 Caroline Spaner1
Caroline Spaner1 Jessica Durkee-Shock2Andrew Weng3Ryan Stubbins4Alina S. Gerrie5
Jessica Durkee-Shock2Andrew Weng3Ryan Stubbins4Alina S. Gerrie5 Stefania Pittaluga6
Stefania Pittaluga6 Jeffrey I. Cohen2
Jeffrey I. Cohen2 Luke Y. C. Chen4,7*
Luke Y. C. Chen4,7*- 1Department of Medicine, University of British Columbia, Vancouver, BC, Canada
- 2Laboratory of Infectious Diseases, National Institute of Allergy and Infectious Disease, National Institutes of Health (NIH), Bethesda, MD, United States
- 3Terry Fox Laboratory, BC Cancer, Vancouver, BC, Canada
- 4Division of Hematology, University of British Columbia, Vancouver, BC, Canada
- 5Centre for Lymphoid Cancer, BC Cancer, Vancouver, BC, Canada
- 6Centre for Cancer Research, National Institutes of Health (NIH), Bethesda, MD, United States
- 7Division of Hematology, Dalhousie University, Halifax, NS, Canada
Aggressive natural killer cell leukemia (ANKL) is a rare, aggressive hematologic malignancy which often presents as fulminant Epstein-Barr virus (EBV)- driven hemophagocytic lymphohistiocytosis (HLH). ANKL lacks a distinct immunologic and morphologic signature, making early diagnosis particularly challenging. Here we present a case of ANKL in a patient presenting with EBV-HLH. After poor treatment response to the HLH-2004 protocol (etoposide and dexamethasone), bone marrow biopsy demonstrated an atypical CD3-/CD56+ natural killer (NK) cell population with diminished CD7 expression consistent with EBV+ ANKL. Asparaginase-based chemotherapy was initiated but his disease progressed and he died from multiorgan failure. This case highlights the diagnostic challenges of ANKL given the lack of standardized diagnostic criteria, the importance of considering T/NK cell malignancies in the differential diagnosis of EBV-HLH, and adds to the literature on this rare disease.
Introduction
Aggressive natural killer cell leukemia (ANKL) is a rare disease characterized by neoplastic proliferation of natural killer (NK) cells in the blood, bone marrow, liver, and spleen (1, 2). Approximately 90% of cases are driven by Epstein Barr Virus (EBV) (3). ANKL is classified by the World Health Organization as a “mature T/NK cell neoplasm” (4). However, it lacks a unique immunophenotypic or molecular diagnostic signature, making early diagnosis particularly challenging. Most patients are of East Asian descent, and the prognosis is poor, with a median survival of 2 months. Moreover, ANKL commonly presents as fulminant EBV-driven hemophagocytic lymphohistiocytosis (HLH), which can be very difficult to distinguish from other discrete causes of EBV-HLH. HLH is a cytokine storm syndrome characterized by pathologic activation of T lymphocytes and macrophages, resulting in marked immune dysregulation, hyperinflammation, and multiorgan failure (5, 6). HLH can be caused by genetic mutations affecting lymphocyte cytotoxicity (primary HLH) or in the setting of underlying illness (secondary HLH), such as malignancy, infection and autoimmune disease.
The differential diagnosis for a patient presenting with EBV-HLH includes distinct malignant and non-malignant etiologies (7–10). Non-neoplastic causes include acute infection. Neoplastic causes include B cell lymphomas, due to the tropism of EBV to B cells; T- and NK-cell lymphomas, including ANKL, extranodal NK/T-cell lymphoma (ENKL), chronic active EBV disease (CAEBV), and systemic EBV positive T-cell lymphoma of childhood (11). There is significant clinicopathologic overlap between these entities. As well, the tumor burden in ANKL can be low, making it even more difficult to elucidate a precise diagnosis in an acutely ill patient. Less than 500 cases of this rare disease have been reported in the literature, and the vast majority of these are from Asia. We present the case of an adolescent who presented in British Columbia, Canada, with florid EBV-HLH and had a fatal outcome.
Case report
A 17-year-old previously healthy male of Chinese descent presented to the emergency department of a community hospital with a 5-day history of headache and fever up to 39.4°C. Physical examination on admission was significant for palpable hepatosplenomegaly without lymphadenopathy. Initial blood work (Table 1) showed pancytopenia with reactive lymphocytes and pelgeroid neutrophils seen on peripheral blood film. No blasts or large lymphoid cells were reported. Liver enzymes including ALT, AST, and bilirubin were elevated, and he had a prolonged aPTT but normal INR and fibrinogen. Ferritin was markedly elevated at 10,614 ug/L (normal range 15-300 ug/L), with elevated LDH, C-reactive peptide (CRP), and fibrin D-dimer. CXCL9 was elevated at 6,395 pg/mL (normal range ≤657 pg/mL), as was soluble interleukin 2 receptor (sIL2r) (16,426 U/mL, normal range 241-846 U/mL). Monospot test was positive, with Epstein-Barr virus (EBV) serology positive for reactive IgG, nonreactive IgM, and a viral load by plasma PCR of 498,000 copies/mL (Table 1). Autoimmune serology, HIV, and viral hepatitis panels were negative.
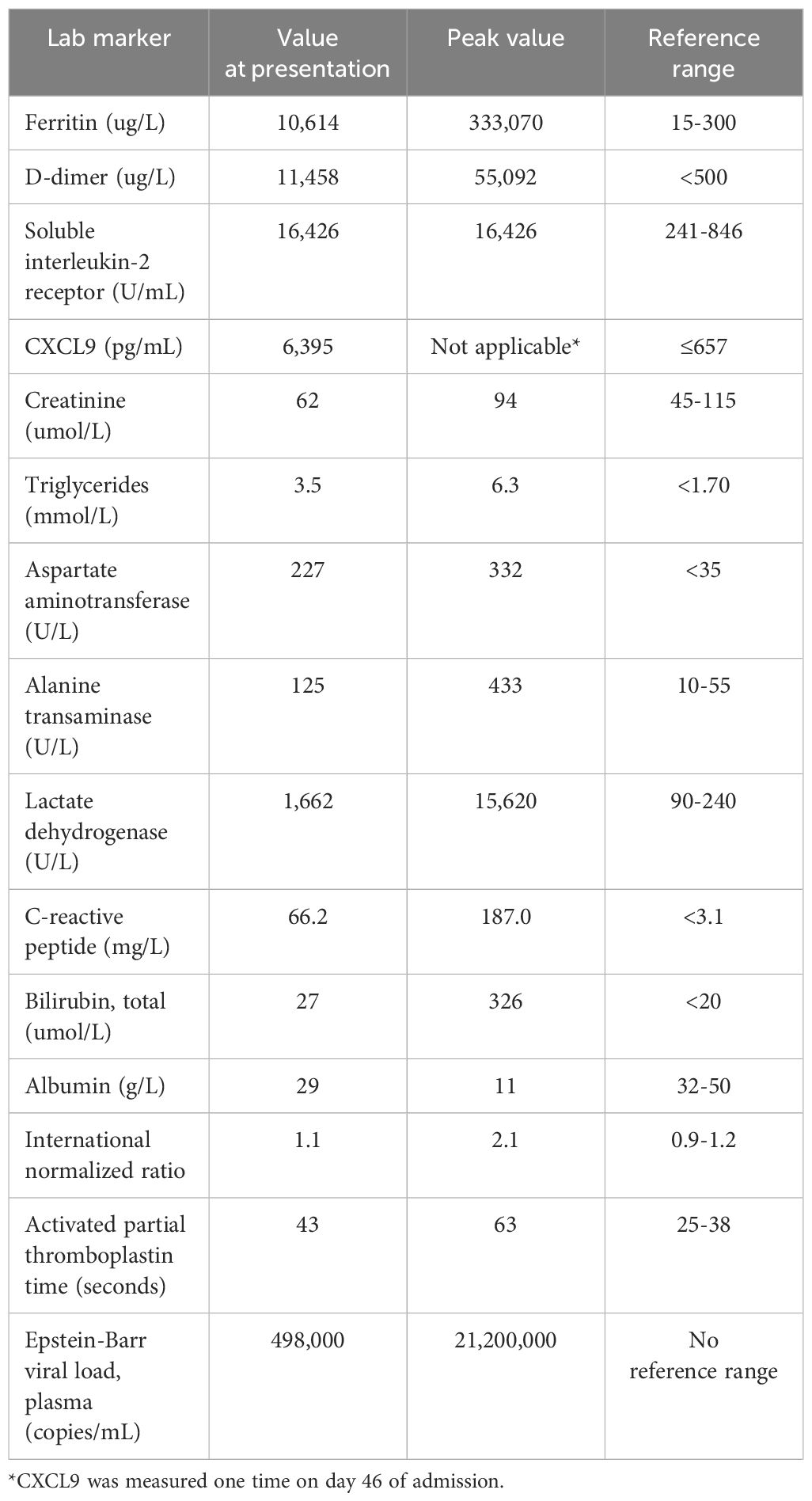
Table 1 Inflammatory markers at time of diagnosis and peak values during course of disease.
A working diagnosis of non-neoplastic EBV-HLH was made based on the HLH-2004 criteria. He was treated initially with dexamethasone 10mg/m2, with the goal of avoiding cytotoxic chemotherapy. However, his fever worsened and his EBV viral load increased to 8,910,000 copies/mL by day 5 of admission so he was transferred to our tertiary care hospital. Etoposide 150 mg/m2 was added, and he received intravenous immunoglobulin (IVIG) 0.5 g/kg, and rituximab 375 mg/m2 targeting EBV-infected B cells (12). Cyclosporine is not typically used initially for management of HLH in our center as it is felt to have limited benefit (13). His HLH parameters remained persistently elevated, and ruxolitinib 10 mg twice daily was then initiated for refractory HLH (14, 15). His biochemical and clinical parameters subsequently improved.
Bone marrow biopsy and aspiration performed on day 2 of admission revealed increased histiocytes and occasional hemophagocytosis, and an abnormal population of large, atypical cells with round to irregular, occasionally convoluted nuclei (Figure 1). Cellularity was 80%, with trilineage hematopoiesis present. Flow cytometry demonstrated an atypical CD3-/CD56+ NK cell population with diminished CD7 expression accounting for 27% of the lymphocyte population (Figure 2) (16). The CD4/8 ratio was 1:1 and no aberrant T cells were noted. Additional immunohistochemistry showed frequent scattered large, atypical lymphoid cells positive for CD56 (Figure 3A), cytoplasmic CD3, CD2, CD56, CD7 (small subset), granzyme B and TIA1, and negative for perforin, BF1, TCR gamma, TCR delta, CD4, and CD8. A number of these large, atypical cells stained positively for Epstein-Barr encoded RNA (EBER) by in situ hybridization (Figure 3B). PCR for T-cell receptor beta showed a polyclonal pattern. Given the atypical cytotoxic NK cell population, marked atypia of the NK cells on CD56 immunostain, and less atypia among CD4+/CD8+ T cells, or CD20+ B cells, a diagnosis of aggressive NK cell leukemia (ANKL) was favored.
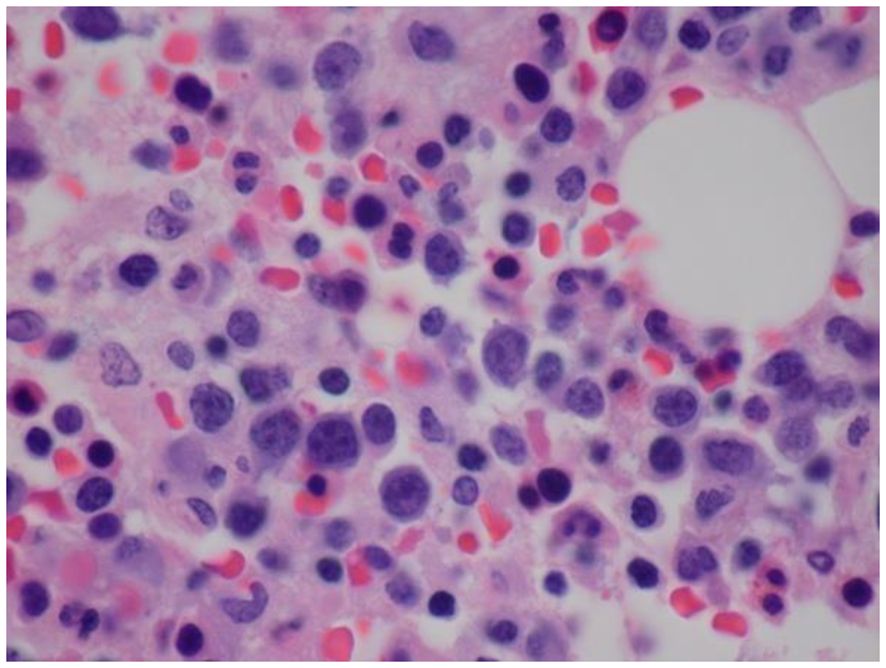
Figure 1 Histopathology of bone marrow demonstrating increased histiocytes and an abnormal population of large, atypical cells with round to irregular, occasionally convoluted nuclei, condensed chromatin, multiple distinct nucleoli and limited cytoplasm (Hematoxylin and Eosin (H&E) staining).
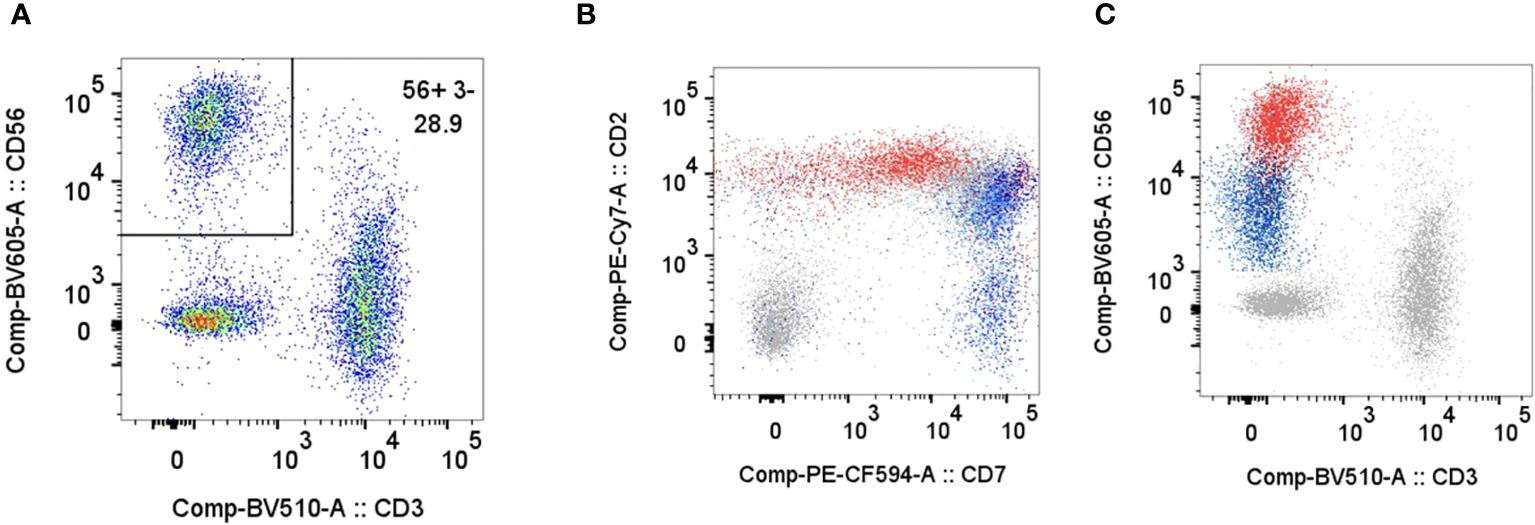
Figure 2 Flow cytometry on bone marrow aspirate specimen. Plots for CD3/CD56 (A), CD7/CD2 (B), CD3/CD56 (C) show a CD56+/CD3- NK cell population comprising 28.9% of the gated lymphocytes (CD45+, Low SSC, CD14-). The patient’s NK cells demonstrate brighter CD56, slightly brighter CD2, and dimmer CD7 compared to normal NK cells. Plots in B and C show an overlay of the patient's gated lymphocytes in grey including abnormal NK cells in red, and NK cells from normal patients in blue.

Figure 3 (A) Immunohistochemical staining of bone marrow with anti-CD56 monoclonal antibody. (B) In situ hybridization of bone marrow for Epstein-Barr virus encoded RNA (EBER), with numerous positive cells, of varying size.
Genetic testing for germline variants through Blueprint Genetics Comprehensive Immune and Cytopenia Panel for HLH demonstrated microdeletion of 22q.11.2 of unclear significance. No germline pathogenic variants consistent with primary HLH were found. The specimens was sent for further review at the National Institutes of Health (NIH), which confirmed the diagnosis of ANKL.
Because his HLH parameters continued to worsen, the patient was started on an asparagine-based SMILE chemotherapy regimen (dexamethasone, ifosfamide, pegaspargase, etoposide) for ANKL prior to a definitive diagnosis (17). He did not receive methotrexate, typically part of SMILE regimen, due to liver dysfunction. The goal of treatment was for disease control in order to allow for allogeneic stem cell transplant (18, 19).
His HLH parameters temporally improved, but then worsened at the 3-week mark of cycle 1 with marked hyperbilirubinemia, hypotension, and an increase in EBV viral load to 21 million copies/mL (Table 1). Accordingly, he was restarted on etoposide and ruxolitinib. On day 14 of cycle 2 of SMILE, his ferritin and EBV viral load continued to worsen with declining liver function. He was started on gemcitabine and oxaliplatin for disease control. Emapalumab, an interferon gamma inhibitor, was also given (20). The emapalumab was started quite late in his course as this medication requires special access in Canada and is not routinely available. He had transient clinical and biochemical responses to emapalumab with defervescence and improvement in ferritin (Figure 4).
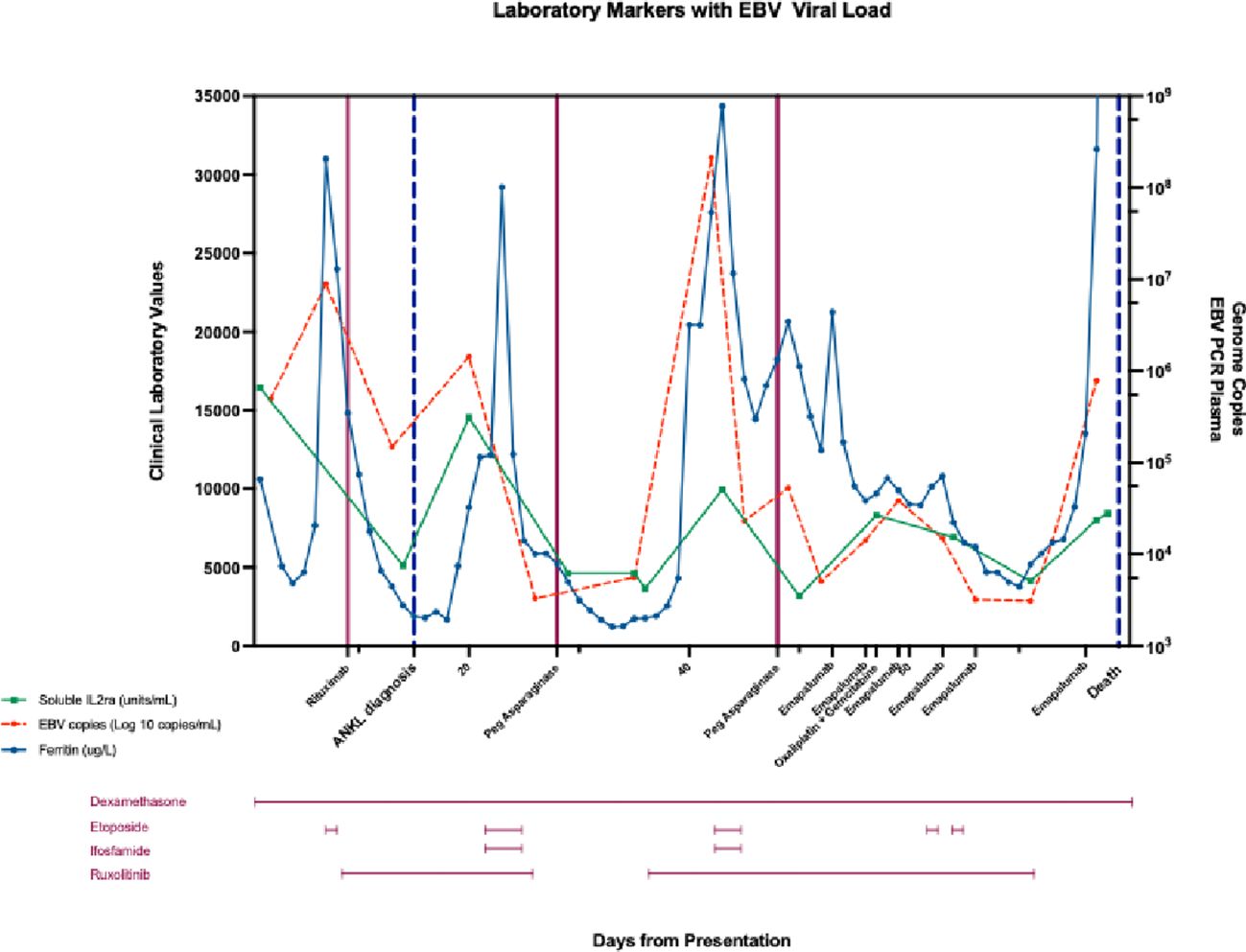
Figure 4 Changes in soluble interleukin 2 receptor (sIL2r, U/mL), ferritin (ug/L), and plasma EBV copies (Log 10 copies/mL) from day of admission (day 1), day of diagnosis (day 15), until day of death (day 78) in response to treatment: dexamethasone (day 1-78), etoposide (day 6, 22-24, day 42-44, day 61, day 64), gemcitabine (day 16), ifosfamide (day 22-24, day 42-44), oxaliplatin (day 57), pegaspargase (day 28, day 48), rituximab (day 9), ruxolitinib (day 10-26, day 35-70), and emapalumab (day 53, 56, 59, 63, 66).
His course was complicated by neutropenic infections with polymicrobial bacteremia (Klebsiella pneumoniae, Escherichia coli, Roseomonas mucosa, Stenotrophomonas maltophilia, and vancomycin-resistant Enterococcus faecium). Allo-HSCT was considered and a donor was identified, however it was not pursued given his worsening organ function, bacteremia, and inability to achieve partial or complete remission. He passed away due to severe lactic acidosis and multiorgan failure 78 days after his initial presentation.
Discussion
ANKL is a rare and aggressive hematologic malignancy which lacks a specific immunophenotypic or molecular diagnostic signature, making early diagnosis and treatment challenging. The presentation of ANKL varies, but typically presents as fulminant HLH with multi-organ failure, while the presence of B-symptoms (fevers, night sweats, unintentional weight loss) and lymphadenopathy often signifies leukemic disease (16). Several factors likely contribute to the immune dysregulation leading to HLH in ANKL and other NK cell disorders. First, the CD56bright subset of neoplastic NK cells secretes high levels of interferon gamma (IFN-γ), resulting in activation of macrophages and histiocytes, leading to the pathologic immune activation seen in HLH (21). In a subset of patients with extranodal T/NK cell lymphoproliferative disorders, a recurrent somatic mutation, ECSIT-T419C activates the NF-kB signaling pathway (22). This results in the downstream activation of tumor necrosis factor (TNF), IFN-γ, and interleukin-1β (IL-1β), suggesting that the aberrant production of these cytokines may be the driver of HLH seen in NK-cell malignancies, including ANKL.
The differential diagnosis of ANKL includes EBV extranodal NK/T cell lymphoma, which carries overlapping morphological and genetic features with ANKL, but has a less aggressive presentation. Even with early recognition, the prognosis is guarded, but the low burden of neoplastic cells in some cases, lack of standardized diagnostic criteria, and nonspecific morphological features can delay precise diagnosis and classification (2). This case highlights important lessons around the diagnosis of ANKL. First, morphology and immunophenotyping are important clues early in the disease presentation. The morphological finding of NK cells in blood and bone marrow should not be dismissed as simply reactive. Peripheral blood immunophenotyping is a widely available test for helping to rapidly distinguish between different subtypes of EBV-HLH. Increased CD8 positive T cells are observed in 80% of non-neoplastic EBV-HLH, whereas patients with T/NK CAEBV will often have an increase population of aberrant CD4 positive T cells (7, 23). In this case, the increased NK cells on flow cytometry were highly suggestive of ANKL, and this rare diagnosis was confirmed with immunohistochemistry and consultation with a center of excellence in this disorder.
The differential diagnosis of EBV-HLH is broad and includes nonneoplastic EBV-HLH often driven by monogenic inborn errors of immunity, T or NK cell CAEBV, systemic EBV positive T-cell lymphoma of childhood, and T/NK cell lymphoproliferative disorders, including ENKL, and ANKL (7, 10, 11). However, there are significant clinicopathologic similarities between these diseases. The degree of marrow infiltrate in ANKL can vary from minimal to severe, which can be confused with non-neoplastic EBV-HLH in the bone marrow, and acute presentations of NK-CAEBV can overlap with ANKL (4). In differentiating ANKL from ENKL, nasal type, the former tends to be associated with gain of 1q and loss of 7p15.1-pp22.3 and 17p13.1 alterations compared to the latter, but this is not always the case (24). In our case, an initial diagnosis of non-neoplastic EBV-HLH was made on the absence of immunologic or pathologic evidence of malignancy, and lack of an expanded atypical CD4+ T cell population or prior history of chronic, high-grade EBV viremia suggestive of NK cell CAEBV. While further immunologic studies revealed an atypical cytotoxic CD56+/CD7- NK cell population, the pathologic diagnosis of ANKL was challenging due to its rarity, and L-asparagine based chemotherapy was initiated prior to confirmatory diagnosis given the aggressive course and high mortality with delayed treatment.
Outcomes are poor in ANKL, but once diagnosed, the goals of therapy are to control the EBV-HLH and proceed to allogeneic stem cell transplantation (allo-SCT) whenever possible. A Korean study of 21 patients demonstrated overall response rates of 33% to 40% with L-asparaginase-based regimens (25). Remissions are short-lived with median progression-free survival (PFS) 3.9 months. However, some patients can achieve durable remission with allo-SCT; in one study, for patients who achieved even-free survival at twelve months, the OS was 85.2% at 5 years (19). Novel therapies for better control of EBV-HLH leading into transplant are needed. JAK inhibition has shown promise in HLH, particularly in pediatric HLH, but provided only transient partial response in this case. Likewise, emapalumab provided transient improvement in fever and HLH parameters but was initiated relatively late in the course of disease, due in large part to access issues in Canada. Emapalumab was approved by the United States Food and Drug Administration (FDA) based on a single arm study in primary HLH where patients received anti-IFN γ early in the course of their disease (20). However, recent anecdotal data suggests that it may not be as effective when given late in patients with malignancy-associated HLH (26). The immune checkpoint inhibitor nivolumab has shown promise in relapsed and refractory EBV-HLH but has limited utility in patients wherein allo-SCT is the definitive therapy, as nivolumab causes severe graft-vs-host disease and a prolonged wash out period is required (27).
Conclusion
This case highlights the diagnostic challenges in ANKL. EBV+ T/NK cell malignancies must be considered in patients with EBV-HLH, and these patients require early disease control and allogeneic stem cell transplantation.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
CS: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. JD: Software, Visualization, Writing – review & editing. AW: Data curation, Writing – review & editing. RS: Writing – review & editing. AG: Writing – review & editing. SP: Writing – review & editing. JC: Data curation, Writing – review & editing. LC: Conceptualization, Investigation, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. LC’s research is supported by a philanthropic gift from the Hsu & Taylor Family to the UBC & VGH Hospital Foundation. This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis. (2013) 8:95. doi: 10.1186/1750-1172-8-95
3. Ko YH, Park S, Kim K, Kim SJ, Kim WS. Aggressive natural killer cell leukemia: is Epstein-Barr virus negativity an indicator of a favorable prognosis? Acta Haematol. (2008) 120:199–206. doi: 10.1159/000193225
4. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo I, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. (2022) 36:1720–48. doi: 10.1038/s41375-022-01620-2
5. Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev. (2016) 30:411–20. doi: 10.1016/j.blre.2016.05.001
6. Setiadi A, Zoref-Lorenz A, Lee CY, Jordan MB, Chen LYC. Malignancy-associated haemophagocytic lymphohistiocytosis. Lancet. (2022) 9:e217–e27. doi: 10.1016/S2352-3026(21)00366-5
7. El-Mallawany NK, Curry CV, Allen CE. Haemophagocytic lymphohistiocytosis and Epstein-Barr virus: a complex relationship with diverse origins, expression and outcomes. Br J Haematol. (2022) 196:31–44. doi: 10.1111/bjh.17638
8. Quintanilla-Martinez L, Ko YH, Kimura H. ES. J. EBV-positive T-cell and NK-cell lymphoproliferative diseases of childhood. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press (2017) p. 355–62.
9. Bollard CM, Cohen JI. How I treat T-cell chronic active Epstein-Barr virus disease. Blood. (2018) 131:2899–905. doi: 10.1182/blood-2018-03-785931
10. Dojcinov SD, Quintanilla-Martinez L. How I diagnose EBV-positive B- and T-cell lymphoproliferative disorders. Am J Clin Pathol. (2022) 159(1):14–33. doi: 10.1093/ajcp/aqac105
11. Quintanilla-Martinez L, Swerdlow SH, Tousseyn T, Barrionuevo C, Nakamura S, Jaffe ES. New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders. Virchows Arch. (2022) 482(1):227–44. doi: 10.1007/s00428-022-03414-4
12. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. (2013) 162:376–82. doi: 10.1111/bjh.12386
13. Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
14. Hansen S, Alduaij W, Biggs CM, Belga S, Luecke K, Merkeley H, et al. Ruxolitinib as adjunctive therapy for secondary hemophagocytic lymphohistiocytosis: A case series. Eur J Haematol. (2021) 106:654–61. doi: 10.1111/ejh.13593
15. Zhang Q, Zhao Y-Z, Ma H-H, Wang D, Cui L, Li W-J, et al. A study of ruxolitinib response–based stratified treatment for pediatric hemophagocytic lymphohistiocytosis. Blood. (2022) 139:3493–504. doi: 10.1182/blood.2021014860
16. Jiang NG, Jin YM, Niu Q, Zeng TT, Su J, Zhu HL. Flow cytometric immunophenotyping is of great value to diagnosis of natural killer cell neoplasms involving bone marrow and peripheral blood. Ann Hematol. (2013) 92:89–96. doi: 10.1007/s00277-012-1574-3
17. Yamaguchi M, Kwong YL, Kim WS, Maeda Y, Hashimoto C, Suh C, et al. Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-Cell Tumor Study Group study. J Clin Oncol. (2011) 29:4410–6. doi: 10.1200/JCO.2011.35.6287
18. Hamadani M, Kanate AS, DiGilio A, Ahn KW, Smith SM, Lee JW, et al. Allogeneic hematopoietic cell transplantation for aggressive NK cell leukemia. A center for international blood and marrow transplant research analysis. Biol Blood Marrow Transpl. (2017) 23:853–6. doi: 10.1016/j.bbmt.2017.01.082
19. Fujimoto A, Ishida F, Izutsu K, Yamasaki S, Chihara D, Suzumiya J, et al. Allogeneic stem cell transplantation for patients with aggressive NK-cell leukemia. Bone Marrow Transpl. (2021) 56:347–56. doi: 10.1038/s41409-020-01009-8
20. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382:1811–22. doi: 10.1056/NEJMoa1911326
21. Mizuno S, Akashi K, Ohshima K, Iwasaki H, Miyamoto T, Uchida N, et al. Interferon-gamma prevents apoptosis in Epstein-Barr virus-infected natural killer cell leukemia in an autocrine fashion. Blood. (1999) 93:3494–504. doi: 10.1182/blood.V93.10.3494.410k14_3494_3504
22. Wen H, Ma H, Cai Q, Lin S, Lei X, He B, et al. Recurrent ECSIT mutation encoding V140A cy hyperinflammation and promotes hemophagocytic syndrome in extranodal NK/T cell lymphoma. Nat Med. (2018) 24:154–64. doi: 10.1038/nm.4456
23. Goubran M, McGinnis E, Stubbins RJ, Nicolson H, Pourshahnazari P, Belga S, et al. A young woman with persistent sore throat, Epstein-Barr virus, lymphadenopathy, and aberrant CD4 + CD7- T-cells. Am J Hematol. (2023) 98:824–9. doi: 10.1002/ajh.26838
24. Nakashima Y, Tagawa H, Suzuki R, Karnan S, Karube K, Ohshima K, et al. Genome-wide array-based comparative genomic hybridization of natural killer cell lymphoma/leukemia: different genomic alteration patterns of aggressive NK-cell leukemia and extranodal Nk/T-cell lymphoma, nasal type. Genes Chromosomes Cancer. (2005) 44:247–55. doi: 10.1002/gcc.20245
25. Jung KS, Cho S-H, Kim SJ, Ko YH, Kang E-S, Kim WS. l-asparaginase-based regimens followed by allogeneic hematopoietic stem cell transplantation improve outcomes in aggressive natural killer cell leukemia. J Hematol Oncol. (2016) 9:41. doi: 10.1186/s13045-016-0271-4
26. Johnson WT, Epstein-Peterson ZD, Ganesan N, Pak T, Chang T, Dao P, et al. Emapalumab as salvage therapy for adults with Malignancy-associated hemophagocytic lymphohistiocytosis. Haematologica. (2024). doi: 10.3324/haematol.2023.284179
Keywords: hemophagocytic lymphohistiocytosis, aggressive natural killer cell leukemia, EBV - Epstein-Barr virus, PEG-asparaginase, T/NK cell malignant lymphoma
Citation: Spaner C, Durkee-Shock J, Weng A, Stubbins R, Gerrie AS, Pittaluga S, Cohen JI and Chen LYC (2024) Case report: Aggressive natural killer cell leukemia and refractory hemophagocytic lymphohistiocytosis in an adolescent. Front. Hematol. 3:1413794. doi: 10.3389/frhem.2024.1413794
Received: 07 April 2024; Accepted: 18 June 2024;
Published: 10 July 2024.
Edited by:
Valentina Giudice, University of Salerno, ItalyReviewed by:
Concetta Micalizzi, Giannina Gaslini Institute (IRCCS), ItalyNeha Rastogi, Medanta The Medicity Hospital, India
Danilo De Novellis, Ospedali Riuniti San Giovanni di Dio e Ruggi d’Aragona, Italy
Copyright © 2024 Spaner, Durkee-Shock, Weng, Stubbins, Gerrie, Pittaluga, Cohen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luke Y. C. Chen, bGNoZW4yQGJjY2FuY2VyLmJjLmNh