 Javier Sanmartín-Martínez
Javier Sanmartín-Martínez Valerie R. Wiersma
Valerie R. Wiersma Anna E. Marneth
Anna E. Marneth- 1Department of Laboratory Medicine, Laboratory of Hematology, Research Institute for Medical Innovation, Radboud University Medical Center, Nijmegen, Netherlands
- 2Department of Hematology, University Medical Center Groningen (UMCG), University of Groningen, Groningen, Netherlands
Although the involvement of glycan structures in diseases has long been recognized, their detailed and high-throughput investigation has only recently been made possible due to technological advancements. For this reason, glycosylation is a generally understudied phenomenon, however it could provide critical information on the pathobiology of many disorders by virtue of its widespread abundance and critical role in protein function. Here, we focus on myeloid malignancies, conditions for which the survival rates are often poor and curative therapeutic options are generally limited. We review the current literature on (1) N-glycosylation of major hematopoietic growth receptors found mutated in myeloid malignancies, (2) chemoresistance through intracellular glycan-related processes, and (3) mechanisms by which altered N-glycosylation contributes to interactions between myeloid blasts and bone marrow stromal cells leading to niche hijacking. For each topic, we describe the related pathobiology and its (potential) clinical implications. The combination of glycoproteomic and genomic information is expected to result in a deeper molecular understanding of the pathobiology of these diseases, which could subsequently be used for improving prognostication and therapeutic strategies.
Introduction
Even though the genomic revolution has fundamentally changed our understanding of human biology, offering a more intricate perspective on the pathophysiology of nearly all human diseases, considerable gaps remain in our understanding of protein function and interindividual variability. In order to gain further understanding into disease progression, recent efforts have aimed to elucidate the role of post-translational modifications in protein functionality. Key among these modifications for its widespread abundance and range of functions is glycosylation: the covalent attachment of oligosaccharides/glycans to proteins, often through nitrogen (N-) or oxygen (O-) linkages. Unlike protein biosynthesis, which occurs following a template provided by gene transcription and subsequent protein translation, glycan biosynthesis is a non-template driven and hence more flexible process guided by enzymes whose function it is to add or remove sugar moieties from proteins or lipids. Studying these dynamic pathways could reveal some of the unknown mechanisms of disease, thus providing an interesting avenue for the identification of novel biomarkers and the development of novel therapeutics (1).
The N-glycosylation pathway
N-glycans (glycans attached to a nitrogen atom on proteins) are synthesized in the endoplasmic reticulum (ER) as a glycan tree containing fourteen sugar moieties (2). These structures are transferred in its entirety to the asparagine residue of the consensus sequence asparagine-X-serine/threonine (Asn-X-Ser/Thr) in proteins, where X can be any amino acid except for proline (2). Subsequently, two sugar moieties (glucoses) are trimmed, followed by binding of calreticulin or calnexin, chaperones involved in glycoprotein folding and quality control (3–5). When the glycoprotein is properly folded, additional glucose and mannose sugar moieties are trimmed in the ER and the Golgi, consisting of two N-acetylglucosamine (GlcNAc) residues and three mannose (Man) sugars (6). This core structure can subsequently be extended in the Golgi by adding sugar moieties. Based on the extent of mannose trimming and extension by other sugars, this process yields three distinct types of N-glycans: 1) oligomannose (or high-mannose) type, in which only Man residues extend the core; 2) complex type, in which antennae initiated by GlcNAc extend the core; and 3) hybrid type, in which Man extends one arm of the core and one or two GlcNAc-initiated antennae extend the other arm (7). Of note, plasma membrane and secreted proteins are heavily glycosylated, whereby these sugars form the first line of contact in cell-cell interactions, thus conferring glycans a prominent role in processes such as growth receptor signaling and immune surveillance. As such, dysregulations in N-glycan processing can contribute to malignant transformation of tissues by increased proliferative signaling and immune evasion (8, 9). Glycan-binding molecules known as lectins are commonly used to determine the abundance and alterations of N-glycosylation in cancer.
Abundant N-glycan structures in myeloid malignancies
Given the prominent roles of growth receptors in the development of myeloid malignancies, cancers affecting our bone marrow and blood cells, it is of special interest to study the involvement of N-glycosylation in their development and progression. The major myeloid malignancies that will be discussed in this review include acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), and myelodysplastic syndromes (MDS). Both AML and MPN are characterized by hyperproliferation of hematopoietic stem cells (HSCs) and/or myeloid progenitor cells (blasts) in the bone marrow (8). In AML, this is typically accompanied by a block in differentiation leading to accumulation of blasts in the bone marrow and blood, and a reduction of mature, functional myeloid cells (9, 10). In MPN, myeloid differentiation is still largely occurring, resulting in an increase of mature myeloid blood cells (11). A complicating factor in MPN is the development of bone marrow fibrosis which eventually impedes hematopoiesis resulting in reduced numbers of matured myeloid blood cells (12). In MDS, abnormal myeloid proliferation and differentiation results in the development of dysplastic features, cytopenias, and dysfunctional myeloid blood cells (13). Despite the novel therapeutics that have been developed in recent years, gaps in knowledge on patients’ prognoses and the limited therapeutics with curative potential remain a major challenge. Some of these challenges may be overcome by delving into glycoprotein biology, still an underdeveloped area of research in the field of myeloid malignancies.
Aberrant glycosylation is a common feature of cancer cells, including myeloid malignancies. Multiple glycan isoforms have been shown to be abundant in myeloid malignancies (Figure 1), one of those being oligomannosidic N-glycans. This glycan isoform has been found as a ubiquitous characteristic of cancer cells of 34 different cell lines, although the relative abundance of oligomannosidic species in the N-glycome varied widely between them (14). Among the cell lines studied were the AML cell lines HL60 and THP1, for which oligomannosidic N-glycans comprised 84% and 38% of the total N-glycome, respectively (14). Another study of 21 AML cell lines showed that 47–61% of all N-glycans were oligomannosidic (15). The surface expression of these epitopes was confirmed through the use of the mannose-binding lectin Concanavalin A (ConA) in flow cytometry experiments (14). Oligomannosidic N-glycans have also been found to be upregulated in a hematopoietic progenitor (HSC/CMP)-like population from MDS patients (n=14) compared to healthy controls (n=10), and significantly higher expressed in high risk versus low risk MDS hematopoietic progenitors (HSC/CMPs) (16). However, oligomannosidic N-glycans were low in leukemic stem cells and HSC/CMP populations from primary AML samples in this study. This may be due to the low sample size (n=9) and the large heterogeneity in AML (16). Altogether, at least in some cases, oligomannosidic structures appear to be a feature of malignant myeloid blasts. Future studies are required to define N-glycan aberrancies in primary AML samples (especially in the leukemia initiating cells) using larger and/or more uniform AML cohorts.
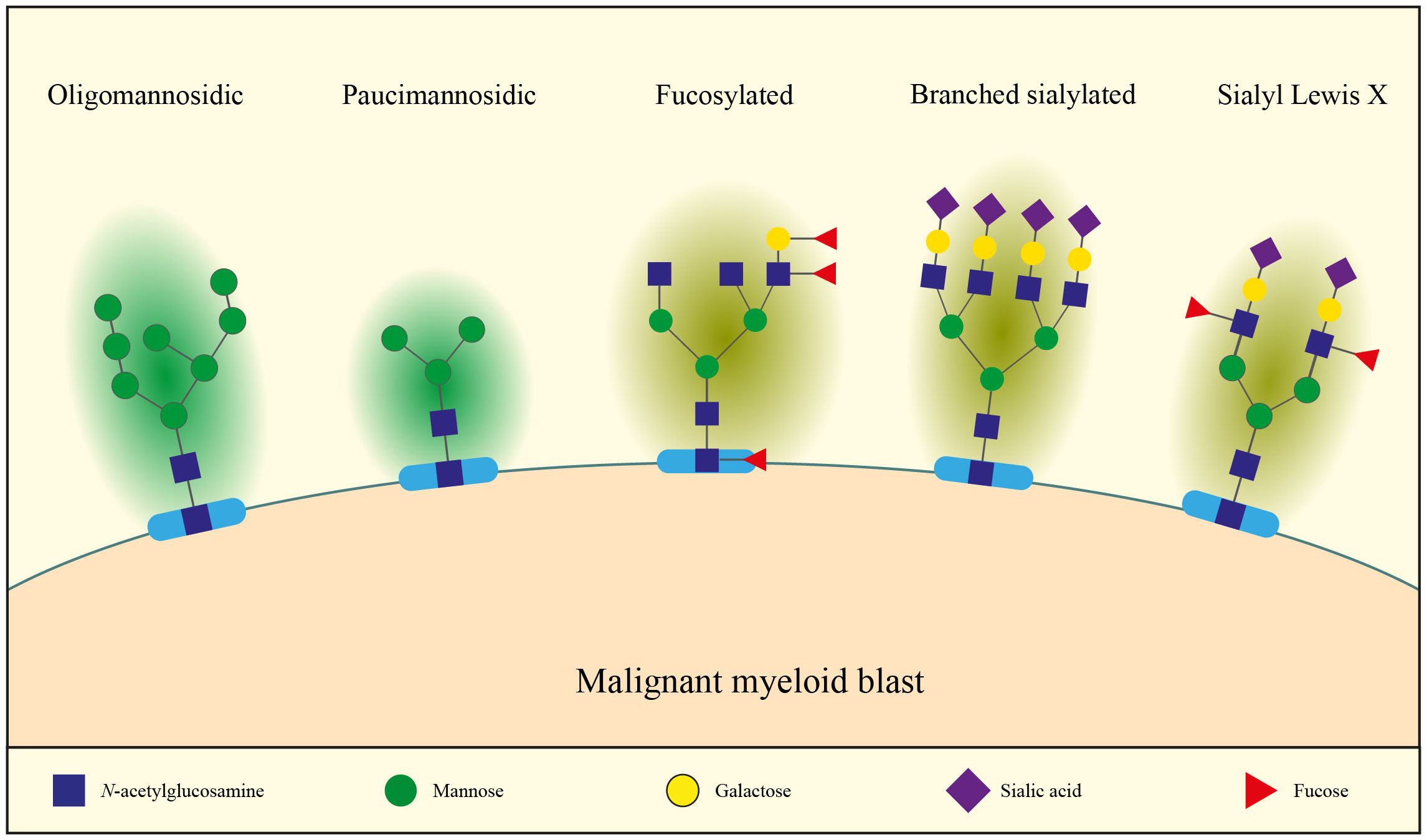
Figure 1 Malignant myeloid blasts present a context-dependent glycan signature with, for example, increased levels of immature oligomannosidic, paucimannosidic, fucosylated and branched sialylated N-glycans, as well as increased sialyl lewis X (see main text for details).
Besides oligomannosidic structures, paucimannosylation (Man2-3GlcNAc2 in particular, Figure 1) was an overrepresented glycan structure in 85% of the studied cancer cell lines (29/34), including AML cells (17). The presence of paucimannosidic structures in AML cell lines was confirmed in a second study, with an abundance of 3–18% of all N-glycans (15). Paucimannose structures also appeared as one of the most pronounced factors driving the discrimination of AML cell lines according to their differentiation arrest in the French-American-British (FAB) classification (15). High paucimannosylation correlated with (myelo)monocytic (FAB M4 and M5) while low paucimannosylation was more common in promyelocytic arrest (FAB M3). It would be of interest to determine whether normal myelomonocytes, monocytes, and promyelocytes show similar N-glycan structures as their AML counterparts or whether these N-glycan structures are acquired upon leukemic transformation.
Fucosylation (Figure 1), which occurs on both N- and O-glycans, has also been shown to be associated with the FAB stratification of AML patients. One such example comes from stainings with alpha-L fucose binding lectin Ulex europaeus agglutinin (UEA) (18). Healthy myeloid progenitor cells were essentially negative upon UEA staining, while monocytes showed an intermediate intensity. In AML, UEA binding also increased from immature (FAB M1 to M2) to the myelomonocytic leukemia (FAB M4) with 10%, 37.5% and 75% of samples testing positive, respectively (18). Additionally, the colony forming cells in FAB M1 and M2 AMLs were UEA-negative while those of FAB M4 AML patients were UEA-positive. Altogether, this suggests an upregulation of alpha-L fucose in monocytic differentiation rather than an oncogenic feature (18). Besides antennary fucosylation, core fucosylation is also frequently found in AML (15). Plasma glycoproteins in MDS and AML patients were shown to contain higher levels of core and terminal fucoses (measured by AAL binding) (19). This suggests that fucosylation may be abundantly present on MDS and AML cells.
An increase in sialylation (Figure 1), resulting in the increased abundance of sialic acid moieties at the external ends of glycans, has also been demonstrated in several cancer forms, including certain myeloid malignancies (16, 20–23). In solid tumors, it has been shown that this could be due to increased branching upon MGAT4/MGAT5 upregulation, resulting in a higher number of antennae which end with sialic acids, hence total sialylation is increased (24–28). In line with these observations, positive correlations between MGAT4 and MGAT5 expression, branching and sialylation were also observed in AML cell lines (15). In particular, branched N-glycans were higher in AML FAB M6 as compared to M4 and M5 (15). Another paper reported higher expression of branched N-glycans on leukemic stem cells and hematopoietic stem cells (HSCs) in MDS compared to controls (16). Besides increased branching, increased sialylation could be due to upregulation of sialyltransferases (ST), the enzymes that transfer a sialic acid residue from CMP-sialic acid to a galactose (gal), N-acetylgalactosamine (GalNAc), or another sialic acid residue on the outer end of N-glycans, O-glycans, and glycolipids. Besides overall increased sialylation, sialyl Lewis X (sLeX, Figure 1) is often highly expressed in cancer, including myeloid malignancies. In addition to sialic acid, sLeX contains an α1–3-linked fucose and β1–4-linked galactose attached to a N-Acetylglucosamine (GlcNAc) (Figure 1). N- and O-glycan mass spectrometry on 21 AML cell lines identified that 1–12% of the total O-glycans and 1–3% of the total N-glycans are decorated with sLeX (15).
To summarize, several forms of N-glycosylation have been found to be abundant on myeloid cancers including high-mannose, paucimannose, fucose containing, branched, sialyl containing and sleX containing N-glycans (Figure 1). Their abundance is context-dependent and may partially depend on cell type, differentiation stage, disease and other factors as described in this review.
In the remainder of this review we will explore the implications of N-glycan forms in the pathophysiology of myeloid malignancies. We describe three hallmarks of cancer that are particularly influenced by N-glycans in the context of myeloid malignancies: (1) oncogenic receptor signaling, (2) chemoresistance, and (3) niche hijacking. Additionally, we will discuss mechanisms contributing to chemoresistance and leukemia-associated N-glycan structures that may be used for therapeutic targeting and/or prognostication. For each subsection, we will describe the pathobiology involved and its (potential) clinical implications.
N-glycosylation in oncogenic receptor signaling
Oncogenic signaling by mutant FLT3 and KIT
Pathobiology
Increased cell proliferation is a hallmark of myeloid malignancies and activating mutations in receptor tyrosine kinases (RTKs), major hematopoietic growth receptors, are frequently found. Two such RTKs are the FMS-like tyrosine kinase 3 (FLT3) and KIT which stimulate proliferation, survival, and myeloid differentiation of early hematopoietic progenitor cells upon binding by their respective ligands: FLT3 ligand and stem cell factor (Figure 2A) (29). FLT3 is the most commonly mutated gene among AML patients, found in 30% of adult (30–32) and 10–15% of pediatric patients (33–35), with two common types of activating mutations; an internal tandem duplication (FLT3-ITD) within the juxtamembrane domain and point mutations or deletions in the tyrosine kinase domain (FLT3-TKD) (36). Among hematopoietic neoplasms, KIT mutations are most commonly found in mastocytosis [>90% of patients (37, 38)] and AML [~10% of patients (39)]. These mutations are primarily located in the second tyrosine kinase domain with D816V being the most common activating mutation, but also juxtamembrane and extracellular mutations close to the transmembrane domain are recurrently found.
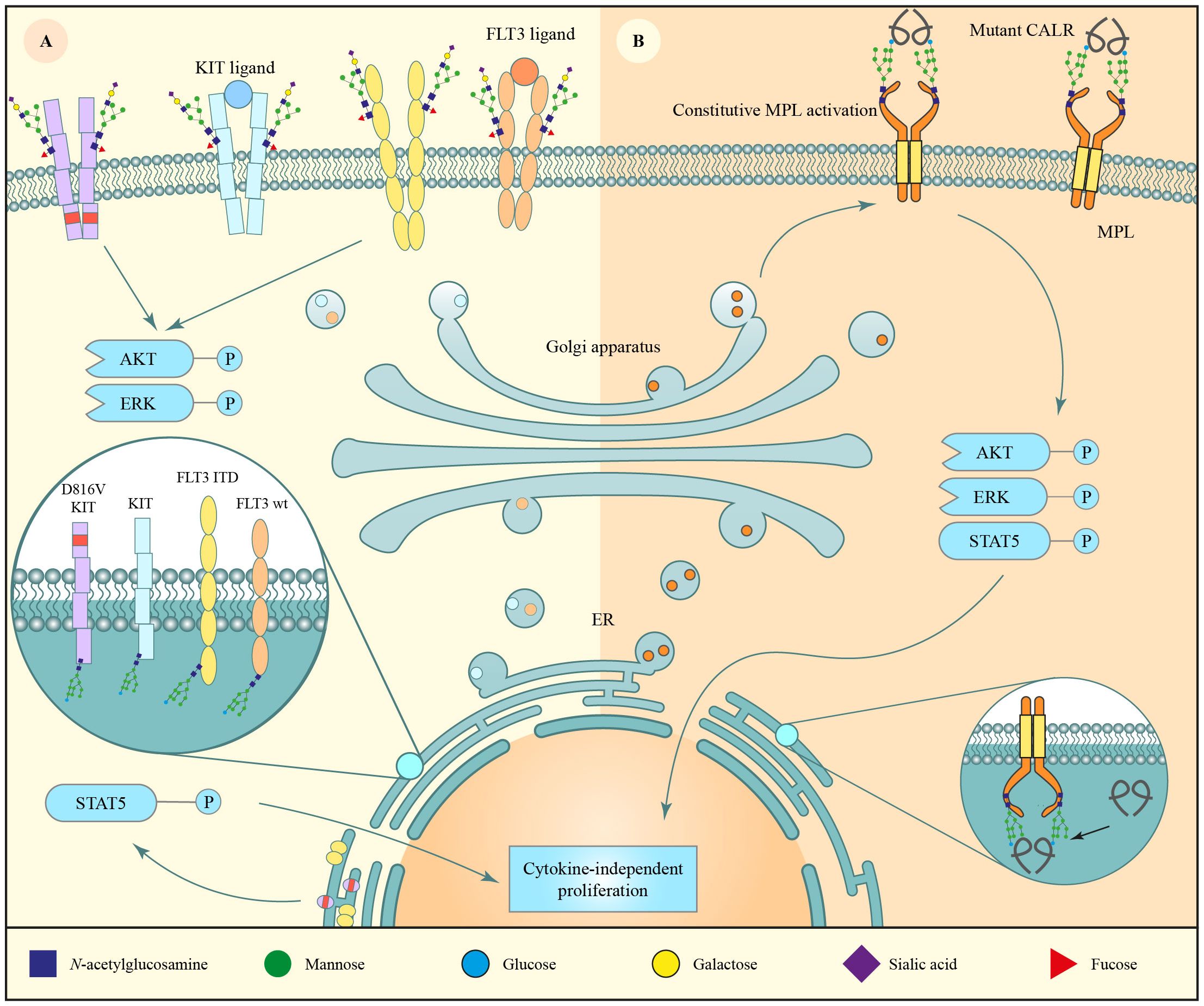
Figure 2 N-glycosylation of major hematopoietic growth receptors found mutated in myeloid malignancies. (A) The receptor tyrosine kinase (RTK) fms-like receptor tyrosine kinase 3 (FLT3) and KIT are often mutated in myeloid disorders, e.g. presenting with an internal tandem duplication (FLT3-ITD) or D816V mutation, respectively. The wild-type isoform of the receptors undergo regular N-glycan maturation and are expressed on the cell surface where, upon FLT3 ligand binding, they activate the AKT/ERK pathways. FLT3-ITD and KIT D816V are constitutively phosphorylated, which likely causes retention of the immature N-glycoproteins in the ER, where they activate oncogenic STAT5 signaling, leading to increased proliferation. (B) Mutant calreticulin (CALR) binds the immature N-glycan form of the thrombopoietin receptor MPL in the endoplasmic reticulum. Subsequently, the complex is transported to the cell membrane, where MPL is constitutively activated by mutant CALR, leading to cytokine-independent proliferation and development of myeloproliferative neoplasms (MPN).
FLT3 and KIT are synthesized as 110 kDa proteins, which are N-glycosylated in the ER to form 125–130 kDa immaturely N-glycosylated isoforms. The proteins are subsequently folded in the ER prior to export to the Golgi apparatus where the immature N-glycans mature to more complex N-glycans leading to 145–150 kDa protein isoforms that are translocated to the plasma membrane (40–43). Aberrant glycosylation of these receptors, as seen in their mutated isoforms, disrupts this maturation process which ultimately impacts their localization and downstream signaling (Figure 2A) (44–50). Proportionally, wildtype FLT3 and KIT are predominantly expressed as the large 150 kDa mature N-glycoprotein, whereas similar amounts of 130 and 150 kDa FLT3-ITD are expressed. FLT3-TKD isoform ratios fall between those two extremes depending on the mutation (40, 41, 44, 51) and KIT D816V is primarily expressed as the 125 kDa immature N-glycan isoform (42–44). Correspondingly, wildtype FLT3 and KIT are more abundant on the plasma membrane as compared to FLT3-ITD, the common FLT3-TKD mutant D835Y, and KIT D816V which largely localize to the ER/Golgi apparatus (40, 41, 43). This difference in localization between wiltype and mutant FLT3/KIT results in altered downstream signaling (Figure 2A) (44–50). ER stalling of FLT3-ITD has been suggested to originate from inefficient folding and enhanced chaperone dependence as a consequence of its constitutive autophosphorylation (41).
Notably, wildtype FLT3 and both ITD and D835Y TKD mutant FLT3 activate intracellular pAKT and pERK signaling, whereas only FLT3-ITD seems to strongly activate pSTAT5 (40, 47, 51). This strong pSTAT5 activation, originating from the immature ER-localized N-glycan form (40, 47), has been suggested to be particularly important for AML carcinogenesis, since FLT3-ITD causes a stronger myeloproliferative disorder in mice as compared to FLT3-TKD D835Y (40, 52). Similarly, the KIT D816V signal transduction pathways are markedly different from wildtype KIT signaling with lower activation of pERK and stronger activation of pSTAT5 (44–47).
The effects of N-glycosylation on FLT3-ITD-associated pathobiology were studied through the use of the N-glycan biosynthesis inhibitor tunicamycin (47). Tunicamycin inhibits UDP-GlcNAc: Dolichyl-phosphate GlcNAc-1-phosphate transferase, a key ER enzyme involved in the early steps of N-glycan biosynthesis (53). Inhibition of N-glycan biosynthesis with tunicamycin strongly reduced trafficking of FLT3-ITD and KIT D816V from the ER to the plasma membrane without affecting their stability (47). Hence, inhibition of N-glycan biosynthesis led to increased accumulation of FLT3-ITD in the ER, enhanced pSTAT5 signaling, and reduced pAKT and pERK (47, 54). However, sole signaling of ER-resident FLT3-ITD, without signaling from plasma membrane-bound FLT3-ITD, may not be sufficient for oncogenic transformation. Indeed, the use of inhibitors interfering early in the N-glycosylation pathway [2-deoxyglucose (2-DG) and fluvastatin], which keep FLT3-ITD in the ER, has shown promising results (Figures 3A, B) (55, 56) as discussed below. The same may hold true for KIT D816V.
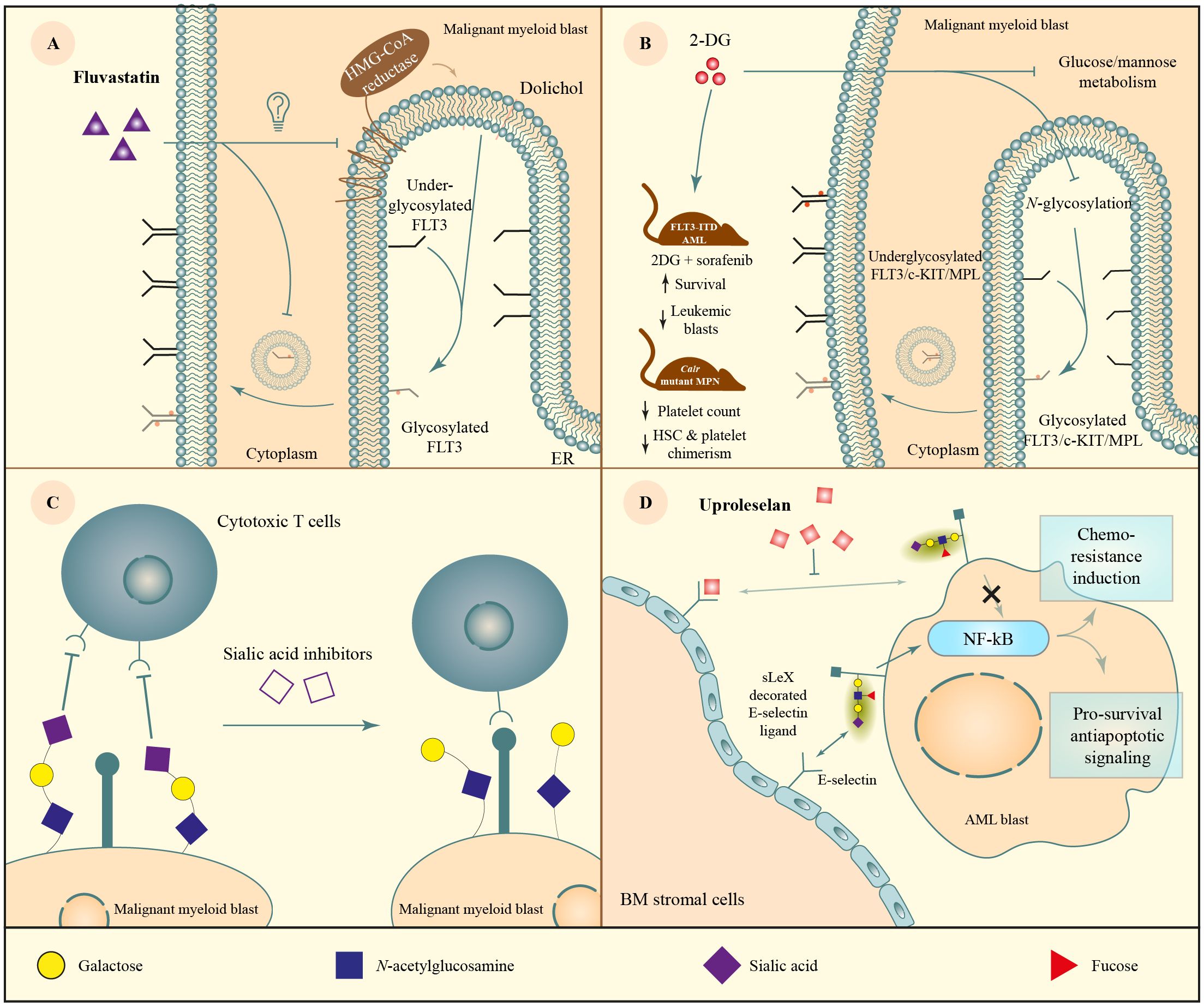
Figure 3 Therapeutic targeting of N-glycosylation. (A) Fluvastatin, a 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitor was expected to reduce N-glycan biosynthesis, however somehow it interferes with ER to plasma membrane transportation or maturation of immaturely N-glycosylated FLT3-ITD and KIT D816V, thereby contributing to reduced pro-proliferative cell signaling of these mutated receptor tyrosine kinases (RTKs). (B) 2-deoxyglucose (2-DG), a glucose analog that inhibits a variety of processes including N-glycosylation through the dysregulation of glucose and mannose metabolism. The compound has been tested in mouse models of AML (FLT3-ITD) and MPN (mutant Calr) where hallmarks of the malignancy were reduced such as leukemic blasts (AML), as well as platelet count and Calr mutant chimerism in long-term hematopoietic stem cells (LT-HSCs) and platelets. (C) The use of sialic acid inhibitors is being investigated for its potential in assisting immunotherapy. Sialic acids displayed on the cell membrane block the interactions necessary for immune surveillance. Inhibiting their biosynthesis consequently reducing their membrane expression has been suggested to increase tumor killing by cytotoxic T cells. (D) The E-selectin inhibitor uproleselan mimics the carbohydrate ligand of E-selectin, resulting in competitive inhibition of its ligand signaling in leukemic blasts. E-selectin ligands activate the NF-κB signaling pathway resulting in pro-survival and antiapoptotic effects which confer chemotherapy resistance. Uproleselan interferes with these processes.
Clinical implications
Several early acting N-glycan inhibitors have been tested in preclinical in vivo models of FLT3-ITD and KIT D816V, including statins and 2-DG. Statins are classically used for lowering cholesterol and reducing cardiovascular risks by blocking 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG COA reductase, Figure 3A), the rate-limiting step in the mevalonate pathway. Besides cholesterol, the mevalonate pathway also produces dolichol, the base for biosynthesis of N-glycosylation. Hence, statins were hypothesized to inhibit N-glycan biosynthesis and, at least partially, inhibit FLT3-ITD and KIT D816V-mediated signaling and cell growth (55). Indeed, cell surface expression of these oncogenes and downstream pAKT and pERK were downregulated upon treatment with statins. pSTAT5 levels were upregulated, similar to when cells were treated with tunicamycin (47, 54, 55). Surprisingly, however, instead of accumulation of unglycosylated FLT3-ITD, statin treatment resulted in the accumulation of immature high-mannose FLT3-ITD (55). Nevertheless, statins strongly reduced cell growth of FLT3-ITD and KIT D816V mutant cell lines and/or AML patient samples, an effect that could not be reversed by cholesterol replenishment according to the authors (data not shown) (55). Finally, statins reduced BA/F3-FLT3-ITD cell growth in mice and significantly prolonged survival of the mice (55). The addition of N-glycan biosynthesis inhibitors to therapy regimens may be useful for FLT3 and KIT mutant myeloid malignancies. Although a window of opportunity seems to exist to use N-glycan biosynthesis inhibitors in myeloid malignancies, strong inhibition will likely cause toxicity.
2-DG is a glucose analog in which the 2-hydroxyl group is replaced by a hydrogen. Because of its structural similarity to D-glucose and D-mannose, 2-DG’s mode of action involves inhibition of glucose- and mannose-related cellular processes including N-glycan biosynthesis and glycolysis (Figure 3B) (57, 58). 2-DG has been shown to reduce cell viability of leukemic cell lines. Cells carrying FLT3-ITD or KIT D816V mutations were more susceptible to 2-DG than FLT/KIT wild type leukemic cells (54). FLT3-ITD and KIT D816V are major oncogenic drivers in these cells, are N-glycosylated, and their cell surface signaling is reduced upon inhibition of N-glycan biosynthesis. Both 2-DG and statins that inhibit early N-glycosylation exert similar effects on these cells (55, 56). Altogether, these results support that although the treatments are general inhibitors of N-glycosylation, it is likely that the observed effects are (at least in part) through inhibition of N-glycosylation of the mutated receptor.
Since 2-DG-induced apoptosis in FLT3-ITD and KIT D816V cell lines could be largely rescued by supplementing D-mannose, the authors concluded that reduced N-glycosylation rather than reduced glycolysis is likely the main contributor to its cytotoxicity (54). It should be noted, however, that mannose does not only feed into glycosylation pathways, but can be converted into glucose intracellularly, thereby possibly feeding into the glycolysis pathway as well. Moreover, since FLT3-ITD mutant as compared to wildtype cells have increased glycolytic activity, they may be more sensitive to inhibition of glycolysis by 2-DG (56).
In addition, reduced oncogene expression may also contribute to the antileukemic effects of 2-DG. Specifically, reduced N-glycosylation of FLT3-ITD and KIT D816V decreased their cell surface expression, limiting downstream pERK signaling, likely contributing to apoptosis in mutated cells (54). Furthermore, 2-DG reduced mRNA and protein expression of KIT D816V and FLT3-ITD in AML cells (54). Moreover, 2-DG has been shown to reduce protein levels of the anti-apoptotic MCL-1 protein even before alteration of FLT3-ITD or KIT D816V expression is detected, which may also contribute to its antileukemic effect (54). Besides its effectiveness in vitro, 2-DG treatment reduced the growth of KIT D816V positive Kasumi-1 cells in xenograft mouse models (54). However, growth of FLT3-ITD positive MOLM-14 cells was not reduced in xenograft mouse models with single agent treatment at the same dose, whereas 2-DG had synergistic effects with the chemotherapeutic cytarabine in the MOLM-14 engrafted mice and with the FLT3 kinase inhibitor sorafenib in BA/F3-FLT3-ITD engrafted mice (54, 56). Another important observation was that 2-DG was also active on FLT3 inhibitor quizartinib resistant cells (resistant due to a secondary D835 mutation in the FLT3 activation loop), raising the question whether 2-DG treatment could enhance the effect of FLT3 inhibitors (54). Altogether, 2-DG is a pleiotropic compound that may rely on multiple modes of action, among them inhibiting N-glycosylation, in FLT3-ITD and KIT D816V mutant AML.
Oncogenic receptor signaling in MPN and CML
Pathobiology
Mutations directly affecting the N-glycosylation site of receptors could contribute to the development of myeloid malignancies. A missense mutation in the macrophage receptor colony-stimulating factor 3 receptor (CSF3R) has been described in two MPN and a chronic myeloid leukemia (CML) patient. This mutation disrupts the N-glycan site at asparagine 610, which is essential for ligand dependency, thus causing constitutive, ligand-independent CSF3R signaling (59).
Other mutations may result in myeloid malignancies through glycosylation-dependent mechanisms without directly affecting N-glycan sites. For example, the thrombopoietin receptor also known as myeloproliferative leukemia protein (MPL) is a master regulator of megakaryopoiesis and is expressed on HSCs and megakaryocyte lineage cells. Overstimulation of MPL due to somatic activating mutations in MPL or its downstream kinase Janus Kinase 2 (JAK2) are disease-initiating in MPN. Furthermore, mutations in Calreticulin (CALR) drive overstimulation of MPL signaling in a glycosylation-dependent manner (Figure 2B). Specifically, in wildtype cells, CALR is mainly localized to the ER due to its C-terminal KDEL sequence, where it functions as a chaperone to fold N-glycosylated proteins (60). Here, CALR binds monoglucosylated N-glycans shortly after the outer two glucoses of the glycan tree have been trimmed. Due to heterozygous frameshift mutations in CALR, as found in MPN (61, 62), mutant CALR does not contain the ER-retaining KDEL sequence that is present in wildtype CALR. Additionally, mutant CALR acquires the ability to strongly bind the glycoprotein MPL, which has four N-glycan sites (63, 64). This mutant CALR-MPL complex subsequently travels to the plasma membrane leading to overactivation of MPL signaling (Figure 2B) and a concomitant increase in megakaryopoiesis and platelet counts. It is hypothesized that mutant CALR-MPL-induced oncogenic signaling mostly originates from the cell surface, as opposed to FLT3-ITD and KIT D816V signaling which is already initiated in the ER (65). Importantly, both the N-glycan sites of MPL and glycan-binding lectin sites of CALR are required for the oncogenic interaction between mutant CALR and MPL (65–68). Additionally, N-glycosylation of MPL is required for its cell surface expression (49). Hence, inhibition of N-glycosylation may be a therapeutic vulnerability in MPN as discussed below (58).
Clinical implications
CSF3R mutations are currently screened when certain myeloid malignancies are suspected. A common site which is routinely screened for in myeloid malignancies is T618, which is in close apposition to the N610 mutation here described. Because of the close proximity between these two sites, it is probably that sequencing information for the latter site is available when the T618 mutation is screened for. Therefore, it should be rather straightforward to investigate this site using existing diagnostic panels.
Similar to FLT3-ITD and KIT D816V, inhibition of ER-related N-glycosylation processes by 2-DG and other, more specific early N-glycan inhibitors have been tested in CALR mutant cell lines and primary MPN samples, showing preferentially reduced growth of CALR mutant cells (58). We hypothesize that in comparison to FLT3-ITD and KIT D816V, inhibition of N-glycan biosynthesis will reduce oncogenic MPL signaling more efficiently since oncogenic MPL signaling occurs predominantly at the cell surface and not from the ER (65), and because N-glycosylation is important for MPL stability but not for FLT3-ITD or KIT D816V (47, 64). Besides CALR-mutant MPN models, N-glycosylation inhibitors similarly hindered oncogenic growth of MPL-expressing Jak2V617F mutant hematopoietic cells (58). This suggests that reduction of MPL cell surface expression rather than the N-glycan dependent interaction of mutant CALR with MPL was the major player in efficacy of N-glycan inhibition in preclinical MPN models. Indeed, upregulation of N-glycan-related pathways were found both in platelets from CALR as well as JAK2 mutated patients as compared to healthy control platelets, and platelet counts were reduced upon 2-DG treatment in mouse models of both Calr and Jak2-mutant MPN (58). It should be said that normal MPL signaling and megakaryopoiesis will be suppressed upon inhibition of N-glycan biosynthesis as well, but importantly, 2-DG reduced the Calr mutant chimerism as compared to vehicle treatment in competitively transplanted mice, both in platelets as well as in the disease-initiating long-term hematopoietic stem cell population (58). Additional studies are required to elucidate the molecular mechanisms (both glycosylation and glycolysis-related) behind this preferential 2-DG sensitivity of CALR mutant versus wildtype cells.
Impact of N-glycosylation on chemoresistance
Oligomannose N-glycans in AML and their impact on chemosensitivity
Pathobiology
As described above, increased mannosylation is commonly seen in malignant versus healthy cells. Interestingly, in our recent work we demonstrated that the level of oligomannosidic N-glycans correlates with chemosensitivity in AML (Figure 4). Specifically, mass spectrometry analyses revealed that high mannose N-glycans make up to 75% of all glycan structures in cytarabine resistant AML cells, a slight but significant increase when compared to their sensitive counterparts (69%) (69). Additionally, resistant cells showed a two-fold increase in their mannose expression as compared to sensitive cells, indicating that both the fraction of cells expressing oligomannosidic N-glycans and its abundance within the total glycan pool increase in chemoresistant cells. Moreover, somatic variances in mannosidases MAN1B1 and MAN2B1, enzymes that remove mannoses from N-glycans, showed a trend toward increasing sensitivity to the chemotherapeutic cytarabine in AML (70). In line with these results, binding of the oligomannose-recognizing lectin ConA correlated with cytarabine resistance both in cell lines and primary AML material (69). These results suggest that the study of N-glycans may be used for predicting chemotherapy response in AML.
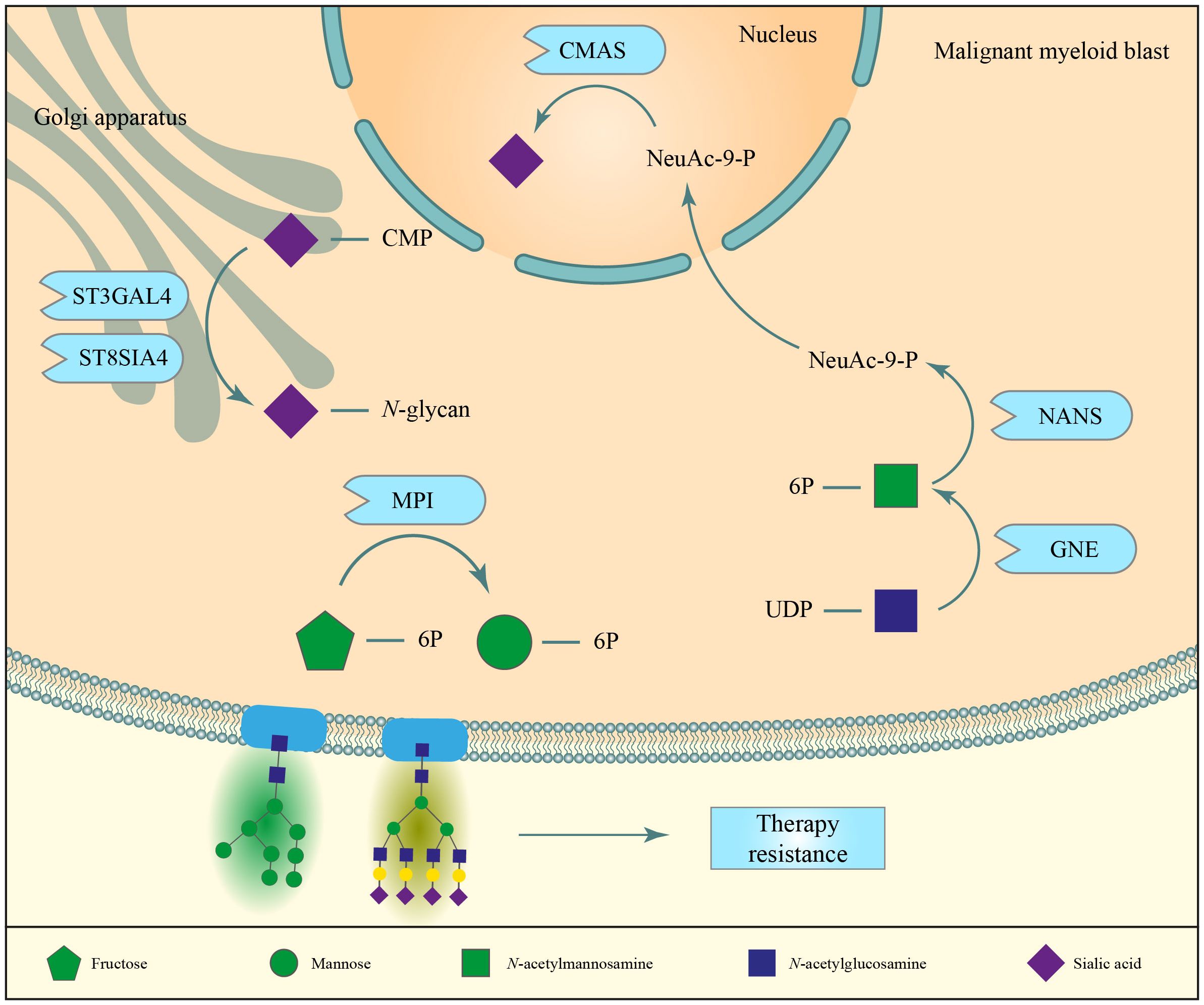
Figure 4 N-glycans contribute to the development of chemoresistance in AML cells. High expression of mannose-6-phosphate isomerase (MPI) correlated with a higher resistance to FLT3 tyrosine kinase inhibitors and cytarabine therapies. The expression of other enzymes associated to N-glycan biosynthesis, namely sialyltransferases, CMAS, NANS and GNE have been correlated to therapy resistance.
Mannose metabolism, and more specifically mannose-6-phosphate isomerase (MPI), have been implied in the resistance of AML cells to FTL3-tyrosine kinase inhibitor (TKI) and the chemotherapeutic cytarabine (Figure 4) (71). MPI is an enzyme contributing to mannose supply for N-glycan biosynthesis and can act in two directions: it can convert mannose-6-phosphate into fructose-6-phosphate and vice versa. In the latter case, the more readily available glucose-6-phosphate is converted to fructose-6-phosphate, which is fed into the mannose metabolism pathway for the production of GDP-mannose, which is used in glycosylation (72). High expression of this enzyme was linked with worse prognosis in AML patients and with gene signatures associated with oxidative phosphorylation and fatty acid oxidation, both of which are known features of chemoresistant AML cells (69, 71). Moreover, it was demonstrated that AML cells treated with MPI inhibitors or knocked out for MPI were more susceptible to treatment with FLT3-TKIs and cytarabine. Altogether, this supports the hypothesis that high MPI expression protects AML cells to cytarabine and TKI treatment (71). The relevance of N-glycosylation in chemoresistance is further highlighted by the observation that the previously discussed N-glycan biosynthesis inhibitor tunicamycin is also capable of sensitizing AML cells to cytarabine treatment (69).
Clinical implications
Since its abundance on cancer cells and correlation with cytarabine chemoresistance in AML, it would be interesting to target oligomannosidic N-glycans. Perhaps, lectins or lectin-containing antibodies could be used to preferentially target aberrant glycan structures on malignant cells for therapeutic purposes (73–75). In this respect, we demonstrated that Galectin-9, a beta-galactoside-binding lectin, induced cell death in AML CD34+ stem cells, but not healthy cord blood derived counterparts (74). Alternatively, inhibition of N-glycan biosynthesis or the administration of mannose mimetics could be implemented to reduce cell proliferation and/or sensitize AML cells to chemotherapy (69). Indeed, the N-glycan biosynthesis inhibitor tunicamycin is capable of sensitizing AML cells to cytarabine. Altogether, these data provide a rationale for reducing or targeting oligomannosidic N-glycan expression in myeloid malignancies.
Sialylated N-glycans and their impact on chemoresistance
Pathobiology
As described in the introduction, increased overall sialylation is a hallmark of several cancer types, also described context-dependently in myeloid malignancies (20, 21, 23, 76). Increased branching and upregulation of sialyltransferases can contribute to increased overall sialylation in cancer. To evaluate the role of sialyltransferases in chemoresistance, the mRNA expression of all 20 human sialyltransferases was determined in 76 diagnostic AML samples and three AML cell lines in their parental state and after long-term culture with increasing concentrations of the chemotherapeutic adriamycin (doxorubicin) (20). ST3GAL4, ST6GALNAC4, ST8SIA4, and overall sialic acid decoration were consistently higher while ST3GAL1, ST3GAL5, and ST8SIA6 were consistently lower expressed in chemotherapy resistant compared to chemosensitive cells (20). Of those, only ST3GAL4 and ST8SIA4 can sialylate N-glycans (although they are not exclusive for N-glycans) and their inhibition was shown to reduce expression of α2–3 sialylation and α2–8 sialylation in adriamycin resistant AML cell lines, respectively.
Knockdown of ST8SIA4 reduced expression of drug efflux pumps P-glycoprotein 1 (also known as multidrug resistance protein 1, CD243) and reduced phosphorylation of PI3K, AKT, and NF-kB (20). Consequently, adriamycin resistant HL-60 AML cells regained sensitivity to pharmacological intervention in vitro and in subcutaneous in vivo mouse models upon downregulation of ST8SIA4 (Figure 4) (20). In addition, after four weeks of growth, tumor volume was 30% lower in mice injected with ST8SIA4 knocked down HL-60 cells as compared to control-transduced cells (20). These results suggest that ST8SIA4 not only affects chemoresistance but also cell proliferation. Similarly, enzymes involved in the biosynthesis of sialic acid and its transport to the Golgi apparatus for its addition to an N-glycan tree, such as CMAS, SLC35A1, NANS and GNE, were found to associate with suppression of cytokine-independent proliferation and reduced survival of the AML FAB M6 cell line TF-1 (77). Knockout of either CMAS or SLC35A1 induced sensitivity toward MEK/ERK inhibitors, albeit inducing resistance to JAK inhibitors (77). Furthermore, the expression of α2,3-linked sialic acids on TF-1 cells negatively correlated with the sensitivity toward MEK/ERK inhibitors (77). Thus, altered N-glycan sialylation could serve as a biomarker for therapy sensitivity. The role of sLeX in chemoresistance and niche hijacking (Figure 5, top left) will be discussed in a separate section.
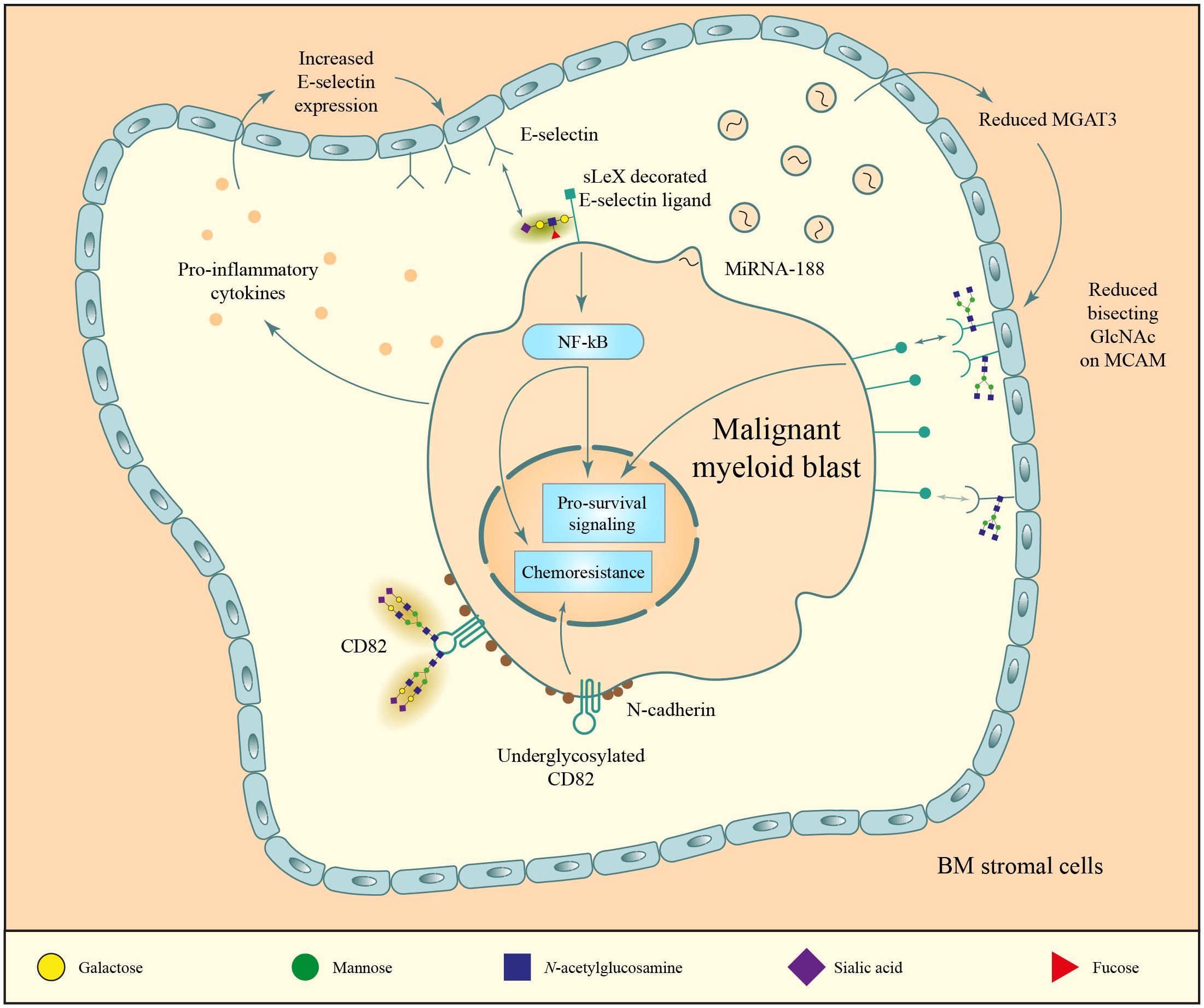
Figure 5 N-glycan mediated environment interactions. 1) malignant myeloid blasts release pro-inflammatory (upper left) and miRNA-containing vesicles (upper right) that increase the expression of E-selectin and decrease the expression of MGAT3, respectively. These changes lead to an increased expression of E-selectin in stromal cells, which bind to the sialyl Lewis X (sLeX)-decorated N-glycans on the blast surface, and to reduced bisecting GlcNAc structures on the MCAM protein of stromal cells, which causes increased MCAM expression that stimulates myeloid blast proliferation through CD13. 2) N-glycosylation of the membrane scaffold CD82 is crucial for proper cluster formation of N-cadherin (bottom part of the figure). Disruptions in these structures lead to aberrant cluster formation and increased binding of N-cadherin to its ligands which supports bone marrow homing, and increased chemotherapy resistance.
Clinical implications
In line with the above data, high ST3GAL4 expression has been linked to dismal prognosis in AML (78). However, N-linked α2–3 sialic acids, produced by ST3GAL4 and ST3GAL6, were downregulated on high risk versus low risk MDS leukemic stem cells, suggesting that there may be different prognostic roles for ST3GAL4 and/or α2–3 sialic acid levels in MDS compared to AML (16). Interestingly, analyses of plasma from MDS and AML patients using a lectin array combined with surface plasmon resonance imaging did suggest that progression from MDS to AML may be accompanied by increased N-linked α2–3 sialylation (19). Further investigations are required to determine whether plasma N-glycosylation levels are a reflection of the N-glycan aberrancies found on the plasma membrane of malignant myeloid cells and whether they are similar between leukemic stem cells and their progeny. With this knowledge, the level of sialylation may be used as biomarker for prognostics in MDS/AML. Similarly, the expression level of sialyltransferases can be potentially used as biomarker for therapy resistance, as described above.
Furthermore, besides its role in therapy resistance, other clinical applications of sialic acids include their targeting for potentiating immunotherapeutic strategies. Sialic acids are known to interfere in the binding of key immune cell receptors with antigens expressed on tumor cells (Figure 3C) (79). This strategy has shown early preclinical success in solid cancer and multiple myeloma models owing to the capability of sialic acids to “mask” cancer cells from the immune system (79, 80).
Roles of N-glycosylation in niche hijacking
sLeX – E-selectin interaction increases AML bone marrow homing, cell survival, and chemotherapy resistance
Pathobiology
In addition to their increased proliferation and effects on chemoresistance, an important feature in the pathobiology of myeloid malignancies is the reprogramming of the blast and healthy HSCs microenvironment, also referred to as their niche. These niches encompass soluble signaling as well as direct cell-to-cell and cell-to-matrix interactions. Malignant blasts have been shown to alter these interactions in a way that promotes tumor survival and resistance to chemotherapy (niche hijacking) (81, 82). A prominent protein mediating the interaction between hematopoietic (stem) cells and their niche is the glycan-binding plasma membrane protein E-selectin. Under physiological conditions, E-selectin is expressed on the luminal and abluminal sides of marrow microvessels and in bone marrow niches (Figure 5, top left). Here, it contributes to HSC recruitment from the vasculature and interactions between hematopoietic (stem) cells and their niche (83, 84). In an MLL-AF9 AML mouse model, pro-inflammatory cytokines secreted by AML blasts caused hyperexpression of E-selectin on bone marrow endothelial cells (85). This resulted in the increased binding affinity of these blasts to E-selectin, whereby E-selectin engagement conferred malignant blasts chemoresistance (85). Hence, AML blasts seem to acquire a secretory profile that hijacks the microenvironment to support their own growth and survival (Figure 5) (85).
To date, three E-selectin ligands have been identified which all play a major role in leukemic blast adhesion and trafficking (86), being cutaneous lymphocyte antigen (CLA), CD43 and hematopoietic cell E-/L-selectin ligand. All three ligands interact with E-selectin via its tetrasaccharide sialylated Lewis X (sLeX/sCD15), whose expression depends on the activity of fructosyltransferases (FUTs) (85, 87). Correspondingly, mouse MLL-AF9-transformed HSPCs with a double knock-out of FUT4 and FUT7 were no longer able to bind E-selectin ex vivo (85).
Interestingly, high expression of sLeX-associated fucosyltransferases FUT3, FUT6, FUT7 and ST3GAL4 have been linked to dismal prognosis in AML in monovariate analyses (88) [and conference abstract (78)]. Conversely, AML patients with high levels of LeX-associated FUT4 (89) had a good prognosis upon chemotherapy, although they had higher rates of relapse upon allogeneic stem cell transplantation (88). Multivariate analyses of the impact of FUT3, FUT4, FUT6, and FUT7 expression confirmed the independent poor prognostic impact of high FUT6 and low FUT4 expression in AML (88). Altogether, these data reveal an essential role for the sLeX-E-selectin axes in AML blast survival, chemotherapy resistance, and patient prognosis. Nonetheless, the specific prognostic impact of glycosyltransferases involved in sLeX generation should still be confirmed in independent AML datasets.
Clinical implications
The association between E-selectin interactions of AML blasts and survival/chemoresistance argues for the use of E-selectin blockers in the clinic. Indeed, the use of E-selectin antagonists (uproleselan/GMI-1271, Figure 3D) has been studied in a phase I/II clinical trial (NCT02306291) as an adjuvant to mitoxantrone, etoposide and cytarabine or fludarabine, cytarabine and idarubicin chemotherapy in AML with promising early results. Adding uproleselan to these treatment regimens was well tolerated and effective, as evidenced by its low mucositis rates, and its high remission rates in older patients and secondary AML patients that had undergone previous therapy with hypomethylating agents (90). This treatment strategy is now being investigated in a phase III clinical trial which includes patients aged 18 to 75 with relapsed or refractory AML (NCT03616470) (91). Moreover, uproleselan is also being investigated as a consolidating agent after doxorubicin and cytarabine chemotherapy for its potential to prevent relapses in older AML patients (NCT03701308).
Reduced CD82 N-glycosylation on AML cells supports N-cadherin clustering, bone marrow homing, and increased chemotherapy resistance
Pathobiology
Another relevant protein mediating the interaction between hematopoietic stem cells and their microenvironment is N-cadherin, a member of the classic cadherins family which mediates calcium-dependent cell-cell adhesions and is expressed in both bone marrow stromal cells and a subpopulation of hematopoietic progenitor cells (92). N-cadherin regulates reserve HSC maintenance and could also indirectly promote chemoresistance by increasing homing of leukemic blasts to the bone marrow, which shields them from toxic insults (93, 94). Indeed, a leukemic stem cell subpopulation with high N-cadherin expression was enriched in patient samples obtained after chemotherapy treatment compared to those obtained prior to therapy (95). Furthermore, N-cadherin+ HSCs were found to be more effective leukemia inducers than their negative counterparts when engrafted into mice (96). More recent reports have established a key role for CD82 in this process, regulated by its posttranslational modifications. Specifically, CD82 lacking N-glycosylation sites resulted in an increased molecular packing of N-cadherin in the plasma membrane (Figure 5, bottom) which promoted bone marrow homing of AML cells when injected into NSG mice (97). The relevance of CD82 for cell migration has also been highlighted in other cancer types, namely colorectal and ovarian cancer, where a causative link has been suggested between glycosylation at the asparagine 157 residue of CD82 and metastasis development through activation of the Wnt/β-catenin pathway (98, 99).
Clinical implications
Given its importance in bone marrow homing and chemoresistance, it would be interesting to elucidate the interplay between CD82 and N-cadherin in more detail. Subsequently, such molecular mechanisms may be exploited for sensitizing AML cells to (chemo)therapy or reducing AML self-renewal and proliferation. The N-cadherin antagonist ADH-1 has been tested in a murine model of multiple myeloma with promising early results, leading to a significant reduction in tumor burden with minimal toxicity (100). It would be interesting to test this compound in myeloid malignancies.
Reduced abundance of bisecting N-Acetylglucosamine (GlcNAc) on stromal cells increases binding to and stimulates proliferation of CD13-expressing MDS/AML cells
Pathobiology
Bone marrow stromal cells from MDS/AML patients were found to have a lower abundance of bisecting GlcNAc N-glycans (Figure 5, top right). Correspondingly, the glycosyltransferase that attaches these bisecting GlcNAc, MGAT3, was expressed at a lower level (101). Co-culture of normal bone marrow stromal cells with two different AML cell lines reduced the levels of bisecting GlcNAcs on the stromal cells (101). This reduction was a result of decreased MGAT3 protein levels due to microRNA miR-188–5p (Figure 5, top right), found in high quantities in exosomes from one of the cell lines and in primary MDS/AML samples (101). One protein particularly affected by this decrease in MGAT3 is the adhesion protein melanoma cell adhesion molecule (MCAM, CD146) on stromal cells, whose cell surface expression was found to be increased when its bisecting GlcNAc content was reduced (101). Stromal MCAM can bind CD13 expressed on myeloid blasts, activating the intracellular ERK signaling pathway (Figure 5, top right) and promoting malignant transformation (101).
Clinical implications
Given the hypothesis that reduced expression of bisecting GlcNAc N-glycans on MCAM increases MCAM expression and leads to increased proliferation of CD13-expressing AML blasts, it would be interesting to determine which AML subtypes express CD13 and have increased stromal MCAM expression. Subsequently, blocking the MCAM-CD13 interaction therapeutically may be beneficial for the respective AML patients. The use of MCAM blocking antibodies (like ABX-MA1) in AML may be of interest, which have also proven to inhibit tumor growth and metastasis of human melanoma (102) and osteosarcoma (103).
Conclusions and perspectives
In conclusion, the intricate relationship between N-glycosylation and the pathogenesis of myeloid malignancies is allowing us to gain a deeper understanding of how the disease develops and identify new areas of therapeutic interest. As we continue to map aberrant N-glycan structures which are more prevalent on malignant as compared to control cells, or more abundant on chemoresistant versus chemosensitive cells, opportunities arise to establish biomarkers for chemotherapy resistance and risk stratification. In addition, it will allow identification of therapeutic targets since glycosylation is present on the plasma membrane and therefore easily accessible. In this respect, targeting glycosylation may reduce cancer cell growth and improve therapy sensitivity, consequently increasing treatment responses as discussed in this review. Furthermore, novel immunotherapeutic options are emerging for myeloid malignancies, for which the targeting of glycosylation may also improve effectiveness.
With recent advancements in mass spectrometry, it has become possible to decipher glycan structures. While previously mass spectrometry only allowed for quantification of sugar moieties in glycan structures, it is now also possible to determine how sugar molecules are attached to one another. This structural information may help in the development and improvement of glycan-binding structures for diagnostic and therapeutic purposes. Due to technological advancements, we expect that the field as well as the clinical applications will develop substantially in the coming years. Thus, in the future we may see the implementation of glycosylation-related parameters in prognostication and personalized therapies.
Author contributions
JM: Writing – original draft, Writing – review & editing, Conceptualization, Visualization. VW: Writing – review & editing, Conceptualization, Funding acquisition, Visualization. AM: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Supervision, Visualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. JM and AM acknowledge funding from the Dutch Cancer Society Koningin Wilhelmina Fonds (KWF, Young Investigator grant 14764) and Radboudumc Hypatia Track (R0007046). VW would like to acknowledge funding from the Dutch Cancer Society Koningin Wilhelmina Fonds (KWF, Young Investigator grant 10709).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang W, Özdemir V. Special issue: glycomics and personalized glycomedicine. Omics. (2019) 23:599–600. doi: 10.1089/omi.2019.0177
2. Bieberich E. Synthesis, processing, and function of N-glycans in N-glycoproteins. Adv Neurobiol. (2014) 9:47–70. doi: 10.1007/978-1-4939-1154-7_3
3. Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci USA. (1994) 91:913–7. doi: 10.1073/pnas.91.3.913
4. Deprez P, Gautschi M, Helenius A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell. (2005) 19:183–95. doi: 10.1016/j.molcel.2005.05.029
5. Grinna LS, Robbins PW. Glycoprotein biosynthesis. Rat liver microsomal glucosidases which process oligosaccharides. J Biol Chem. (1979) 254:8814–8. doi: 10.1016/S0021-9258(19)86771-6
6. Ruddock LW. Molinari M. N-glycan processing in ER quality control. J Cell Sci. (2006) 119:4373–80. doi: 10.1242/jcs.03225
7. Stanley P, Moremen KW, Lewis NE, Taniguchi N, Aebi M. N-Glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al eds. Essentials of Glycobiology. 4th ed. Cold Spring Harbor (NY (2022) p. 103–16.
8. Zeidner JF, Roy D, Perl A, Gojo I. Myeloid Malignancies. Am Cancer Society’s Oncol Pract. (2018), 397–421. doi: 10.1002/9781118592168
9. Akinduro O, Weber TS, Ang H, Haltalli MLR, Ruivo N, Duarte D, et al. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat Commun. (2018) 9:519. doi: 10.1038/s41467-017-02376-5
10. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. (2022) 140:1345–77. doi: 10.1182/blood.2022016867
11. Thapa B, Fazal S, Parsi M, Rogers HJ. Myeloproliferative Neoplasms. Treasure Island, FL: StatPearls (2024).
12. Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. (2015) 5:e366. doi: 10.1038/bcj.2015.95
14. Chatterjee S, Ugonotti J, Lee LY, Everest-Dass A, Kawahara R, Thaysen-Andersen M. Trends in oligomannosylation and alpha1,2-mannosidase expression in human cancers. Oncotarget. (2021) 12:2188–205. doi: 10.18632/oncotarget.v12i21
15. Blöchl C, Wang D, Madunić K, Lageveen-Kammeijer GSM, Huber CG, Wuhrer M, et al. Integrated N- and O-glycomics of acute myeloid leukemia (AML) cell lines. Cells. (2021) 10(11), 3058. doi: 10.3390/cells10113058
16. van Spronsen MF, Horrevorts S, Cali C, Westers TM, Van Gassen S, Saeys Y, et al. Dysregulation of developmental and cell type-specific expression of glycoconjugates on hematopoietic cells: a new characteristic of myelodysplastic neoplasms (MDS). Leukemia. (2023) 37:702–7. doi: 10.1038/s41375-022-01784-x
17. Chatterjee S, Lee LY, Kawahara R, Abrahams JL, Adamczyk B, Anugraham M, et al. Protein paucimannosylation is an enriched N-glycosylation signature of human cancers. PROTEOMICS. (2019) 19:1900010. doi: 10.1002/pmic.201900010
18. Delwel R, Touw I, Bot F, Lowenberg B. Fucose binding lectin for characterizing acute myeloid leukemia progenitor cells. Blood. (1986) 68:41–5. doi: 10.1182/blood.V68.1.41.bloodjournal68141
19. Chrastinová L, Pastva O, Bocková M, Kovářová H, Ceznerová E, Kotlín R, et al. Linking aberrant glycosylation of plasma glycoproteins with progression of myelodysplastic syndromes: a study based on plasmonic biosensor and lectin array. Sci Rep. (2023) 13:12816. doi: 10.1038/s41598-023-39927-4
20. Ma H, Zhou H, Song X, Shi S, Zhang J, Jia L. Modification of sialylation is associated with multidrug resistance in human acute myeloid leukemia. Oncogene. (2015) 34:726–40. doi: 10.1038/onc.2014.7
21. Zhang X, Dong W, Zhou H, Li H, Wang N, Miao X, et al. alpha-2,8-sialyltransferase is involved in the development of multidrug resistance via PI3K/akt pathway in human chronic myeloid leukemia. IUBMB Life. (2015) 67:77–87. doi: 10.1002/iub.1351
22. Blochl C, Wang D, Mayboroda OA, Lageveen-Kammeijer GSM, Wuhrer M. Transcriptionally imprinted glycomic signatures of acute myeloid leukemia. Cell Biosci. (2023) 13:31. doi: 10.1186/s13578-023-00981-0
23. Skacel PO, Edwards AJ, Harrison CT, Watkins WM. Enzymic control of the expression of the X determinant (CD15) in human myeloid cells during maturation: the regulatory role of 6-sialytransferase. Blood. (1991) 78:1452–60. doi: 10.1182/blood.V78.6.1452.bloodjournal7861452
24. Silva MC, Fernandes Â, Oliveira M, Resende C, Correia A, de-Freitas-Junior JC, et al. Glycans as immune checkpoints: removal of branched N-glycans enhances immune recognition preventing cancer progression. Cancer Immunol Res. (2020) 8:1407–25. doi: 10.1158/2326-6066.CIR-20-0264
25. Peng W, Zhu R, Zhou S, Mirzaei P, Mechref Y. Integrated transcriptomics, proteomics, and glycomics reveals the association between up-regulation of sialylated N-glycans/integrin and breast cancer brain metastasis. Sci Rep. (2019) 9:17361. doi: 10.1038/s41598-019-53984-8
26. Takamatsu S, Oguri S, Minowa MT, Yoshida A, Nakamura K, Takeuchi M, et al. Unusually high expression of N-acetylglucosaminyltransferase-IVa in human choriocarcinoma cell lines: a possible enzymatic basis of the formation of abnormal biantennary sugar chain. Cancer Res. (1999) 59:3949–53.
27. Potapenko IO, Haakensen VD, Luders T, Helland A, Bukholm I, Sorlie T, et al. Glycan gene expression signatures in normal and Malignant breast tissue; possible role in diagnosis and progression. Mol Oncol. (2010) 4:98–118. doi: 10.1016/j.molonc.2009.12.001
28. de-Souza-Ferreira M, Ferreira EE, de-Freitas-Junior JCM. Aberrant N-glycosylation in cancer: MGAT5 and beta1,6-GlcNAc branched N-glycans as critical regulators of tumor development and progression. Cell Oncol (Dordr). (2023) 46:481–501. doi: 10.1007/s13402-023-00770-4
29. Lyman SD, James L, Vanden Bos T, de Vries P, Brasel K, Gliniak B, et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. (1993) 75:1157–67. doi: 10.1016/0092-8674(93)90325-K
30. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
31. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
32. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
33. Manara E, Basso G, Zampini M, Buldini B, Tregnago C, Rondelli R, et al. Characterization of children with FLT3-ITD acute myeloid leukemia: a report from the AIEOP AML-2002 study group. Leukemia. (2017) 31:18–25. doi: 10.1038/leu.2016.177
34. Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. (2006) 108:3654–61. doi: 10.1182/blood-2006-03-009233
35. Zwaan CM, Meshinchi S, Radich JP, Veerman AJ, Huismans DR, Munske L, et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood. (2003) 102:2387–94. doi: 10.1182/blood-2002-12-3627
36. Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. (2020) 111:312–22. doi: 10.1111/cas.14274
37. Lanternier F, Cohen-Akenine A, Palmerini F, Feger F, Yang Y, Zermati Y, et al. Phenotypic and genotypic characteristics of mastocytosis according to the age of onset. PloS One. (2008) 3:e1906. doi: 10.1371/journal.pone.0001906
38. Kristensen T, Vestergaard H, Møller MB. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J Mol Diagn. (2011) 13:180–8. doi: 10.1016/j.jmoldx.2010.10.004
39. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. Lancet. (2018) 392:593–606. doi: 10.1016/S0140-6736(18)31041-9
40. Schmidt-Arras D, Böhmer SA, Koch S, Müller JP, Blei L, Cornils H, et al. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood. (2009) 113:3568–76. doi: 10.1182/blood-2007-10-121426
41. Schmidt-Arras DE, Böhmer A, Markova B, Choudhary C, Serve H, Böhmer FD. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol. (2005) 25:3690–703. doi: 10.1128/MCB.25.9.3690-3703.2005
42. Shi X, Sousa LP, Mandel-Bausch EM, Tome F, Reshetnyak AV, Hadari Y, et al. Distinct cellular properties of oncogenic KIT receptor tyrosine kinase mutants enable alternative courses of cancer cell inhibition. Proc Natl Acad Sci U S A. (2016) 113:E4784–93. doi: 10.1073/pnas.1610179113
43. Bougherara H, Subra F, Crepin R, Tauc P, Auclair C, Poul MA. The aberrant localization of oncogenic kit tyrosine kinase receptor mutants is reversed on specific inhibitory treatment. Mol Cancer Res. (2009) 7:1525–33. doi: 10.1158/1541-7786.MCR-09-0138
44. Sun J, Pedersen M, Ronnstrand L. The D816V mutation of c-Kit circumvents a requirement for Src family kinases in c-Kit signal transduction. J Biol Chem. (2009) 284:11039–47. doi: 10.1074/jbc.M808058200
45. Bibi S, Arslanhan MD, Langenfeld F, Jeanningros S, Cerny-Reiterer S, Hadzijusufovic E, et al. Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: possible new targets of therapy. Haematologica. (2014) 99:417–29. doi: 10.3324/haematol.2013.098442
46. Park H, Kim D, Koh Y, Yoon SS. The upregulation of Pim kinases is essential in coordinating the survival, proliferation, and migration of KIT D816V-mutated neoplastic mast cells. Leuk Res. (2019) 83:106166. doi: 10.1016/j.leukres.2019.106166
47. Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. (2009) 36:326–39. doi: 10.1016/j.molcel.2009.09.019
48. Takahashi S. Mutations of FLT3 receptor affect its surface glycosylation, intracellular localization, and downstream signaling. Leuk Res Rep. (2020) 13:100187. doi: 10.1016/j.lrr.2019.100187
49. Hu X, Chen F. Targeting on glycosylation of mutant FLT3 in acute myeloid leukemia. Hematology. (2019) 24:651–60. doi: 10.1080/16078454.2019.1666219
50. Duan C, Fukuda T, Isaji T, Qi F, Yang J, Wang Y, et al. Deficiency of core fucosylation activates cellular signaling dependent on FLT3 expression in a Ba/F3 cell system. FASEB J. (2020) 34:3239–52. doi: 10.1096/fj.201902313RR
51. Janke H, Pastore F, Schumacher D, Herold T, Hopfner KP, Schneider S, et al. Activating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute myeloid leukemia. PloS One. (2014) 9:e89560. doi: 10.1371/journal.pone.0089560
52. Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. (2005) 105:4792–9. doi: 10.1182/blood-2004-11-4430
53. Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. (1979) 18:2186–92. doi: 10.1021/bi00578a008
54. Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V, et al. Antileukemic Activity of 2-Deoxy-d-Glucose through Inhibition of N-Linked Glycosylation in Acute Myeloid Leukemia with FLT3-ITD or c-KIT Mutations. Mol Cancer Ther. (2015) 14:2364–73. doi: 10.1158/1535-7163.MCT-15-0163
55. Williams AB, Li L, Nguyen B, Brown P, Levis M, Small D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood. (2012) 120:3069–79. doi: 10.1182/blood-2012-01-403493
56. Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang J, et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia. (2017) 31:2143–50. doi: 10.1038/leu.2017.45
57. Pajak B, Siwiak E, Sołtyka M, Priebe A, Zieliński R, Fokt I, et al. 2-deoxy-d-glucose and its analogs: from diagnostic to therapeutic agents. Int J Mol Sci. (2019) 21(1), 234. doi: 10.3390/ijms21010234
58. Jutzi JS, Marneth AE, Ciboddo M, Guerra-Moreno A, Jiménez-Santos MJ, Kosmidou A, et al. Whole-genome CRISPR screening identifies N-glycosylation as a genetic and therapeutic vulnerability in CALR-mutant MPNs. Blood. (2022) 140:1291–304. doi: 10.1182/blood.2022015629
59. Spiciarich DR, Oh ST, Foley A, Hughes SB, Mauro MJ, Abdel-Wahab O, et al. A novel germline variant in CSF3R reduces N-glycosylation and exerts potent oncogenic effects in leukemia. Cancer Res. (2018) 78:6762–70. doi: 10.1158/0008-5472.CAN-18-1638
60. Wiersma VR, Michalak M, Abdullah TM, Bremer E, Eggleton P. Mechanisms of translocation of ER chaperones to the cell surface and immunomodulatory roles in cancer and autoimmunity. Front Oncol. (2015) 5:7. doi: 10.3389/fonc.2015.00007
61. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. (2013) 369:2379–90. doi: 10.1056/NEJMoa1311347
62. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. (2013) 369:2391–405. doi: 10.1056/NEJMoa1312542
63. Elf S, Abdelfattah NS, Chen E, Perales-Patón J, Rosen EA, Ko A, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discovery. (2016) 6:368–81. doi: 10.1158/2159-8290.CD-15-1434
64. Albu RI, Constantinescu SN. Extracellular domain N-glycosylation controls human thrombopoietin receptor cell surface levels. Front Endocrinol. (2011) 2. doi: 10.3389/fendo.2011.00071
65. Masubuchi N, Araki M, Yang Y, Hayashi E, Imai M, Edahiro Y, et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia. (2020) 34:499–509. doi: 10.1038/s41375-019-0564-z
66. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. (2016) 127:1325–35. doi: 10.1182/blood-2015-11-681932
67. Elf S, Abdelfattah NS, Baral AJ, Beeson D, Rivera JF, Ko A, et al. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood. (2018) 131:782–6. doi: 10.1182/blood-2017-08-800896
68. Han L, Schubert C, Köhler J, Schemionek M, Isfort S, Brümmendorf TH, et al. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J Hematol Oncol. (2016) 9:45. doi: 10.1186/s13045-016-0275-0
69. Zhang T, Marsman G, Pereira-Martins DA, Wuhrer M, Huls GA, Wiersma VR. Concanavalin A staining: a potential biomarker to predict cytarabine sensitivity in acute myeloid leukemia. Front Hematol. (2024) 2. doi: 10.3389/frhem.2023.1302328
70. Xu H, Muise ES, Javaid S, Chen L, Cristescu R, Mansueto MS, et al. Identification of predictive genetic signatures of Cytarabine responsiveness using a 3D acute myeloid leukaemia model. J Cell Mol Med. (2019) 23:7063–77. doi: 10.1111/jcmm.14608
71. Woodley K, Dillingh LS, Giotopoulos G, Madrigal P, Rattigan KM, Philippe C, et al. Mannose metabolism inhibition sensitizes acute myeloid leukaemia cells to therapy by driving ferroptotic cell death. Nat Commun. (2023) 14:2132. doi: 10.1038/s41467-023-37652-0
72. Ichikawa M, Scott DA, Losfeld ME, Freeze HH. The metabolic origins of mannose in glycoproteins. J Biol Chem. (2014) 289:6751–61. doi: 10.1074/jbc.M113.544064
73. Hamorsky KT, Kouokam JC, Dent MW, Grooms TN, Husk AS, Hume SD, et al. Engineering of a lectibody targeting high-mannose-type glycans of the HIV envelope. Mol Ther. (2019) 27:2038–52. doi: 10.1016/j.ymthe.2019.07.021
74. Choukrani G, Visser N, Ustyanovska Avtenyuk N, Olthuis M, Marsman G, Ammatuna E, et al. Galectin-9 has non-apoptotic cytotoxic activity toward acute myeloid leukemia independent of cytarabine resistance. Cell Death Discovery. (2023) 9:228. doi: 10.1038/s41420-023-01515-w
75. Ryva B, Zhang K, Asthana A, Wong D, Vicioso Y, Parameswaran R. Wheat germ agglutinin as a potential therapeutic agent for leukemia. Front Oncol. (2019) 9:100. doi: 10.3389/fonc.2019.00100
76. Glavey SV, Manier S, Natoni A, Sacco A, Moschetta M, Reagan MR, et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood. (2014) 124:1765–76. doi: 10.1182/blood-2014-03-560862
77. Lee DH, Kang SH, Choi DS, Ko M, Choi E, Ahn H, et al. Genome wide CRISPR screening reveals a role for sialylation in the tumorigenesis and chemoresistance of acute myeloid leukemia cells. Cancer Lett. (2021) 510:37–47. doi: 10.1016/j.canlet.2021.04.006
78. Leonti AR, Pardo L, Alonzo TA, Gerbing RB, Eidenschink Brodersen L, Ries RE, et al. Transcriptome profiling of glycosylation genes defines correlation with E-selectin ligand expression and clinical outcome in AML. Blood. (2019) 134:3772. doi: 10.1182/blood-2019-124525
79. Daly J, Sarkar S, Natoni A, Henderson R, Swan D, Carlsten M, et al. Hypersialylation protects multiple myeloma cells from NK cell-mediated immunosurveillance and this can be overcome by targeted desialylation using a sialyltransferase inhibitor. Blood. (2019) 134:138. doi: 10.1182/blood-2019-126613
80. Bull C, Boltje TJ, Balneger N, Weischer SM, Wassink M, van Gemst JJ, et al. Sialic acid blockade suppresses tumor growth by enhancing T-cell-mediated tumor immunity. Cancer Res. (2018) 78:3574–88. doi: 10.1158/0008-5472.CAN-17-3376
81. Hira VVV, Van Noorden CJF, Carraway HE, Maciejewski JP, Molenaar RJ. Novel therapeutic strategies to target leukemic cells that hijack compartmentalized continuous hematopoietic stem cell niches. Biochim Biophys Acta Rev Cancer. (2017) 1868:183–98. doi: 10.1016/j.bbcan.2017.03.010
82. Ferraro F, Celso CL, Scadden D. Adult stem cels and their niches. Adv Exp Med Biol. (2010) 695:155–68. doi: 10.1007/978-1-4419-7037-4_11
83. Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. (2012) 18:1651–7. doi: 10.1038/nm.2969
84. Greenberg AW, Kerr WG, Hammer DA. Relationship between selectin-mediated rolling of hematopoietic stem and progenitor cells and progression in hematopoietic development. Blood. (2000) 95:478–86. doi: 10.1182/blood.V95.2.478
85. Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. (2020) 11:2042. doi: 10.1038/s41467-020-15817-5
86. Spertini C, Baïsse B, Bellone M, Gikic M, Smirnova T, Spertini O. Acute myeloid and lymphoblastic leukemia cell interactions with endothelial selectins: critical role of PSGL-1, CD44 and CD43. Cancers. (2019) 11:1253. doi: 10.3390/cancers11091253
87. Ales E, Sackstein R. The biology of E-selectin ligands in leukemogenesis. Adv Cancer Res. (2023) 157:229–50. doi: 10.1016/bs.acr.2022.07.001
88. Dai Y, Cheng Z, Pang Y, Jiao Y, Qian T, Quan L, et al. Prognostic value of the FUT family in acute myeloid leukemia. Cancer Gene Ther. (2020) 27:70–80. doi: 10.1038/s41417-019-0115-9
89. Mondal N, Dykstra B, Lee J, Ashline DJ, Reinhold VN, Rossi DJ, et al. Distinct human alpha(1,3)-fucosyltransferases drive Lewis-X/sialyl Lewis-X assembly in human cells. J Biol Chem. (2018) 293:7300–14. doi: 10.1074/jbc.RA117.000775
90. DeAngelo DJ, Jonas BA, Liesveld JL, Bixby DL, Advani AS, Marlton P, et al. Phase 1/2 study of uproleselan added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia. Blood. (2022) 139:1135–46. doi: 10.1182/blood.2021010721
91. DeAngelo DJ, Erba HP, Jonas BA, O’Dwyer M, Marlton P, Huls GA, et al. A phase III trial to evaluate the efficacy of uproleselan (GMI-1271) with chemotherapy in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol. (2019) 37:TPS7066–TPS. doi: 10.1200/JCO.2019.37.15_suppl.TPS7066
92. Puch S, Armeanu S, Kibler C, Johnson KR, Muller CA, Wheelock MJ, et al. N-cadherin is developmentally regulated and functionally involved in early hematopoietic cell differentiation. J Cell Sci. (2001) 114:1567–77. doi: 10.1242/jcs.114.8.1567
93. Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. (2013) 19:1513–7. doi: 10.1038/nm.3364
94. Wein F, Pietsch L, Saffrich R, Wuchter P, Walenda T, Bork S, et al. N-cadherin is expressed on human hematopoietic progenitor cells and mediates interaction with human mesenchymal stromal cells. Stem Cell Res. (2010) 4:129–39. doi: 10.1016/j.scr.2009.12.004
95. Zhi L, Wang M, Rao Q, Yu F, Mi Y, Wang J. Enrichment of N-Cadherin and Tie2-bearing CD34+/CD38–/CD123+ leukemic stem cells by chemotherapy-resistance. Cancer Letters. (2010) 296:65–73. doi: 10.1016/j.canlet.2010.03.021
96. Qiu S, Jia Y, Xing H, Yu T, Yu J, Yu P, et al. N-Cadherin and Tie2 positive CD34+CD38−CD123+ leukemic stem cell populations can develop acute myeloid leukemia more effectively in NOD/SCID mice. Leuk Res. (2014) 38:632–7. doi: 10.1016/j.leukres.2014.03.007
97. Marjon KD, Termini CM, Karlen KL, Saito-Reis C, Soria CE, Lidke KA, et al. Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene. (2016) 35:4132–40. doi: 10.1038/onc.2015.449
98. Zhu L, Chen Y, Du H, Cong Y, Yan W, Ma K, et al. N-glycosylation of CD82 at Asn157 is required for suppressing migration and invasion by reversing EMT via Wnt/beta-catenin pathway in colon cancer. Biochem Biophys Res Commun. (2022) 629:121–7. doi: 10.1016/j.bbrc.2022.08.079
99. Li J, Xu J, Li L, Ianni A, Kumari P, Liu S, et al. MGAT3-mediated glycosylation of tetraspanin CD82 at asparagine 157 suppresses ovarian cancer metastasis by inhibiting the integrin signaling pathway. Theranostics. (2020) 10:6467–82. doi: 10.7150/thno.43865
100. Mrozik KM, Cheong CM, Hewett D, Chow AW, Blaschuk OW, Zannettino AC, et al. Therapeutic targeting of N-cadherin is an effective treatment for multiple myeloma. Br J Haematol. (2015) 171:387–99. doi: 10.1111/bjh.13596
101. Feng J, Wang Y, Li B, Yu X, Lei L, Wu J, et al. Loss of bisecting GlcNAcylation on MCAM of bone marrow stoma determined pro-tumoral niche in MDS/AML. Leukemia. (2023) 37:113–21. doi: 10.1038/s41375-022-01748-1
102. Mills L, Tellez C, Huang S, Baker C, McCarty M, Green L, et al. Fully human antibodies to MCAM/MUC18 inhibit tumor growth and metastasis of human melanoma. Cancer Res. (2002) 62:5106–14.
Keywords: N-glycosylation, acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), chronic myeloid leukemia (CML), chemoresistance, niche interactions, receptors
Citation: Sanmartín-Martínez J, Wiersma VR and Marneth AE (2024) The sweet symphony of N-glycans in myeloid malignancies. Front. Hematol. 3:1415618. doi: 10.3389/frhem.2024.1415618
Received: 10 April 2024; Accepted: 13 June 2024;
Published: 08 July 2024.
Edited by:
Valentina Giudice, University of Salerno, ItalyReviewed by:
Ivo Paul Touw, Erasmus University Rotterdam, NetherlandsÂngela Fernandes, University of Porto, Portugal
Copyright © 2024 Sanmartín-Martínez, Wiersma and Marneth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna E. Marneth, YW5uYS5tYXJuZXRoQHJhZGJvdWR1bWMubmw=
†ORCID: Javier Sanmartin Martinez, orcid.org/0009-0000-0683-8750
Valerie R. Wiersma, orcid.org/0000-0001-8012-0376
Anna E. Marneth, orcid.org/0000-0001-9065-0581