 Christie T. M. Short
Christie T. M. Short Helen C. O’Neill
Helen C. O’Neill Jonathan K. H. Tan
Jonathan K. H. Tan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Hematol., 26 November 2024
Sec. Hematopoiesis and Stem Cells
Volume 3 - 2024 | https://doi.org/10.3389/frhem.2024.1396672
This article is part of the Research TopicNiche Contributions to Clonal ExpansionView all 3 articles
Introduction: Haematopoietic stem cell transplantation (HSCT) is commonly used to treat patients with haematological disorders. Myeloablative conditioning is an important preparation for patients receiving haematopoietic stem cells (HSC) or haematopoietic stem and progenitor cells (HSPC). While widely successful, HSCT is still associated with high rates of mortality. The recovery time between complete myeloablation and haematopoietic recovery is a large factor in the recovery rate. Successful engraftment of HSC is also directly correlated with the number of HSC niches available. This highlights the importance of the haematopoietic niche and its recovery from myeloablation as an important therapeutic target.
Methods: This murine model study specifically considers changes in spleen tissue architecture and cellular composition involving stromal and vascular cells that occur following lethal irradiation.
Results: Spleen recovered fully between 4- and 8-weeks after irradiation due to reconstitution by HSPC from bone marrow. Specific temporal changes in spleen architecture were identified, and these were linked to the cell types that constitute the white pulp, red pulp and marginal zones. Mesenchymal stromal cells returned before endothelial cells, and reticular cell types recovered more quickly in spleen following irradiation. Losses in gp38+ fibroblastic reticular cells and MAdCAM-1+ marginal reticular cells were associated with loss of the white pulp in the first 4 weeks following irradiation. White pulp was restored following recovery of supporting reticular cells.
Discussion: This study tests how spleen regeneration following a lethal dose of irradiation can be influenced by co-infusion of bone marrow HSPC together with either neonatal spleen stromal cells, or cells of the stromal STX3 line. Both the infusion of neonatal spleen stromal cells and STX3 stromal cells hastened recovery of both mesenchymal and vascular compartments. Following neonatal spleen stromal cell infusion, endothelial cells increased early, but a delay in structural reformation of distinct red and white pulp areas was found. Results from this study show that spleen regeneration can be influenced and even hastened through cellular therapy. Neonatal spleen stromal cells, co-infused together with HSPC following irradiation conditioning, represent a potential therapeutic opportunity for hastening spleen regeneration.
Haematopoietic stem cell transplantation (HSCT) is utilised to treat patients with haematological disorders like leukaemia. Myeloablative conditioning is routinely used to prepare patients for transplantation. While HSCT is widely successful, it is still associated with high rates of mortality. The recovery time between complete myeloablation and haematopoietic recovery is a large factor in recovery rate. Successful engraftment of transplanted HSC is directly correlated with the number of HSC niches available (1). The recovery of the haematopoietic niche following irradiation or myeloablation is therefore an important therapeutic target.
The bone marrow HSC niche has diverse functionality, allowing maintenance of a quiescent population of primitive HSC, subsequent HSC differentiation and haematopoietic cell development. The majority of active HSC reside in perisinusoidal niches in bone marrow, with rapid availability of oxygen, nutrients and cellular support (2). In bone marrow niches, the majority of cell types are osteoblasts in the endosteal niche, and endothelial cells in the perivascular niche. Leptin receptor+ (LepR+) mesenchymal stromal cells and endothelial cells are responsible for the maintenance and regulation of the bone marrow niche (3). Distinct domains around sinusoids regulate HSPC, evidenced by their spatial distribution throughout the bone marrow in proximity to arterioles, sinusoids and the endosteum.
In the treatment of haematopoietic malignancies, myeloablative irradiation causes damage to bone marrow and destroys HSC niche space, particularly sinusoidal endothelial cells (4). Irradiation damage to bone marrow stromal cells can be permanent inhibiting their ability to interact with other niche cells and their capacity to maintain and support HSC (1, 5). The success of HSC transplantation is correlated with the number of niches available per unit time (6). Thus, the damage caused to supporting cells in the niche may influence the efficacy of HSCT. It has been shown that transplantation of bone marrow endothelial cells can augment haematopoietic recovery following myeloablation and HSC transplantation (7). In these studies, bone marrow endothelial structure was disrupted at 10-15 days post irradiation. Within 5 days of transplantation of bone marrow endothelial progenitors, accelerated recovery of bone marrow cellularity, sinusoidal vessels, HSC regeneration and mature blood cell counts was reported (7). It was also shown that transfer of whole bone marrow cells supplemented with bone marrow endothelial cells into conditioned recipients resulted in long-term, multi-lineage engraftment and increased host survival (8).
The spleen also has capacity to support haematopoiesis and hosts a small population of distinct tissue-resident HSC (9). Murine HSPC were first localised in proximity with the endothelium of red pulp sinuses in postnatal murine spleen (2), and splenic red pulp was found to retain haematopoietic activity. Extramedullary haematopoiesis in spleen is dependent on the synthesis of CXCL12 by TCF21+ perisinusoidal stromal cells, and stem cell factor (SCF) by both endothelial cells and TCF21+ perisinusoidal stromal cells (9). Other studies have shown that macrophages may also be responsible for shaping and maintaining the niche in spleen (10). While irradiation of spleen has been used to treat haematopoietic malignancies, the importance of splenic stroma in haematopoiesis has not been investigated as fully as in bone marrow. The spleen is primed for myelopoiesis and is capable of more expedient production of myeloid cells than bone marrow. HSC in spleen are also quicker to enter the cell cycle than their bone marrow counterparts (11). HSC engraftment is dependent on available niche space in both spleen and bone marrow (1), so that hastening the recovery of spleen and the splenic niche after irradiation could increase the effectiveness of HSCT. Here, the recovery potential of spleen following irradiation conditioning has been analysed, and the impact of two distinct cellular therapies investigated.
An irradiation model for HSCT was established using control mice to assess damage to the multiple distinct regions present in spleen using immunofluorescent microscopy. The presence of distinct cell types was assessed and quantified by flow cytometry. Spleen recovery was then compared following irradiation using two distinct cell therapy approaches. The first involved adoptive transfer of neonatal spleen stromal cells. Since development of neonatal spleen has been described as dependent on the interaction of primitive lymphoid tissue inducer (LTi) cells with mesenchymal lymphoid tissue organiser (LTo) cells (12), infusion of neonatal spleen stromal cells into irradiated animals was hypothesised to accelerate spleen recovery. The second approach involved transfer of cells of the STX3 spleen stromal line, previously established in this lab (13). STX3 is a cloned continuous stromal cell line that supports haematopoiesis (13, 14), and progeny clones of STX3 reflect perivascular reticular cells of mesenchymal origin resembling osteoprogenitors (15). These two distinct approaches have been tested with a view to improve regeneration of spleen and its HSC niches after myeloablative irradiation. Previously combined delivery of bone marrow stromal cells and haematopoietic cells into irradiated hosts was shown to improve the regeneration of bone marrow HSC niches (7, 16).
Specific, pathogen-free female C57BL/6JArc (C57BL/6J) mice were obtained from the Animal Resource Centre (Perth, WA, Australia). C57BL6J Nzeg-enhanced (e) GFP mice were derived and kindly provided by Klaus Matthaei (John Curtin School of Medical Research, Canberra, Australia). Adult female mice were used at ages 6- to 16-weeks. Neonatal female and male mice were used between 1- and 8-days of age. Adult euthanasia was performed by cervical dislocation, and neonatal euthanasia was achieved through hypothermia. Mice were housed in the Animal Holding Facility (Bond University, Gold Coast, Australia) and handled according to protocols approved by the Animal Ethics Committee of the University of Queensland (Brisbane, QLD, Australia) under protocol: BOND/027/18. Mice were irradiated at the University of Queensland Biological Resources Centre at the Translational Research Institute (Brisbane, Queensland, Australia). Mice were housed for one week before irradiation which was delivered as a split dose of 4.5 Gy initially, followed by a second dose of 5 Gy after 4 hours (9.5 Gy total). Animals were then transferred immediately to the Animal Holding Facility at Bond University.
For adoptive cell transfer through the tail vein, animals were placed beneath a heat lamp in a cylindrical restraint. A 26G insulin syringe was used to deliver an infusion of bone marrow rescue cells (105 cells in 200µL PBS) within 8 hours of irradiation. Further adoptive cell transfers involved either neonatal spleen stromal cells prepared from C57BL6J Nzeg-eGFP mice, or STX3 spleen stromal cells (105 cells in 200µL PBS). Mice were sustained under observation on antibiotic water (neomycin sulfate (1.1g/L) and polymyxin B sulfate (106 U/L); Sigma-Aldrich Corporation; St. Louis, Missouri, USA) following irradiation.
Cardiac perfusion with fixative was performed in some animals prior to harvest of spleen. This applied to adult irradiated animals previously infused with a combination of rescue bone marrow cells and neonatal spleen stromal cells from C57BL6J Nzeg-eGFP mice. Animals were anaesthetised using a Darvall Stinger Streamline Isoflurane vaporiser (Darvall Vet; Gladesville, NSW, Australia). They were placed in the induction chamber under a heat lamp and exposed to isofluorane set at 5% for induction and 3% for surgery of the upper thorax. The underlying membrane was separated from the skin and opened to cut the ribcage and expose the heart. A small portion of the liver was excised, and a dish placed under it to collect fluid flow-through. A 25G butterfly needle was inserted into the left ventricle at an inferior and lateral angle and 8mL of PBS was delivered over 3 minutes. Following this, 8mL of formalin solution (10%, neutral buffered) was delivered over 3 minutes. Following successful cardiac perfusion and animal sacrifice, splenectomy was performed.
Cells of the STX3 spleen stromal cell line were cultured as a monolayer in supplemented Dulbecco’s Modified Eagle Medium (sDMEM) (Sigma-Aldrich Corporation) at 37°C in 5% CO2 air and 97% humidity as described previously (13). Cells were released through scraping and repetitive pipetting at 90% confluence, and passaged by 1:3 dilution. For adoptive transfer, cells were harvested, washed and resuspended in PBS.
Bone marrow cells were prepared from 8- to 10-week-old C57BL/6J female mice. The femur and tibia of both hind legs were dissected and the epiphyses cut off to expose the diaphyses. Central bone marrow was ejected into a 14mL conical tube containing 2mL ice-cold PBS using a 23G × 1 ¼” syringe, followed by repetitive pipetting to dissociate cells and form a single cell suspension. Cells were pelleted for 5 minutes at 200G and 4°C, and then resuspended in 1mL red blood cell lysis buffer (eBioscience Incorporated; San Diego, CA, USA) for 5 minutes at room temperature. Cells were washed in medium and cell number calculated by staining dead cells with trypan blue (12% in PBS) (Sigma-Aldrich Corporation) and counting on a haemocytometer using a Leitz Diavert Inverted microscope (Leica Microsystems GmbH; Wetzlar, Germany).
Spleen stromal cells were prepared from neonatal C57BL6J Nzeg-eGFP mice for adoptive transfer as a cell therapy. The spleen stromal cell fraction was also isolated from adult mice undergoing HSCT for quantification of stromal subsets by flow cytometry.
Spleens were physically dissociated using a 1mL syringe plunger (Terumo Corporation; Shibuya, Tokyo, Japan) and cells resuspended in a 14mL conical centrifuge tube containing 10mL DMEM and 2% FCS (Sigma-Aldrich Corporation), before placement on ice for 1 minute to settle. Supernatant was removed using a 70µm reversible cell strainer (STEMCELL Technologies; Vancouver, British Colombia, Canada), and stromal fragments collected on the strainer. Enzymic digestion of stromal fragments involved addition of 2mL of Collagenase IV mix (DMEM, 2% FCS, collagenase from Clostridium histolyticum (1mg/mL), Deoxyribonuclease I from bovine pancreas (40µg/mL) (Sigma-Aldrich Corporation)) to release stromal cells attached to the grid of the strainer. Collected cells were incubated with rotation for 10 minutes at 37°C on a Bio RS-24 Mini-rotator (speed = 3) (Biosan: Riga, Latvia). The cell suspension was pipetted rapidly to disperse cell fragments before incubation in an additional 4mL of Collagenase D solution (DMEM, 2% FCS, Collagenase D (1mg/mL) and Deoxyribonuclease I (40µg/mL)) for another 10 minutes at 37°C with gentle rotation. Cells were washed with 8mL of ice-cold medium (DMEM with 2% FCS), and centrifuged for 5 minutes at 200G and 4°C. Supernatant was discarded, the pellet resuspended in medium, filtered through a 100µm cell strainer (Greiner Bio-One; Kremsmünster, Austria), and cells counted.
Antibody staining and flow cytometry was used to characterise and quantify stromal cell populations in spleen. Spleen stromal cells were prepared through enzymic digestion and resuspended in staining buffer (1% FCS, 0.1% sodium azide in PBS), and aliquoted at 106 cells/well into a 96-well U-bottom plate (TPP Techno Plastic Products AG; Trasadingen, Switzerland), sedimented for 5 minutes at 200G and 4°C, and supernatant discarded. Cells were resuspended in 10μL antibody cocktail prepared by diluting antibody 1:100 in staining buffer, followed by incubation for 10 minutes at 4°C protected from light. Wells were washed once with 150μL staining buffer before supernatant was discarded and secondary reagents added (1:400 dilution) as needed for a further 10-minute incubation. After a final wash, samples were resuspended into 150μL staining buffer and transferred into 12 × 75mm labelled Falcon tubes (Becton Dickinson; Franklin Lakes, New Jersey, USA.).
To discriminate live cells, 40μg/mL propidium iodide (PI; Sigma-Aldrich Corporation) was prepared and 2μL added to samples. Flow cytometry was performed on a BD FACSAria™ Fusion flow cytometer (Becton Dickinson) using single-colour controls (SCC) to adjust compensation for spectral overlap. For all antibody-based SSC, cells were used for staining. Where possible, fluorescence-minus-one controls (FMOC) were used to set gates to distinguish staining. These antibody cocktails contain all but one antibody to define the negative population boundaries for the excluded antibody. Acquired data was analysed using FlowJo v10.2 software (FlowJo LLC; Ashland, Oregon, USA).
Fluorochrome conjugated antibodies used to detect both endothelial and mesenchymal stromal cells included CD45-APC-eFluor® 780 (30-F11), MAdCAM-1-biotin (MECA-367) and CD105-PE-Cy7 (MJ7/18) from eBioscience; and TER-119-BV421 (TER-119), gp38-PE (8.1.1), CD31- Alexa Fluor® 647 (MEC13.3) from BioLegend (San Diego, CA, USA). Secondary conjugates used were Streptavidin (SA)-BV510 from BioLegend.
Changes in spleen structure and architecture were determined using immunofluorescence staining of tissue sections to distinguish known cells and regions of spleen. Spleens were harvested and frozen in Tissue-Tek ® O.C.T Compound (Sakura Finetek Japan Co., Ltd; Tokyo, Japan). Frozen spleens were sectioned at 6-8µm thickness (-12 to -14˚C) using a Leica CM1800 Cryostat (Leica Microsystems GmbH) and applied to Sodalime white glass (UberFrost) Printer Slides (InstrumeC; Victoria, Australia). Sections were fixed in acetone for 5 minutes at room temperature, then washed 3 times in PBS for 3 minutes at room temperature. Tissues were blocked before addition of primary antibodies. This involved Fc block (CD16/32 [1:100]: BioLegend) unless purified antibodies were used, in which case blocking was achieved using FACS buffer only. Incubations were performed inside an incubation chamber away from light. Primary antibodies were applied and incubated for 8 hours at 4°C. Slides were washed 3 times in PBS for 3 minutes at room temperature, before application of secondary antibodies, which were incubated for 1 hour at 4°C. Slides were washed again 3 times in PBS at room temperature, then a coverslip applied using Dako Fluorescence Mounting Medium (Agilent Technologies; Santa Clara, California, USA). Antibodies used included MOMA-1-biotin (MOMA-1) from Abcam (Cambridge, United Kingdom); FDC-M1-purified (FDC-M1) from BD Pharmingen (San Diego, CA, USA); B220-Alexa Fluor® 647 (RA3-6B2), CD29-PE (HMβ1-1), CD31-Alexa Fluor® 647 (MEC13.3), CD31-PE (390), CD105-Alexa Fluor® 488 (MJ7/18), CD105-PE (MJ7/18), F4/80-biotinylated (BM8), F4/80-FITC (BM8), gp38-PE (8.1.1) and MAdCAM-1-Alexa Fluor® 488 (MECA-367) from BioLegend; and CD11b-biotin (M1/70), CD3ϵ-APC (145-2C11), MAdCAM-1-biotin (MECA-367) and TER-119-biotin (TER-119) from eBioscience. Secondary conjugates used were streptavidin (SA)-Alexa Fluor® 488 (BioLegend); SA-APC and SA-PE (eBioscience); and Donkey anti-Rat IgG-Alexa Fluor® 488 (Life Technologies; Carlsbad, CA, USA). All antibodies were screened and tested to ensure no background staining before use.
Spleen sections were imaged using a Nikon Eclipse Ti2-E fluorescent microscope (Nikon Corporation; Tokyo, Japan) commonly at 10X magnification. Images were processed in NiS Elements AR software (Nikon Eclipse Ti2-E; Nikon Corporation) and FIJI v2.0.0 open-source software (Laboratory for Optical and Computational Instrumentation; Maddison, Michigan, USA) including the QuickFigures plugin (17, 18).
All experiments used biological triplicates as a minimum and individual experiments compared one control and one test sample as a minimum. Statistical analyses were performed in GraphPad Prism v9 software (GraphPad; San Diego, CA, USA) and power and sample sizes were calculated in G*Power software (Heinrich Heine University Düsseldorf, Düsseldorf, Northrhine-Westphalia, Germany). Data are expressed as mean ± SEM. Outliers were identified using the ROUT method (Q = 5%). Significance was assessed using unpaired t-tests with Welch’s corrections. Significantly different values are indicated as (*) when p ≤ 0.05, (**) when p ≤ 0.005, (***) when p ≤ 0.0005, (****) when p ≤ 0. 0001 and non-significant (ns) when p > 0.05. Graphs identify only paired comparisons which are significantly different.
To understand the capacity of spleen to regenerate following irradiation, spleen structure based on cellularity was analysed in young adult mice following total body irradiation as a lethal dose (split dose of 9.5Gy), and transfer of rescue bone marrow cells. Spleen stromal cells were stained through immunocytochemistry at multiple time points for 8 weeks following irradiation.
Distinct subsets of stromal and haematopoietic cells are located in the white pulp, red pulp and marginal zone areas of the spleen. Staining for these cells can be used to monitor the integrity of spleen and its cellularity. In red pulp, staining detected CD11b+ myeloid cells, CD105+ red pulp sinusoids, CD31+ mature endothelial cells, F4/80+ red pulp macrophages and TER-119+ erythroid. In white pulp B220+ B cells, CD3+ T cells, FDC-M1+ follicular dendritic cells and gp38+ fibroblastic reticular cells were distinguished. The marginal zone was delineated by MOMA-1+ marginal metallophilic reticular macrophages and MAdCAM-1+ marginal reticular cells. These markers were used to define the regions of spleen and any loss and recovery of region integrity following irradiation.
Structural integrity of the spleen compartments was lost through whole body lethal irradiation, but this loss resolved after 8 weeks in mice given a rescue bone marrow infusion (Figures 1–3). At 1-week post-irradiation, major loss of the white pulp was evident through loss of CD3 staining for T cells and gp38 staining for fibroblastic reticular cells (Figure 1). Myeloid cells were still visible in the red pulp by staining for CD11b, but red pulp was disorganised in comparison with control spleen tissue. Additionally, clusters of myeloid cells were observed in some parts of the tissue that appear to correlate with the marginal zone. By 4-weeks post-irradiation, some recovery of the white pulp regions was evident through organisation of gp38+ fibroblastic reticular cells and appearance of CD3+ T cells (Figure 1). These cells provide a necessary role for support of CD3+ T cells. By 4 weeks, spleen repair was also evident with morphological appearance resembling control spleen. However, it is notable that the white pulp areas remained weakly populated by T cells and fibroblastic reticular cells. After 8-weeks, structural morphology had returned, and cellular losses experienced through irradiation appear to have fully resolved.
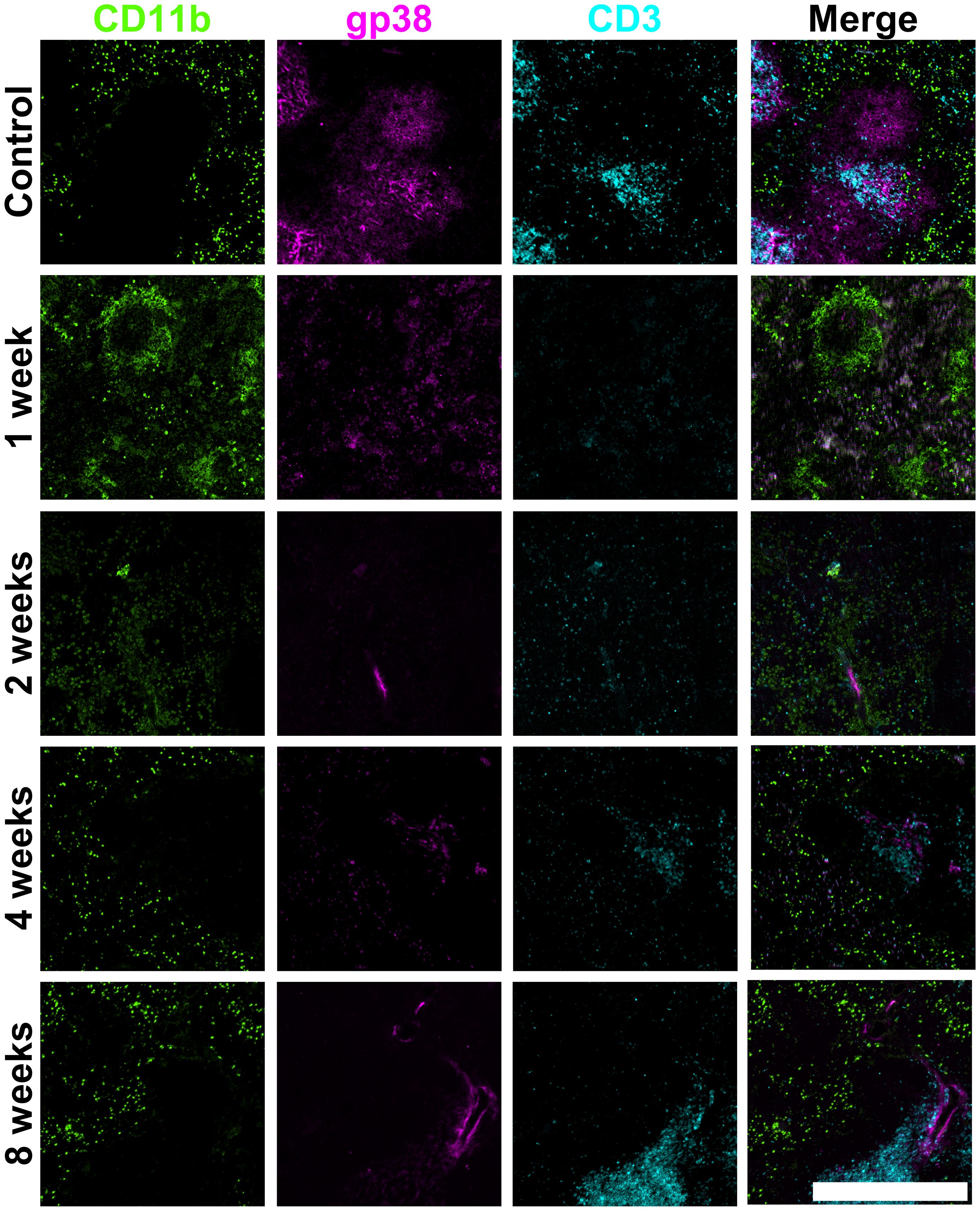
Figure 1. Changes in spleen red and white pulp following irradiation. Frozen spleen sections were prepared from a female adult C57BL/6J mouse that received a split dose of lethal irradiation (9.5Gy total) and 105 syngeneic bone marrow rescue cells at 8 hours post-irradiation. Cryosections were prepared and fixed with acetone. Antibodies specific for CD11b (biotinylated; secondary conjugate SA-Alexa Fluor® 488), gp38 (PE) and CD3 (APC) were used for staining. Scale bar represents 500µm.
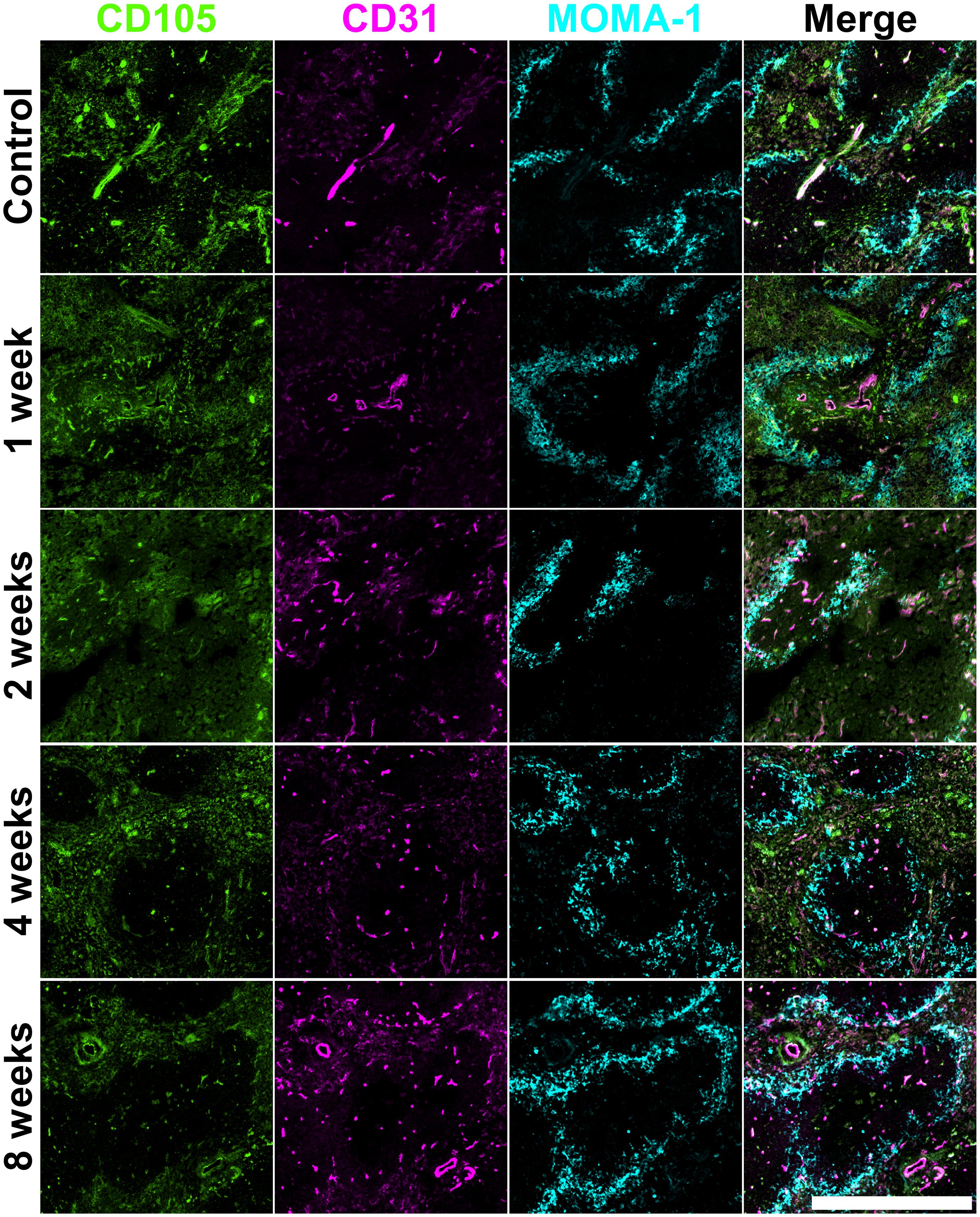
Figure 2. Vasculature change in irradiated adult spleen. Frozen spleen sections were prepared from a female adult C57BL/6J mouse lethally irradiated (9.5Gy total) and given 105 syngeneic bone marrow rescue cells at 8 hours post-irradiation. Cryosections were prepared and fixed with acetone. Antibodies specific for CD105 (Alexa Fluor® 488), CD31 (PE) and MOMA-1 (biotinylated; secondary conjugate SA-APC) were used for staining. Scale bar represents 500µm.
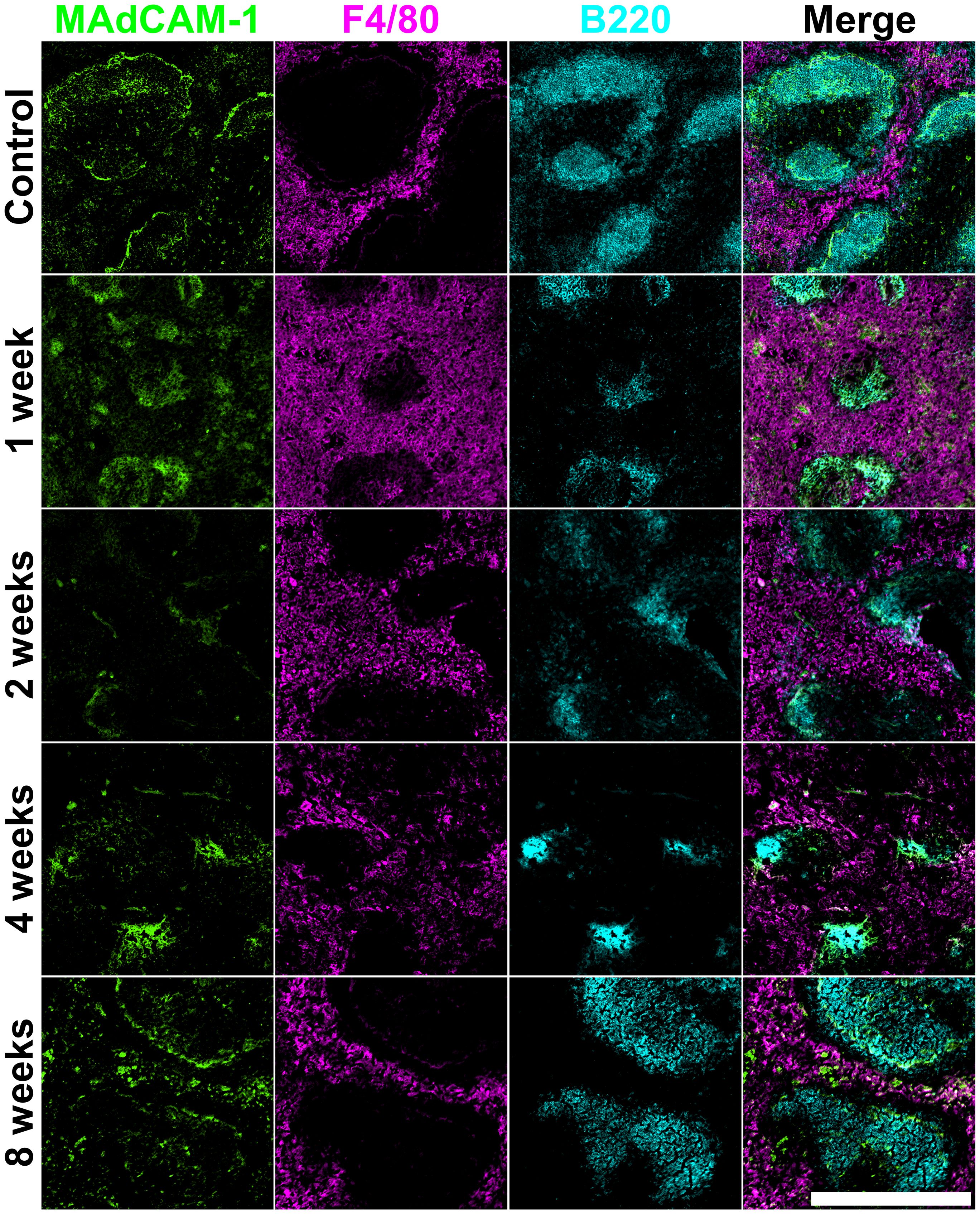
Figure 3. Architecture of irradiated adult spleen. Frozen spleen sections were prepared from a female adult C57BL/6J mouse lethally irradiated (9.5Gy total) and given 105 syngeneic bone marrow rescue cells at 8 hours post-irradiation. Cryosections were prepared and fixed with acetone. Antibodies specific for MAdCAM-1 (Alexa Fluor® 488), F4/80 (PE) and B220 (Alexa Fluor® 647) were used for staining. Scale bar represents 500µm.
A second set of stainings was used to analyse the impact of irradiation on vasculature. At 1-week post-irradiation, some loss of structure in relation to CD105+ red pulp staining was observed, with loss of definition of white pulp evident at 2-weeks post-irradiation (Figure 2). By comparison, structure dependent on mature CD31+ endothelial cells was largely unaffected, and the staining pattern resembled that of control spleen. The area of staining for MOMA-1+ marginal metallophilic reticular macrophages appeared to be slightly increased in relation to control spleen and the white pulp areas demarcated by MOMA-1 staining of marginal metallophilic macrophages appeared to remain intact. Some loss of cells in these compartments was evident at 2-weeks post-irradiation, but by 4-weeks the delineation of white pulp areas and the marginal zone was restored, although these areas appeared smaller than in control spleens (Figure 2). Spleen structure in relation to CD105, CD31 and MOMA-1 staining cells was rescued completely by 8-weeks post-irradiation.
Further staining identified changes in marginal zones identified by staining of marginal reticular cells with MAdCAM-1+, red pulp macrophages through F4/80 staining and the B220+ B cell areas of the white pulp follicles. By 1-week post-irradiation, MAdCAM-1+ marginal reticular cells became reduced in number and bordered smaller white pulp areas (Figure 3). Clear borders of marginal reticular cells were not observed again until 8-weeks post-irradiation. Increased staining for F4/80+ red pulp macrophages was observed at 1-week post-irradiation, coupled with a proportional decrease in white pulp areas as shown by smaller areas of B220+ B cell follicles. This suggests loss of B cells and possibly an influx of inflammatory myeloid cells. By 2-weeks, F4/80+ red pulp macrophages continued to occupy the red pulp, and the area of white pulp was depleted. Quite small B cell follicles were evident at 4-weeks, and organisation of the surrounding red pulp and interfacing marginal zone was unclear (Figure 3). By 8-weeks post-irradiation, B cells refilled the follicles and splenic structure appeared to have regenerated and was comparable to control spleen.
A final staining involved FDC-M1 staining of follicular dendritic cells, which support B cell development in white pulp. TER-119 staining of red blood cells was used to delineate red pulp regions (Appendix A.1). At 1-week post-irradiation, an increase in staining for FDC-M1 was evident with distribution of follicular dendritic cells into the red pulp region. Across the 8-week recovery period, red pulp maintained TER119+ red blood cells. The tissue also maintained structural integrity of red pulp comparable with control spleen, although white pulp areas became smaller in size and more disorganised with respect to FDC-M1 staining of follicular dendritic cells. By 4-weeks after irradiation, spleen white pulp areas were very small in size, yet still showed staining for underlying follicular dendritic cells (Appendix A.1). The proportional increase in red pulp could be a result of an influx of red blood cells and increased vasculature. Like other staining panels, the structure of the spleen was completely regenerated and resembled control spleen by 8-weeks.
All figures show how an irradiation dose of 9.5Gy sustains immediate damage to multiple cell types and regions in the spleen. White pulp areas reduce in size and are impacted the most, while the splenic structure transforms to accommodate new vasculature in the red pulp. Marginal metallophilic reticular macrophages increase in number immediately following irradiation, and then decrease over time to control numbers. Cells forming and inhabiting the red pulp increase in number between 1- and 4-weeks post-irradiation with corresponding loss of white pulp. Spleen structure at 8-weeks post-irradiation is comparable with normal spleen, indicating that most regeneration is achieved between weeks 4 to 8.
To further assess damage to spleen following irradiation, flow cytometry was used to quantify cell subset changes. The rationale was that changes to particular cell types after irradiation would reflect the effect of the lethal irradiation dose and could be used alongside immunofluorescence staining showing structure to interpret damage to the spleen and haematopoietic niches. It will also inform the availability and provision of cells for recovery. Antibodies were combined in a staining panel specific to endothelial cells and mesenchymal cells, since HSC are known to reside in perisinusoidal niches containing these two main cell types. A gating strategy was developed and is shown in Figure 4A. This identifies the procedure to gate out singlets (FSC, SSC-H, SSC-W) and live cells (PI), and then to gate out erythrocytes (TER119-) and to select stromal cells (CD45-) (Figure 4A). Representative plots of each timepoint are included in Appendix A.2. As spleen weight varies with time post-irradiation, changes to endothelial and mesenchymal cell compartments was analysed by comparing cell frequencies between timepoints of 1-, 2-, 4- and 8-weeks, in line with spleen structural analyses.
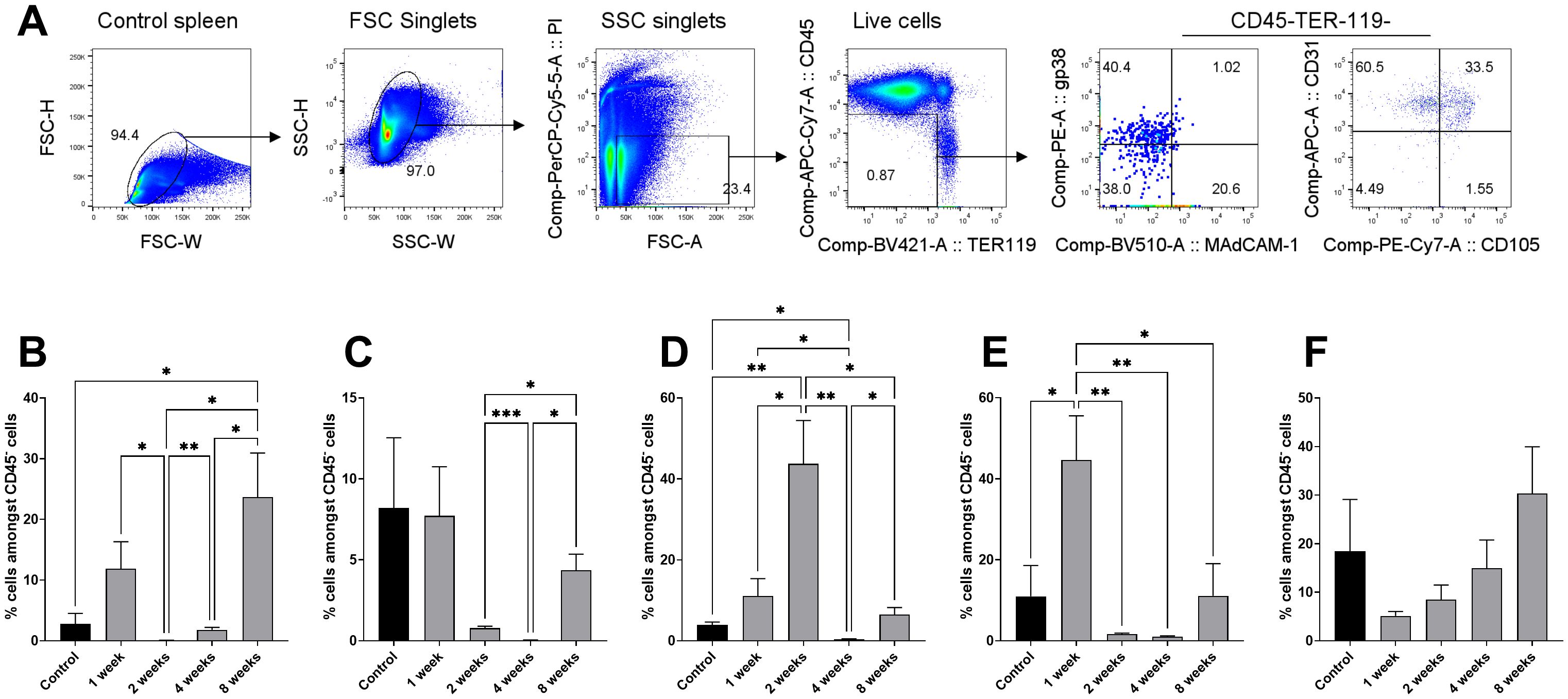
Figure 4. Stromal cell subset changes in spleen following irradiation. Spleen cells were prepared from 9-week-old female C57BL/6J mice at 1-, 2-, 4- and 8-weeks following total body irradiation (split dose of 9.5 Gy) followed by adoptive transfer of 105 syngeneic rescue bone marrow cells at 8 hours post-irradiation. Stromal cells were dissociated through enzymatic digestion and then cells stained with antibodies specific for CD45 (APC-eFluor® 780), TER-119 (Brilliant Violet 421), MAdCAM-1 (Brilliant Violet 510), gp38 (PE), CD31 (Alexa Fluor® 647) and CD105 (PE-Cy7) and analysed flow cytometrically. (A) Gating strategy is shown to remove singlets (FSC, SSC-H, SSC-W) and live cells (PI-), erythrocytes (TER119-). Distinct populations of CD45- stromal cells were then gated based on staining. (B) gp38+ fibroblastic reticular cells; (C) MAdCAM-1+ marginal reticular cells; (D) CD31+ mature endothelial cells; (E) CD31+CD105+ vascular endothelial cells; (F) CD105+ red pulp sinusoids. All error bars represent SEM. Significantly different values are indicated as (*) when p ≤ 0.05, (**) when p ≤ 0.005, (***) when p ≤ 0.0005 and non-significant (ns) when p > 0.05.
The first analysis involved assessment of gp38+ fibroblastic reticular cells and MAdCAM-1+ marginal reticular cells in irradiated mice (Figures 4B, C), as a first representation of the tissue response to irradiation damage. Frequencies of CD31+ mature endothelial cells, CD31+CD105+ vascular endothelial cells and CD105+ red pulp sinusoids were also analysed (Figures 4D, E, F). For fibroblastic reticular cells, no significant change in cell frequency amongst gated CD45- cells was detected at 1-week post-irradiation in comparison to control spleen (11.89% ± 12.54% and 2.787± 2.79% ± 2.79%, respectively; p = 0.0895) (Figure 4B). However, significant differences were observed between the timepoints tested. Cell frequency reduced at the 2-week timepoint (p = 0.0322) to 0.07% ± 0.05%, increasing marginally at 4-weeks (p = 0.0059) to 1.80% ± 1.10% (Figure 4B). This increased again at 8 weeks (p = 0.0392) to 23.68% ± 16.23%, which was significantly higher than control spleen (p = 0.0434) (Figure 4B). This rapid loss with delayed recovery of fibroblastic reticular cells is reflected by staining patterns for gp38 in spleen sections shown in Figure 1.
For marginal reticular cells, no differences were found between control spleen and the timepoints tested (Figure 4C). However, marginal reticular cell frequency dropped between 2- and 4-weeks post-irradiation from 0.78% ± 0.32% to 0.03% ± 0.04% (p = 0.0007) (Figure 4C). This increased at the 8-week timepoint to 4.36% ± 2.22% (p = 0.0120), though this was lower than controls (8.20% ± 8.67%; p = 0.4454) (Figure 4C). Section staining for MAdCAM-1 in Figure 3 shows this loss at 4 weeks.
Endothelial cells were investigated as a representation of vascular niches involved in HSPC regulation (Figures 4D–F). A significant (p = 0.0074) increase in CD31+ mature endothelial cell frequency was observed at 2-weeks compared to controls (43.72% ± 30.29% and 3.89% ± 1.27%, respectively), which was also a significant increase over 1-week post-irradiation (11.09% ± 12.05%; p = 0.0193) (Figure 4D). This marked increase was followed by a significant loss at 4 weeks (0.38% ± 0.29%; p = 0.0049), which was then significantly recouped at 8-weeks (6.49% ± 3.84%; p = 0.0233) (Figure 4D).
For vascular endothelial cells, frequency was highest at 1-week compared to controls (44.65% ± 30.89% and 10.97% ± 15.23%, respectively; p = 0.0299) (Figure 4E). This high frequency was followed by a significant loss at 2-weeks (1.64% ± 0.71%; p = 0.0056) and then at 4-weeks (1.01% ± 0.51%; p = 0.0052) but return to levels comparable with controls at 8-weeks (11.08% ± 17.81%; p = 0.9917) (Figure 4E). CD105+ cells, indicative of angiogenic vasculature or red pulp sinusoidal endothelial cells, decreased at 1-week post-irradiation, before gradually increasing over the following timepoints tested, but these frequencies were not significantly different to controls (Figure 4F). These changes in endothelial cell numbers post-irradiation are also reflected by staining shown in Figure 2.
Two potential cell therapies for augmentation of spleen recovery were explored and compared. In the first group, 105 cells of the STX3 spleen stromal line were infused with 105 bone marrow cells into irradiated hosts. In the second group, irradiated animals received an infusion of 105 neonatal spleen stromal cells along with 105 rescue bone marrow cells. Neonatal spleen stromal cells were prepared from C57BL6J Nzeg-eGFP mice by isolation of the stromal cell fraction, and then enzymatic digestion to ensure dissociation of cells. Their eGFP expression allowed detection of donor stromal cells. Fixation of spleen through cardiac perfusion before euthanasia was found to preserve eGFP expression. Results of the irradiation recovery experiment outlined in Figures 1-3 show that spleen recovery is evident by 4-weeks post-irradiation. Transplantation of HSC into irradiated mice shows that engraftment in spleen is evident as early as 6 days post-irradiation, and increases steadily until significant engraftment is achieved by 21 days (Cao et al., 2004). Thus, for the two cell therapy experiments, spleen regeneration was monitored over a shorter time period out to 20-days, and host spleens were analysed at 5-, 10-, 15- and 20-days following irradiation.
It was hypothesised that acceleration of spleen regeneration would be evident by reduced loss of specific cell subsets determined through flow cytometry. We also hypothesised that spleen architecture would recover sooner given the infusion of additional cells available to support spleen development, and that this would be evident through section staining. The plasticity of the spleen, its ability to increase haematopoiesis during stress, and its ability to reduce cell production and to return to steady-state homeostasis, also supports a study which aims to hasten spleen regeneration through an infusion of stromal cells.
Following neonatal spleen stromal cell transfer, staining for eGFP identified uptake of stromal cells into spleen between 5- and 10-days after infusion (Figure 5). Spleen vasculature changes were assessed through staining for CD105+ red pulp sinusoids and CD31+ endothelial cells. Spleen showed a notable loss of organised structure at 5-days post-irradiation with loss of white pulp areas, and retention of CD105 staining of red pulp sinusoids (Figure 5). This is consistent with control irradiated spleen seen at 1-week in Figure 2. By 20-days post-irradiation, these losses were recouped, despite a delay in structural organisation. CD31+ central arterioles were preserved at 1-week, although CD31dim red pulp endothelial cells were lost by 10-days but began to recover after 15 days with more normal follicle definition by 20-days (Figure 5). Infused eGFP+ spleen stromal cells and CD105+ red pulp sinusoids clearly co-localised with CD31+ central arterioles and CD105+ red pulp sinusoids from 10- to 20-days (Figure 5).
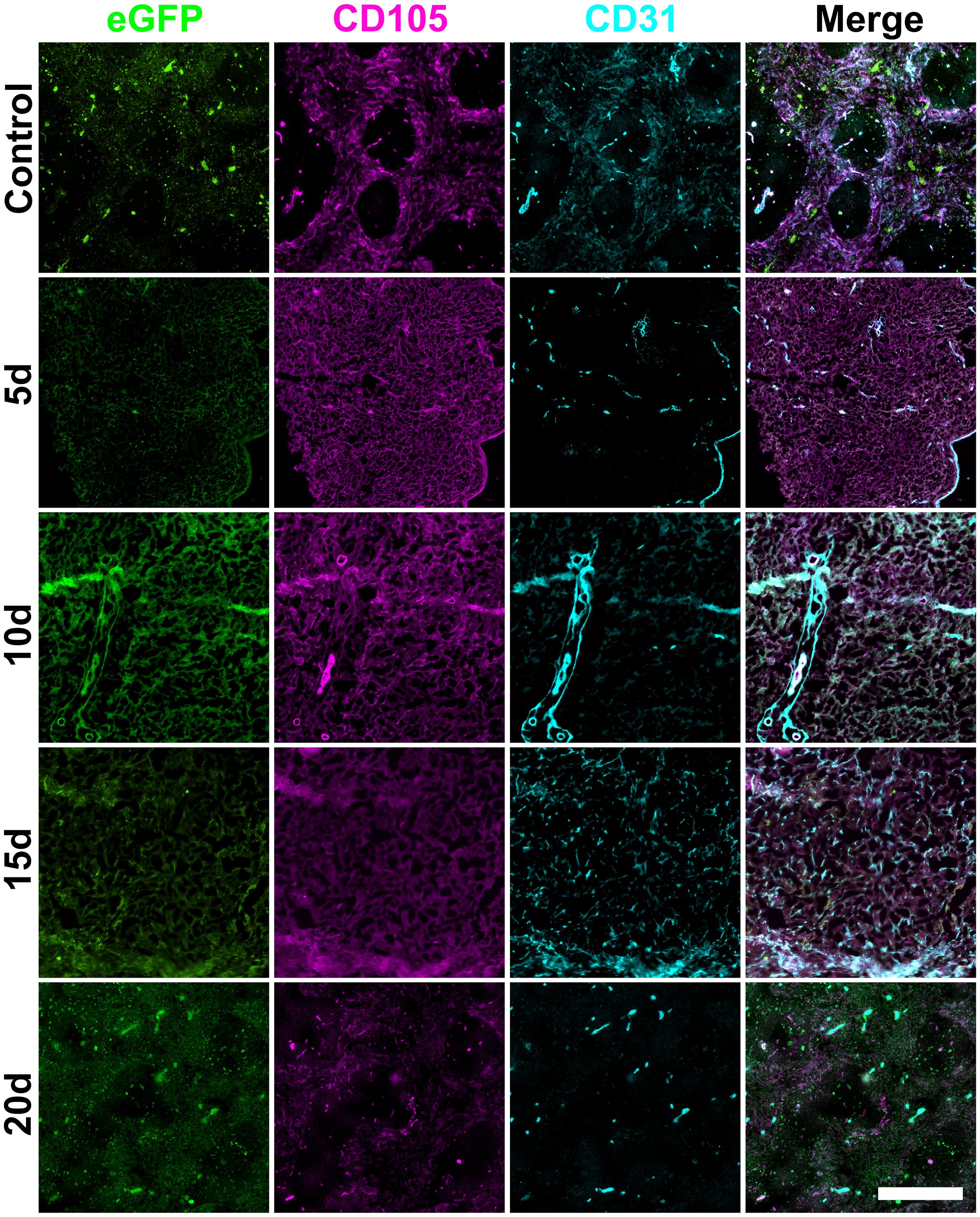
Figure 5. Changes in splenic vasculature following neonatal spleen cell infusion. Female adult C57BL/6J mice following lethal irradiation were given a rescue transfer of 105 syngeneic bone marrow cells and 105 neonatal C57BL6J Nzeg-eGFP enzymatically digested spleen stromal cells. Frozen spleen sections were prepared from animals sacrificed at 5-, 10-, 15- and 20-days post-irradiation and adoptive cell transfer. Cryosections were fixed with acetone. Staining involved antibodies specific for CD105 (PE) and CD31 (Alexa Fluor® 647). Scale bar represents 400µm.
Changes in mesenchymal and reticular cell subsets were examined in irradiated spleens given cellular therapy at 5-, 10-, 15- and 20-day timepoints. However, following neonatal spleen stromal cell transfer staining for only the 15-day timepoint was successfully achieved (Figure 6). In these experiments, eGFP staining identified influx of infused stromal cells into the irradiated spleen (Figure 6A). Also notable was the appearance of gp38+ fibroblastic reticular cells at 15-days (Figure 6A), as seen in Figure 1 with irradiated spleens given only rescue bone marrow. This staining is localised in T cell areas, and does not overlap with F4/80 staining in the red pulp. Furthermore. eGFP staining across all regions coincided with CD29 staining and identifies the influx of spleen stromal cells and could identify the origin of both myeloid and fibroblastic reticular cells (Figures 6A, B). MAdCAM-1+ marginal reticular cell areas were reduced in size in spleens of animals given neonatal stromal cells by 15-days, although these areas were few in number and small in size (Figure 6B) compared to control spleen (Figure 2). The earlier recovery of irradiated spleen given an infusion of neonatal spleen stromal cells was also reflected in the earlier recovery of T and B cell areas by 15-days compared with 4-weeks for T cells and 8-weeks for B cells (Appendix A.3) compared with controls shown in Figures 1, 3. These results suggest that cellular therapies can contribute to the regeneration of spleen vasculature, most notably evident by recovery of CD105+ red pulp sinusoids in animals given neonatal spleen stromal cells.
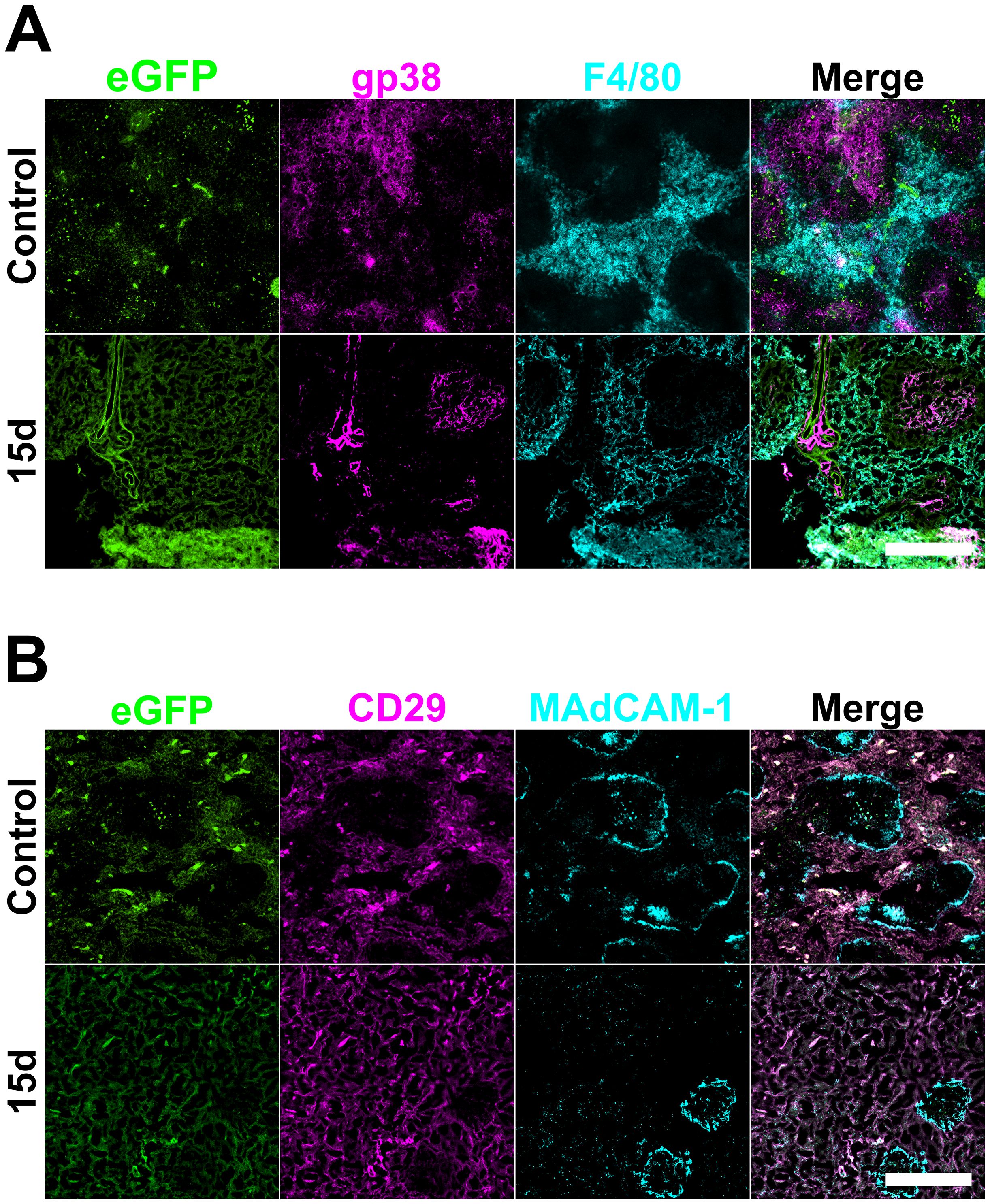
Figure 6. Mesenchymal architecture of spleen following neonatal spleen cell transfer. Female adult C57BL/6J mice following lethal irradiation were given a rescue transfer of 105 syngeneic bone marrow cells and 105 neonatal C57BL6J Nzeg-eGFP enzymatically digested spleen stromal cells. Frozen spleen sections were prepared from animals sacrificed at 5-, 10- and 15-days post-irradiation and adoptive cell transfer. Cryosections were fixed with acetone. Spleen sections from only 15-day timepoints are shown. Staining involved antibodies specific for (A) gp38 (PE) and F4/80 (biotinylated, followed by SA-APC), and for (B) CD29 (PE) and MAdCAM-1 (biotinylated, streptavidin-APC). Scale bars represent 400µm.
Following STX3 spleen stromal cell transfer (Figure 7), recovery of CD105+ red pulp sinusoids was restrained by loss of distinct red pulp structure observed by 5-days post-irradiation which was not restored by 20-days post-irradiation. This was also seen with control mouse spleen (Figure 2), and with mice given neonatal spleen stromal cells (Figure 5). STX3-treated spleens showed mature CD31+ endothelial cells located in central arterioles which appeared to remain unaffected by irradiation, with structures present across the 5- to 20-day time course (Figure 7), resembling control irradiated spleens (Figure 2).
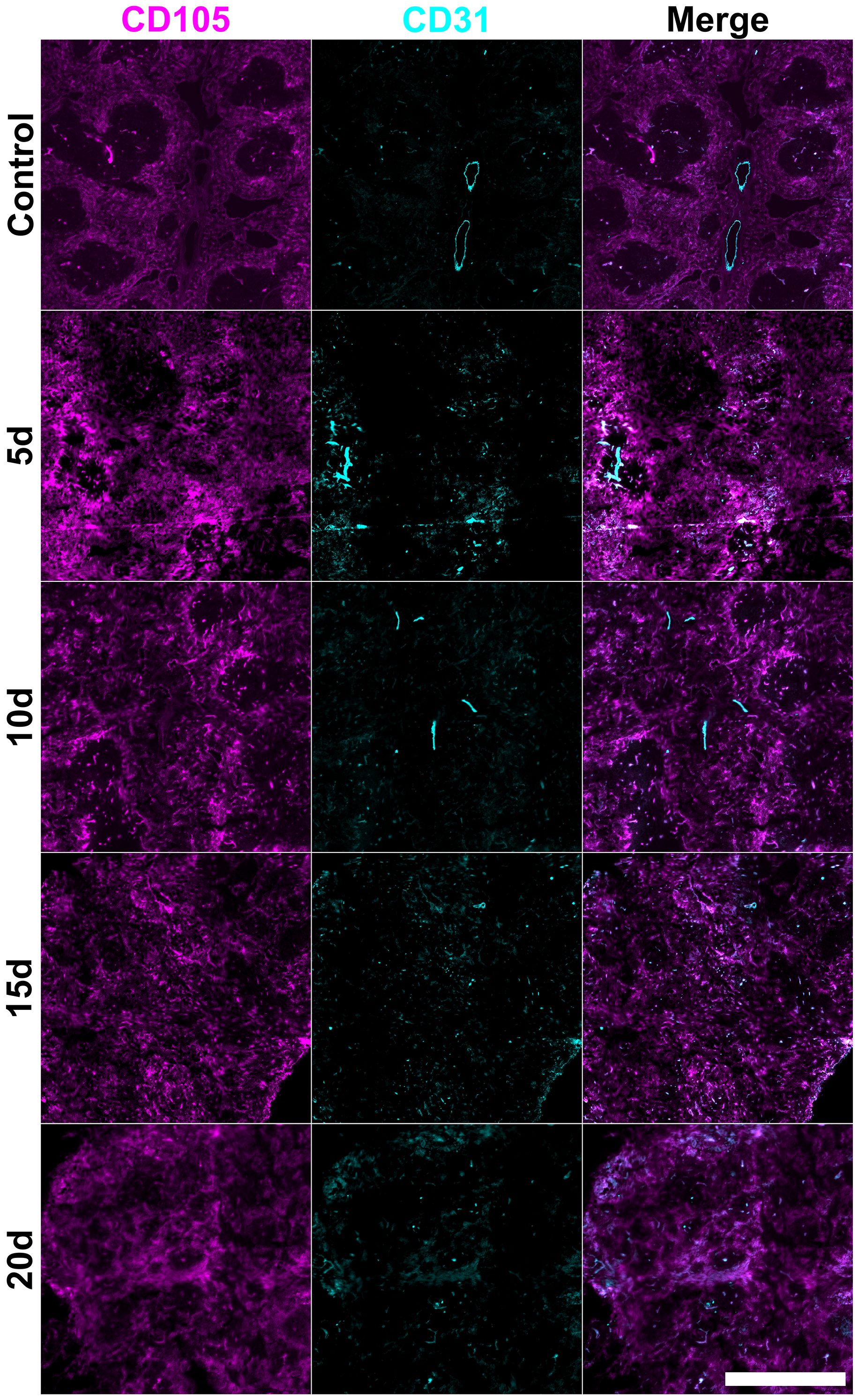
Figure 7. Changes in splenic vasculature following STX3 stromal cell transfer. Female adult C57BL/6J mice following lethal irradiation and rescue transfer of 105 syngeneic bone marrow along with 105 cultured STX3 spleen stromal cells. Frozen spleen sections were prepared from animals sacrificed at 5-, 10-, 15- and 20-days post-irradiation and cell transfer. Cryosections were fixed with acetone and stained with antibodies specific for CD105 (PE) and CD31 (Alexa Fluor® 647). Scale bar represents 500µm.
Following STX3 spleen stromal cell transfer, mesenchymal gp38+ cells and also CD29+ cells were visible at all tested timepoints and were not restricted in location to white pulp (Figure 8) as was seen in controls given no cellular therapy (Figure 1). Widespread gp38+ staining was observed from 5-days to 15-days post-irradiation, and showed some alignment with white pulp by 20-days (Figure 8). Comparatively, in STX3-treated spleens, MAdCAM-1+ marginal reticular cells formed distinct structures between 5- and 15-days post-irradiation, although this was lost at 20-days post-irradiation (Figure 8). These marginal zone structures were more defined than those seen post-irradiation in control irradiated spleen (Figure 3).
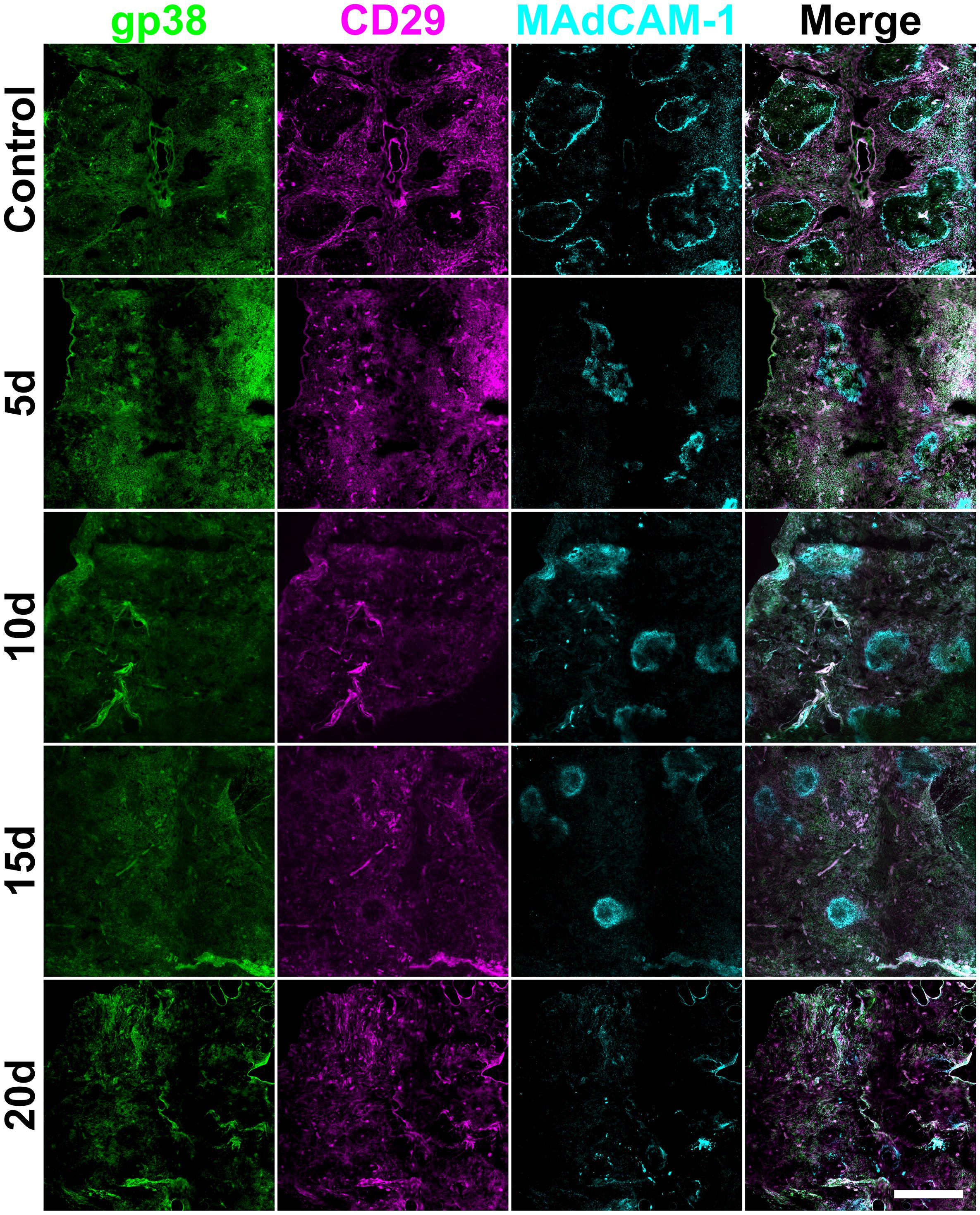
Figure 8. Splenic mesenchymal architecture following STX3 stromal cell transfer. Female adult C57BL/6J mice following lethal irradiation and rescue transfer of 105 syngeneic bone marrow and 105 cultured STX3 spleen stromal cells. Frozen spleen sections were prepared from animals sacrificed at 5-, 10-, 15- and 20-days post-irradiation and cell transfer. Cryosections were fixed with acetone and stained with antibodies specific for gp38 (Alexa Fluor® 488), CD29 (PE) and MAdCAM-1 (biotinylated; followed by SA-APC). Scale bar represents 500µm.
Recovery of white pulp regions of T and B cells also lends support to these findings. B cell follicles are detectable by 10-days in hosts given STX3 stromal cell therapy (Appendix A.4) which is earlier than in hosts given neonatal spleen stromal cells (15-days) (Appendix A.3), or in recovering control mice (between 4- and 8-weeks) (Figure 3). T cell areas recover by 10-days (Appendix A.4) which is also earlier than in mice given neonatal spleen stromal cells (15-days) (Appendix A.3) or in control mice between 4- and 8- weeks (Figure 1).
Flow cytometry was then used to identify cell subsets and to quantitate changes in subset representation following irradiation and cellular therapy. Since cell subsets were compared across treatments at several different time points, absolute numbers of cells were quantified. Representative gating for each therapy is included in Figure 9A; representative plots for all timepoints are included in Appendices A.5 and A.6. Comparison of the two cellular therapies showed that infusion of neonatal spleen cells led to a significantly greater number of gp38+ fibroblastic reticular cells at 15 days post-irradiation compared to STX3 spleen stromal cells (2927.0 ± 1505.0 compared to 781.5 ± 1082.0; p = 0.0194) (Figure 9B).
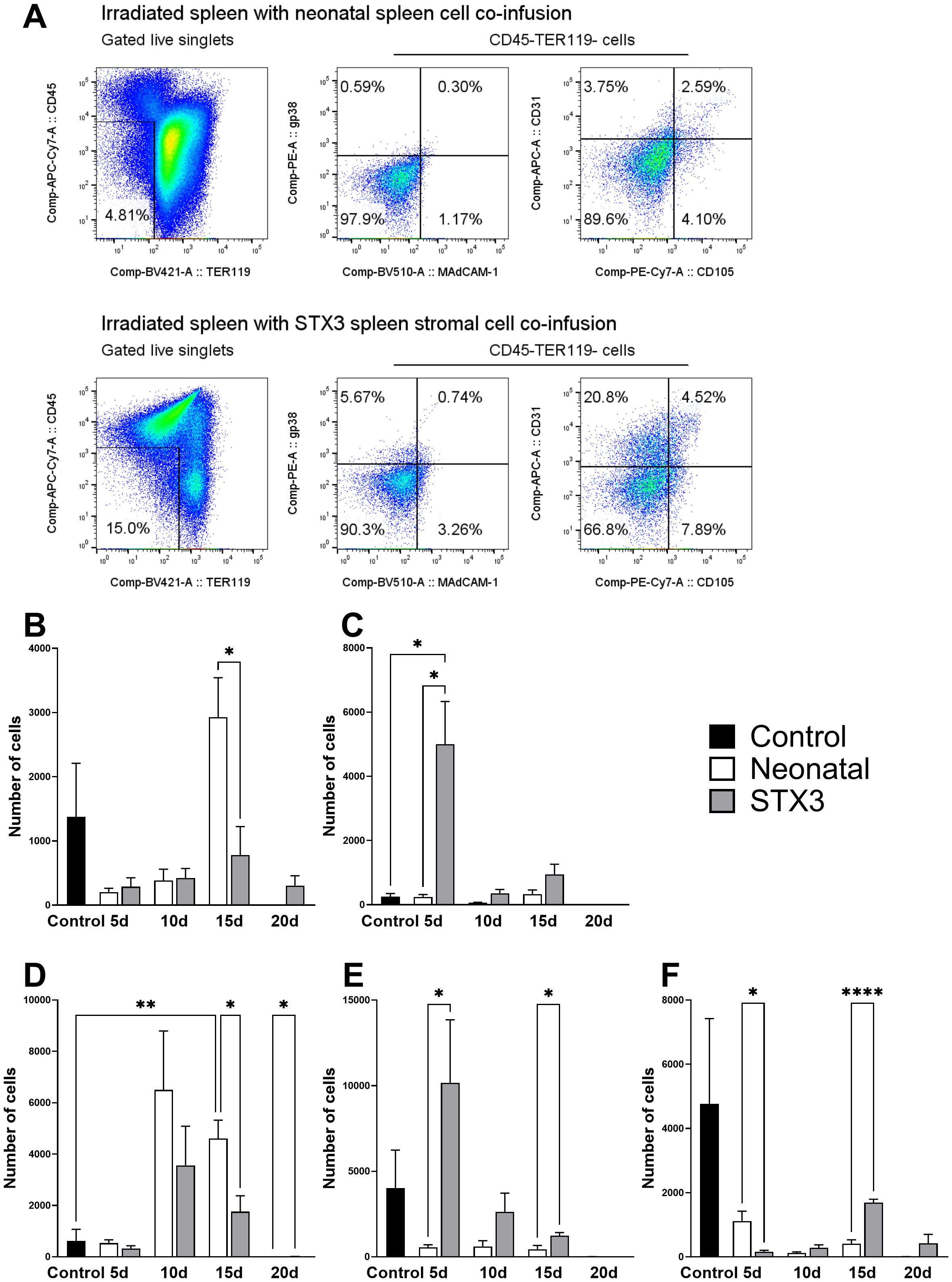
Figure 9. The impact of neonatal spleen cell transplantation and STX3 spleen stromal cell transplantation on mesenchymal and vascular subsets. Spleen cells were prepared from adult female C57BL/6J mice 5-, 10-, 15- and 20-days after receiving total body irradiation (split dose of 9.5 Gy). The host received adoptive transfer of 105 syngeneic rescue bone marrow cells at 8-hours post-irradiation and either 105 C57BL6J Nzeg-eGFP spleen cells enzymatically digested spleen stromal cells, or 105 cultured STX3 stromal cells. Host spleens were isolated and cells prepared and stained for subset identification using flow cytometry and the antibodies outlined in Figure 4. Distinct populations of CD45- stromal cells were gated based on staining as shown in Figure 4A. (A) Representative plots of each cellular therapy tested. Data shows total cell numbers for each cell subset investigated: (B) gp38+ fibroblastic reticular cells; (C) MAdCAM-1+ marginal reticular cells; (D) CD31+ mature endothelial cells; (E) CD31+CD105+ vascular endothelial cells; (F) CD105+ red pulp sinusoids. All error bars represent SEM. Significantly different values are indicated as (*) when p ≤ 0.05, (**) when p ≤ 0.005, (****) when p ≤ 0.0001 and non-significant (ns) when p > 0.05.
An early significant increase in MAdCAM-1+ marginal reticular cells was observed at 5-days in mice given STX3 spleen stromal cells (4996.0 ± 3270.0; p = 0.0162) compared with neonatal spleen stromal cell transfer, which was also significantly higher than control spleen (251.3 ± 197.5; p = 0.0162) (Figure 9C). In terms of mature CD31+ endothelial cells, a significant increase in the number of cells was observed at 15-days post-irradiation following infusion of neonatal spleen stromal cells (4605.0 ± 1754.0) (Figure 9D), compared with controls (p = 0.0017). This increase at 15-days was also significantly higher than observed for STX3-treated spleens (1760.0 ± 1513; p = 0.0134) (Figure 9D).
STX3 spleen stromal cell transfer regenerated significantly more (p = 0.0474) CD31+CD105+ vascular endothelial cells (10169.0 ± 9009.0) than did neonatal spleen stromal cells (552.5 ± 387.4) in the first 5 days post-irradiation (Figure 9E). This was also observed at 15-days post irradiation (2623 ± 2686 compared to 443.7 ± 546.4; p = 0.0203) (Figure 9E). Finally, infusion of either neonatal spleen stromal cells and STX3 spleen stromal cells showed loss of CD105+ red pulp sinusoids following irradiation, although these numbers were not significantly different to controls (Figure 9F). Neonatal spleen stromal cell transfer showed an earlier and higher (1119.0 ± 747.0) reconstitution of CD105+ red pulp sinusoids than did STX3 spleen cell transfer at 5-days (159.4 ± 111.0; p = 0.0249) (Figure 9F). At the 15-day timepoint, however, mice given STX3 spleen stromal cells displayed a significant increase (1692.0 ± 235.6; p < 0.0001) in red pulp sinusoids over mice given neonatal spleen stromal cells.
Together, these results indicate that both neonatal spleen stromal cell infusion and STX3 spleen stromal cell infusion following irradiation contribute to earlier recovery of splenic stromal cells. STX3 spleen stromal cell infusion also contributed more significantly to regeneration of vascular endothelial cells and red pulp sinusoids than did neonatal spleen stromal cell infusion. The earlier rescue of mature CD31+ endothelial cells following neonatal spleen stromal cell transfer suggests that these cells can be very effective in splenic regeneration following irradiation. Future studies could test the role of specific neonatal spleen stromal subsets in regeneration.
This study specifically considers changes in spleen structure and cellularity involving stromal and vascular cells that occurs following lethal irradiation used as a conditioning regimen during HSCT. Spleen tissue recovers fully between 4- and 8-weeks after irradiation due to reconstitution of hematopoietic cells by HSPC from bone marrow. Specific temporal changes have been identified and linked to loss and reformation of cell structures that constitute the main compartments of spleen identifiable as white pulp, red pulp and marginal zones. This study goes further to test how spleen regeneration following damage from a lethal dose of irradiation can be influenced by co-infusion of neonatal spleen stromal cells or the cells of the splenic stromal cell line STX3 along with rescue bone marrow HSPC. Vascular or endothelial cell compartments are rescued sooner following co-infusion of neonatal spleen cells, while both mesenchymal and vascular compartments recover more quickly following infusion of STX3 cells. Following neonatal spleen stromal cell infusion, endothelial cell numbers increased early, but a delay in structural reformation to give distinct red and white pulp areas was identified. This is interpreted to mean a temporal change in reorganisation impacted by stromal cell content in the recovering spleen.
The impact of irradiation on HSC niches in spleen has not been well investigated, but the expectation is that the radiation sensitivity of endothelial and mesenchymal cells in spleen would be consistent with that of bone marrow. Most HSC in bone marrow reside adjacent to sinusoidal blood vessels (2). Endothelial cells are a main component of perivascular niches, and in postnatal mice, spleen HSPC are localised with the endothelium of red pulp sinuses (2). Irradiation used as part of HSCT conditioning regimes damages bone marrow and ablates HSC niches, in particular the sinusoidal blood vessels and the sinusoidal endothelial cells. Irradiation can permanently damage bone marrow stromal cells, inhibiting their capacity to maintain and support HSC (5). The interaction of bone marrow niche cells and HSC rapidly reconstitutes haematopoiesis (1), thus the success of haematopoietic recovery is connected with the recovery of the bone marrow stromal microenvironment involving both endothelial and mesenchymal cells.
To identify irradiation damage to spleen architecture, we measured changes in spleen stromal and endothelial cell content over a period of 8-weeks following lethal irradiation and infusion with HSPC from bone marrow. Irradiation damage to the spleen under this regime is not permanent and complete regeneration of spleen tissue was achieved between 4- and 8-weeks. Mesenchymal stromal cells returned before endothelial cells, and several reticular cell types including gp38+ fibroblastic reticular cells and MAdCAM-1+ marginal reticular cells were the first to recover in spleen following irradiation (Figures 1–3). T and B cells located in white pulp areas are situated atop a physical scaffold of fibroblastic reticular cells and follicular dendritic cells that release cytokines and chemokines to facilitate efficient communication between immune cells (19, 20). Selective loss of white pulp reticular cells is associated with profound immunodeficiency. This is demonstrated by losses in gp38+ fibroblastic reticular cells and MAdCAM-1+ marginal reticular cells seen here (Figures 1, 3). This was associated with loss of white pulp in the first 4 weeks following irradiation (Figures 1, 3). Restoration of reticular cell structures is essential to reinstate T and B cells in these regions. Full immune functionality is evident by the full restoration of T and B cell areas of white pulp by 8-weeks. This coincides with restoration of supporting reticular cells (Figures 1, 3).
Here we show that infusion of neonatal spleen cells, enzymatically dissociated to release all stromal cells in the tissue, hastens spleen recovery following irradiation for HSCT. This was best demonstrated by an increase in the frequency of fibroblastic reticular cells at 15-days after irradiation (Figure 9B) and mature endothelial cells present in spleen at 10-days after treatment (Figure 9D). Neonatal spleen stromal cell infusion, coupled with bone marrow HSPC transfer into irradiated mice, provides a burst of proliferative cells that shifts the regeneration dynamic towards earlier recovery of these stromal and endothelial cell subsets compared with that observed in control irradiated spleen given only HSPC from bone marrow. Notably, spleens from irradiated animals treated with neonatal spleen cells showed increased cell viability at 15-days post-irradiation compared with STX3-treated spleens, which may reflect the increases seen in gp38+ fibroblastic cell and CD31+ endothelial cell numbers (Figures 9B, D). Variable spleen size, observed following irradiation and cellular therapy, can limit direct comparisons of the treatments explored. While further analysis could correct this by comparing cell proportionality rather than absolute numbers of cells, results still are subject to gating strategies and may not accurately reflect changes in cell populations. Direct comparison of the changes in cell populations following application of two distinct stromal cell therapies may therefore be limited by the low number of some stromal cell types and the difficulty in the isolation of viable cells. Despite these limitations, data shown here does however inform of the changing dynamics of cell repopulation of spleen following irradiation.
The co-infusion of STX3 spleen stromal cells showed greater increases in frequencies of marginal reticular cells, vascular endothelial cells and red pulp sinusoids than did neonatal spleen stromal cell transfer (Figures 9C–F). Microscopic imaging of spleen shows loss of CD105+ red pulp sinusoid structure over 20-days following STX3 stromal cell infusion (Figure 6). However, in flow cytometric analysis, increased CD105+ red pulp sinusoid frequency was observed at 15-days (Figure 9F). STX3 cells express CD105 (21), and increased expression of this marker may be more indicative of STX3 localisation in the spleen than specific regeneration of red pulp sinusoids. Overall, infusion of STX3 spleen stromal cells had a greater impact on the regeneration of the spleen than did neonatal spleen stromal cell infusion.
An important caveat to our results is that under the current experimental protocol we cannot be certain how many infused stromal cells homed to spleen. Neonatal spleen stromal cells from C57BL/6J Nzeg-eGFP mice did however localise in spleens between 5- and 10-days based on detection of eGFP staining. Cells delivered intravenously are known to localise in the capillary beds of the lungs and reside there for 2-days post-transplantation (22). However, transplanted mesenchymal stem cells can also home to several sites other than the lungs, such as liver, heart and, of relevance to this study, spleen (23). Additionally, the spleen is the most frequent site of engraftment following HSC transplantation, with over one third of HSC homing to spleen (23). This study shows that it is possible to influence the recovery of spleen through infusion of neonatal spleen stromal cells which boosts stromal and endothelial cell numbers. However, we also show evidence that increased numbers of stromal cells can delay the reorganisation of structure and the formation of the white pulp compared with control irradiated spleen. In future, the dynamics of enhanced recovery will be better investigated through infusion of specific stromal cell subsets, and through optimisation of the number and type of cells infused. Inclusion of timepoint-matched analyses of irradiated spleens without infusion of either cell therapy explored here, along with more intermediate timepoints, would allow greater understanding of the impact of these therapies on splenic regeneration kinetics. In addition, the use of other techniques to track transplanted cells, as well as analysis of changes in extracellular matrix components and growth factors involved in splenic regeneration could further augment the interpretation of results.
In summary, the results of this study show that the regenerative capacity of spleen can be hastened through cellular therapy. Neonatal spleen cell transfer or fractionated spleen stromal cells, co-infused together with HSPC following irradiation conditioning, represent a potential therapeutic opportunity to explore the dynamics of spleen regeneration in more detail.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Animal Ethics Committee of the University of Queensland. The study was conducted in accordance with the local legislation and institutional requirements.
CS: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. HO’N: Conceptualization, Formal analysis, Supervision, Writing – review & editing. JT: Conceptualization, Data curation, Formal analysis, Methodology, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by funding provided by the National Health and Medical Research Council of Australia (New Investigator grant #APP11867) and the Cutmore Bequest to Bond University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2024.1396672/full#supplementary-material
1. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. (2003) 425:836–41. doi: 10.1038/nature02041
2. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. (2005) 121:1109–21. doi: 10.1016/j.cell.2005.05.026
3. Comazzetto S, Murphy MM, Berto S, Jeffery E, Zhao Z, Morrison SJ. Restricted hematopoietic progenitors and erythropoiesis require SCF from leptin receptor+ Niche cells in the bone marrow. Cell Stem Cell. (2019) 24:477–486.e6. doi: 10.1016/j.stem.2018.11.022
4. Mauch P, Constine L, Greenberger J, Knospe W, Sullivan J, Liesveld JL, et al. Hematopoietic stem cell compartment: acute and late effects of radiation therapy and chemotherapy. Int J Radiat Oncol - Biol - Phys. (1995) 31:1319–39. doi: 10.1016/0360-3016(94)00430-S
5. Abbuehl JP, Tatarova Z, Held W, Huelsken J. Long-term engraftment of primary bone marrow stromal cells repairs niche damage and improves hematopoietic stem cell transplantation. Cell Stem Cell. (2017) 21:241–255.e6. doi: 10.1016/j.stem.2017.07.004
6. Abkowitz JL, Robinson AE, Kale S, Long MW, Chen J. Mobilization of hematopoietic stem cells during homeostasis and after cytokine exposure. Blood. (2003) 102:1249–53. doi: 10.1182/blood-2003-01-0318
7. Salter AB, Meadows SK, Muramoto GG, Himburg H, Doan P, Daher P, et al. Endothelial progenitor cell infusion induces hematopoietic stem cell reconstitution in vivo. Blood. (2009) 113:2104–7. doi: 10.1182/blood-2008-06-162941
8. Poulos MG, Ramalingam P, Gutkin MC, Llanos P, Gilleran K, Rabbany SY, et al. Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J Clin Invest. (2017) 127:4163–78. doi: 10.1172/JCI93940
9. Inra CN, Zhou BO, Acar M, Murphy MM, Richardson J, Zhao Z, et al. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature. (2015) 527:466–71. doi: 10.1038/nature15530
10. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. (2012) 487:325–9. doi: 10.1038/nature11260
11. Coppin E, Florentin J, Vasamsetti SB, Arunkumar A, Sembrat J, Rojas M, et al. Splenic hematopoietic stem cells display a pre-activated phenotype. Immunol Cell Biol. (2018) 96:772–84. doi: 10.1111/imcb.12035
12. Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. (2009) 183:2205–12. doi: 10.4049/jimmunol.0804324
13. Ni K, O’Neill H. Spleen stromal cells support haemopoiesis and in vitro growth of dendritic cells from bone marrow. Br J Haematology. (1999) 105:58–67. doi: 10.1111/j.1365-2141.1999.01294.x
14. Despars G, O’Neill HC. Heterogeneity amongst splenic stromal cell lines which support dendritic cell hematopoiesis. In Vitro Cell Dev Biol - Anim. (2006) 42:208–15. doi: 10.1290/0602016.1
15. O’Neill HC, Lim HK, Periasamy P, Kumarappan L, Tan JKH, O’Neill TJ. Transplanted spleen stromal cells with osteogenic potential support ectopic myelopoiesis. PloS One. (2019) 14:e0223416. doi: 10.1371/journal.pone.0223416
16. Zhang Y, Adachi Y, Suzuki Y, Minamino K, Iwasaki M, Hisha H, et al. Simultaneous injection of bone marrow cells and stromal cells into bone marrow accelerates hematopoiesis in vivo. Stem Cells. (2004) 22:1256–62. doi: 10.1634/stemcells.2004-0173
17. Mazo G. QuickFigures: A toolkit and ImageJ PlugIn to quickly transform microscope images into scientific figures. PloS One. (2021) 16:e0240280. doi: 10.1371/journal.pone.0240280
18. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. (2012) 9:676–82. doi: 10.1038/nmeth.2019
19. Den Haan J, Mebius R, Kraal G. Stromal cells of the mouse spleen. Front Immunol. (2012) 3:201. doi: 10.3389/fimmu.2012.00201
20. Junt T, Scandella E, Ludewig B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol. (2008) 8:764–75. doi: 10.1038/nri2414
21. Lim HK, Periasamy P, O’Neill HC. In vitro murine hematopoiesis supported by signaling from a splenic stromal cell line. Stem Cells Int. (2018) 2018:9896142. doi: 10.1155/2018/9896142
22. Fisher B, Packard BS, Read EJ, Carrasquillo JA, Carter CS, Topalian SL, et al. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol. (1989) 7:250–61. doi: 10.1200/jco.1989.7.2.250
Keywords: haematopoiesis, haematopoietic stem cell niche, spleen, bone marrow, irradiation, regeneration, spleen regeneration
Citation: Short CTM, O’Neill HC and Tan JKH (2024) Exploring the regenerative capacity of the spleen following irradiation. Front. Hematol. 3:1396672. doi: 10.3389/frhem.2024.1396672
Received: 06 March 2024; Accepted: 04 November 2024;
Published: 26 November 2024.
Edited by:
Isabel Beerman, National Institute on Aging (NIH), United StatesReviewed by:
Perpetua Pinto-do-Ó, Universidade do Porto, PortugalCopyright © 2024 Short, O’Neill and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen C. O’Neill, aG9uZWlsbEBib25kLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.