 Liliia Ibneeva
Liliia Ibneeva Tatyana Grinenko
Tatyana Grinenko- 1Institute for Clinical Chemistry and Laboratory Medicine, University Hospital and Faculty of Medicine, Technische Universität Dresden, Dresden, Germany
- 2Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine at Shanghai, Ruijin Hospital Affiliated to Jiao Tong University School of Medicine, Shanghai, China
Quiescence is a fundamental state of adult hematopoietic stem cells (HSCs) characterized by their residence in the G0 phase of the cell cycle. Despite being quiescent, HSCs retain their capacities for self-renewal and multipotency, enabling them to produce all blood lineages. Recent discoveries have shown that HSCs can dive into an even deeper state of quiescence with a very low division rate in steady-state conditions, known as dormancy. Dormant HSCs (dHSCs) have the most superior stem cell properties among HSCs, placing them at the top of the hematopoietic hierarchy. In this review, we argue that quiescence and dormancy are not synonyms in the context of HSCs. Specifically, dHSCs constitute a unique reserve pool of HSCs, mobilized only under stress conditions to protect the HSC compartment throughout life. While HSC quiescence is well-studied, the molecular features of HSC dormancy remain less well-defined. We will discuss the available methods for dHSC isolation and summarize the latest findings on the roles of niche factors, transcription factors, chromatin regulators, and cell cycle-related proteins in maintaining HSC dormancy. Additionally, we will explore whether insights from the quiescent HSC research can be applied to dHSCs. Lastly, we will assess the therapeutic potential of utilizing or targeting dHSCs to improve stem cell transplantation outcomes and treat hematological diseases, opening new avenues for research and clinical applications in regenerative medicine and oncology.
1 Introduction
Hematopoietic stem cells (HSCs) are rare cells that continuously regenerate the entire hematopoietic system, producing billions of blood cells. In vertebrates, the primary definitive HSCs originate from the hemogenic endothelium on the ventral wall of the dorsal aorta within the aorta-gonad-mesonephros region via a process termed endothelial-to-hematopoietic transition. Following their emergence, HSCs migrate to the fetal liver before ultimately colonizing the bone marrow (BM), which then becomes the predominant site of adult hematopoiesis (1). Adult HSCs primarily reside within the distinct environment of the BM, termed the ‘BM niche’. This niche is populated by specialized cells that provide HSCs and progenitors with vital factors necessary for their maintenance, survival, and differentiation (2).
Two defining qualities of HSCs are self-renewal and multipotency. Self-renewal refers to the ability of HSCs to replicate themselves to maintain their population, while multipotency denotes their capability to differentiate into all types of mature blood cells (3). According to the hematopoietic tree model, HSCs evolve into mature cells through intermediate stages involving hematopoietic progenitors with varying differentiation and self-renewal potentials (4). These include multipotent progenitors (MPPs), which retain the capacity to produce all blood lineages (5), lineage-committed progenitors, and unipotent progenitors, the latter being capable of producing only a single type of cells. While the classical model of HSC differentiation posits that HSCs undergo distinct step-wise progenitor phases, recent evidence challenges this view, suggesting that HSCs may independently determine their lineage-specific fate (6, 7). For instance, it was demonstrated that HSCs are able to directly differentiate into myeloid progenitors without undergoing cell division upon transplantation into non-irradiated mice (8). Moreover, HSCs could bypass numerous downstream progenitor stages to produce mature erythrocytes (9). Additionally, another study provided evidence that megakaryocytes (Mks) might also originate directly from HSCs (10). Specifically, a single-cell in vitro culture of murine HSCs showed that they could produce Mks without undergoing cell division. Therefore, despite significant advances in understanding the differentiation process, the full scope and nature of it is yet to be firmly established. Nevertheless, the immense differentiation capacity of HSCs is crucial for maintaining hemostasis and responding to injury and disease.
Intriguingly, even though blood is a constantly renewing tissue, most of HSCs remain non-cycling or quiescent. In homeostasis, most HSCs reside in the G0 phase of the cell cycle and only a small fraction of HSCs enters the cell cycle to undergo lineage commitment or self-renewal divisions (11). HSCs in the G0 phase of the cell cycle, or quiescent HSCs, can be identified by their DNA content (2N), lower RNA content than in cells in the G1 phase, the absence of expression of proliferation markers, such as Ki67, or by label incorporation assay (12). The simplicity and accessibility of methods for detecting HSCs in the G0 phase of the cell cycle lead to the thorough investigation of HSC quiescence, revealing the key molecular players associated with this state (13–15). HSC quiescence is critical for the long-term sustenance of HSCs, protecting them from the accumulation of replication-associated DNA damage (16). Studies reveal that disruption of HSC quiescence results in excessive replication, leading to impaired capacity for self-renewal and loss of repopulation capacity in transplantation assays (17, 18). Overly proliferation leads to HSC premature exhaustion (19), emphasizing the importance of quiescence for maintaining the “stemness” of HSCs.
For a long time, the terms quiescent and dormant HSCs have been used interchangeably (12, 14, 15, 20). However, the seminal studies by the Trumpp group defined dormant HSCs (dHSCs) as a distinct subpopulation of quiescent HSCs, which are characterized by a deeper state of quiescence and very low overall biosynthetic activity. While quiescent HSCs represent about 70% of the total HSC population (21, 22), the dHSC subset constitutes only 15-30% of the HSC pool (23–25). By definition, dHSCs have extremely low in vivo divisional history, reflected by their chromosome label retention in long-term label retention assays (Figure 1A). Importantly, dHSCs demonstrate the highest long-term repopulation capacity in transplantation assay and are less responsive to activating stimuli than quiescent HSCs. Yet, dHSCs can reversibly enter the cell cycle in response to severe stress signals, such as bacterial or viral infection or blood loss (24, 26, 27). Thus, dHSCs are a unique subpopulation of HSCs, which resides at the apex of the hematopoietic hierarchy and serves as a reserve pool of HSCs during the entire life (23, 24, 28). The inability to maintain the dormant state may expose dHSCs to replication stress and promote the accumulation of somatic mutations, increasing the risk of their exhaustion or malignant transformation (29). Conversely, dHSCs failing to enter the cell cycle under stress conditions may limit blood cell production, potentially resulting in bone marrow failure (30). Therefore, tight control of HSC dormancy is vital for effective and healthy hematopoiesis.
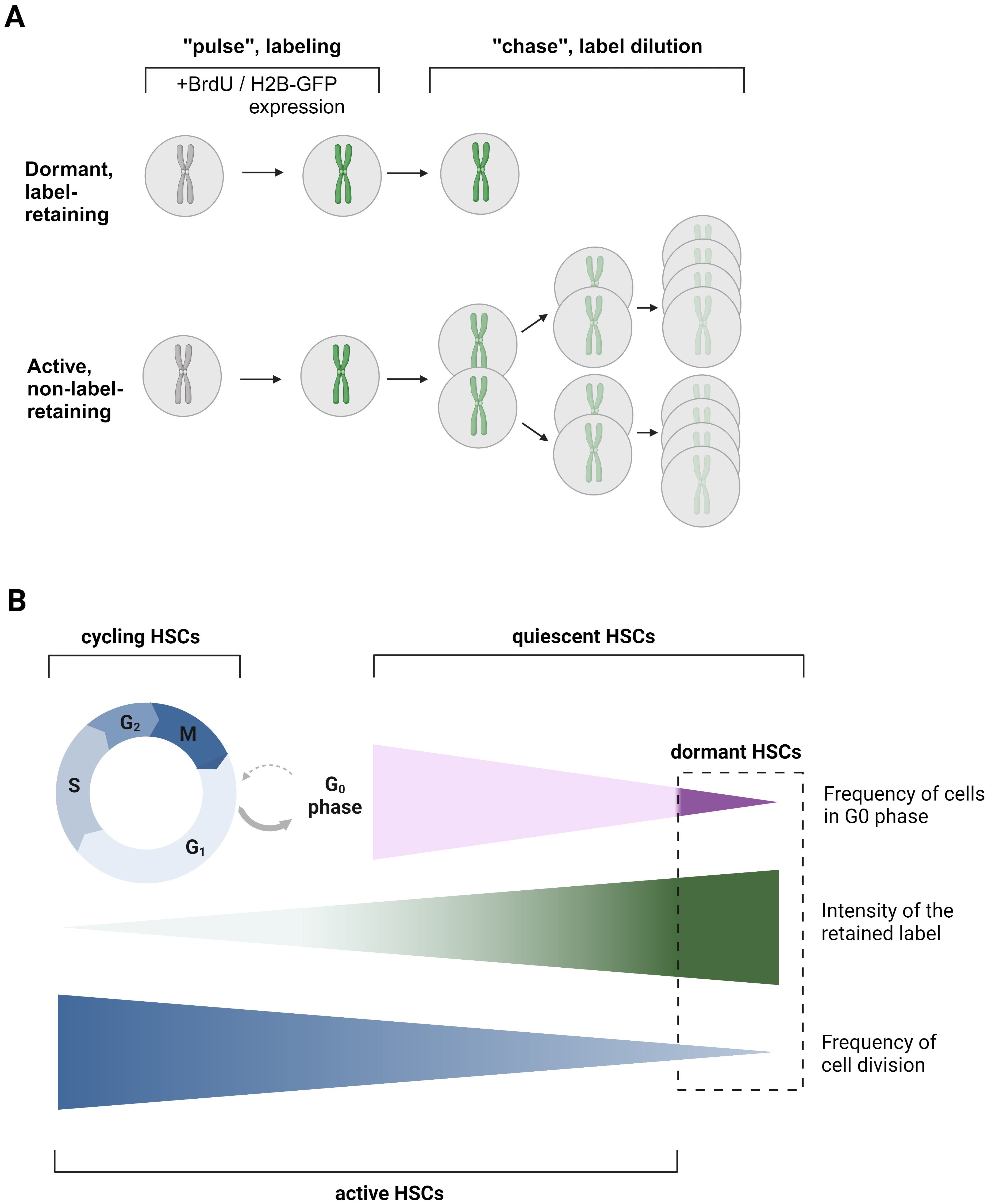
Figure 1. Label-retention assay as a classical approach for dHSC detection. (A) The principle of label retention assay. During the “pulse” or labeling phase, cells either incorporate BrdU, or the expression of H2B-GFP is induced by the Tet-on system through the administration of doxycycline (dox). In the subsequent ‘chase’ or label dilution phase, the previously introduced label is diluted out with each cell division after the withdrawal of BrdU/Dox or by turning off H2B-GFP expression in the Tet-off system through dox administration. This dilution allows for the distinction between dormant (non-dividing) and active (dividing) cells. BrdU – bromodeoxyuridine. (B) Scheme explaining the differences in terms quiescent HSCs, dHSCs, aHSCs, and cycling HSCs. Quiescent HSCs are those HSCs, which are in the G0 phase of the cell cycle. dHSCs, dormant, HSCs, are the most quiescent, label-retaining HSCs. aHSCs, active HSCs, are those HSCs that have undergone or are undergoing cell division. Cycling HSCs are those HSCs that are in G1-S-G2-M phases of the cell cycle. The figure was created using Biorender.com.
While quiescent HSCs are extensively studied (13, 15), the nature of dormancy in HSCs remains less explored. The advent of reporter mice based on retinoic acid-induced gene Gprc5c and the discovery of the surface marker CD38 for the identification of dHSCs shed light on the mechanisms supporting dHSCs’ function (24, 25). In this review, we aim to provide the differentiation between the states of quiescence and dormancy in HSCs. We will discuss the latest discoveries that have established the unique features of HSC dormancy and offer insights into the extent to which our understanding of quiescent HSCs can be applied to dHSCs.
2 Identification of dormant HSCs
In the previous paradigm, it was believed that all HSCs periodically enter and exit the cell cycle, with the entire HSC pool undergoing turnover within weeks (31). However, later studies have firmly established the presence of the subpopulation of dHSCs within the quiescent HSC compartment that shows almost no divisional activity under steady-state conditions (23, 28, 32). To identify these dHSCs, long-term label retention assays have been employed. Typically, an initial “pulse” phase includes HSC labeling followed by a “chase” phase, where this label is progressively diluted over an extended period of time due to cell divisions (Figure 1A). One prevalent approach utilizes a thymidine analogue, bromodeoxyuridine (BrdU), which integrates into the DNA during the S-phase of the cell cycle within the labeling period. As cells divide post-labeling, the BrdU is diluted among progeny. Rapidly dividing HSCs will dilute the BrdU quickly (named active HSCs, aHSCs), while dHSCs will retain the BrdU label for more than 70 days. The presence of two subpopulations of HSCs, dHSCs and aHSCs, has been predicted by mathematical modeling based on BrdU dilution kinetics. While the one-population model failed to explain the experimental data, the two-population model describes it with great accuracy, strongly suggesting the presence of dHSCs (23, 32). According to Wilson et al., dHSCs are predicted to divide approximately every 145 days in mice, resulting in only about 5 divisions during a murine lifetime, whereas aHSCs likely divide every 36 days. Importantly, Ki67-Hoechst 33342 staining revealed not only that nearly all dHSCs reside in the G0 phase but also that 83% of aHSCs are in the G0 phase too (Figure 1B) (23). It is worth noting that the percentage of total HSCs in the G0 phase in this study is higher than in previously cited work (21) due to the use of the HSC phenotype (Lineage- с-Kit+ Sca-1+ CD48- CD150+ CD34-), enriching for quiescent HSCs. Therefore, those aHSCs are highly quiescent, although at least a part of them are transcriptionally cell-cycle primed (24).
Nevertheless, the BrdU assay has limitations: antibody-mediated BrdU recognition requires cell fixation, and therefore, it cannot be used to study viable dHSCs. Additionally, BrdU has been suggested to cause an injury response in the periphery, which induces a positive feedback loop leading to dHSC activation (23). This can explain why dHSCs are successfully labeled in only a two-week period. Therefore, an advanced system involving transgenic mouse strain R26rtTA/Col1A1H2B-GFP has been established (23, 28). It allows ubiquitous expression of chromatin structural protein histone 2B fused with a green fluorescent protein (H2B-GFP) under the control of an inducible tetracycline-mediated system (using either Tet-on or Tet-off systems), resulting in the labeling of chromosomes. Mirroring the BrdU approach, labeled cells dilute the H2B-GFP with each division, allowing the identification of dHSCs as those HSCs that retain this label longer than 70 days (Figure 1A).
In contrast to a BrdU assay-based prediction (23), H2B-GFP dilution data argued that dHSCs count their divisions and divide only 4 times before entering permanent quiescence (33). This conclusion, however, has been challenged by Morcos and colleagues, who demonstrated that the H2B-GFP assay cannot be used for accurately determining the number of HSC divisions due to H2B-GFP short degradation time of 4-6 weeks, inhomogeneous initial labeling, and background fluorescence due to leaky H2B-FP expression. Furthermore, performing two consecutive rounds of pulse and chase revealed that HSCs from older mice, following the second pulse, diluted the label at a rate similar to that of younger mice after their first pulse. Therefore, it has been inferred that dHSCs undergo continuous cell cycle activity without losing their potential after 4 divisions (34). However, the H2B-GFP assay allows the labeling of the most quiescent dHSCs (25). The transplantation of H2B-GFPhi HSCs revealed that dHSCs have significantly higher repopulation potential and self-renewal capacity compared with aHSCs with the same surface marker phenotype, which have significantly diluted the H2B-GFP label. The H2B-GFPlo HSCs also failed to reconstitute lethally irradiated mice in serial transplantation assay (23, 35). This superiority in quiescence, self-renewal, and reconstitution capability positions dHSCs at the top of the hematopoietic hierarchy.
3 dHSCs act as a reserve pool of HSCs
Due to their very infrequent cycling, dHSCs do not significantly contribute to daily blood cell production under steady-state conditions. Instead, dHSCs come into action upon receiving the signals of severe hematopoietic stress. Post in vivo treatment with either chemotherapeutic agent 5-fluorouracil (5-FU) or the granulocyte colony-stimulating factor (G-CSF), known for mobilizing HSCs from BM, dHSCs begin cycling as evidenced by the loss of BrdU or H2B-GFP label (23). Another significant factor affecting dHSCs is interferon alpha (IFNα), a cytokine produced by immune cells in response to challenges like viruses, bacteria, and tumors. A single high-dose injection of IFNα or the viral RNA mimetic polyinosinic:polycytidylic acid (polyI:C) - which similarly triggers the IFNα response - can promote dHSC activation in vivo (24, 26). A component of the outer wall of Gram-negative bacteria, lipopolysaccharide (LPS), is also capable of inducing dHSC proliferation (24, 27). Importantly, once the stressor is removed and conditions return to normal, dHSCs rapidly revert to their dormant state to preserve the HSC pool (23). Nevertheless, chronic stress conditions, such as repetitive cycles of polyI:C injections can lead to irreversible HSC exhaustion - loss of stem cell function due to reaching the limit of their self-renewal capacity (36). Frequent divisions can also favor the accumulation of somatic DNA mutations, potentially resulting in oncogenic transformation (37). Besides, dormancy protects HSCs from chemotherapeutic agents. When HSC dormancy is disrupted before chemotherapeutic interventions - as observed with repeated polyI:C injections followed by two rounds of 5-FU treatment - it can lead to fatal anemia, likely due to the eradication of all dHSCs (26). Thus, dormancy not only preserves the dHSCs from replication stress, ensuring their functionality, but also positions them as a reserve pool of HSCs throughout an individual’s lifetime.
4 Molecular features of dHSCs
Prior research using label retention assays and reporter mice to identify dHSCs has elucidated some of their unique molecular signatures. In this section, we will primarily rely on the studies comparing dHSCs with aHSCs, as the latter represent the best available approximation to quiescent HSCs thus far (Figure 1B). Consistent with their biological function, dHSCs exhibit transcriptional profiles demonstrating markedly reduced biosynthetic activity, including diminished metabolism, downregulated DNA replication machinery, decreased mRNA processing and lower rates of ribosomal biogenesis (23, 24) (Figure 2). Furthermore, dHSCs are characterized by very low levels of CD34 mRNA (23) and enriched in MolO gene expression signature, which indicates the most functional HSCs (38). Concurrently, tumor suppressor with deubiquitinase activity Cyld (cylindromatosis) exhibited high mRNA transcript levels in dHSCs but not in MPPs. Tesio and colleagues demonstrated that CYLD inhibits dHSC cycling by deactivating the pathway involving tumor necrosis factor receptor-associated factor 2 (TRAF2) and p38MAPK. Mechanistically, CYLD removes polyubiquitin chains from TRAF2, thereby decreasing p38MAPK activity and preventing cell proliferation (39).
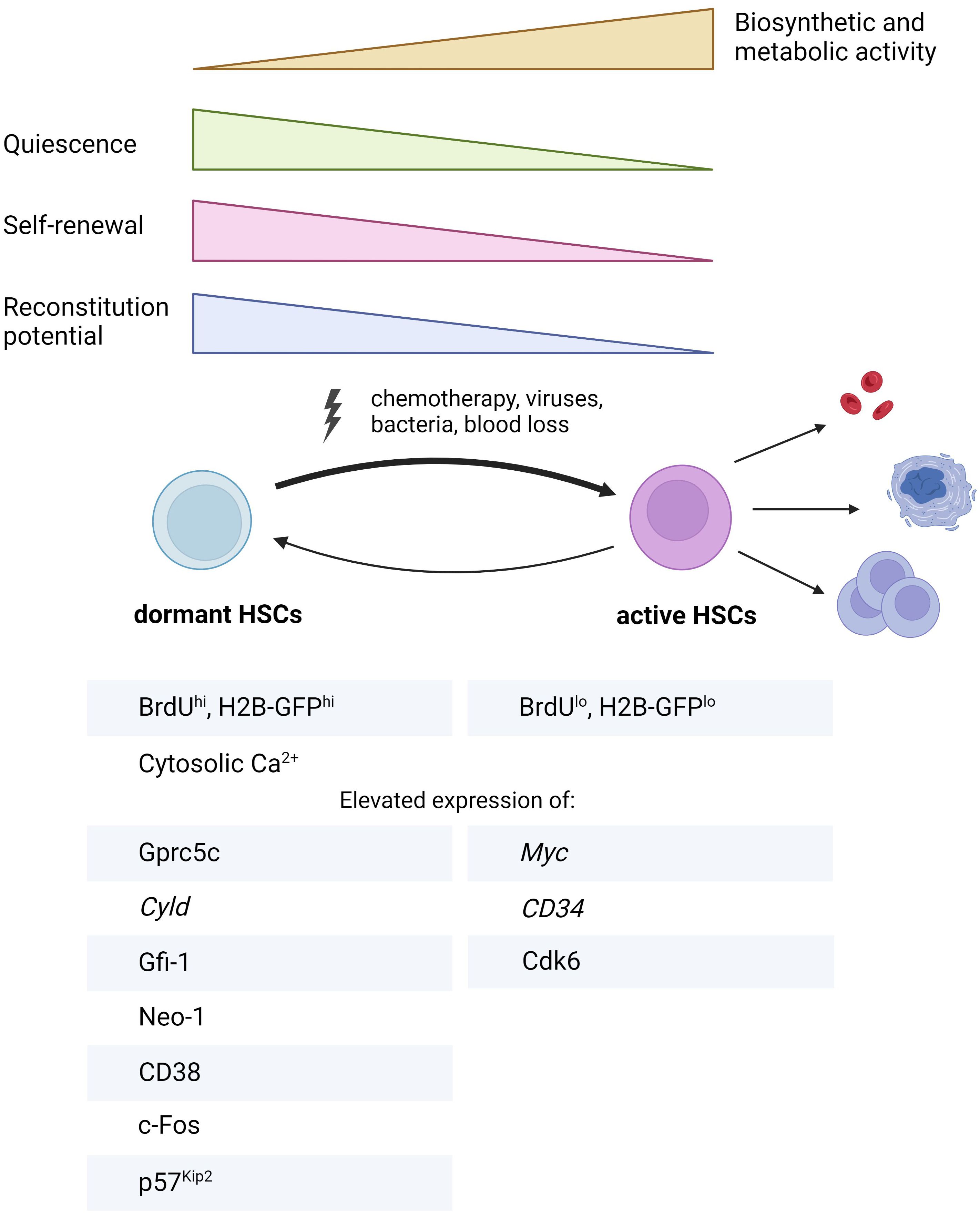
Figure 2. Summary of key characteristics distinguishing dHSCs from aHSCs. BrdU, bromodeoxyuridine, Cyld, cylindromatosis; Cdk6, cyclin-dependent kinase 6; Gfi-1, growth factor independent 1; Gprc5c, orphan G protein-coupled receptor, class C group 5; H2B-GFP, histone 2B-green fluorescent protein; Neo-1, neogenin 1. The figure was created using Biorender.com.
Notably, Cabezas and colleagues established a critical role of retinoic acid (RA)-induced signaling in the maintenance of HSC dormancy. They found that dHSCs are enriched in retinoic acid-induced gene Gprc5c mRNA (orphan G protein-coupled receptor, class C, group 5) compared to aHSCs and MPPs. Subsequently, a transgenic reporter mouse based on EGFP expression under Gprc5c promoter (Gprc5c-EGFP mouse) was developed, enabling the detection of dHSCs. By utilizing this mouse, authors showed that lack of dietary vitamin A in mice results in the disruption of HSC quiescence, whereas administering the active metabolite of vitamin A, all-trans-RA, maintains dHSC quiescence under stress conditions both in vitro and in vivo (24). In the follow-up study, they discovered the non-classical RA signaling axis, which supports HSC stemness (40). According to their data, all-trans RA is converted by Cyp26b1 enzyme into 4-oxo-RA metabolite, which in turn binds to RA receptor beta (RARβ). The latter is responsible for activating a transcriptional program enhancing HSC identity. Although this study primarily concentrated on HSCs rather than dHSCs, Cyp26b1 transcript was identified as being enriched in dHSCs in the previous study from this group (24), suggesting that Cyp26b1/4-oxo-RA/RARβ axis might be relevant for dHSC function too. Another research group unveiled the role of another class of RA receptors - RXRs (retinoid X receptor), in the maintenance of HSC quiescence and function. While the individual deletions of either Rxra or Rxrb do not markedly impact HSC fitness, the double deletion of both Rxra and Rxrb results in HSC activation, subsequently inducing myeloproliferative-like disease (41). Importantly, a study by Zhang and colleagues disclosed that Gprc5c protein is also expressed in human HSCs (hHSCs) (42). Their work also suggests that the Gprc5c+ hHSCs can be categorized as dHSCs, resolving a long-standing question regarding the existence of dHSCs in humans.
BM niche has a complex three-dimensional structure involving multiple components: osteoblasts, which form the endosteal niche; endothelial cells that create a sinusoidal and arteriolar vascular niche; mesenchymal stem cells - MSCs, known for their secretory functions; other cells like Mks, immune cells (regulatory T-lymphocytes, macrophages) and both sensory and sympathetic neurons, each contributing uniquely to the niche’s complexity (2). For many years, it was widely believed that quiescent HSCs resided in the endosteal niche (20). However, recent insights have revealed that they predominantly occupy perisinusoidal niches in the BM (43, 44). In agreement with this, the interaction of neogenin-1 (Neo-1) expressed on dHSCs with netrin-1, which is most likely produced by arteriolar endothelial and periarteriolar stromal cells in the BM niche, has been suggested (27). Although extracellular Ca2+ was once thought to anchor quiescent HSCs near the endosteum (45), the proposed localization of dHSCs in the perivascular niche, alongside the finding emphasizing the significance of intracellular Ca2+ for dHSCs, has shifted this perspective. The role of intracellular Ca2+ for dHSCs has been discovered using a reporter mouse based on the cell cycle inhibitor p27Kip1, which enables the detection of HSCs in the G0 cell cycle phase (mVenus-p27K– mouse). High-throughput small-molecule screening using this reporter mouse revealed that high levels of cytoplasmic Ca2+ are associated with HSC dormancy (46). However, the precise function of Ca2+ in dHSC maintenance remained unknown. Recently, we revealed that cADPR, synthesized through the conversion of NAD+ by CD38, elevates cytoplasmic Ca2+ in dHSCs. This elevation induces the expression of the transcription factor (TF) c-Fos, which subsequently promotes dHSC quiescence in p57Kip2-dependent manner. Thus, we identified CD38 as an important missing mediator of higher levels of intracellular Ca2+ in dHSCs in steady state (25).
Despite significant advancements in the field of dHSCs (Figure 2), progress has been impeded by challenges in identifying and isolating dHSCs. The H2B-GFP label retention assay is time-consuming and necessitates a lengthy chase period, making the study of dHSCs in young mice unfeasible. Furthermore, Gprc5c-EGFP and mVenus-p27K– mouse models are not commercially accessible, and to date, there are no known monoclonal antibodies for FACS recognizing dHSC markers Gprc5c or Neo-1, hindering the straightforward isolation of dHSCs. We discovered CD38 as a surface marker for murine dHSCs, which should facilitate the future investigation of HSC dormancy using simple antibody staining for dHSC FACS isolation. Currently, the upstream regulators responsible for very low metabolic and synthetic activities observed in dHSCs, including additional niche factors, signaling proteins, epigenetic regulators, and transcription factors, are not well characterized. Therefore, the next part of the review will focus on determining whether the molecular pathways known to induce quiescence in HSCs also play a role in regulating the dormancy of dHSCs.
5 Known quiescence regulators in the context of dHSCs
Quiescence is a well-studied cellular state in HSCs, with numerous genes shown to play a role in maintaining this state (13–15). As dHSCs represent a subfraction of quiescent HSCs, the molecular features of these two populations most likely overlap significantly. We propose that each known regulator of HSC quiescence should be individually validated in the context of dHSCs. To identify the top candidates of interest, we analyzed the gene lists comparing transcriptional profiles of active HSCs and dHSCs (24), including our own (25). We listed the most promising candidates below, however, in the future, they need to be validated at the protein level using label-retention assay, reporter mice or updated dHSC phenotype (25).
5.1 Niche factors
BM niche produces factors including various cytokines, chemokines, and extracellular matrix components interacting with their specific receptors or adhesion molecules on HSCs to maintain their quiescence in homeostasis or activate them in response to stress ( (8), reviewed extensively in (12)). Binding of the quiescence factors to their respective receptors on HSCs (e.g. stem cell factor (SCF) and c-Kit; angiopoietin-1 (Ang-1) and Tie2; thrombopoietin (TPO) and myeloproliferative leukemia protein (MPL)) triggers signaling cascades inducing cell cycle arrest. Such cascades involve signaling tyrosine kinases, transcription factors, epigenetic regulators, and cell cycle-related proteins inhibiting the cell cycle progression, which will be discussed below. Genes related to cell-to-cell and matrix interactions were found to be increased in H2B-GFPhi dHSCs (25, 47), which might indicate ongoing communication between dHSCs and their surrounding microenvironment, keeping them ready to quickly determine their fate. Remarkably, leaving the dormant state was accompanied by an immediate decrease in the expression of genes associated with niche communication, possibly indicating a migration to different niches. Furthermore, dHSCs are enriched in TGF-β signaling signature genes (25, 47). TGF-β is a well-established regulator secreted by Mks and non-myelinating Schwann cells (48–50), which inhibits the cell cycle progression of HSCs via p57Kip2 (48). Thus, niche-secreted factors most likely play an important role in the support of HSC dormancy.
The localization of dHSCs in the BM is poorly studied. Confocal microscopy imaging revealed that label-retaining dHSCs were found as individual cells homogenously distributed in the BM, indicating the exceptionally small size of their niches (23, 51). It has been identified earlier that H2B-GFPhi dHSCs were enriched in trabecular bone region and were retained there via non-canonical Wnt signaling (52), in contrast to the later study suggesting that dHSCs reside in perivascular niches guided by Netrin-Neo-1 signaling (27). Additionally, recent studies have demonstrated an important role of extracellular matrix component hyaluronic acid in the maintenance of HSC dormancy in mice and humans (42). Moreover, we have found that CD38 ecto-enzymatic activity on the neighboring cells can promote hHSC quiescence (18). Therefore, BM niche is an essential component of HSC dormancy regulation and requires further investigation.
5.2 Cell cycle regulators
HSCs transit into the G1 cell cycle phase upon receiving the mitogenic stimuli from the niche cells. This transition is tightly controlled by the so-called restriction point, which is mediated through the protein complexes consisting of cyclin-dependent kinases (CDKs) and corresponding cyclins. In the early G1 phase, before the restriction point, cyclin D is produced in response to mitogen stimulation and then forms complexes with either CDK4 or CDK6. Cyclin D-CDK4/6 complex inactivates the tumor suppressor Retinoblastoma protein (Rb) by phosphorylation, which mediates the release of E2F transcription factors from Rb-E2F complex, subsequently leading to the activation of S-phase-related transcriptional program. Rb phosphorylation marks the cell’s transition through the restriction point into the late G1 phase. Here, E2F transcription factor activity leads to the expression of cyclin A and cyclin E, which form complexes with CDK2. The resulting cyclin A/E-CDK2 complex hyperphosphorylates Rb, leading to its full inactivation and further transition into S-phase (15, 53). In the absence of mitogenic stimuli, cells enter the reversible state of cell cycle arrest or quiescence. It is further enhanced by cyclin-dependent kinase inhibitors (CKIs) represented by two families, which suppress the activity of CDKs halting the transition through the cell cycle. The members of Cip/Kip CKIs family: p21Cip, p27Kip1, and p57Kip2, suppress the activity of CDK4, CDK6, and CDK2. The members of INK4 family of CKIs: p16INK4A, p15INK4B, p18INK4C, and p19INK4D, have a narrower function inhibiting only CDK4/6 (54).
The roles of cell cycle-related proteins in HSC cell cycle regulation discovered using knock-out mice are extensively discussed in (15, 53, 55). Briefly, cyclins D1/D2/D3 and Cdk2/4/6 likely play redundant roles in HSCs since single deletions of Cyclin D members or a Cdk do not result in significant hematopoietic phenotypes, in contrast to the severe abnormalities observed during embryogenesis with combined deficiencies of either Cyclins D1-3, Cdk2&4 or Cdk4&6. While full knock-out Cdk6-/- mice do not exhibit any changes in the frequencies of HSCs in the G0 cell cycle phase in a steady state, they fail to start proliferating efficiently upon polyI:C administration (56). In humans, long-term HSCs display lower levels of Cdk6 protein than short-term HSCs, which are primed for activation (57). As for CKIs, p57Kip2 is recognized as a primary mediator of HSC quiescence in homeostasis (17, 18). p27Kip1 deficiency does not affect HSC and MPP compartments but rather progenitors, causing their increased proliferation (58). p21Cip is not required for HSC quiescence in homeostasis (59) but becomes critical during the stress response, as it is a target gene of p53 (60). INK4 family CKI p16INK4A is dispensable in young mice (61); however, in old mice, it is associated with age-related HSC decline (62). Surprisingly, p18INK4C deficiency leads to increased cycling of HSCs due to enhanced self-renewal capacity, which in turn results in improved functionality (63). The relevance of other INK4 family members for HSC quiescence, p15INK4B and p19INK4D, has to be defined in further research.
Within the context of dHSCs, it has been shown that they are characterized by low protein levels of CDK6 (24) along with high levels of p57Kip2 CKI (25). Monitoring p27Kip1 activity has been suggested for defining dHSCs using mVenus-p27K– reporter mouse (46). It should be noted, however, that this reporter allows only 50% enrichment for label-retaining dHSCs (46). The deletion p21Cip did not alter the frequency of H2B-GFPhi HSCs (28), indicating its nonessential role for dHSCs. The potential contributions of other CKIs to the deeply quiescent state typical for dHSCs remain to be fully investigated.
5.3 Transcription factors
The expression levels of Myc oncogene, which promotes cell cycle activity via repression of p21Cip and is also necessary for HSC differentiation (64, 65), are low in dHSCs (24). While other TFs have been shown to support HSC quiescence, their role remains unexplored for dHSCs. Gfi-1 (growth factor independent 1) is a zinc-finger transcriptional repressor recruiting histone-modifying enzymes to suppress the transcription of target genes. Gfi-1 is also essential for lymphopoiesis and myelopoiesis (66). Studies using transgenic mice revealed that deletion of Gfi-1 leads to increased proliferation of either LSK (Lin- Sca-1+ c-Kit+ (67)) compartment and HSCs (LSK Flt3- (68), LSK CD48- CD150+ (69)) via decreased p21Cip mRNA expression (68), with subsequent loss of their functional capacity in competitive repopulation assay. Additionally, low levels of Gfi-1 transcripts are associated with an inferior prognosis for acute myeloid leukemia (AML) patients (70) and can lead to early transformation into blast crisis in chronic myeloid leukemia (CML) (71). Gfi1b is a paralogue of Gfi-1 and a transcriptional repressor. The absence of Gfi1b leads to increased HSC proliferation without compromising their repopulation capacity (72). In the bulk RNAseq dataset comparing the transcriptome of dHSCs, aHSCs, and MPPs (24), Gfi1b expression is similar in all three populations, whereas Gfi-1 expression is elevated in dHSCs compared to other populations, suggesting that Gfi-1 can indeed regulate the quiescent state of dHSCs. Indeed, loss of Gfi-1 leads to decreased frequency of H2B-GFP-retaining HSCs, indicating that it is indispensable for dHSCs (28).
Egr1 (Early growth response 1) is an immediate-early response (IER) gene highly expressed in HSCs (LSK Flt3-) in homeostasis, whereas its expression is downregulated upon stimulation with chemotherapy and G-CSF. The lack of Egr1 causes enhanced HSC proliferation along with their egress from BM to PB. Despite this, Egr1-/- HSCs demonstrate comparable regenerative capacity to that of wild-type HSCs in the first two rounds of transplantation into recipient mice, although Egr1-/- HSCs exhibit a more rapid loss of function in subsequent serial transplantation. Mechanistically, it has been suggested that Egr1 supports HSC quiescence via decreased levels of Bmi1, an important epigenetic regulator, whose role will be discussed later, and Cdk4 along with increased levels of p21Cip (73). Upon HSC activation, Egr1 is suppressed by Cdk6 (56). As for dHSCs, it has been proposed that signaling mediated by niche-derived netrin-1 via its receptor Neo-1 leads to an increase in Egr1 levels and a decrease in Cdk6 levels, thereby supporting HSC dormancy (27).
Intriguingly, we found that Fos, another member of IER genes, is enriched in dHSCs (25). c-Fos is recognized as a crucial positive regulator of myeloid differentiation (74) and demonstrates transient expression in stimulated cells, leading to cell cycle progression (75, 76). A previous study has reported that the IFNα/β inducible c-Fos overexpression suppresses the in vitro proliferation and colony formation of Lin- Sca-1+ cells (77). However, this population mainly contains progenitor cells and a very small amount of dHSCs. Therefore, the role of c-Fos in the regulation of dHSC cell cycle remained unexplored. In our work, we have shown for the first time that c-Fos is regulated by the enzymatic activity of CD38 and supports the dormancy of adult HSC under physiological conditions via p57Kip2 cell cycle inhibitor (25). In line with our finding, another study reported that in contrast to adult dHSCs, the expression of Fos is downregulated in highly proliferative fetal HSCs (78). While adult HSCs attain quiescence approximately four weeks post-birth (79), the specific timing and factors facilitating the emergence of the dHSC population are yet to be identified and represent an area for future investigation.
Other TFs supporting HSC quiescent state were identified (12). Among these are tumor suppressor p53, which acts via Gfi1 and Necdin (80); FoxO family TF Foxo3a, which suppresses the production of ROS and increases levels of p27Kip1 and p57Kip2 (81); homeobox family TF Pbx1, whose loss leads to reduction in p57Kip2 (82); another homeobox family TF Meis1, which decreases the levels of Ccnd1 (83, 84); nuclear receptor superfamily TF Nurr1, which likely upregulates p18INK4C (85); interferon regulatory factor, IRF2, acting via upregulation of p27Kip1 and p57Kip2 (86); and basic leucine zipper family TF HLF (hepatic leukemia factor) exerting its function via upregulation of IRF2 (87). Among these TFs, only HLF was suggested as a potential TF that regulates dHSCs, as its expression was exclusively elevated in CD150hi dHSCs (47). Analysis of RNAseq datasets for dHSCs has not revealed any enrichment of other aforementioned TF genes in dHSCs, indicating that their specific roles in the context of dHSCs need to be individually validated.
5.4 Metabolism
The role of metabolism in the maintenance of dHSCs is fundamental to their functionality. By their nature, dHSCs reside in a state of deep quiescence accompanied by markedly low levels of metabolic and synthetic processes. Additionally, most HSCs reside in a hypoxic BM microenvironment with O2 levels ranging between 1 to 4% (88), subjecting dHSCs to chronic hypoxia. Signaling pathway enrichment analysis has indicated a downregulation of TCA (tricarboxylic acid) cycle-related genes and mitochondrial respiratory chain complex genes in dHSCs (24, 25). Approximately 60% of label-retaining dHSCs exhibited low mitochondrial membrane potential (MMP), as evidenced by TMRE (tetramethylrhodamine ethyl ester) staining, compared to more actively dividing HSCs. The transcriptional profiles of MMPlo HSCs closely resemble those of dHSCs, highlighting low mitochondrial activity as a key determinant of HSC dormancy. This low MMP is responsible for diminished levels of reactive oxygen species (ROS), thereby protecting them from apoptosis and accumulation of DNA damage. Interestingly, dHSCs also demonstrate lower glycolytic rates compared to their cell cycle-primed counterparts, maintaining a minimal level of metabolism essential for their sustenance. Lysosomes have been suggested as principal mediators of this reduced metabolic activity, with quiescent HSCs enriched in large lysosomes, which have a lower degradation capability and engulf mitochondria, thus limiting their metabolic function. Inhibition of lysosomal activity, followed by transplantation assay, revealed that reduced lysosomal function enhances HSC repopulation capacity (89). Therefore, the metabolic profile of dHSCs is not just a passive outcome of their dormancy but actively contributes to the maintenance of their stem cell identity.
The further characterization of dHSC metabolism is challenged by their rarity, with approximately 700-1000 cells present in the entire murine BM (23, 24), making techniques like the Seahorse assay not feasible. However, the development of low-input metabolomics tools should facilitate future research in identifying other metabolic contributors in dHSC biology [reviewed in (90)].
5.5 Epigenetic regulators
Epigenetic regulation modulates gene expression via various mechanisms, including DNA methylation, histone modifications, and non-coding RNAs, such as microRNAs and long non-coding RNAs. The minimal levels of metabolic activity and biosynthesis observed in dHSCs, along with blocked differentiation programs, imply a predominance of closed chromatin states and an abundance of repressive epigenetic marks. The epigenetic state of dHSCs remains largely unexplored primarily due to their scarcity. Nevertheless, few candidates have been suggested as potentially key players in dHSC biology. Bmi1, a component of the Polycomb repressive complex 1 (PRC1) that induces a repressive chromatin state, is critical for the self-renewal capacity of HSCs (91). Despite its consistent expression levels in both dHSCs and active HSCs (24), Bmi1 likely plays an important role in dHSC self-renewal. A study by Qiu and colleagues suggested the potential roles of SmarcA4, a component of SWI-SNF chromatin remodeling complex, and Mllt3/AF9 (myeloid/lymphoid or mixed-lineage leukemia; translocated to, 3), a chromatin reader component of the super elongation complex (SEC), in CD150hi label-retaining dHSCs (47). Later studies have demonstrated that SmarcA4 might be responsible for HSC quiescence upon stress (92), whereas Mllt3 has been shown to be a crucial regulator of hHSC stemness (93). Furthermore, a marked increase in the expression of Satb1, a chromatin organizer, was found in dHSCs (24). Previous studies have shown that a lack of Satb1 leads to a loss of HSC quiescence and enhanced differentiation (94), underscoring its potentially important role in dHSC regulation. Finally, advancements in low-input and single-cell epigenomics hold promise for uncovering other players in maintaining the epigenetic landscape of dHSCs (95, 96).
6 Discussion
In this review, we argue that separating dormant and quiescent HSCs is not a question of mere semantics; in fact, distinguishing between them is crucial, highlighting dHSCs as a unique subset. Cabezas-Wallscheid et al. have demonstrated that HSC transition through gradual intermediate states of quiescence before reentering the cell cycle rather than through a binary on/off switch, with dHSCs being the most quiescent subpopulation (24). Another intermediate state, the Galert state, has been first identified for muscle stem cells (MuSCs), implying that such MuSCs are activation-primed but still quiescent. This state, marked by activation of mTORC1 expression, manifests after the distant muscle injury and endows MuSCs with enhanced regeneration potential. Similarly, HSCs exhibit a Galert state in response to muscle injury, indicating at least two distinct quiescent states. Galert stem cells revert to the G0 state after stressor removal and regeneration (97). It is unknown whether some of the HSCs in a steady state are predisposed to switching into Galert upon stress. Nevertheless, this demonstrates a dynamic quiescence landscape.
dHSCs add another level of complexity to this model. While their ability to switch into the Galert state remains unexplored, they are characterized by a deeper quiescence than other HSCs in the G0 cell cycle phase, with markedly reduced biosynthesis and metabolism. These properties are tightly linked with the enhanced functionality of dHSCs, serving as a reservoir for HSCs that are activated upon severe stress. The underlying reason for the difference between the extremely low division rate of dHSCs and more prone to entering the cell cycle quiescent HSCs in homeostasis remains unclear (98). Future studies will reveal whether this difference is mediated by distinct mechanisms keeping quiescent HSCs and dHSCs in the G0 phase or by differing mechanisms controlling the exit from the G0 phase for both populations.
dHSCs hold promise for the development of therapeutic strategies. Identifying specific surface markers for human dHSCs could enable the enrichment of highly potent donor HSCs, enhancing the success of transplantation outcomes. Future studies are essential to explore strategies for mobilizing dHSCs without depleting their reserve. Hematological cancers often lead to healthy HSC dysfunction and resultant pancytopenias (99), as these HSCs become trapped in dormancy by cancer cell byproducts. Developing therapies to activate dHSCs in the context of cancer could unlock further clinical potential. Notably, many hematologic malignancies, such as chronic lymphocytic leukemia, AML, and multiple myeloma, exhibit high levels of CD38 expression (100). The coculture of hHSCs with CD38+ tumor cells, in the presence of inhibitor targeting CD38 or cADPR antagonist, revealed that inhibition of CD38 enzymatic activity led to the cell cycle entrance of hHSCs. Moreover, human bone marrow imaging showed that quiescent hHSCs colocalized with CD38+ cells in healthy patients (25). We propose that, through a paracrine mechanism, a tumor microenvironment enriched with the products of CD38 ecto-enzymatic activity may suppress the cell cycle of healthy hHSCs, leading to cancer-related pancytopenia. Therefore, inhibiting CD38-mediated cADPR production might support healthy hematopoiesis in patients with hematologic malignancies.
Exploring the regulatory mechanisms behind dHSC quiescence can provide crucial insights into the survival tactics of dormant cancer stem cells, particularly within AML. AML is characterized by a cellular hierarchy dominated by leukemia stem cells (LSCs) that share several stem-like features with dHSCs, such as self-renewal capabilities, the potential to differentiate into blast cells, and notably, a resistance to chemotherapy attributed to their dormant state. This dormancy is a key factor in the persistence of LSCs and a significant cause of AML relapse (101). By comparing the dormancy mechanisms of dHSCs and LSCs, it would be possible to identify novel therapeutic targets to disrupt the persistence of LSCs. Consequently, this could lead to more effective leukemia treatments and a decrease in the rates of relapse in AML.
Author contributions
LI: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. TG: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Deutsche Forschungsgemeinschaft (GR 4857/2-1 and GR 4857/3-1) and Mildred-Scheel-Nachwuchszentrum fellowship to TG.
Acknowledgments
All figures were created using Biorender.com. ChatGPT 4o (OpenAI) was used for language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Costa G, Kouskoff V, Lacaud G. Origin of blood cells and HSC production in the embryo. Trends Immunol. (2012) 33:215–23. doi: 10.1016/j.it.2012.01.012
2. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. (2019) 20:303–20. doi: 10.1038/s41580-019-0103-9
3. Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. (2010) 2:640–53. doi: 10.1002/wsbm.v2:6
4. Cheng H, Zheng Z, Cheng T. New paradigms on hematopoietic stem cell differentiation. Protein Cell. (2020) 11:34–44. doi: 10.1007/s13238-019-0633-0
5. Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. (2013) 13:102–16. doi: 10.1016/j.stem.2013.05.014
6. Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. (2017) 19:271–81. doi: 10.1038/ncb3493
7. Kucinski I, Campos J, Barile M, Severi F, Bohin N, Moreira PN, et al. A time- and single-cell-resolved model of murine bone marrow hematopoiesis. Cell Stem Cell. (2024) 31:244–59 e10. doi: 10.1016/j.stem.2023.12.001
8. Grinenko T, Eugster A, Thielecke L, Ramasz B, Kruger A, Dietz S, et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat Commun. (2018) 9:1898. doi: 10.1038/s41467-018-04188-7
9. Singh RP, Grinenko T, Ramasz B, Franke K, Lesche M, Dahl A, et al. Hematopoietic Stem Cells but Not Multipotent Progenitors Drive Erythropoiesis during Chronic Erythroid Stress in EPO Transgenic Mice. Stem Cell Rep. (2018) 10:1908–19. doi: 10.1016/j.stemcr.2018.04.012
10. Roch A, Trachsel V, Lutolf MP. Brief report: single-cell analysis reveals cell division-independent emergence of megakaryocytes from phenotypic hematopoietic stem cells. Stem Cells. (2015) 33:3152–7. doi: 10.1002/stem.2106
11. Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol. (1997) 25:445–53.
12. Chen Z, Guo Q, Song G, Hou Y. Molecular regulation of hematopoietic stem cell quiescence. Cell Mol Life Sci. (2022) 79:218. doi: 10.1007/s00018-022-04200-w
13. Li J. Quiescence regulators for hematopoietic stem cell. Exp Hematol. (2011) 39:511–20. doi: 10.1016/j.exphem.2011.01.008
14. Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. (2014) 141:4656–66. doi: 10.1242/dev.106575
15. Gudmundsson KO, Du Y. Quiescence regulation by normal haematopoietic stem cells and leukaemia stem cells. FEBS J. (2023) 290:3708–22. doi: 10.1111/febs.v290.15
16. Kosan C, Godmann M. Genetic and epigenetic mechanisms that maintain hematopoietic stem cell function. Stem Cells Int. (2016) 2016:5178965. doi: 10.1155/2016/5178965
17. Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. (2011) 9:262–71. doi: 10.1016/j.stem.2011.06.014
18. Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, et al. p57Kip2 and p27Kip1 Cooperate to Maintain Hematopoietic Stem Cell Quiescence through Interactions with Hsc70. Cell Stem Cell. (2011) 9:247–61. doi: 10.1016/j.stem.2011.07.003
19. Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. (2005) 202:1599–611. doi: 10.1084/jem.20050967
20. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol. (2010) 10:201–9. doi: 10.1038/nri2726
21. Hur J, Choi JI, Lee H, Nham P, Kim TW, Chae CW, et al. CD82/KAI1 maintains the dormancy of long-term hematopoietic stem cells through interaction with DARC-expressing macrophages. Cell Stem Cell. (2016) 18:508–21. doi: 10.1016/j.stem.2016.01.013
22. Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci U S A. (1999) 96:3120–5. doi: 10.1073/pnas.96.6.3120
23. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. (2008) 135:1118–29. doi: 10.1016/j.cell.2008.10.048
24. Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB, et al. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell. (2017) 169:807–23.e19. doi: 10.1016/j.cell.2017.04.018
25. Ibneeva L, Singh SP, Sinha A, Eski SE, Wehner R, Rupp L, et al. CD38 promotes hematopoietic stem cell dormancy. PloS Biol. (2024) 22:e3002517. doi: 10.1371/journal.pbio.3002517
26. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. (2009) 458:904–8. doi: 10.1038/nature07815
27. Renders S, Svendsen AF, Panten J, Rama N, Maryanovich M, Sommerkamp P, et al. Niche derived netrin-1 regulates hematopoietic stem cell dormancy via its receptor neogenin-1. Nat Commun. (2021) 12:608. doi: 10.1038/s41467-020-20801-0
28. Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. (2009) 27:84–90. doi: 10.1038/nbt.1517
29. Yamashita M, Dellorusso PV, Olson OC, Passegue E. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat Rev Cancer. (2020) 20:365–82. doi: 10.1038/s41568-020-0260-3
30. Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. (2012) 11:36–49. doi: 10.1016/j.stem.2012.05.013
31. Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA, et al. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. (2007) 449:238–42. doi: 10.1038/nature06115
32. van der Wath RC, Wilson A, Laurenti E, Trumpp A, Lio P. Estimating dormant and active hematopoietic stem cell kinetics through extensive modeling of bromodeoxyuridine label-retaining cell dynamics. PloS One. (2009) 4:e6972. doi: 10.1371/journal.pone.0006972
33. Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell. (2016) 167:1296–309 e10. doi: 10.1016/j.cell.2016.10.022
34. Morcos MNF, Zerjatke T, Glauche I, Munz CM, Ge Y, Petzold A, et al. Continuous mitotic activity of primitive hematopoietic stem cells in adult mice. J Exp Med. (2020) 217. doi: 10.1084/jem.20191284
35. Bernitz JM, Rapp K, Daniel MG, Shcherbinin D, Yuan Y, Gomes A, et al. Memory of divisional history directs the continuous process of primitive hematopoietic lineage commitment. Stem Cell Rep. (2020) 14:561–74. doi: 10.1016/j.stemcr.2020.03.005
36. Bogeska R, Mikecin AM, Kaschutnig P, Fawaz M, Buchler-Schaff M, Le D, et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell. (2022) 29:1273–84.e8. doi: 10.1016/j.stem.2022.06.012
37. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. (2015) 520:549–52. doi: 10.1038/nature14131
38. Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell. (2015) 16:712–24. doi: 10.1016/j.stem.2015.04.004
39. Tesio M, Tang Y, Mudder K, Saini M, von Paleske L, Macintyre E, et al. Hematopoietic stem cell quiescence and function are controlled by the CYLD-TRAF2-p38MAPK pathway. J Exp Med. (2015) 212:525–38. doi: 10.1084/jem.20141438
40. Schonberger K, Obier N, Romero-Mulero MC, Cauchy P, Mess J, Pavlovich PV, et al. Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell. (2022) 29:131–48.e10. doi: 10.1016/j.stem.2021.10.002
41. Menendez-Gutierrez MP, Porcuna J, Nayak R, Paredes A, Niu H, Nunez V, et al. Retinoid X receptor promotes hematopoietic stem cell fitness and quiescence and preserves hematopoietic homeostasis. Blood. (2023) 141:592–608. doi: 10.1182/blood.2022016832
42. Zhang YW, Mess J, Aizarani N, Mishra P, Johnson C, Romero-Mulero MC, et al. Hyaluronic acid–GPRC5C signalling promotes dormancy in haematopoietic stem cells. Nat Cell Biol. (2022) 24:1038–48. doi: 10.1038/s41556-022-00931-x
43. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. (2015) 526:126–30. doi: 10.1038/nature15250
44. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. (2016) 532:323–8. doi: 10.1038/nature17624
45. Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. (2006) 439:599–603. doi: 10.1038/nature04247
46. Fukushima T, Tanaka Y, Hamey FK, Chang CH, Oki T, Asada S, et al. Discrimination of dormant and active hematopoietic stem cells by G0 marker reveals dormancy regulation by cytoplasmic calcium. Cell Rep. (2019) 29:4144–58.e7. doi: 10.1016/j.celrep.2019.11.061
47. Qiu J, Papatsenko D, Niu X, Schaniel C, Moore K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. (2014) 2:473–90. doi: 10.1016/j.stemcr.2014.01.016
48. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. (2009) 113:1250–6. doi: 10.1182/blood-2008-04-146480
49. Zhang H, Kozono DE, O’Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, et al. TGF-beta inhibition rescues hematopoietic stem cell defects and bone marrow failure in fanconi anemia. Cell Stem Cell. (2016) 18:668–81. doi: 10.1016/j.stem.2016.03.002
50. Hinge A, Filippi MD. Deconstructing the complexity of TGFbeta signaling in hematopoietic stem cells: quiescence and beyond. Curr Stem Cell Rep. (2016) 2:388–97. doi: 10.1007/s40778-016-0069-x
51. Sacma M, Pospiech J, Bogeska R, de Back W, Mallm JP, Sakk V, et al. Haematopoietic stem cells in perisinusoidal niches are protected from ageing. Nat Cell Biol. (2019) 21:1309–20. doi: 10.1038/s41556-019-0418-y
52. Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. (2012) 150:351–65. doi: 10.1016/j.cell.2012.05.041
53. Tesio M, Trumpp A. Breaking the cell cycle of HSCs by p57 and friends. Cell Stem Cell. (2011) 9:187–92. doi: 10.1016/j.stem.2011.08.005
54. Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. (2003) 36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x
55. Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. (2011) 195:709–20. doi: 10.1083/jcb.201102131
56. Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan AS, Prchal-Murphy M, Heller G, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. (2015) 125:90–101. doi: 10.1182/blood-2014-06-584417
57. Laurenti E, Frelin C, Xie S, Ferrari R, Dunant C, Zandi S, et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. (2015) 16:302–13. doi: 10.1016/j.stem.2015.01.017
58. Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat Med. (2000) 6:1235–40. doi: 10.1038/81335
59. van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, et al. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. (2007) 25:836–43. doi: 10.1634/stemcells.2006-0631
60. Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. (2007) 39:99–105. doi: 10.1038/ng1937
61. Stepanova L, Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood. (2005) 106:827–32. doi: 10.1182/blood-2004-06-2242
62. Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. (2006) 443:421–6. doi: 10.1038/nature05159
63. Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. (2004) 6:436–42. doi: 10.1038/ncb1126
64. Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. (2004) 18:2747–63. doi: 10.1101/gad.313104
65. Laurenti E, Varnum-Finney B, Wilson A, Ferrero I, Blanco-Bose WE, Ehninger A, et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell. (2008) 3:611–24. doi: 10.1016/j.stem.2008.09.005
66. Beauchemin H, Moroy T. Multifaceted actions of GFI1 and GFI1B in hematopoietic stem cell self-renewal and lineage commitment. Front Genet. (2020) 11:591099. doi: 10.3389/fgene.2020.591099
67. Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. (2004) 23:4116–25. doi: 10.1038/sj.emboj.7600419
68. Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. (2004) 431:1002–7. doi: 10.1038/nature02994
69. Xie X, Patnana PK, Frank D, Schutte J, Al-Matary Y, Kunstner A, et al. Dose-dependent effect of GFI1 expression in the reconstitution and the differentiation capacity of HSCs. Front Cell Dev Biol. (2023) 11:866847. doi: 10.3389/fcell.2023.866847
70. Hönes JM, Botezatu L, Helness A, Vadnais C, Vassen L, Robert F, et al. GFI1 as a novel prognostic and therapeutic factor for AML/MDS. Leukemia. (2016) 30:1237–45. doi: 10.1038/leu.2016.11
71. Kok CH, Watkins DB, Leclercq T, D’Andrea RJ, Hughes TP, White DL. Low GFI1 expression in white blood cells of CP-CML patients at diagnosis is strongly associated with subsequent blastic transformation. Leukemia. (2013) 27:1427–30. doi: 10.1038/leu.2013.47
72. Khandanpour C, Sharif-Askari E, Vassen L, Gaudreau MC, Zhu J, Paul WE, et al. Evidence that growth factor independence 1b regulates dormancy and peripheral blood mobilization of hematopoietic stem cells. Blood. (2010) 116:5149–61. doi: 10.1182/blood-2010-04-280305
73. Min IM, Pietramaggiori G, Kim FS, Passegue E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. (2008) 2:380–91. doi: 10.1016/j.stem.2008.01.015
74. Lord KA, Abdollahi A, Hoffman-Liebermann B, Liebermann DA. Proto-oncogenes of the fos/jun family of transcription factors are positive regulators of myeloid differentiation. Mol Cell Biol. (1993) 13:841–51. doi: 10.1128/mcb.13.2.841-851.1993
75. Kovary K, Bravo R. The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol Cell Biol. (1991) 11:4466–72. doi: 10.1128/mcb.11.9.4466-4472.1991
76. Miao GG, Curran T. Cell transformation by c-fos requires an extended period of expression and is independent of the cell cycle. Mol Cell Biol. (1994) 14:4295–310. doi: 10.1128/mcb.14.6.4295-4310.1994
77. Okada S, Fukuda T, Inada K, Tokuhisa T. Prolonged expression of c-fos suppresses cell cycle entry of dormant hematopoietic stem cells. Blood. (1999) 93:816–25. doi: 10.1182/blood.V93.3.816
78. McKinney-Freeman S, Cahan P, Li H, Lacadie SA, Huang HT, Curran M, et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell. (2012) 11:701–14. doi: 10.1016/j.stem.2012.07.018
79. Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. (2006) 116:2808–16. doi: 10.1172/JCI28310
80. Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. (2009) 4:37–48. doi: 10.1016/j.stem.2008.11.006
81. Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. (2007) 1:101–12. doi: 10.1016/j.stem.2007.02.001
82. Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell. (2008) 2:484–96. doi: 10.1016/j.stem.2008.03.004
83. Ariki R, Morikawa S, Mabuchi Y, Suzuki S, Nakatake M, Yoshioka K, et al. Homeodomain transcription factor Meis1 is a critical regulator of adult bone marrow hematopoiesis. PloS One. (2014) 9:e87646. doi: 10.1371/journal.pone.0087646
84. Unnisa Z, Clark JP, Roychoudhury J, Thomas E, Tessarollo L, Copeland NG, et al. Meis1 preserves hematopoietic stem cells in mice by limiting oxidative stress. Blood. (2012) 120:4973–81. doi: 10.1182/blood-2012-06-435800
85. Sirin O, Lukov GL, Mao R, Conneely OM, Goodell MA. The orphan nuclear receptor Nurr1 restricts the proliferation of haematopoietic stem cells. Nat Cell Biol. (2010) 12:1213–9. doi: 10.1038/ncb2125
86. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med. (2009) 15:696–700. doi: 10.1038/nm.1973
87. Komorowska K, Doyle A, Wahlestedt M, Subramaniam A, Debnath S, Chen J, et al. Hepatic leukemia factor maintains quiescence of hematopoietic stem cells and protects the stem cell pool during regeneration. Cell Rep. (2017) 21:3514–23. doi: 10.1016/j.celrep.2017.11.084
88. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. (2014) 508:269–73. doi: 10.1038/nature13034
89. Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, et al. Restraining lysosomal activity preserves hematopoietic stem cell quiescence and potency. Cell Stem Cell. (2020) 26:359–76.e7. doi: 10.1016/j.stem.2020.01.013
90. Zhang YW, Schonberger K, Cabezas-Wallscheid N. Bidirectional interplay between metabolism and epigenetics in hematopoietic stem cells and leukemia. EMBO J. (2023) 42:e112348. doi: 10.15252/embj.2022112348
91. Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. (2004) 21:843–51. doi: 10.1016/j.immuni.2004.11.004
92. Nitta E, Yamashita M, Suda T. Chromatin remodeling factor SmarcA2 contributes to maintain hematopoietic stem cell quiescence. Exp Hematology. (2013) 41. doi: 10.1016/j.exphem.2013.05.150
93. Calvanese V, Nguyen AT, Bolan TJ, Vavilina A, Su T, Lee LK, et al. MLLT3 governs human haematopoietic stem-cell self-renewal and engraftment. Nature. (2019) 576:281–6. doi: 10.1038/s41586-019-1790-2
94. Will B, Vogler TO, Bartholdy B, Garrett-Bakelman F, Mayer J, Barreyro L, et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nat Immunol. (2013) 14:437. doi: 10.1038/ni.2572
95. Fu Z, Jiang S, Sun Y, Zheng S, Zong L, Li P. Cut&tag: a powerful epigenetic tool for chromatin profiling. Epigenetics. (2024) 19:2293411. doi: 10.1080/15592294.2023.2293411
96. Hu Y, Shen F, Yang X, Han T, Long Z, Wen J, et al. Single-cell sequencing technology applied to epigenetics for the study of tumor heterogeneity. Clin Epigenetics. (2023) 15:161. doi: 10.1186/s13148-023-01574-x
97. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. (2014) 510:393–6. doi: 10.1038/nature13255
98. Johnson C, Belluschi S, Laurenti E. Beyond “to divide or not to divide”: Kinetics matters in hematopoietic stem cells. Exp Hematol. (2020) 92:1–10.e2. doi: 10.1016/j.exphem.2020.11.003
99. Miraki-Moud F, Anjos-Afonso F, Hodby KA, Griessinger E, Rosignoli G, Lillington D, et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc Natl Acad Sci. (2013) 110:13576–81. doi: 10.1073/pnas.1301891110
100. Konen JM, Fradette JJ, Gibbons DL. The good, the bad and the unknown of CD38 in the metabolic microenvironment and immune cell functionality of solid tumors. Cells. (2019) 9:52. doi: 10.3390/cells9010052
Keywords: hematopoietic stem cells, dormancy, quiescence, bromodeoxyuridine, H2B-GFP, label retention assay, retinoic acid signaling, CD38
Citation: Ibneeva L and Grinenko T (2024) Dissecting dormancy and quiescence in hematopoietic stem cells. Front. Hematol. 3:1401713. doi: 10.3389/frhem.2024.1401713
Received: 15 March 2024; Accepted: 05 September 2024;
Published: 25 September 2024.
Edited by:
Louis Pelus, Indiana University Bloomington, United StatesReviewed by:
Andres Garcia-Garcia, University of Basel, SwitzerlandRoi Gazit, Ben Gurion University of the Negev, Israel
Copyright © 2024 Ibneeva and Grinenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatyana Grinenko, Z3JpMTMxMDFAcmpoLmNvbS5jbg==; Liliia Ibneeva, bGlsaWlhLmlibmVldmFAdW5pa2xpbmlrdW0tZHJlc2Rlbi5kZQ==