 patients (6.7%) carried a pathogenic/likely pathogenic germline variant (41). Germline variants can synergize with CH in the risk of progressing to a hematologic neoplasm flagging the need to account for germline-CH interactions in risk models (39).
3.3 CCUS and longitudinal blood counts
Cargo and colleagues described the association between the mutation pattern of CCUS and longitudinal blood counts (30). Similar to patients with MDS, patients with CCUS were found to have a significantly lower white cell count and platelet concentration than patients with idiopathic cytopenia of unknown significance (ICUS). Patients with mutations in chromatin modifier genes and transcription factor genes had progressive decreases in platelet and hemoglobin concentrations that were not appreciated in non-mutated patients. These were most pronounced in patients with ASXL1 mutations, with projected transfusion dependence in 4 years. Patients with TP53 mutations did not have a difference in hemoglobin or platelets but did have significantly lower white blood cell count, in particular neutrophil and monocyte counts. DNMT3A mutations had no effect on blood counts over the study period, supporting previous reports of mutation being low risk.
4 CCUS: risk stratification
CCUS has a variable risk of evolving into a myeloid neoplasm informed by clonal metrics, the gene mutation complement and mCA. Variant allele frequency (VAF) ≥10% and number of mutations ≥2, are associated with a positive predictive value of 0.86 and 0.88 for subsequent diagnosis of a MN (17). Specific genes and co-mutation patterns have also been observed to correlate with an increased risk of progression to a myeloid neoplasm. Spliceosome genes (SF3B1, SRSF2, U2AF1), JAK2, and RUNX1 mutations have positive predictive values for MN ranging from 0.88 to 0.97 irrespective of additional co-mutations (17). Applying unsupervised clustering to precursor states and myeloid neoplasms to identify genetically defined groups revealed a CH-like and MN-like group with the first largely composed of individuals with isolated DNMT3A mutations and the second of individuals with mutations in splicing factors, TP53, or DTA genes in combination with other mutations or isolated mutations in TET2, ASXL1 and less common mutations implicated in myeloid neoplasms. Comparing the CH-like and MN-like group with ICUS there was a significant difference in overall survival. Within the CCUS cohort, there was a marked difference in the cumulative incidence of progression to a myeloid neoplasm between the CH-like and MN-like groups (29).
In a small retrospective cohort, TET2 (39%), SRSF2 (26%) and ASXL1 (20%) were the most frequently mutated genes in CCUS that go on to evolve to MDS/AML. Acquisition of a mutation in a transcription factor or cell-signaling gene was observed at the time of progression from CCUS to a MN in 43% of patients (15). Large-scale longitudinal sequencing of the Lifelines cohort allowed prospective study of CH evolution. In contrast to TET2 mutated CH which was observed to acquire new gene mutations over time, DNTM3A and TP53 mutations did not increase the risk of additional gene mutations. The most common DNMT3A mutations did not significantly increase the risk of myeloid neoplasms. By contrast, JAK2, spliceosome and TP53 mutations were confirmed as “high risk” mutations associated with progression to myeloid malignancy as previously noted in retrospective studies (42).
Notably, genome wide association studies (GWAS) have identified germline variants associated with DNMT3A- and TET2-mutant CH that shed light on the predisposition to CH generally, but perhaps also suggest an inherited risk for low versus higher risk CH. For DNMT3A CH, the loci include 14q32.13-TCL1A, 5p15.33-TERT, 3q25.33-SMC4, 6q21-CD164, 11q22.3-ATM, 1q42.12-PARP1, 18q12.3-SETBP1, 22q12.1-CHEK2 and for TET2 CH, 5p15.33-TERT, 14q32.13-TCL1A and 7q32.2-TMEM209. As noted above, clone size is associated with the risk of progression to a myeloid neoplasm and germline variants associating with clone size have also been identified. (43). As regards the impact of mCA on CH disease progression, Gao and colleagues reported that mCA are an independent risk factor for progression of CH to a hematologic neoplasm (MDS, MPN, AML, CML, CLL) (35). Germline variants associated with increased risk for gene mutations and mCA show considerable overlap. Remarkably, germline variants portending increased risk for DNMT3A-CH reduce mCA risk (43) providing insight into clonal trajectories that are at least partially inherited.
Leeks and colleagues leveraged a large data set from the United Kingdom Biobank (UKB) to build a systematic risk prognostication tool for CHIP/CCUS (44). Using easily accessible clinical and laboratory features, they were able to generate a 10-year molecularly-based prognostic model for myeloid neoplasms. Key prognostic factors included age, mutation status (including mutation number and VAF), and blood incidences (red cell distribution width, mean corpuscular volume, and presence of cytopenia). Splicing factors and acute myeloid leukemia related genes (IDH1, IDH2, FLT3, and RUNX1) provided increased prognostic value for MN, while a single DNMT3A mutation conferred a smaller risk. The vast majority of patients in this cohort with CHIP/CCUS (~90%) had a 1% 10-year cumulative risk of myeloid neoplasm. Leeks and colleagues created the clonal hematopoiesis risk score (CHRS) with three risk categories: high risk (CHRS ≥ 12.5), intermediate risk (CHRS 10 to 12), and low risk (CHRS ≤ 9.5). The goal of these risk categories was to highlight a group of patients who have a low cumulative risk of developing a myeloid neoplasm (<2% incidence over 10 years). They found that the 10-year cumulative incidence of MN was 52.2%, 7.83%, and 0.67%% in the high, intermediate, and low risk CHIP/CCUS groups, respectively. This model was validated against an independent CCUS cohort (Pavia) and CCUS/CHIP cohort (DFCI/BWH). The performance of CHRS was not altered by the inclusion of TP53 as a high risk mutation and did not include information about mCA.
Concurrently, Gu and colleagues used the UK Biobank to develop three time-dependent disease-specific regression models for prognostication of MDS, AML, and MPN (33). They analyzed data from 454,340 participants to understand the genetic landscape of individuals who developed myeloid neoplasm within a 15 year follow-up period. Similar to prior studies, high risk mutations such as JAK2, SRSF2, SF3B1, and IDH2 were common in patients who developed MN while mutations in DNMT3A, TET2, and ASXL1 were more prevalent in patients with CH who did not develop MN. All three prognostic models were validated against two CCUS cohorts (Leeds and Pavia) with good performance. Based on these data, Gu and colleagues generated a web-based tool called MN-Predict to calculate the year-by-year risk of progression to myeloid neoplasm over 15 years. A strength of the MN-Predict is its ability to provide risk stratification for each myeloid neoplasm subtype as blood count parameters such as cytopenias may have opposite prognostic value in risk of MDS vs MPN. Like CHRS, MN-Predict does not incorporate information about mCA. Pertinent to both CHRS and MN-predict, limitations of the UK Biobank include younger median age (non-CHIP/CCUS 57 years and CHIP/CCUS 62 years) and predominately white (93.9%) racial/ethnic background.
The International Prognostic Scoring System–Molecular (IPSS-M) was developed as a clinical prognostic tool to provide individualized predictions of leukemia-free and overall survival for patients with MDS and incorporates molecular genetic mutations in addition to clinical and cytogenetic parameters (45). When the IPSS-M risk group was calculated for a cohort of patients with CCUS, 80% of patients were categorized as very low to low risk and as part of an exploratory analysis, the authors noted a trend towards differential OS comparing very low/low and all other patients with CCUS (34). A real-world analysis of IPSS-M applied to CCUS also found evidence of its predictive power to discriminate myeloid neoplasm-free and overall survival concluding that larger validation studies are warranted (46).
5 CCUS versus MDS
A single, academic institution comparison of the molecular and clinical characteristics of CCUS and low risk MDS (LR-MDS) patients highlighted considerable overlap. 187 patients were considered, 75 with CCUS and 112 with LR-MDS. 61% of the CCUS patients had >1 mutation, compared to 51% of the LR-MDS group. 83% of the evaluable CCUS patients with somatic mutations had at least one mutation with VAF >=20%. Within the limits of the short median follow-up of the CCUS group, 16 months, there was a 17% rate of transformation to MDS/AML and there was no significant difference in median OS between the CCUS and low risk MDS groups (47). Another study compared 222 cases of CCUS with 698 cases of MDS applying the IPSS-M. Similar to the prior study, within the limits of a short median follow-up of 18 months, there was no difference in overall survival between the patients with CCUS and MDS categorized as having very low and low risk disease by the IPSS-M (34).
This raises the question of how CCUS is distinct from MDS. MDS is a clonal hematopoietic disorder characterized by cytopenia(s), dysplasia in one or more myeloid lineages, and an increased risk of evolution to AML (48). CCUS is currently differentiated from MDS by the absence of 1) overt dysplasia (>=10% dysplasia in the erythroid, myeloid or megakaryocytic lineage), 2) increased blasts and/or 3) MDS-defining cytogenetic findings (17). As regards the first of these distinctions, gene-specific relationships between VAF and dysplasia have been observed, with certain mutations leading to overt dysplasia at low VAF (DTA gene + other, SF3B1+/- other, TP53 +/- other) whereas other mutations reach clonal dominance (SRSF2 +/- other, DTA-DTA, or DTA single) before leading to dysplasia (29). In other words, for specific genotypes, morphologic dysplasia is a late manifestation of mutant clones and thus, uninformative earlier in the clonal trajectory. Thus, it has been proposed that CCUS with certain mutation patterns may provide presumptive evidence of MDS independent of morphologic criteria (17, 29) and additional genetic subgroups within MDS are anticipated (20).
6 Therapeutic targets
The identification of myeloid neoplasm precursor conditions has ushered in clinical trials focused on CCUS/CHIP. Several approaches are being taken and strategies include repurposing medications for MDS, targeting specific genetic subtypes of predisease via actionable mutations and interventions aimed at mitigating dysregulated inflammatory signaling.
6.1 DNA methylation regulators: TET2
TET2 is a tumor suppressor that catalyzes the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) on the path to unmethylated cytosine (49, 50). TET2 mutations are early events in leukemogenesis (51) that combined with additional mutations increase the risk of myeloid neoplasms (52–54). Inactivating mutations in TET2 result in increased HSC self-renewal and frequency (55, 56). Ascorbate promotes the activity of TET2 in hematopoietic stem cells and is specifically enriched in HSC/MPP at 2-20 fold increased levels compared to other hematopoietic progenitors and declines with differentiation. In experimental models, ascorbate deficiency leads to decreased 5hmC in HSC/MPP, increased HSC frequency and reconstitution capacity which can be reversed by dietary ascorbate. Moreover, the effects of ascorbate deficiency on HSC phenocopies inactivating mutations of Tet2. Ascorbate depletion additionally accelerated leukemogenesis in a model of heterozygous Tet2 deleted leukemia whereas repletion prolonged survival (57). Thus, ascorbic acid is currently being investigated in patients with CCUS (NCT03418038; NCT03682029).
The final safety and efficacy results of the phase 2, single site trial of high-dose intravenous ascorbic acid in patients with TET2 mutant CCUS was recently reported by Xie and colleagues (NCT03418038). The treatment consisted of high dose intravenous ascorbic acid administered three times weekly over twelve weeks. The primary endpoint was hematologic improvement, as defined by the MDS IWG 2018 criteria, at week 20. Secondary endpoints included safety and adverse event grading. Ten patients were enrolled. The median number of mutations was 3, suggesting higher risk disease (see section 4). None of the subjects met criteria for hematologic improvement. Interestingly, 3 patients displayed improvement in cytopenias per CTCAE grading and correlative studies suggested a connection between DNA hypomethylation at specific enhancer sites and cytopenia improvement (58).
6.2 Epigenetic regulators: IDH1/2
Recurrent mutations in isocitrate dehydrogenase 1 and 2 (IDH1, IDH2) are found in myeloid neoplasms and predisease (59, 60). These enzymes function in the Krebs cycle to catalyze the formation of α-ketoglutarate (α-KG) from isocitrate. Mutant IDH1/2 generate the oncometabolite 2-HG from α-KG (61, 62) which competes for binding to α-KG dependent enzymes such as TET2 and several lysine demethylases resulting in aberrant epigenetic regulation (63) and a differentiation block (64). 2-HG has also been demonstrated to perturb T cell metabolism and function impairing the antitumor T cell response (65, 66) and indicating that IDH1/2 mutations affect tumor biology through both cell-intrinsic and -extrinsic mechanisms. Clonal hematopoiesis with IDH1 and IDH2 precede AML with the same mutations (67). IDH1/2 mutations have been identified as a high risk feature in clonal hematopoiesis suggesting that therapeutic strategies targeting mutant IDH1/2-driven biology in the future could alter the natural history of mutant predisease preventing or slowing progression to myeloid neoplasms.
Small molecule inhibitors of mutant IDH2, enasidenib, and mutant IDH1, ivosidenib and olutasidenib, are approved for the treatment of IDH1/2 mutant AML. These inhibitors suppress oncometabolite production, promote differentiation and overall are well tolerated (68). Enasidenib is currently being investigated in a phase I study for the treatment of IDH2-mutated CCUS with the primary objective being the potential of enasidenib to induce hematologic improvement and secondary objectives including assessing for a reduction in the burden of IDH2 mutation as well as AML- and MDS-free survival (NCT05102370). Ivosidenib is under investigation in individuals with IDH1-mutated CCUS with the primary objective also being hematologic improvement. (NCT05030441).
6.3 Dysregulated inflammatory signaling
6.3.1 IL1β
Inflammaging is a term that has been coined to refer to the increase in systemic inflammation observed in aging people and murine models; for example, increased circulating levels of the pro-inflammatory cytokines TNFa, IL1-β, IL-6 (13, 69). Inflammatory cytokines are elevated across the spectrum of CH ranging from CHIP to MDS with driver-gene specific associations. TET2 mutated CHIP is associated with elevated IL1-β whereas JAK2 and SF3B1 mutated CHIP correlate with elevated IL-18 (37). Proteomic profiling of individuals with and without CH shows enrichment of signatures related to inflammation and immune response to infection in individuals with CH. These same pathways were also enriched individuals with CH and anemia (70). Increased inflammatory mediators are reported to favor the clonal expansion of Tet2-deficient HSPC (71–73). IL-1 signaling mediates the expansion of Tet2+/- clonal hematopoiesis in aging (74). Genetic deletion of the IL-1 receptor, pharmacologic inhibition of IL-1 signaling or antibiotics (suppression of the bacterial microbiome) in a model of the aging hematopoietic system, results in mitigation of age associated inflammatory signatures and myeloid biased differentiation (75).
Canakinumab is a monoclonal antibody targeting IL1-β which was investigated in the CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) trial in subjects with high cardiovascular risk randomized to receive placebo or the investigational agent. CV events were reduced by Canakinumab, particularly in subjects with TET2 mutation, but there was no survival benefit. An increase in infections was observed in patients randomized to Canakinumab (76). An exploratory analysis of the CANTOS study showed that the investigational agent improved anemia, particularly in individuals with CH mutations and anemia, in concert with reduced enrichment of proteomic signatures related to inflammation and immune response to infection (77). Canakinumab is currently being studied in MDS with the initial experience indicating it is safe in this population (NCT04239157; (78)). Canakinumab is also being studied in a randomized, placebo-controlled trial in high risk CCUS patients as a therapeutic intervention to prevent progression to a myeloid neoplasm with secondary endpoints including the effect of canakinumab on hematologic overall response rates and cardiovascular episodes compared to controls as well as quality of life (NCT05641831).
6.3.2 TGFβ-SMAD
With initial observations suggesting overlap between high risk forms of clonal hematopoiesis and low risk MDS as regards inflammation, cardiovascular risk, cytopenias and survival (citation) a logical step is to explore the impact of LR-MDS therapy on clonal hematopoiesis. TGFβ-SMAD signaling regulates hematopoiesis (79) and is overactivated in MDS. Preclinical work targeting overactivated TGFβ-SMAD signaling reported improved hematopoiesis including attenuated anemia in models of MDS (80, 81). Luspatercept, is a ligand trap that contains the extracellular domain of the activin receptor II (a member of the TGFβ receptor superfamily) connected to the human IgG1 Fc (81). Luspatercept was approved in August 2023 for first-line treatment of transfusion dependent LR-MDS based on the results of the COMMANDS trial (82). A subgroup analysis of the COMMANDS trial queried the impact of luspatercept in the subset of patients with clonal hematopoiesis related mutations in TET2, DNMT3A, ASXL1 and SF3B1. Interestingly, luspatercept improved other blood counts concomitant with anemia, including WBC count. It also led to downregulation of inflammatory signatures and proinflammatory cytokines/chemokines as well as significantly decreased hepcidin and NT-proBNP. These findings create a rationale for including patients with CCUS in future investigations with luspatercept (83).
6.3.3 JAK-STAT
VEXAS lies at the crossroads of clonal hematopoiesis and severe inflammatory disease mediated by aberrant, UBA1-mutant hematopoietic stem progenitor cells and innate immune effector cells. Characterized by cytopenias, with vacuolization of hematopoietic precursors on pathology, frequent manifestations of VEXAS are fever and other constitutional complaints, rash (including neutrophilic dermatosis), pulmonary infiltrates, chondritis, arthritis, vasculitis, thrombosis. The initial report of VEXAS also offered the first glimpse of upregulated innate immune signaling pathways and potential targets to abort severe end organ damage. Transcriptomics of isolated innate immune effector cells showed upregulated TNFα, IL-6, IFNγ signaling (23). A recently described, rare disease with an estimated frequency of less than 1/10,000 (84) to date, the majority of available treatment data is retrospective. An international, retrospective analysis of patient outcomes following treatment with JAK inhibitors reported that 15 of 30 patients (50%) had a clinical response. Response rates were highest in patients treated with ruxolitinib compared to other JAK inhibitors. Response to ruxolitinib was irrespective of an accompanying myeloid neoplasm. The median duration of clinical response was not reached with a median follow-up of 6.9 months. A significant increase in hemoglobin and platelet count was observed in patients treated with ruxolitinib. However, in two patients who underwent serial monitoring of mutant UBA1 variant allele frequency, expansion of the UBA1-mutant clone was measured (85).
6.4 Hypomethylating agents
The efficacy of azacitadine in the treatment of autoimmune disorders associated with MDS or CMML has previously been reported (86). More recently, the potential for azacitadine to mitigate autoinflammation driven by UBA1-mutant clonal hematopoiesis has also been reported. A retrospective study analyzed 19 male patients with myeloid dysplasia and autoinflammatory disease including 11 with VEXAS. Time to next treatment was examined. The median time to next treatment was 21.9 months for azacitadine (87). Notably, complete molecular remission of VEXAS following treatment with azacitadine has been observed (88). An open-label, single-arm multicenter, phase II study of Azacitadine in patient with MDS/CMML and steroid dependent/refractory systemic autoinflammatory disease (NCT02985190) reported the first prospective results of VEXAS (in association with a myeloid neoplasm), treated with Azacitadine and observed that 75% of patients with VEXAS responded with a rapid decrease in inflammatory disease accompanied by reduced steroid requirement (89). UBA1-mutant clonal hematopoiesis, with or without an associated myeloid neoplasm, thus highlights the immunomodulatory dimension to azacitadine activity.
7 Discussion
The genomic revolution led to the identification of “predisease” in the hematopoietic tissue. The recent inclusion by the ICC and WHO of predisease and MDS-defining genetic alterations reflect our evolving diagnostic understanding of myeloid neoplasms and their precursors. Several studies have demonstrated that CCUS and low risk MDS have similar outcomes, suggesting that in the future, additional molecular genetic mutations and/or co-mutation patterns will join the ranks of MDS-defining genetic alterations.
Cohort studies have shown that CCUS is often present in cases of unexplained cytopenias. The mutation profile of CCUS overlaps between CHIP and MDS. Mutations in transcription factor or cell-signaling genes have been observed in cases of progression from CCUS to MN. Clonal metrics such as VAF ≥10%, number of mutations ≥2, and presence of high risk mutations have high positive predictive value for progression to MN. Advances in risk stratification of CH suggest individuals at high risk of developing myeloid neoplasm can be identified years in advance of overt disease, based on clinical and genetic metrics. These patients may benefit from referral to Hematology for observation and possible enrollment in clinical trials (Figure 1).
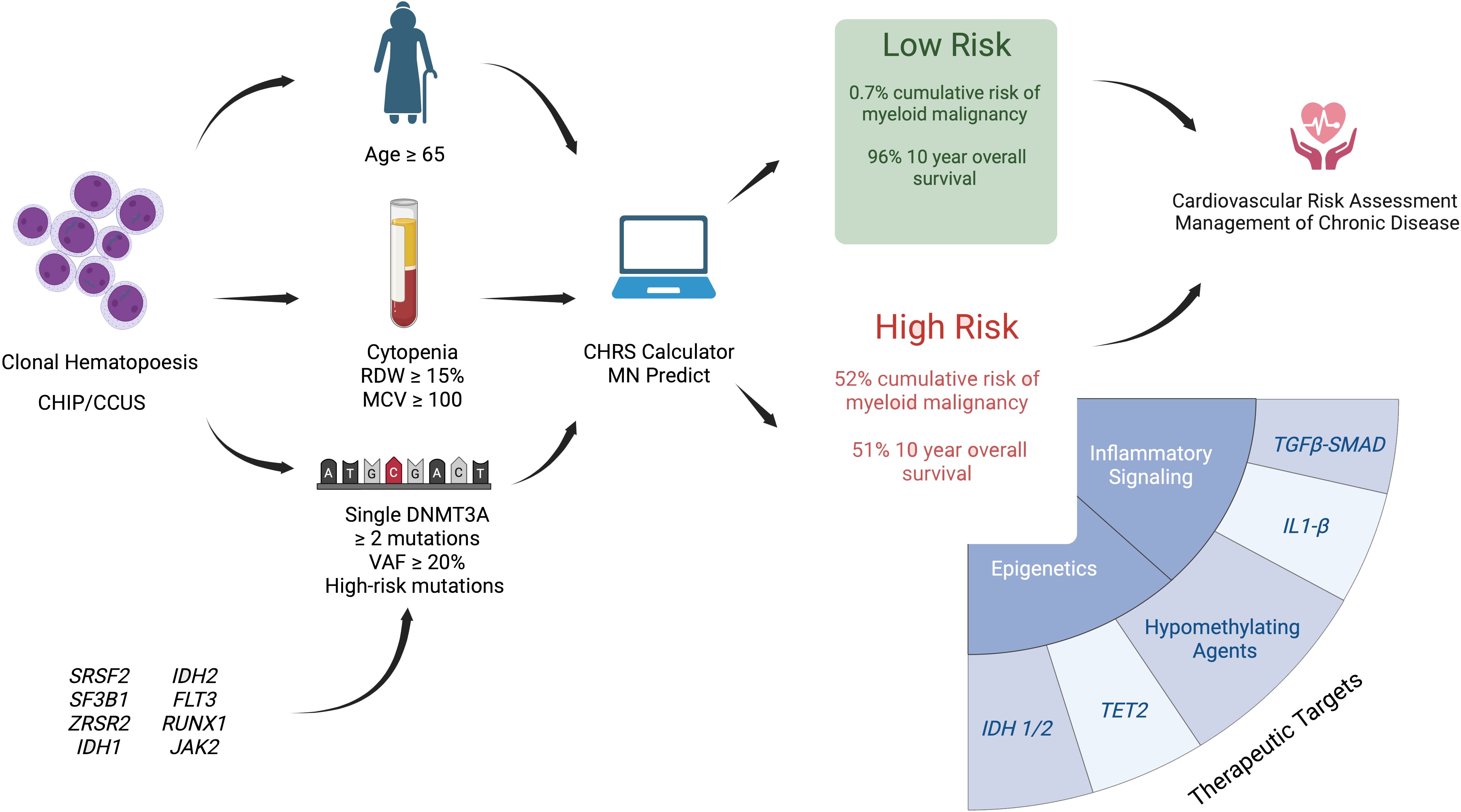
Figure 1 Illustration of proposed risk stratification models for clonal hematopoiesis and potential future interventions for high-risk CCUS. Presence of clonal hematopoiesis may be detected through tumor profiling in solid cancers, hereditary cancer predisposition testing, cell free circulating tumor DNA testing, and evaluation of cytopenia. Risk stratification is based on patient’s age, presence of CCUS vs CHIP, blood parameters including degree of cytopenia, RDW, and MCV, as well as molecular characteristics of CH (presence of DNMT3A mutation, number of mutations, presence of high risk mutations, and VAF ≥ 20%). Two web based clonal hematopoiesis risk calculators have been published - CHRS (44) and MN Predict 33 #174, but remain to be validated. The potential of the IPSS-M, developed for MDS prognostication, to risk stratify CCUS is under investigation. Clinical trials for CCUS include interventions aimed at modification of epigenetic regulation and inflammatory signaling. Cardiovascular risk assessment and management of chronic disease is indicated in all CHIP/CCUS. Created with Biorender.com.
Two risk stratification models – CHRS and MN-Predict – have been developed to improve the prognostication of CHIP/CCUS. These models use clinical, laboratory, and molecular parameters to provide either year-to-year or 10-year probability of MN. The CHRS calculator is designed to identify a small group of individuals at high risk of progression to MN who may warrant referral to hematology. The MN-Predict calculator incorporates metabolic parameters in addition to blood indices to provide a more granular year-by-year risk of progression to each MN subtype. Although tested against external CHIP/CCUS cohorts, these models have yet to undergo prospective validation or head-to-head comparison. Both models were developed using the UK Biobank data set which has several limitations including the cohort’s relatively young age, lack of bone marrow assessments at enrollment, and low incidence of CHIP/CCUS. Unlike the IPPS-M for MDS, these models omit the presence of chromosomal abnormalities which may further enhance prognostic value.
Multiple approaches are under investigation with the aim of interrupting the progression of CCUS. Therapeutic strategies have focused on addressing aberrant epigenetic regulation or dysregulated inflammatory signaling. Hematologic response rate and evaluation of clonal metrics have emerged as useful primary-end points in CCUS clinical trials, although a standardized response assessment is still needed. Further evidence-based recommendations and guidelines are needed for the risk stratification and management of CCUS.
Author contributions
CT: Writing – original draft, Writing – review & editing. AZ: Writing – review & editing. LM: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
AM received research funding (institutional) from Celgene/BMS, Abbvie, Astex, Pfizer, Kura, Medimmune/AstraZeneca, Boehringer-Ingelheim, Incyte, Takeda, Novartis, Shattuck Labs, Geron, Foran, and Aprea. AMZ participated in advisory boards, and/or had a consultancy with and received honoraria from AbbVie, Pfizer, Celgene/BMS, Jazz, Incyte, Agios, Servier, Boehringer-Ingelheim, Novartis, Astellas, Daiichi Sankyo, Geron, Taiho, Seattle Genetics, Otsuka, BeyondSpring, Takeda, Ionis, Amgen, Janssen, Genentech, Epizyme, Syndax, Gilead, Kura, Chiesi, ALX Oncology, BioCryst, Notable, Orum, Mendus, Zentalis, Schrodinger, Regeneron, Syros, and Tyme. AMZ served on clinical trial committees for Novartis, Abbvie, Gilead, Syros, BioCryst, Abbvie, ALX Oncology, Kura, Geron and Celgene/BMS. LM consults for Inventiva and has consulted for Rigel.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CH, clonal hematopoiesis; CCUS, clonal cytopenia of undetermined significance; CHIP, clonal hematopoiesis of indeterminate potential; VAF, variant allele fraction; MDS, myelodysplastic syndromes; MDS/MPN, myelodysplastic/myeloproliferative neoplasms; CMUS, clonal monocytosis of undetermined significance; CCMUS, clonal cytopenia and monocytosis of undetermined significance; AML, acute myeloid leukemia; CMML, Chronic myelomonocytic leukemia; VEXAS, Vacuoles E1 enzyme X-linked Autoinflammatory Somatic syndrome; MN, myeloid neoplasm; ICUS, idiopathic cytopenia of unknown significance; CHRS, clonal hematopoiesis risk score; mCAs, mosaic chromosomal abnormalities; IPSS-M, International Prognostic Scoring System–Molecular.
References
1. Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood. (1995) 86:3118–22. doi: 10.1182/blood.V86.8.3118.3118
2. Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. (1996) 88:59–65. doi: 10.1182/blood.V88.1.59.59
3. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. (2012) 44:1179–81. doi: 10.1038/ng.2413
4. Forsberg LA, Rasi C, Razzaghian HR, Pakalapati G, Waite L, Thilbeault KS, et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet. (2012) 90:217–28. doi: 10.1016/j.ajhg.2011.12.009
5. Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, Mchugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. (2012) 44:642–50. doi: 10.1038/ng.2271
6. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
7. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
8. Xie M, Lu C, Wang J, Mclellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and Malignancies. Nat Med. (2014) 20:1472–8. doi: 10.1038/nm.3733
9. Mckerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. (2015) 10:1239–45. doi: 10.1016/j.celrep.2015.02.005
10. Mitchell E, Spencer Chapman M, Williams N, Dawson KJ, Mende N, Calderbank EF, et al. Clonal dynamics of hematopoiesis across the human lifespan. Nature. (2022) 606:343–50. doi: 10.1038/s41586-022-04786-y
11. Stonestrom AJ, Menghrajani KN, Devlin SM, Franch-Exposito S, Ptashkin RN, Patel SY, et al. High-risk and silent clonal hematopoietic genotypes in patients with nonhematologic cancer. Blood Adv. (2024) 8:846–56. doi: 10.1182/bloodadvances.2023011262
12. Mendez LM, Patnaik MM. Clonal Hematopoiesis: Origins and determinants of evolution. Leuk Res. (2023) 129:107076. doi: 10.1016/j.leukres.2023.107076
13. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
14. Winter S, Gotze KS, Hecker JS, Metzeler KH, Guezguez B, Woods K, et al. Clonal hematopoiesis and its impact on the aging osteo-hematopoietic niche. Leukemia. (2024). 38:936–46. doi: 10.1038/s41375-024-02226-6
15. Cargo CA, Rowbotham N, Evans PA, Barrans SL, Bowen DT, Crouch S, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. (2015) 126:2362–5. doi: 10.1182/blood-2015-08-663237
16. Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. (2015) 126:2355–61. doi: 10.1182/blood-2015-08-667063
17. Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. (2017) 129:3371–8. doi: 10.1182/blood-2017-01-763425
18. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
19. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Cytopenia levels for aiding establishment of the diagnosis of myelodysplastic syndromes. Blood. (2016) 128:2096–7. doi: 10.1182/blood-2016-07-728766
20. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
21. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of hematolymphoid tumors: myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
22. Cargo C, Cullen M, Taylor J, Short M, Glover P, Van Hoppe S, et al. The use of targeted sequencing and flow cytometry to identify patients with a clinically significant monocytosis. Blood. (2019) 133:1325–34. doi: 10.1182/blood-2018-08-867333
23. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
24. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicenter case series of 116 French patients. Br J Dermatol. (2022) 186:564–74. doi: 10.1111/bjd.20805
25. Malcovati L. VEXAS: walking on the edge of Malignancy. Blood. (2023) 142:214–5. doi: 10.1182/blood.2023020772
26. Kusne Y, Fernandez J, Patnaik MM. Clonal hematopoiesis and VEXAS syndrome: survival of the fittest clones? Semin Hematol. (2021) 58:226–9. doi: 10.1053/j.seminhematol.2021.10.004
27. Gutierrez-Rodrigues F, Kusne Y, Fernandez J, Lasho T, Shalhoub R, Ma X, et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood. (2023) 142:244–59. doi: 10.1182/blood.2022018774
28. Wu Z, Gao S, Gao Q, Patel BA, Groarke EM, Feng X, et al. Early activation of inflammatory pathways in UBA1-mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep Med. (2023) 4:101160. doi: 10.1016/j.xcrm.2023.101160
29. Galli A, Todisco G, Catamo E, Sala C, Elena C, Pozzi S, et al. Relationship between clone metrics and clinical outcome in clonal cytopenia. Blood. (2021) 138:965–76. doi: 10.1182/blood.2021011323
30. Cargo C, Bernard E, Beinortas T, Bolton KL, Glover P, Warren H, et al. Predicting cytopenias, progression, and survival in patients with clonal cytopenia of undetermined significance: a prospective cohort study. Lancet Haematol. (2024) 11:e51–61. doi: 10.1016/S2352-3026(23)00340-X
31. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. (2014) 28:241–7. doi: 10.1038/leu.2013.336
32. Niroula A, Sekar A, Murakami MA, Trinder M, Agrawal M, Wong WJ, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med. (2021) 27:1921–7. doi: 10.1038/s41591-021-01521-4
33. Gu M, Kovilakam SC, Dunn WG, Marando L, Barcena C, Mohorianu I, et al. Multiparameter prediction of myeloid neoplasia risk. Nat Genet. (2023) 55:1523–30. doi: 10.1038/s41588-023-01472-1
34. Huber S, Baer C, Hutter S, Wossidlo N, Hoermann G, Pohlkamp C, et al. Genomic landscape of CCUS compared to MDS and its implications on risk prediction. Leukemia. (2024). doi: 10.1038/s41375-024-02273-z
35. Gao T, Ptashkin R, Bolton KL, Sirenko M, Fong C, Spitzer B, et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. (2021) 12:338. doi: 10.1038/s41467-020-20565-7
36. Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TE, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. (2020) 367:1449–54. doi: 10.1126/science.aay9333
37. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal hematopoiesis in 97,691 whole genomes. Nature. (2020) 586:763–8. doi: 10.1038/s41586-020-2819-2
38. Mustjoki S, Young NS. Somatic mutations in “Benign” Disease. N Engl J Med. (2021) 384:2039–52. doi: 10.1056/NEJMra2101920
39. Liu J, Tran D, Chan ICC, Wiley B, Watson C, Batachi-Boyou AL, et al. Genetic determinants of clonal hematopoiesis and progression to hematologic Malignancies in 479,117 individuals. Blood. (2023) 142:811. doi: 10.1182/blood-2023-182423
40. Wen S, Hu F, Nag A, Tachmazidou I, Deevi SVV, Taiy H, et al. Ancestry-specific genetic determinants of clonal haematopoiesis: a comparative analysis of 136,401 admixed Americans and 419,228 Europeans. Blood. (2023) 142(Supplement 1):816. doi: 10.1182/blood-2023-173484
41. Molteni E, Bono E, Galli A, Elena C, Ferrari J, Fiorelli N, et al. Prevalence and clinical expression of germ line predisposition to myeloid neoplasms in adults with marrow hypocellularity. Blood. (2023) 142:643–57. doi: 10.1182/blood.2022019304
42. Van Zeventer IA, De Graaf AO, Salzbrunn JB, Nolte IM, Kamphuis P, Dinmohamed A, et al. Evolutionary landscape of clonal hematopoiesis in 3,359 individuals from the general population. Cancer Cell. (2023) 41:1017–1031 e4. doi: 10.1016/j.ccell.2023.04.006
43. Kar SP, Quiros PM, Gu M, Jiang T, Mitchell J, Langdon R, et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat Genet. (2022) 54:1155–66. doi: 10.1038/s41588-022-01121-z
44. Weeks LD, Niroula A, Neuberg D, Wong W, Lindsley RC, Luskin M, et al. Prediction of risk for myeloid Malignancy in clonal hematopoiesis. NEJM Evid. (2023) 2:1–15. doi: 10.1056/EVIDoa2200310
45. Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. (2022) 1:EVIDoa2200008. doi: 10.1056/EVIDoa2200008
46. Nanaa A, Al-Kali A, Jevremovic D, Nguyen PL, Greipp P, He R, et al. IPSS-M - use for predicting survival and progression in patients with ccus - a retrospective multi-institutional study. ASH annual meeting and exposition. Blood. (2023) 142:4590. doi: 10.1182/blood-2023-191048
47. Li M, Binder M, Lasho T, Ferrer A, Gangat N, Al-Kali A, et al. Clinical, molecular, and prognostic comparisons between CCUS and lower-risk MDS: a study of 187 molecularly annotated patients. Blood Adv. (2021) 5:2272–8. doi: 10.1182/bloodadvances.2020003976
48. Cazzola M. Myelodysplastic syndromes. N Engl J Med. (2020) 383:1358–74. doi: 10.1056/NEJMra1904794
49. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
50. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. (2011) 333:1300–3. doi: 10.1126/science.1210597
51. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. (2012) 4:149ra118. doi: 10.1126/scitranslmed.3004315
52. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid Malignancies. Blood. (2009) 114:144–7. doi: 10.1182/blood-2009-03-210039
53. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. (2009) 360:2289–301. doi: 10.1056/NEJMoa0810069
54. Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. (2015) 27:502–15. doi: 10.1016/j.ccell.2015.03.009
55. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U.S.A. (2011) 108:14566–71. doi: 10.1073/pnas.1112317108
56. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. (2011) 20:11–24. doi: 10.1016/j.ccr.2011.06.001
57. Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, et al. Ascorbate regulates hematopoietic stem cell function and leukemogenesis. Nature. (2017) 549:476–81. doi: 10.1038/nature23876
58. Xie Z, Mccullough K, Lasho TL, Fernandez JA, Finke CM, Amundson MR, et al. Phase II study assessing safety and preliminary efficacy of high dose ascorbic acid in patients with TET2 mutant clonal cytopenias. ASH annual meeting. Blood. (2023) 142:4611. doi: 10.1182/blood-2023-186595
59. Mardis ER, Ding L, Dooling DJ, Larson DE, Mclellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. (2009) 361:1058–66. doi: 10.1056/NEJMoa0903840
60. Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. (2010) 28:2348–55. doi: 10.1200/JCO.2009.27.3730
61. Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. (2010) 207:339–44. doi: 10.1084/jem.20092506
62. Agrawal M, Niroula A, Cunin P, Mcconkey M, Shkolnik V, Kim PG, et al. TET2-mutant clonal hematopoiesis and risk of gout. Blood. (2022) 140:1094–103. doi: 10.1182/blood.2022015384
63. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
64. Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. (2014) 14:329–41. doi: 10.1016/j.stem.2013.12.016
65. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
66. Notarangelo G, Spinelli JB, Perez EM, Baker GJ, Kurmi K, Elia I, et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science. (2022) 377:1519–29. doi: 10.1126/science.abj5104
67. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. (2018) 24:1015–23. doi: 10.1038/s41591-018-0081-z
68. Issa GC, Dinardo CD. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. (2021) 11:107. doi: 10.1038/s41408-021-00497-1
69. Rea IM, Gibson DS, Mcgilligan V, Mcnerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. (2018) 9:586. doi: 10.3389/fimmu.2018.00586
70. Woo J, Lu D, Lewandowski A, Xu H, Serrano P, Healey M, et al. Effects of IL-1beta inhibition on anemia and clonal hematopoiesis in the randomized CANTOS trial. Blood Adv. (2023) 7:7471–84. doi: 10.1182/bloodadvances.2023011578
71. Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp Hematol. (2018) 59:60–5. doi: 10.1016/j.exphem.2017.11.002
72. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–849 e5. doi: 10.1016/j.stem.2018.10.013
73. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukemic myeloproliferation in a Tet2-deficient host. Nature. (2018) 557:580–4. doi: 10.1038/s41586-018-0125-z
74. Caiado F, Kovtonyuk LV, Gonullu NG, Fullin J, Boettcher S, Manz MG. Aging drives Tet2+/- clonal hematopoiesis via IL-1 signaling. Blood. (2023) 141:886–903. doi: 10.1182/blood.2022016835
75. Kovtonyuk LV, Caiado F, Garcia-Martin S, Manz EM, Helbling P, Takizawa H, et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood. (2022) 139:44–58. doi: 10.1182/blood.2021011570
76. Ridker PM, Everett BM, Thuren T, Macfadyen JG, Chang WH, Ballantyne C, et al. Anti-inflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
77. Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR, et al. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun. (2018) 9:1900. doi: 10.1038/s41467-018-04234-4
78. Garcia-Manero G, Adema V, Urrutia S, Ma F, Yang H, Ganan-Gomez I, et al. Clinical and biological effects of canakinumab in lower-risk myelodysplastic syndromes (MDS): results from a phase 2 clinical trial. ASH annual meeting. Blood. (2022) 140:2078–80. doi: 10.1182/blood-2022-167703
79. Blank U, Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood. (2015) 125:3542–50. doi: 10.1182/blood-2014-12-618090
80. Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. (2008) 112:3434–43. doi: 10.1182/blood-2008-02-139824
81. Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. (2014) 20:408–14. doi: 10.1038/nm.3512
82. Platzbecker U, Della Porta MG, Santini V, Zeidan AM, Komrokji RS, Shortt J, et al. Efficacy and safety of luspatercept versus epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): interim analysis of a phase 3, open-label, randomized controlled trial. Lancet. (2023) 402:373–85. doi: 10.1016/S0140-6736(23)00874-7
83. Hasan M, Vodala S, Hayati S, Garcia-Manero G, Gandhi AK, Surangani RNVS. Clonal hematopoiesis-related mutations are associated with favorable clinical benefit following luspatercept treatment in patients with lower-risk myelodysplastic syndrome. ASH annual meeting. Blood. (2023), 142:3214. doi: 10.1182/blood-2023-178666
84. Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA. (2023) 329:318–24. doi: 10.1001/jama.2022.24836
85. Heiblig M, Ferrada MA, Koster MJ, Barba T, Gerfaud-Valentin M, Mekinian A, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood. (2022) 140:927–31. doi: 10.1182/blood.2022016642
86. Fraison JB, Mekinian A, Grignano E, Kahn JE, Arlet JB, Decaux O, et al. Efficacy of Azacitidine in autoimmune and inflammatory disorders associated with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Res. (2016) 43:13–7. doi: 10.1016/j.leukres.2016.02.005
87. Bourbon E, Heiblig M, Gerfaud Valentin M, Barba T, Durel CA, Lega JC, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. (2021) 137:3682–4. doi: 10.1182/blood.2020010177
88. Sockel K, Gotze K, Ganster C, Bill M, Georgi JA, Balaian E, et al. VEXAS syndrome: complete molecular remission after hypomethylating therapy. Ann Hematol. (2024) 103:993–7. doi: 10.1007/s00277-023-05611-w
89. Mekinian A, Zhao LP, Chevret S, Desseaux K, Pascal L, Comont T, et al. A Phase II prospective trial of azacitidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia. (2022) 36:2739–42. doi: 10.1038/s41375-022-01698-8
Keywords: CCUS, clonal hematopoiesis, ChIP, clonal evolution, VEXAS
Citation: Taborda CC, Zeidan AM and Mendez LM (2024) Clonal cytopenia of undetermined significance: definitions, risk and therapeutic targets. Front. Hematol. 3:1419323. doi: 10.3389/frhem.2024.1419323
Received: 18 April 2024; Accepted: 17 June 2024;
Published: 12 July 2024.
Edited by:
Bruno António Cardoso, Catholic University of Portugal, PortugalReviewed by:
Ioanna Triviai, Max Planck Institute for Molecular Genetics, GermanyHugh Young Rienhoff Jr., Institute for Further Study, United States
Copyright © 2024 Taborda, Zeidan and Mendez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lourdes M. Mendez, bG91cmRlcy5tZW5kZXpAeWFsZS5lZHU=; Amer M. Zeidan, YW1lci56ZWlkYW5AeWFsZS5lZHU=