 Sonia Morè1
Sonia Morè1 Laura Corvatta
Laura Corvatta Massimo Offidani
Massimo Offidani- 1Clinica di Ematologia, Azienda Ospedaliero Universitaria delle Marche, Ancona, Italy
- 2Medicina Interna, Ospedale E. Profili, Fabriano, Italy
Systemic light-chain (AL) amyloidosis is a monoclonal plasma cell disease characterized by the deposition of amyloidogenic monoclonal light-chain fragments in organs, causing their dysfunction. Clinical manifestations could be very aspecific, but the most frequent ones are proteinuria with or without renal failure or heart failure, with the kidney and the heart being the first two involved organs. Histological diagnosis with Congo red staining is the gold standard, but typing the amyloid with immunohistochemistry or mass spectrometry of the Congo red positive tissue is necessary to establish if an AL or ATTR amyloidosis could be diagnosed. Staging AL amyloidosis before treatment could help physicians to prognosticate the disease. Recently, staging systems were set separately for different involved organs, using biomarkers. Autologous stem cell transplant after a daratumumab-based induction treatment is the cornerstone of therapy in younger and fit patients, with the goal of reaching a deep and rapid disease hematological and organ response. Novel therapies, borrowed from a therapeutical model of multiple myeloma, are studied to optimize AL amyloidosis outcomes. In this review, we make an overview of diagnostic procedures, staging system, and therapies of AL amyloidosis.
1 Introduction
AL amyloidosis, also called “primary” amyloidosis, is the most common form of systemic amyloidosis, affecting approximately 10 per million per year (1). AL amyloidosis is characterized by a clonal expansion of either differentiated plasma cells or, less frequently, mature B cells, leading to the production of immunoglobulin free light chains (FLCs), or a fragment, which are excessively secreted (2). Of the two classes of light chains, κ and λ, each consisting of an N-terminal variable Ig domain attached to a C-terminal constant Ig domain, λ light chains are twice as likely to cause systemic AL amyloidosis (3). However, the mechanism that leads to fibril formation in AL amyloidosis is still poorly understood as excess FLC production is also observed in monoclonal gammopathy of undetermined significance (MGUS), multiple myeloma (MM), and Waldenstrom macroglobulinemia, with only a fraction of FLCs capable of forming amyloid deposits in vivo (4). The diagnosis of AL amyloidosis involves establishing the presence of monoclonal protein, which is often a small clone (<10%) that can cause organ damage by organ deposition of light chains. MGUS is often incidentally detected on workup for other conditions and prevalence increases with age. Its incidence is reported to be 3.2% for individuals who are more than 50 years old, increasing to 5.3% in those 70 years or older (5, 6). DNA sequencing studies have shown germline gene mutations on the variable λ region that reduce the thermodynamic stability of the protein and could account for the propensity of λ light chains to form amyloid deposits. More specifically, only a small fraction of the 29–30 functional V λ segments contributed significantly to the development of amyloidosis, with just five segments (IGLV1–44, 2–14, 3–21, 3–1, and 6–57) being collectively responsible for approximately 70% of the cases in AL amyloidosis, with the expression of the IGVL1–44 gene increasing five times the odds of developing cardiac amyloidosis (7). Amyloidogenic light chains are also more likely to undergo endoproteolysis, resulting in the release of amyloidogenic light-chain fragments prone to improper aggregation (8). These light-chain fragments then aggregate and get tangled into amyloid fibrils that deposit in end organs, usually the kidney, heart, gastrointestinal tract, liver, and peripheral nervous system, leading to organ dysfunction (9). A few cases of concomitant AL and ATTR amyloidosis that arise from independent pathological mechanisms are described in the literature. However, the existence of one does not exclude the existence of the other, thus needing great expertise in the diagnostic process (10–12).
2 Diagnosis
The symptoms of amyloidosis can vary greatly, depending on the organs involved in the disease, hence, the patient can present with various unspecific symptoms that could be misinterpreted, thus delaying the diagnosis; a survey showed that 40% of patients with AL amyloidosis remain undiagnosed 1 year after the onset of symptoms (13). AL amyloidosis can clinically start with constitutional symptoms, such as fatigue or weight loss/gain. Based on the specific organ involved, clinical presentation could certainly vary. Cardiac amyloidosis can cause arrhythmias, heart failure with volume overload, dyspnea, fatigue, orthopnea, elevated troponins, and even sudden cardiac death. Peripheral edema, nephrotic syndrome, uremia-related encephalopathy, or thrombosis could be clinical manifestations of renal AL amyloidosis. Less frequent clinical manifestations could be orthostatic hypotension, lightheadedness, bladder/bowel dysfunction, gastroparesis (nerve involvement), macroglossia, hoarseness, periorbital purpura, bilateral carpal tunnel syndrome, or obstructive sleep apnea (soft tissue involvement). Gastrointestinal tract involvement could start with low appetite, bloating, malabsorption, diarrhea or constipation, dyspepsia, nausea and emesis, and bleeding. Jaundice, ascites, hepatic rupture, portal hypertension, or, rarely, Budd–Chiari syndrome could be clinical manifestations of hepatic AL amyloidosis. Pulmonary involvement could manifest with cough or dyspnea (14). By the time the patient develops these symptoms, organ damage has already occurred and is often irreversible. However, as cardiac and renal amyloidosis can be detected by performing NT-proBNP and albuminuria and a monoclonal component can be detected at least 4 years before diagnosis, it is essential for hematologists to screen for patients with amyloidosis presenting with MGUS by adding NT-proBNP and 24-h proteinuria to the routine panel (6). Recently, owing to the increase in aging of the population, ATTRwt (wild-type transthyretin) amyloidosis is becoming more prevalent and is predicted to become the most common form of amyloidosis in the following years. This form is caused by protein oxidative modifications and failures in the repair mechanisms leading to native TTR dissociation and aggregation into fibrils that deposit in end organs, especially the heart (15). It is therefore crucial to differentiate between ATTR amyloidosis (either wild type or its inherited, autosomal dominant, variant version) and AL amyloidosis with only cardiac involvement. Thus, when suspecting cardiac amyloidosis by either a suggestive echocardiogram, EKG, or cardiac magnetic resonance, it is important to evaluate the presence of monoclonal proteins (by either an abnormal serum-free light-chain ratio, positive serum or urine immunofixation, and electrophoresis). If a monoclonal protein is detected, the patient must undergo a tissue biopsy of either the heart or the fat pad, as histological diagnosis remains the gold standard for amyloidosis, with Congo red staining binding to the amyloid fibrils that appear with characteristic apple-green birefringence under polarized light microscopy. However, a positive biopsy does not differentiate between the different types of amyloidosis and subsequently subtyping of amyloid fibrils must be performed by immunohistochemistry or mass spectroscopy (16). If the monoclonal component is absent but the patient has a suggestive history and imaging, a bone scintigraphy can be carried out and, if positive [with a grade ≥2 myocardial uptake (Perugini score) of 99mTc-PYP, DPD, and HDP], the patient can be diagnosed with ATTR cardiac amyloidosis, thus avoiding endomyocardial biopsy. Patients with AL amyloidosis can also rarely have a cardiac positivity on technetium scintigraphy (17), often correlating with poor outcomes. Genetic testing should be performed to differentiate ATTRm from ATTRwt causes of ATTR-CM (9). It is really important to consider other causes of infiltrative cardiomyopathy, such as sarcoidosis, hemochromatosis, post-radiation fibrosis, Loffler syndrome, or endocardial fibroelastosis, to make an accurate differential diagnosis. In some cases, the detection of the clone could be very difficult. Studies of novel tools, such as mass spectrometry (MS) on serum and next-generation flow cytometry analysis of the bone marrow, are ongoing to validate methods to better detect plasma cell clones (18, 19).
3 Staging
Once the diagnosis of AL amyloidosis is established, the extent of the organ damage must be assessed and the physician has to stage the disease. The Mayo Clinic group first established a simple and reliable staging system in 2004 that stratifies the disease into one of three stages according to the troponin T (TnT) and NT-proBNP values, with the specified thresholds of TnT < 0.035 µg/L and NTproBNP < 332 ng/L. The disease was classified as stage I, II, or III if TnT and NT-proBNP were both low, only one was high, or both were elevated, respectively (20).
In 2015, a European group proposed a modification of the Mayo 2004 model, to better discriminate very-high-risk patients, dividing stage III into two subgroups—IIIa and IIIb—according to the NT-proBNP threshold of 8,500 pg/mL (21). The Mayo model was revised in 2012, incorporating different thresholds for TnT and NT-proBNP and including difference in FLC (dFLC) as an additional marker for disease burden. One point was attributed to TnT ≥ 0.025 ng/mL, NT-proBNP ≥ 1,800 pg/mL, and dFLC ≥ 180 mg/L, thus stratifying the disease into stages I–IV, respectively (22). Recently, the Boston University has offered a new staging system using BNP (with a threshold > 81 pg/mL) instead of NTpro BNP, allowing for centers without access to NT-proBNP or TnT to accurately stage AL amyloidosis. Three stages were thus developed based on a BNP > 81 pg/mL and troponin I (TnI) > 0.1 ng/mL, whereby the disease was classified into stages I, II, and III when both markers were lower than prespecified thresholds, only one was elevated, or both were elevated, respectively, with stage III further divided into IIIa and IIIb depending on BNP below or above 700 pg/mL (23) The staging systems are summarized in Table 1.

Table 1 Staging systems.
4 Response assessment
Attaining optimum depth and speed of disease response being the goal of treatment, the evaluation of response has a crucial role in the patient’s journey. In 2012, Palladini et al. gathered data from 816 patients in both Europe and the United States and identified and validated with criteria both hematological and cardiac responses to first-line treatment in AL amyloidosis, based on their association with survival and as a surrogate for endpoint in clinical trials. Four hematologic response categories were defined: amyloid complete response (aCR; negative serum and urine and normal FLC ratio), very good partial response [VGPR; difference between involved and uninvolved FLCs (dFLC) < 40 mg/L], partial response (PR; dFLC decrease 50%), and no response (NR) (24). However, more recently, the criteria requirement of normal FLC ratio in order to define CR was updated to include abnormal FLC ratio (FLCr) inverted in favor of the non-amyloidogenic FLC, meaning that an abnormal FLCr can still qualify as CR when the uninvolved FLC (uFLC) is greater than the involved FLC (iFLC) (25). Furthermore, for those with a low presenting FLC difference (dFLC) of 20–50 mg/L, a target response should be a dFLC. Manwani et al. introduced the “stringent dFLC response”, demonstrating as the involved–uninvolved dFLC < 10 mg/L predicted overall survival (OS) in 915 prospectively evaluated patients with low presenting baseline dFLC (20–50 mg/L) treated with bortezomib-based therapy (26). Several conditions have been identified as criteria to evaluate a therapeutic shift towards a second-line therapy: if there is no change in serum involved FLC after the first cycle, if there is a lower than PR after two to three cycles, and if treatment is not tolerated. Muchtar et al. introduced the “absolute iFLC response”, considering that, in patients who achieve normal FLC ratio, high baseline involved FLC value (iFLC) continues to predict a worse outcome compared to those with a normal iFLC. The authors demonstrated that iFLC should be reduced as low as possible, preferably to ≤2 mg/dL, rather than considering the FLC ratio. This has been associated with higher organ response rate and improved progression-free survival (PFS) and OS (27). Many efforts have been made to develop an algorithm to predict a bad probability to response to therapy in order to rapidly change treatment trying to deepen disease response. A UK consensus demonstrated that only 5% of patients with a baseline dFLC of >400 mg/L and no response at 1 month improved their response, with these two circumstances being possible predicting factors (28).
Organ response to therapy should be assessed separately for the heart, kidney, liver, and nervous system by measuring specific organ biomarkers in blood and urine (29). As for cardiac response, NTproBNP was used as a surrogate biomarker for response and progression, defined as a change of 30% and 300 ng/L, respectively (with a required baseline NTproBNP above 650 ng/L) (24). With regard to renal response, it is defined as at least 30% decrease in proteinuria or a drop below 0.5 g/24 h, in the absence of renal progression defined as a >25% decrease in eGFR (30). Hematologic and organ response are summarized in Table 2.
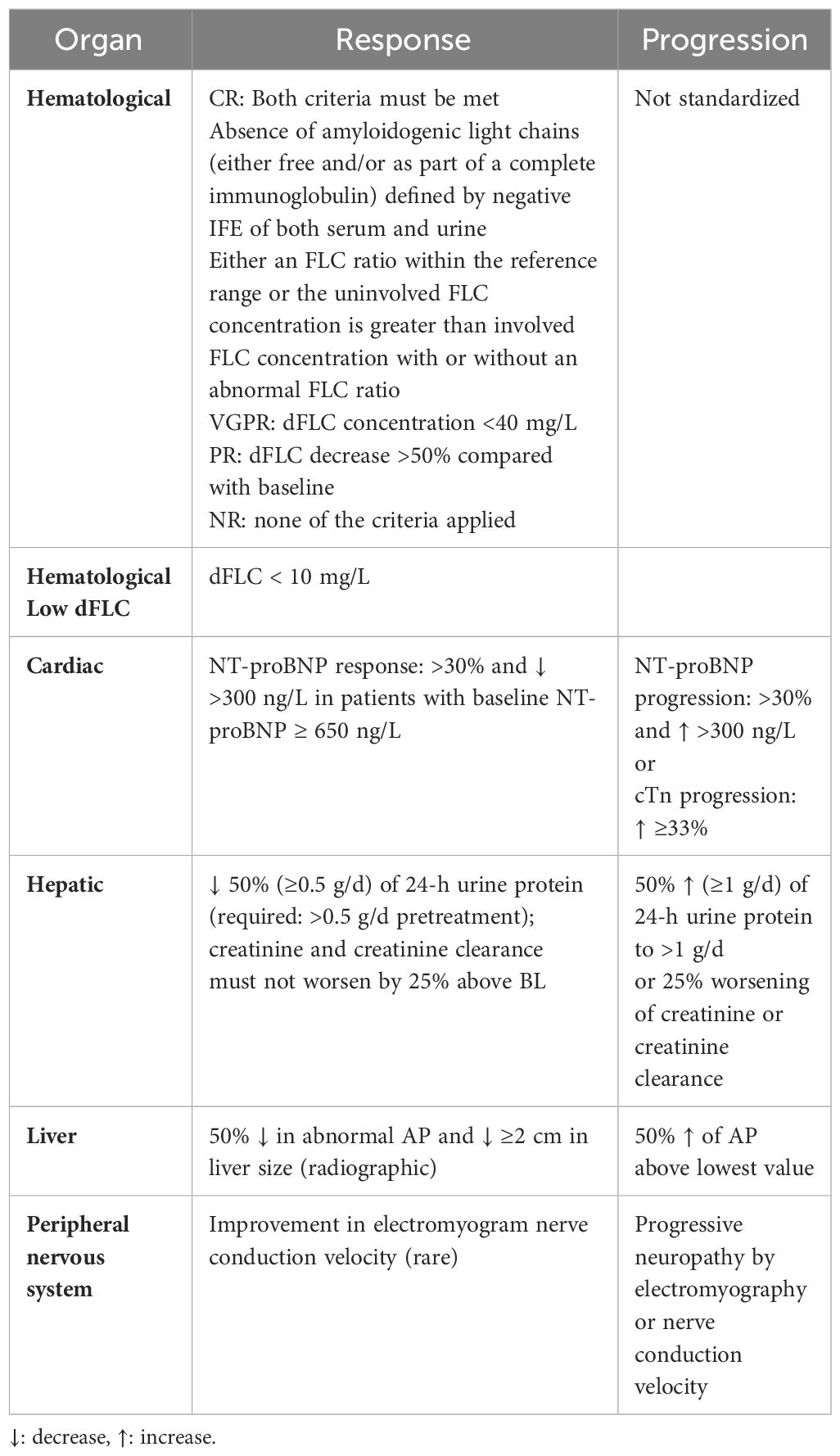
Table 2 Response evaluation.
Recently, there has been an effort to overcome the binary organ response and to introduce graded response criteria. In an abstract presented at the 2021 ASH meeting, Muchtar and colleagues offered new response criteria based on reduction in proteinuria, as greater reduction in proteinuria following successful therapy showed an improvement in both renal survival and OS. Four renal response categories were formulated based on the reduction level in pretreatment 24-h proteinuria in the absence of renal progression: renal complete response (renCR, 24-h proteinuria ≤200 mg/24 h); renal very good partial response (renVGPR, >60% reduction in 24-h proteinuria); renal partial response (renPR, 31%–60% reduction in 24-h proteinuria); and renal no response (renNR, 30% or less reduction). Renal response was assessed at landmark stages (6, 12, and 24 months from treatment initiation) and as best renal response. A renal response as early as 6 months after therapy initiation was able to predict time to renal replacement therapy (RRT) and OS based on renal response depth was observed as early as 12 months from therapy initiation and improved with time (31). Similarly, a new study validated graded cardiac response based on reduction in NT-proBNP. Four cardiac response categories were formulated based on the reduction level in pretreatment 24-h NT-proBNP: cardiac complete response (carCR, NT-proBNP ≤ 350 pg/mL or BNP ≤ 80 pg/mL); cardiac very good partial response (carVGPR, >60% reduction in NT-proBNP); cardiac partial response (carPR%, 31%–60% reduction in NT-proBNP); and cardiac no response (carNR, 30% or less reduction). The study showed a correlation between the depth of hematological response and cumulative cardiac progression and between a deep cardiac response and OS, thus emphasizing the role of graded cardiac response criteria in better assessing cardiac improvement compared with the traditional binary response system (32). Because the natriuretic peptides are dependent on renal function, other methods for cardiac response assessment have been explored, such as echocardiographic strain measurement (33–35), extracellular volume measurement using cardiac magnetic resonance (36), and functional assessment such as the 6-min walk test (37, 38), but none of them demonstrated superiority over natriuretic peptides. However, a complete HR does not necessarily immediately translate into organ response. Recently, the role of minimal residual disease (MRD) in predicting patients’ response to treatment has become more important. In the study conducted by Sidana et al., patients with MRD negativity (sensitivity of 10-5) were more likely to have achieved cardiac response (67% vs. 22%, p = 0.04) and obtained a better 1-year PFS at a median follow-up of 14 months (100% vs. 64%, p = 0.006) (39). Kastritis and colleagues also showed better organ response in MRD-negative patients compared to MRD-positive ones (86% vs. 77%), both renal (88% vs. 87.5%) and cardiac (100% vs. 73%). MRD negativity also led to lower rates of hematologic relapse (0% vs. 21%, p = 0.029) (40). Similarly, Palladini et al. demonstrated that undetectable MRD (measured by NGF, sensitivity of 10–5) was associated with higher rates of renal and cardiac responses (90% vs. 62%, p = 0.006 and 95% vs. 75%, p = 0.023, respectively). Furthermore, hematological progression was more frequent in the setting of persistently positive MRD (0% vs. 25% at 1 year, p = 0.001), indicating a subset of patients who would benefit from further/more intensive treatment (41). Bomsztyk et al. recently prospectively demonstrated that the measurement of clonal FLC by mass spectrometry (MS) can identify an FLC clone in patients presenting with a normal FLC ratio and in the majority of patients in conventional CR post-treatment. After treatment, significantly better OS has been demonstrated in patients with detectable residual monoclonal FLC by FLC-MS than patients with a conventional CR/VGPR but FLC-MS positive, sustained at both 6 and 12 months (42). MRD detection could be assessed by NGF, which can detect MRD in patients with AL amyloidosis; otherwise, CR and persistent MRD could explain persistent organ dysfunction and predict hematologic progression (39–41, 43–45). However, technique standardization, time-point evaluation, and the clinical applicability of these methods are lacking and limit their use. NGS has been demonstrated in small recent studies to be feasible in patients with AL amyloidosis, representing the track of clonotypic light-chain sequences as a promising approach for detecting and maintaining profound therapeutic responses (46). NGS has shown to be a sensitive method for detecting MRD in AL amyloidosis and could be utilized in future studies (47).
There is uncertainty about the timing and depth of free light-chain reduction that could define suboptimal response and the need to switch to second-line therapy. Thus, identifying early suboptimal response in patients during induction regimens could be crucial to optimize outcomes. A recent Australian retrospective study evaluated the impact of disease response after two cycles of induction therapy on subsequent hematological and organ response. The failure to achieve hematological PR after 2 months of bortezomib-based therapy defined a high probability to therapeutic failure, suggesting a chance to alternatively use salvage therapy after the 2-month response assessment. For patients with advanced stage IIIa/b cardiac disease, the failure of early response has been shown to be a factor that may lead to worse OS (48).
5 Therapeutic approach
5.1 Newly diagnosed AL amyloidosis
The goal for therapy in patients with AL amyloidosis is to obtain a deep and durable response in the shortest time possible. Systemic therapy should normalize or nearly normalize serum FLCs’ circulating amount and their consecutive deposition in involved organs by directly targeting the clone that produces them. The more rapid the therapeutic action is, the more efficiently progressive organ failure should be stopped; organ recovery has been associated with improved survival outcomes in patients with AL amyloidosis (33, 49). Recently, novel therapeutic approaches are under study to remove amyloid fibril deposition in involved organs, trying to revert organ damage (50). Clinicians should also minimize toxicity in therapy by choosing a risk-adapted therapeutic approach that does not lead to mortality or decompensate patients, combining it with the best personalized supportive care.
Guidelines suggest patient optimization before starting treatment as the key to AL amyloidosis management, firstly by monitoring daily blood pressure, pulse rate, and weight to evaluate the clinical status of patients with renal or cardiac involvement. An inpatient start of therapy should be preferable for patients who are unstable, which should be treated with supportive treatments such as diuretics with the aim of 1–2 kg weight loss/week unless hypoxic, preferring bumetanide plus spironolactone combination and avoiding ACE inhibitors and beta blockers. Fluid overload should be carefully evaluated; Holter electrocardiogram in case of baseline arrhythmias, pacing in case of bradycardia, or implantable cardioverter defibrillator for refractory persistent or serious ventricular arrhythmias, should be considered.
The major changes in the patient’s journey for treating AL amyloidosis started in the 1990s with high-dose melphalan and autologous stem cell transplantation (HDM/SCT), which became the standard of care in selected patients with early disease and brought a prolonged duration of hematologic complete response, event-free survival (EFS), and OS in patients achieving a complete remission (51). In the 2000s, the first immunomodulators (IMIDs) were used, reducing the overall 2- to 3-year mortality from 80% to 70%, until 50% 10 years later by the introduction of bortezomib in upfront treatment strategy. Hematological response rate increased in parallel from 30% to 65% to 80% during this journey of continuous development of novel drugs (52). In the 2020s, the use of daratumumab, first in a relapsed/refractory (R/R) setting and then in a frontline setting, changed the history of R/R AL amyloidosis, leading from 48% to 86% VGPR with a median time to response of 1 to 4 weeks and deep and fast renal and cardiac responses in more than one-half of all the patients treated (53, 54). Daratumumab, a monoclonal antibody targeting CD38 on the plasma cells surface, has already demonstrated a high efficacy in patients with MM with a good safety profile and without renal and cardiac toxicity. Andromeda was the pivotal phase 3 trial comparing dara-VCd (up to 24 cycles) with VCd alone (6 cycles) in 388 patients with newly diagnosed systemic AL amyloidosis (excluding stage IIIb disease), with a primary endpoint of hematologic CR and secondary endpoints of organ response and MOD-PFS (55). The Dara-VCd arm compared with VCd alone demonstrated to make CR jump high from 19.2% to 59.5% in a median of 16 days vs. 24 days of the competitor, with significantly better MOD-PFS in the daratumumab group. Eighteen-month organ responses also improved, with cardiac and renal responses in the dara-VCd arm vs. VCd alone being 53% vs. 24% and 58% vs. 26%, respectively (56).
Treatment with daratumumab-based regimens clearly reaches deep responses in 70% to 80% of patients with less-advanced disease, translating into organ responses, improving quality of life and finally better OS. More advanced diseases remain an unmet medical need. Refinement of therapies based on biomarkers and clonal and genetic characteristics of disease; addressing the question of organ improvement; and solving the issue of ongoing maintenance should be the goals of future improvements in therapy.
5.2 Transplant-eligible AL amyloidosis
Autologous stem cell transplant (ASCT)-treated patients had particularly large survival improvement during the last few years, improving the 5-year OS rate from 54% to 76% from 1980 to 2019, with the best improvement described for patients without cardiac involvement (median OS increasing >6 years over this time). Early mortality in ASCT-treated patients decreased from 10% to 2%, with organ failure, particularly the heart, being the most important cause of death followed by sudden heart attack (57).
The first step after having decided to start a systemic therapy is to evaluate the ASCT eligibility of patients with newly diagnosed AL amyloidosis. Sanchorawala et al. recently pointed out ASCT eligibility criteria from the EHA-ISA guidelines: age between 18 and 70 years, at least one vital organ involvement, left ventricular ejection fraction ≥40% and NYHA class <III, oxygen saturation ≥95% in room air and DLCO >50%, supine systolic blood pressure ≥90 mmHg, ECOG performance status ≤2, direct bilirubin <2 mg/dL, NTproBNP <5,000 pg/mL, and troponin I <0.1 ng/mL (or troponin T <0.06 ng/mL or hsTroponin T <75 ng/mL). The presence of medically refractory pleural effusions, cardiac stage IIIb disease, orthostatic hypotension refractory to medical therapy, acquired factor X deficiency with active bleeding, and gastrointestinal active bleeding due to local disease involvement remained as contraindications for ASCT. Stem cell mobilization should be made by G-CSF at 10–16 μg/kg/day in single or split dose, using Plerixafor on demand or planned and preferably avoiding cyclophosphamide. Melphalan conditioning regimen’s dose should be ruled out based on age and cardiac/renal disease risk, which is 200 mg/m2 in the low-risk group (age ≤65 years, cardiac stage I, and eGFR >50 mL/min) and 140 mg/m2 in patients with renal failure (eGFR ≤30 mL/min) considering a chronic dialysis schedule if necessary. A multidisciplinary evaluation should be necessary for patients aged 66–77 years, with cardiac stage II and borderline renal function (eGFR 30–50 mL/min) (9). A single day of melphalan conditioning was associated with a higher probability of post-HSCT CR compared to 2 days of conditioning in a retrospective Mayo Clinic study, which compared the effect of single-day melphalan versus 2 days of melphalan, omitting a day of rest between conditioning and stem cell infusion. Furthermore, post-HSCT CR was higher in patients receiving a full dose of melphalan than in those receiving a reduced dose, supporting a careful patient selection for HSCT, preferring the ASCT strategy in case of eligibility for full-dose melphalan conditioning (58).
Bortezomib-based induction regimens have been the gold standard in systemic AL amyloidosis for years, until the approval of daratumumab-VCd, based on results of the phase 3 Andromeda trial (55), which introduced two to four induction cycles of this drug combination before ASCT in the clinical practice preferably for patients having more than 10% of bone marrow plasmacytosis, a non-stage IIIb cardiac disease, and without a sensory or peripheral neuropathy. The addition of daratumumab to VCd has also recently demonstrated higher rates of deep hematologic response in stage IIIb cardiac AL amyloidosis in real-life retrospective studies. An Italian matched case–control study of 62 newly diagnosed patients showed a 25% cardiac response rate, which is significantly more frequent in the daratumumab cohort than in the standard group (3%) with significantly prolonged PFS (5 vs. 2.4 months in the two groups) and OS (6.7 vs. 4 months in the two groups) (59). Dara-VCd was also described in a Columbia retrospective cohort of patients among the 28% who had stage IIIb cardiac involvement, obtaining an estimated 6-month early mortality that is lower by approximately threefold than historical data (<30% in low- to intermediate-risk patients and 30%–60% in high-risk patients), with a dramatic reduction in Mayo 2004 stage I–IIIa but still substantial in stage IIIb disease (60). Similarly, Oubari et al. published a retrospective multicenter study on 98 patients with stage IIIb AL amyloidosis treated with daratumumab-based or non-daratumumab-based regimens. The authors demonstrated significantly better hematological response at 2 and 6 months, cardiac response, and OS in patients having received daratumumab, showing a clear correlation between depth of response and outcomes (61).
ASCT should be deferred to relapse in case of CR after induction, but prospective studies evaluating differences in terms of outcomes between early ASCT and deferred ASCT are lacking. Data from retrospective studies described similar OS in the two groups of patients, highlighting organ involvement, patient preference, and good treatment response as the first three causes of deferred ASCT (62, 63). Consolidation and continuous lenalidomide maintenance approaches do not have a definitive role, suggesting for a dynamic and personalized approach. Al Saleh et al. firstly demonstrated a significant improvement in PFS with a trend towards improvement in OS in patients receiving post-ASCT consolidation therapy compared to those that do not, primarily in patients failing to achieve ≥VGPR. However, this study has the limitation of being retrospective in nature, lacking the uniformity in selecting patient characteristics and the variability of consolidation regimens before the era of daratumumab (64). For patients having an only organ involvement and failure, the organ transplant could be considered and accurately discussed with the organ transplant multidisciplinary team, evaluating ASCT after solid organ transplant if available. Rajasekariah et al. have retrospectively demonstrated that sequential heart transplant plus ASCT following induction therapy was safe in nine patients with stage IIIa/b cardiac AL amyloidosis without an increase in transplant-related mortality (TRM). Their 3-year OS was 83% and PFS was 56%, which could be considered excellent if compared with the historical cohort, considering that they have received non-daratumumab-based induction regimens (65).
A 25-year longitudinal study on 648 patients treated with VCd and ASCT demonstrated that nearly half of patients obtaining a hematologic CR had no evidence of a recurrent plasma cell dyscrasia at 15 years following ASCT. The authors proposed a risk stratification model based on baseline bone marrow plasma cells’ percentage >10% and difference between involved and uninvolved FLC >180 mg/L as adverse risk factors, which have been identified as factors that are significantly associated with better EFS but not OS in case of ASCT (66). Prospective data lack the role of ASCT versus induction alone, particularly based on a risk stratification of AL amyloidosis, in the era of daratumumab-based therapy. Retrospective data showed no significant difference between ASCT and induction alone in patients with and without t(11;14) (67), with the presence of t(11;14) being a prognostic and predictive biomarker in AL amyloidosis, associated with the higher likelihood of cardiac involvement (68).
The decision to utilize maintenance therapy and its duration is often driven by physician discretion and center policy. Landau et al. showed that each treatment phase deepened the response, potentially suggesting that maintenance following consolidative bortezomib-dexamethasone would do the same or at least maintain remission (69). Ozga et al. retrospectively demonstrated that there was also no survival benefit with the addition of maintenance therapy. It is interesting to note that the median PFS was 4 years longer in the maintenance subgroup, while median OS was 4 years longer in the non-maintenance subgroup, suggesting that maintenance may delay disease recurrence at the expense of cardiotoxicity or myelosuppression related to the long-term use of lenalidomide (70).
5.3 Non-transplant-eligible AL amyloidosis
Patients not eligible for ASCT are treated with chemotherapy-based approaches. Guidelines also recommended a risk-adapted strategy for this group of people. For grade I–IIIa cardiac involvement, Dara-VCd should be considered the first therapeutic option, based on Andromeda results (55). If daratumumab is not available, a bortezomib-based triplet should be started. Bortezomib–melphalan–dexamethasone (BMDex) and VCd have not been compared prospectively, but experts indicate VCd as the preferred regimen in most patients, because of the outpatient management and renal safety. There is no precise consensus on the optimal duration of therapy, but two cycles beyond the best response seem to be the best duration until six to eight cycles as the depth of response can improve. For stage IIIb cardiac involvement, a modified Dara-VCd regimen or the single-agent daratumumab should be recommended.
For these patients being not eligible for ASCT, there are no data about a consolidation strategy. Consolidation therapy is not routinely recommended, but it could be considered to deepen induction response in case of MRD persistence and lack of organ response. Routine maintenance therapy is not actually recommended, even if patients in the Andromeda study received monthly daratumumab maintenance for up to 24 cycles. However, this trial did not randomize patients to receive maintenance therapy or not, and data from this part of the study are not available yet.
As for special populations, the same treatment strategies could be adopted and customized. In case of mild amyloidosis-related neuropathy, bortezomib should be reduced or the single-agent daratumumab should be preferred (52). Regimens without bortezomib should be started in case of severe neuropathy, such as daratumumab monotherapy, lenalidomide–dexamethasone (71–73), melphalan–dexamethasone (74), carfilzomib–dexamethasone (75), or venetoclax if available (NCT05996406 ongoing trial). Giving the hematological toxicity as thrombocytopenia and the increased bleeding/clotting risk of patients with AL amyloidosis, IMIDs use needs a careful assessment in patients at high bleeding risk due to complexity of balancing bleeding vs. clotting risk. Patients with advanced liver dysfunction should avoid hepatotoxic drugs: cases of bortezomib-, lenalidomide-, or cyclophosphamide-induced liver toxicity have been reported, even if they are not considered as hepatotoxic drugs. Daratumumab could be considered as a safe option in terms of liver function; thus, daratumumab–dexamethasone should be the preferred option in this case. In case of renal AL amyloidosis requiring dialysis, lenalidomide should be avoided because it is the only IMID that requires a dose adjustment for renal failure. High-cutoff hemodialysis (HCO-HD) therapy should be associated with chemo-immunotherapy to remove free light chain from the renal filter, based on data on cast nephropathy. HCO-HD and chemotherapy have demonstrated a tendency to improve renal outcomes in MM, but trials on AL amyloidosis are necessary to establish a real recommendation (76).
5.4 Response evaluation after upfront therapy
Achieving optimum depth and speed of disease response being the goal of treatment, the evaluation of response has a crucial role in the patient’s journey. It has to be evaluated in terms of hematological and organ response, every one or two cycles while the patient is on active treatment, 3 months post-ASCT, and every 3 months during the follow-up (14). Organ response has doubled up with daratumumab-based therapies, reaching 53%, 83%, and 50% for cardiac, renal, and hepatic response, respectively (77). Palladini et al. recently updated the hematological response criteria, defining CR as both the absence of serum/urine immunofixation and the normalization of the FLC ratio (25), better defining deep hematologic response that has been shown to lead to deep organ response and subsequently improved OS (24). Organ response to therapy should be assessed separately for the heart, kidney, liver, and nervous system by measuring specific organ biomarkers in blood and urine (29). Muchtar et al. recently confirmed the value of graded cardiac response criteria over the standard binary response system, based on the NT-proBNP changes during therapy. Depth of cardiac response at 6, 12, and 24 months has been demonstrated to significantly correlate with OS both in stage II–IIIa and in stage IIIb, even if stage IIIb was confirmed to have a negative prognostic role on outcomes (78). Graded four-level renal response criteria have been validated based on the reduction of 24-h proteinuria in 737 patients with renal AL amyloidosis involvement, demonstrating the importance of achieving a deep and early renal response to improve renal survival and OS. A deep renal response at 6, 12, and 24 months has been significantly associated with a lower probability of undergoing RRT (31). A similar graduation system has been applied to assess hepatic response to therapy, based on the reduction of alkaline phosphatase, demonstrating its significant correlation with OS (33).
5.5 Novel upfront therapeutic strategies
Novel strategies in treating AL amyloidosis include drugs binding directly to a conserved epitope in misfolded kappa and lambda light chains, to neutralize toxic soluble light chain aggregates and deplete amyloid deposited via macrophage-induced phagocytosis (79, 80). Birtamimab (formerly NEOD001) is an investigational humanized IgG1 monoclonal antibody that was granted orphan drug status by the US FDA and the European Medicines Agency and received FDA Fast Track Designation, based on results of a phase 1/2 clinical trial in patients with AL amyloidosis with persistent organ dysfunction, demonstrating that it is generally safe and well tolerated (81). The phase 3 randomized, double-blind, placebo-controlled VITAL clinical trial studied birtamimab plus standard of care in 260 newly diagnosed and treatment-naive patients with AL amyloidosis. This trial terminated early for futility, failing to reach the first endpoint, which was the time to all-cause mortality or centrally adjudicated cardiac hospitalization ≥91 days after the first study drug infusion. Having a post-hoc analysis of patients with Mayo stage IV AL amyloidosis showed significant improvement in the primary endpoint with birtamimab at month 9 (OS 74% vs. 49% in birtamimab vs. SOC groups, respectively), and the confirmatory AFFIRM-AL phase 3 trial is ongoing. Table 3 shows a summary of novel therapies of ongoing trials.
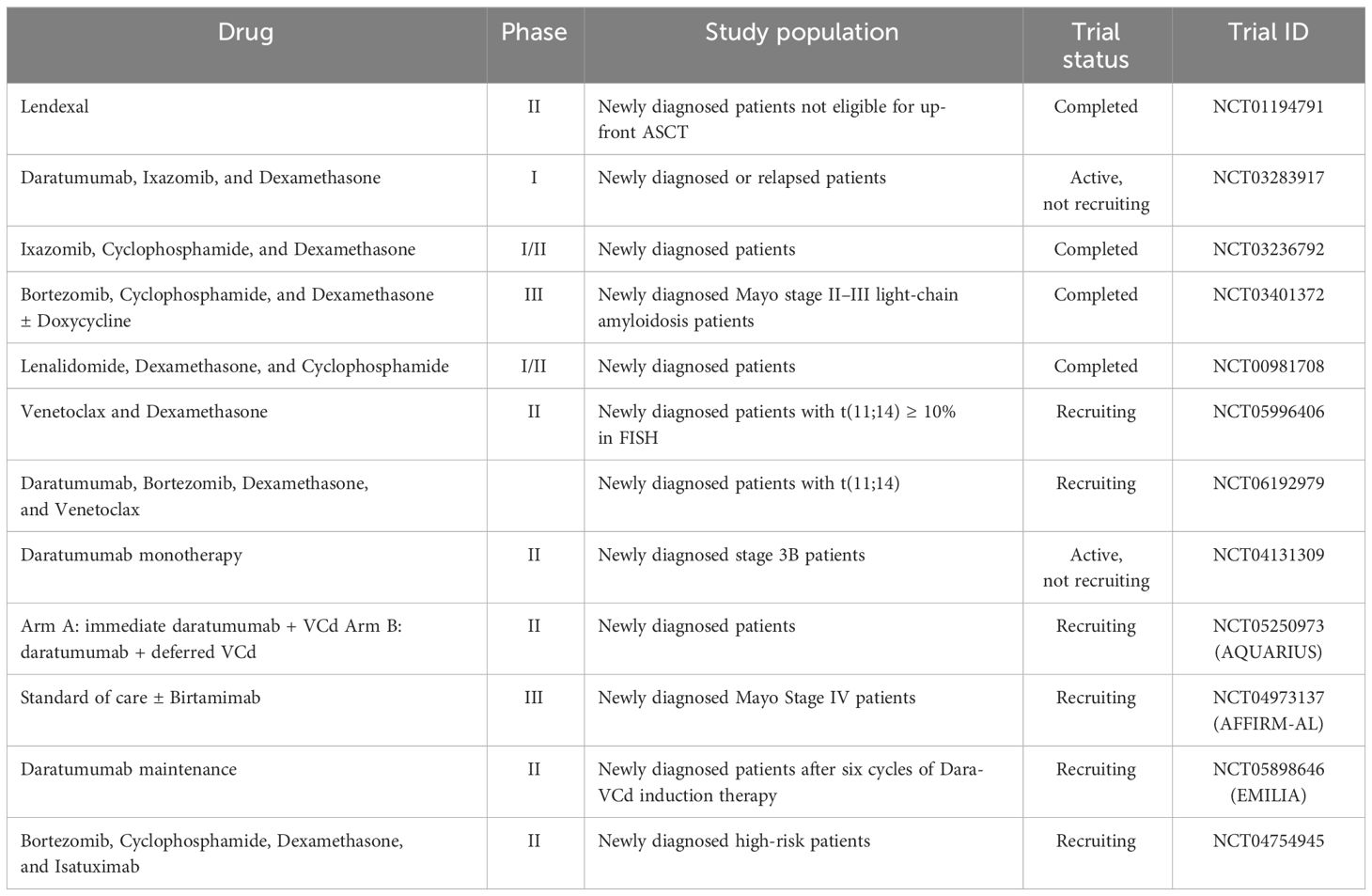
Table 3 Ongoing trials in newly diagnosed AL amyloidosis.
5.6 Relapsed and refractory AL amyloidosis
The management of relapsed/refractory AL amyloidosis (RRAL) represents a challenge since, firstly, it is not well known when to start a second-line therapy although progression criteria have been established in 2005 (82) and, secondly, phase 3 randomized trials in this setting are very few so there is no consensus regarding the best treatment in RRAL. Moreover, if a few patients can undergo organ progression without dFLC increase, hematologic relapse translates into organ progression in most of them after a time that cannot be predicted (83). Achievement of a complete hematologic response (aCR), defined as negative serum and urine immunofixation and normal FLC ratio, is associated with a significantly longer OS if compared with obtaining VGPR (dFLC < 40 mg/L; HR = 2.67), PR (dFLC decrease > 50%; HR = 6.24), or not responding (HR = 12.34) (24). Recently, free light-chain mass spectrometry (FLC-MS) has been demonstrated to detect persistent monoclonal light chains in a significant proportion of patients with CR as per ISA criteria in a study including 487 patients with newly diagnosed AL amyloidosis receiving bortezomib-based regimens upfront (84). Among those who achieved a conventional CR at 6 and 12 months, 26.4% and 39% were FLC-MS negative, respectively. Median OS of patients in CR and FLC-MS negative at 12 months was not reached vs. 108 months in those in CR but FLC-MS positive (p = 0.024). Moreover, 70% of patients who were FLC-MS negative at 12 months achieved a cardiac response vs. 50% of those who were FLC-MS positive (p = 0.015). Multivariate analysis selected FLC-MS negativity at 12 months as an independent factor affecting better outcome, suggesting that FLC-MS assessment could become a new parameter for defining response in amyloidosis AL. The response should be quick as well as deep. In 525 patients who obtained less than VGPR after frontline bortezomib, dFLC at diagnosis > 400 mg/L and no response (<PR) after 1 month were factors significantly associated with no improvement in response. Remarkably, 66% of patients with no risk factors improved their response by 6 months vs. 44% and 5% of those with one and two risk factors, respectively, suggesting that patients with both risk factors should change therapy at 1 month since the likelihood of improved response is low (28). These findings have been confirmed by a recent Australian study (48) in which failure to achieve PR after 2 months of bortezomib-based therapy was associated with a low chance of achieving deep response (only 16% of patients obtained VGPR or better) and a low probability of improving cardiac (6%) and renal (30%) responses. The deeper the organ response, the longer the survival (30, 85), but organ response is a time-dependent variable and time from treatment initiation to maximal response can be 24 months for cardiac response and 29 months for renal response (33). Notably, responses for all organs involved in AL amyloidosis are not always concordant and most cases of heart or kidney non-responses have no clear explanation, suggesting that the biology of the disease could be different in different sites (86). Pavia experience (87) showed that the outcome of patients who required second-line therapy, after a median of 2.7 years, was good with a median OS of 59 months. High-risk dFLC progression, requiring a dFLC > 20 mg/L, a dFLC > 20% of baseline value, and a >50% increase from the value reached at best response, was characterized by a significantly shorter OS (median 46 months vs. not reached in other patients, p = 0.004) and preceded cardiac progression by a median of 6 months in 85% of cases. However, in multivariate analysis, only NT-proBNP progression remained an independent predictor of survival in patients who underwent second-line treatment. In patients with an NT-proBNP progression, median OS was 17 months vs. 62 months in those without NT-proBNP progression, suggesting that patients with high-risk dFLC progression should be considered for rescue therapy before cardiac progression. In the choice of treatment for relapsed AL amyloidosis, several factors mainly related to patients should be considered since previous therapy may have caused severe toxicities and underlying disease may have compromised organ functions. Therefore, the most widely used approach, in terms of further therapy, is to select second-line therapy taking into account patient frailty status, previous regimens to which the patient has been exposed, and duration of response. Retreatment with the same class of drug used in upfront therapy represents an appropriate approach when response is long-lasting (88).
5.7 Immunomodulatory agent-based regimens
The efficacy of lenalidomide plus dexamethasone (Rd) has been evaluated in some small prospective phase 2 studies showing that, in this setting of patients, lenalidomide is much less tolerated than in patients with MM. In the Dispenzieri et al. study, hematologic and organ responses were 75% and 42%, respectively, in patients with symptomatic AL amyloidosis who received at least three Rd cycles, with a starting lenalidomide dose of 25 mg for 21 days. Hematologic toxicity was significant with grade 3–4 thrombocytopenia occurring in 27% of patients and grade 3–4 neutropenia in 45% of them (89). In a similar US study, a lower incidence of hematologic toxicity was documented after lenalidomide dose was reduced to 15 mg/day, which has been considered the maximum tolerated dose in patients with AL amyloidosis (90). A retrospective analysis including 260 patients treated in Germany between 2006 and 2020 evaluated the efficacy and toxicity of the Rd regimen in RRAL amyloidosis (91). Median age was 60 years (34–79) and patients had received a median of two prior lines of therapy including bortezomib (68%) and ASCT (33%). After a median follow-up of 56.5 months, median hematological EFS and OS were 9 and 32 months, respectively. Overall, a worsening in aGFR was documented in 69% of patients, regardless of renal involvement at diagnosis, and 12% of patients required dialysis. Any grade hematologic toxicity developed in 39% of patients, but the most frequent adverse event was infections that occurred in 30% of cases. Patients with gain1q21 and high dFLC (>180 mg/L) had shorter hematological EFS and OS if compared with those of patients without these findings. Several phase 1/2 studies explored pomalidomide plus dexamethasone (Pd) in previously treated patients with AL amyloidosis. In the Mayo Clinic experience (92), 33 patients with a median of two prior regimens and 82% with heart involvement received pomalidomide (2 mg daily for 28 days) plus dexamethasone. Hematologic response was documented in 48% of patients whereas, among those with cardiac and renal involvement, 15% and 8% obtained organ response, respectively. With a median follow-up of 28.1 months, median PFS and OS were 14.1 and 27.9 months, respectively. The most frequent grade 3–4 adverse events were neutropenia (30%) and infections (27%). In another phase 1/2 study (93), the maximum tolerated dose of pomalidomide was established to be 4 mg daily and 27 patients with a median of two prior regimens including bortezomib (77%), ASCT (59%), and lenalidomide (48%) received a median of six cycles with Pd. Hematologic response was documented in 50% of patients and median OS was not reached after a median follow-up of 17 months. Higher hematological response rate (68%) was reported by Palladini et al. (94) in 28 patients already exposed to bortezomib (96%), melphalan (75%), cyclophosphamide (68%), and lenalidomide (25%), probably due to a greater proportion of patients receiving full doses of pomalidomide and dexamethasone. In a large European cohort of 164 patients with a median of three prior lines of therapy (65% refractory to the last line of therapy and 35% relapsed), hematological response was 44% at 6 months. Achieving at least PR at 6 months was associated with a significantly longer OS (median 50 months vs. 27 months, p = 0.033) and PFS (median 37 months vs. 18 months, p < 0.001). Cardiac response was observed in 11% of patients and renal response in 20%. The most frequent severe toxicities were infections (9%), cardiac failure (7%), and creatinine increase (6%) (95).
5.8 Proteasome inhibitor-based regimens
Among mechanisms of action of proteasome inhibitors, making these agents essential in the treatment of MM, inducing endoplasmic reticulum (ER) stress by inhibition of a protein clearance pathway with subsequent activation of unfolded protein response (UPR), resulting in apoptosis, seems to be the most important for AL amyloidosis (96). CAN2007 (97) has been the first prospective phase 1/2 study to evaluate bortezomib monotherapy in relapsed systemic amyloidosis. Among 70 patients enrolled in the study, 18 received bortezomib 1.6 mg/m2 QW, 34 received bortezomib 1.3 mg/m2 BIW, and 18 underwent bortezomib at a lower dose QW or BIW. Overall, 73% and 56% of patients had renal and cardiac involvements, respectively. Hematological response (at least PR) was documented in 68.8% of the 1.6 mg/m2 QW group and in 66.7% of the 1.3 mg/m2 BIW group with 28% and 38% of patients, respectively, discontinuing therapy because of adverse events despite the fact that no grade >3 peripheral neuropathy was reported. After a median follow-up of 51.8 months for all patients, median OS was 62.7 months with a 4-year OS of 67.3%. Remarkably, four patients received long-term bortezomib for between 3.5 and 5.6 years, and at the final analysis, all patients remained progression-free (98).
In the UK phase 1b CATALYST study (99), 10 patients with RRAL amyloidosis and at least one prior line of treatment received carfilzomib at a dose of 20/36 mg/m2 or 20/45 mg/m2 on days 1, 8, and 15 in combination with thalidomide 50 mg daily and dexamethasone. Overall, 80% had previously received cyclophosphamide, bortezomib, and dexamethasone (Cy-Bor-D); 40% cyclophosphamide, thalidomide, and dexamethasone; and 30% ASCT. Hematological response was 60% within and at the end of three cycles of therapy, but no organ response was documented during treatment. Moreover, grade 3 acute kidney injury and creatinine increase were reported in 10% and 10% of patients, respectively, and grade 3 hypertension was reported in 10% of patients. In a previous phase 1/2 study (100) using carfilzomib monotherapy, maximum tolerated dose was established to be 20/36 mg/m2, but considering the whole study population of 28 enrolled patients, 50% of patients developed grade 3–4 cardiac or pulmonary side effects including symptomatic ventricular tachycardia and cardiac arrest in two patients.
TOURMALINE-AL1 (101) represents the first phase 3 trial in relapsed/refratory AL amyloidosis, conducted in 68 sites around the world. Primary endpoints of the trial were hematologic response rate and 2-year vital organ deterioration or mortality rate. Overall, 168 patients were randomized to receive ixazomib (4 mg on days 1, 8, and 15) plus dexamethasone (Ixad) or the physician’s choice consisting of Rd, melphalan–dexamethasone, cyclophosphamide–dexamethasone, and thalidomide–dexamethasone. In the Ixad arm, 41% of patients had received ≥2 prior lines of therapy vs. 40% in the physician’s choice arm and 47% vs. 37% had undergone ASCT, respectively. The best hematological response was obtained by 53% of the Ixad group vs. 51% of the physician’s choice group (p = 0.76); therefore, the primary endpoint was not met. As regards 2-year vital organ deterioration, it was longer with Ixad than with other therapies (median 34.8 months vs. 26.1 months, p = 0.01), and among patients with 2 years of follow-up, 40% and 45% of physician’s choice patients had a vital organ deterioration or mortality event, respectively. Median time to subsequent therapy was 26.5 vs. 12.5 months (p = 0.03), median overall PFS was 11.2 vs. 7.4 months (p = 0.04), and median OS was not reached (with a median follow-up of 45.3 months vs. 40.8 months in the Ixad arm and physician’s choice arm, respectively). The most frequent adverse events of any grade in the Ixad group were diarrhea (34%), rash (33%), cardiac arrythmias (33%), nausea (24%), pneumonia (21%), and peripheral neuropathy (19%).
5.9 Daratumumab-based regimens
The anti-CD38 monoclonal antibody daratumumab, included in most regimens used upfront in newly diagnosed MM, has shown to induce high hematologic response as second-line therapy in patients with AL amyloidosis and, unlike in MM, it was found to be equally effective both as a monotherapy and in combination (102). In a retrospective study from Stanford University (103) including 25 heavily pretreated patients with AL amyloidosis (median prior lines of therapy = 3), hematologic response was 76%, with 36% of patients achieving a CR. Daratumumab was also well tolerated in patients with advanced cardiac involvement. In a phase 2 study conducted in France and Italy (54), 40 pretreated AL patients (65% with renal involvement, 60% with cardiac involvement, and 52% with > 3 prior lines of therapy) received six cycles of IV daratumumab, with the latter administered weekly for the first two cycles and every other week for cycles 3–6. At least VGPR after six cycles, the primary endpoint of trial, was achieved by 47.5% with a median time to hematological response of 1 week. Cardiac and renal response were documented in 17.5% and 20% of patients, respectively. With a median follow-up of 26.4 months, median OS was not reached and median PFS was 24.8 months (2 years; 74.2% and 51.2%, respectively). In a similar US prospective phase 2 study (53), 22 patients with a median of two prior lines of therapy, 68% with renal involvement and 64% with cardiac involvement, received IV daratumumab for 24 months or unacceptable toxicity. Using this long-term administration, at least 86% of patients achieved hematologic VGPR and 41% achieved at least a CR. Moreover, renal response was 67% and cardiac response was 50%; 50% of patients obtaining hematologic CR was MRD negative by multiparametric flow cytometry and median PFS was 28 months. As regards retrospective analyses, 44 patients with RRAL amyloidosis and a median of three prior lines of therapy received daratumumab-based regimens at the Mayo Clinic between 2015 and 2018 (104). The most common regimens administered were daratumumab, pomalidomide, and dexamethasone (DPd) in 36% of patients; daratumumab, lenalidomide, and dexamethasone (DRd) in 32% of patients; and daratumumab, bortezomib, and dexamethasone (DVd) in 18% of patients. Hematologic ORR was 83%, and 80% of patients achieved at least VGPR with an OS at 10 months of 94%. In the larger Italian cohort (105), 72 consecutive pretreated AL patients (median prior lines of therapy = 2) received daratumumab monotherapy (65%), DVd (20%), and DRd (15%). After 16 daratumumab infusions, hematologic response was 83%, renal response was 60%, and cardiac response was 29%. Notably, no significant difference in terms of hematological response rates was observed between patients receiving daratumumab monotherapy and those treated with daratumumab-based regimens. At the last ASH meeting, preliminary data from a multicenter phase 2 study with DPd with daratumumab SQ in patients with RRAL have been presented (106). Nine patients were enrolled (16 planned) and received DPd for 12 cycles with optional continuation of daratumumab and/or pomalidomide if they achieved ≥VGPR after 12 cycles. All evaluable patients for response (five patients) obtained CR and negative serum MS, with 83% obtaining renal response documented after a median of three cycles. There were no grade 3–4 side effects requiring treatment discontinuation. Retreatment with daratumumab in patients with daratumumab failure is feasible, but response rate is low, with only 22% of patients achieving VGPR (107).
6 Venetoclax-based regimens
Among cytogenetic abnormalities, translocation t(11;14) is the most frequently documented in patients with AL amyloidosis (108, 109). This translocation is associated with overexpression of BCL-2 and inhibition of apoptosis (110); patients harboring this abnormality were found to less likely achieve a high rate of hematologic response, particularly if treated with bortezomib-based regimens (111), and were found to have a poorer outcome (112). Venetoclax, the first-in-class oral BCL-2 inhibitor approved for some hematologic malignancies such as acute myeloid leukemia, non-Hodgkin lymphoma, and chronic lymphatic leukemia, has shown to exert a significant activity also in patients with AL amyloidosis and t(11;14). In a retrospective US study (113) including 43 patients with AL who progressed on at least one prior line of therapy, 72% (31 patients) harbored t(11;14). Overall, they have received a median of three prior lines of therapy including bortezomib (98%), cyclophosphamide (79%), daratumumab (58%), lenalidomide (56%), and pomalidomide (28%). Most patients were treated with venetoclax ± steroids (81%) or venetoclax plus bortezomib (23%), with venetoclax dosing being from 100 mg to 800 mg daily. In the whole cohort, hematologic ORR was 68% and at least VGPR was 63%, but stratifying patients according to the presence or absence of t(11;14), ORR and at least VGPR were 81% and 78% vs. 40% and 30% in patients with and without this cytogenetic abnormality, respectively. Overall, after a median follow-up of 14.5 months, median PFS was 31 months and median OS was not reached. However, while median OS was not reached in both groups of patients, median PFS was not reached (12 months = 90%) in patients with t(11;14) and 6.7 months in patients without t(11;14). Venetoclax was well tolerated and the main grade 3–4 side effects were infections (7%), diarrhea (5%), and thrombocytopenia (5%). Similar results were reported in a retrospective experience of Pavia, Athens, and Israel evaluating 26 patients, of whom 88% had t(11;14) (114). Most patients had cardiac (77%) and renal involvement (58%); median prior lines of therapy were 3.5, with 100% and 85% of patients who had previously received bortezomib and daratumumab, respectively; performance status at venetoclax initiation was ≥2 in 53% of patients. Patients were treated with venetoclax ± dexamethasone (69%) or venetoclax with daratumumab (31%) and ORR for all the patients was 88%, with VGPR 35% and CR 35%. After a median follow-up of 33 months, median EFS was 25 months and median OS was 33 months. Infections were the main grade 3–4 side effect (11.5%) and only one patient developed grade 3 diarrhea. At the last EMN Meeting, data from patients treated with venetoclax from the French Amyloidosis Network have been presented (115). Among 51 patients who received venetoclax-based regimens (39 patients with relapsed and 12 patients with newly diagnosed AL amyloidosis), all except 3 had a t(11;14). In relapsed patients, 74.5% achieved at least hematologic VGPR (CR = 56.5%), and of patients exposed/refractory to daratumumab, 56% obtained a CR. Median follow-up was 17.2 months, median PFS was 40 months, and estimated 3-year OS was 68.2%. Therefore, venetoclax monotherapy or venetoclax-based combinations seem to be a very promising approach in patients with t(11;14), even in those exposed to daratumumab. These findings have been recently confirmed by a study evaluating venetoclax-based regimens in 21 patients with daratumumab-refractory AL with t(11;14) (116). The hematologic response was 95%, 53% of patients achieved a CR or better, whereas cardiac and renal responses were 40% and 36%, respectively.
Preliminary antitumor activity has been reported with lisaftoclax (APG-2575) (117), a potent and selective BCL-2 inhibitor, under clinical development in several hematological malignancies. In a study enrolling patients with relapsed/refractory MM and relapsed/refractory AL amyloidosis, five patients with AL received lisaftoclax in combination with pomalidomide and dexamethasone and the ORR was 60%.
6.1 Novel immunotherapeutic approach
Belantamab mafodotin is the first-in-class antibody–drug conjugate (ADC) targeting B-cell maturation antigen (BCMA) approved for relapsed/refractory multiple myeloma (RRMM) who received at least four prior lines of therapy. This immunotherapy has demonstrated efficacy and tolerability also in patients with relapsed refractory AL amyloidosis (118). Twenty-seven patients with a median of three prior lines of therapy (prior exposure to proteasome inhibitors of 100%; immunomodulatory agents, 81%; and anti-CD38 antibody, 81%) received belantamab mafodotin at standard dose of 2.5 mg/kg. A median of five infusions of belantamab mafodotin was administered and, among evaluable patients, hematologic ORR was 80% and VRP/CR was 64%. After a median follow-up of 13 months, 1-year treatment-free survival/death was 69% and 1-year OS was 88%. As regards toxicity, although keratopathy occurred in 70% of patients, only one patient was grade 4 and required treatment cessation. Data on BCMA-targeting bispecific antibodies such as teclistamab, which is approved in relapsed/refractory MM, are very limited considering that in the MajestTec-1 trial, leading to teclistamab approval, patients with amyloidosis were excluded (119). In seven patients with AL amyloidosis with or without concurrent relapsed/refractory MM (median of six prior lines of therapy), teclistamab was found to induce hematologic VGPR or better in 100% of patients. Among four patients evaluable for cardiac organ response and two for renal organ response, three achieved a cardiac response and one achieved a renal response. Fifty-seven percent of patients developed cytokine release syndrome (CRS), all grade 1, and immune effector cell-associated neurotoxicity syndrome (ICANS) did not occur in any patient. Another small series has been presented at the last ASH Meeting (120). Four patients with multi-organ involvement, concurrent MM, and a median of seven prior lines of therapy received teclistamab and achieved 100% hematologic CR with organ response in two out of four patients. Evaluation of anti-BCMA chimeric antigen receptor T-cell therapy (CART) represents a challenge in AL since it is a rare disease, and because it often involves several organs, patients are probably too frail to undergo this immunotherapeutic approach. Moreover, in AL amyloidosis, the activity of anti-BCMA CART has been questioned because of the expression of BCMA that would be lower than that of MM (121). However, data with a novel academic BCMA-CART (HBI0101) are encouraging (122). Nine patients with RRAL amyloidosis had a median of six lines of therapy, all triple-refractory, seven of whom have cardiac involvement (including four with MAYO-stage 3a/3b); six patients received CART cells at a dose of 800 × 106 and the remaining patients received a lower dose. Grade 3 CRS was seen in two out of nine patients, whereas in five out of nine patients, grades 1–2 CRS was seen, and no patients developed ICANS or died because of side effects. Among eight evaluable patients for response, 100% achieved hematologic response, which was CR in five out of eight patients; moreover, five out of eight patients obtained MRD negativity at a level of 10–5. Table 4 summarizes the main ongoing trials in relapsed and refractory AL amyloidosis.
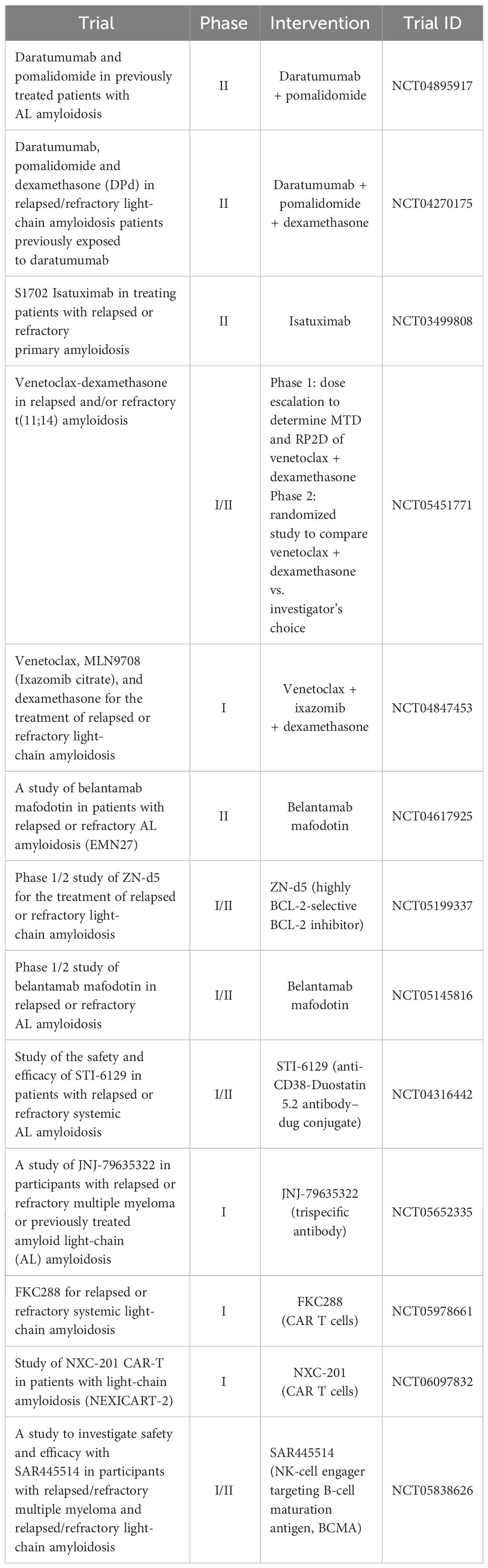
Table 4 Ongoing trials in relapsed refractory AL amyloidosis.
6.2 Thrombotic and hemorrhagic risk and management
Both thromboembolic and hemorrhagic risks are increased in patients with AL amyloidosis, and their management has been poorly emphasized so far. The rate of thromboembolic events is approximately 5%–10% in patients with amyloidosis and lower in patients with cardiac involvement, in whom the main risk factor is atrial myopathy and the impaired contractile function, along with atrial fibrillation and heart failure in general (123, 124). Cardiac thrombi are observed in up to 30% of patients (125–127). Experts at the last ASH Meeting defined nephrotic syndrome and the use of immunomodulatory drugs as factors favoring thrombosis, particularly deep vein thrombosis, renal vein thrombosis, often asymptomatic pulmonary embolus, and rarely cerebral vein thrombosis or arterial ones. Current recommendations advise the use of prophylactic antithrombotic treatment (vitamin K antagonist, low-molecular-weight heparin, or aspirin) in patients receiving IMID-based therapy, borrowing data from MM (128, 129). However, gastrointestinal involvement causing bleeding, factor X deficiency especially if <50%, severe cytopenias, liver amyloidosis involvement, and dysautonomia should be considered as contraindications to starting an antithrombotic therapy (123). Prospective studies are needed to specify when and how to prescribe preventive antithrombotic therapy in this specific population. On the other hand, the bleeding risk increases because of gastrointestinal hemorrhage for amyloid digestive involvement, factor X deficiency, renal failure leading to platelet dysfunction, and increased risk of autonomic dysfunction with orthostatic hypotension and falls. Bleeding is more common in AL amyloidosis than in thromboembolism. Cutaneous and gastrointestinal sites are the most common bleeding sites (130); abnormal coagulation tests have been demonstrated in 14%–32% of cases, and subnormal factor X have been indicated in 14% of cases due to abnormal adsorption, with thrombocytopenia and von Willebrand disease being very uncommon (131–133). Vigilance is needed to recognize these issues and appropriate prophylaxis measures, especially before procedures, can reduce complications and improve outcomes. Further data are needed to better understand mechanisms of bleeding complications and find therapies to manage them.
6.3 Challenging settings
Because organ failure is common in AL amyloidosis, solid organ transplantation could become an urgent need in some particular situations. It is controversial because of the multisystem nature of this disease and its recurrence risk in the allograft (134). For several years, the role of solid organ transplantation in the management of AL amyloidosis remained debatable, but the introduction of newer therapies that can induce very deep and long-lasting hematologic responses changes this perception (135–137). Renal transplant is more commonly performed, but definite selection criteria do not exist. However, it is reasonable to consider either patients with isolated renal AL or patients without severe dysfunction of other involved organs who have achieved at least a VGPR. In some patients, post-renal transplant ASCT could be carried out to improve hematologic response. Ongoing data collection may help to clarify selection criteria for optimal outcomes. Very rare cases of multiorgan transplantation have been described, involving the kidney, heart and liver (138).
Peripheral neuropathy is another difficult clinical setting to manage, which could represent a clinical manifestation of nerve AL amyloidosis or a side effect of therapy (mostly bortezomib). The incidence of peripheral neuropathy in AL amyloidosis varies from 9.6% to 35%, typically symmetrical and length-dependent, affecting the lower limb, and slowly progressing with severe pain. Autonomic neuropathy is a particularly severe complication manifesting with gastroparesis, diarrhea or constipation, impotence, and severe postural hypotension (139). Early diagnosis is very difficult in this neurological AL amyloidosis setting, but starting therapy at the earliest possible time is crucial to obtain a clinical organ response.
7 Conclusion
AL amyloidosis is a rare disease, but in recent years, this diagnosis has become more frequent because of the rising awareness of clinicians and the multidisciplinary management of suspicious clinical signs. It is crucial to make a rapid diagnosis for this disease, preferably a histological one, and to start the correct therapy as soon as possible. The goal of therapy is twofold: (1) to promptly avoid organ failure and (2) to eliminate the underlying plasma cell or B-cell clone to decrease the risk of progressive organ fibril deposition and consequent organ damage. Newer therapies increase the probability of achieving not only a hematological response but also an organ response, and other therapies are ongoing to further improve outcomes.
Author contributions
SM: Conceptualization, Writing – original draft, Writing – review & editing. VM: Conceptualization, Writing – original draft, Writing – review & editing. LC: Writing – original draft, Writing – review & editing. EM: Writing – original draft, Writing – review & editing. AP: Writing – original draft, Writing – review & editing. MO: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FR declared a past co-authorship with the author MO to the handling editor.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. (1992) 79:1817–22. doi: 10.1182/blood.V79.7.1817.1817
2. Misra P, Blancas-Mejia LM, Ramirez-Alvarado M. Mechanistic insights into the early events in the aggregation of immunoglobulin light chains. Biochemistry. (2019) 58:3155–68. doi: 10.1021/acs.biochem.9b00311
3. Bellotti V, Mangione P, Merlini G. Review: Immunoglobulin light chain amyloidosis - The archetype of structural and pathogenic variability. J Struct Biol. (2000) 130:280–9. doi: 10.1006/jsbi.2000.4248
4. Merlini G, Stone MJ, Dc W. Dangerous small B-cell clones Review in translational hematology Dangerous small B-cell clones. Blood. (2013) 108(8):2520–30. doi: 10.1182/blood-2006-03-001164
5. Lewis E, Lee H, Fine N, Miller R, Hahn C, Tay J, et al. Monoclonal gammopathy of undetermined significance in patients with transthyretin amyloidosis (ATTR): analysis using the iStopMM criteria. Clin Lymphoma Myeloma Leuk. (2023) 23:211–7. doi: 10.1016/j.clml.2022.12.012
6. Rubinstein SM, Stockerl-Goldstein K. How to screen for monoclonal gammopathy in patients with a suspected amyloidosis. JACC CardioOncology. (2021) 3:590–3. doi: 10.1016/j.jaccao.2021.07.001
7. Perfetti V, Palladini G, Casarini S, Navazza V, Rognoni P, Obici L, et al. The repertoire of λ light chains causing predominant amyloid heart involvement and identification of a preferentially involved germline gene, IGLV1–44. Blood. (2012) 119:144–50. doi: 10.1182/blood-2011-05-355784
8. Merlini G. AL amyloidosis: From molecular mechanisms to targeted therapies. Hematology. (2017) 2017:1–12. doi: 10.1182/asheducation-2017.1.1
9. Sanchorawala V, Boccadoro M, Gertz M, Hegenbart U, Kastritis E, Landau H, et al. Guidelines for high dose chemotherapy and stem cell transplantation for systemic AL amyloidosis: EHA-ISA working group guidelines. Amyloid. (2022) 29:1–7. doi: 10.1080/13506129.2021.2002841
10. Donnelly JP, Gabrovsek A, Sul L, Cotta C, Rodriguez ER, Tan CD, et al. Evidence of concurrent light chain and transthyretin cardiac amyloidosis in 2 patients. JACC CardioOncology. (2020) 2:127–30. doi: 10.1016/j.jaccao.2020.01.001
11. Abhishek G, John W, Paul S, Abbas AS, Ann HC, Charles S, et al. Coexistence of light chain and transthyretin cardiac amyloidosis. JACC Case Rep. (2024) 29:102285. doi: 10.1016/j.jaccas.2024.102285
12. Vergaro G, Castiglione V, Poletti R, Buda G, Pucci A, Musetti V, et al. Biopsy evidence of sequential transthyretin and immunoglobulin light-chain cardiac amyloidosis in the same patient. JACC Case Rep. (2021) 3:450–4. doi: 10.1016/j.jaccas.2020.12.047
13. Lousada I, Comenzo R, Landau H, Guthrie S, Merlini G. Patient experience with light chain amyloidosis: A survey from the amyloidosis research consortium. J Card Fail. (2015) 21:S63. doi: 10.1016/j.cardfail.2015.06.203
14. Dima D, Mazzoni S, Anwer F, Khouri J, Samaras C, Valent J, et al. Diagnostic and treatment strategies for AL amyloidosis in an era of therapeutic innovation. JCO Oncol Pract. (2023) 19:265–75. doi: 10.1200/OP.22.00396
15. Zhao L, Buxbaum JN, Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry. (2013) 52:1913–26. doi: 10.1021/bi301313b
16. Sethi S, Vrana JA, Theis JD, Leung N, Sethi A, Nasr SH, et al. Laser microdissection and mass spectrometry-based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int. (2012) 82:226–34. doi: 10.1038/ki.2012.108
17. Quarta CC, Zheng J, Hutt D, Grigore SF, Manwani R, Sachchithanantham S, et al. 99mTc-DPD scintigraphy in immunoglobulin light chain (AL) cardiac amyloidosis. Eur Hear J Cardiovasc Imaging. (2021) 22:1304–11. doi: 10.1093/ehjci/jeab095
18. Milani P, Valentini V, Ferraro G, Basset M, Russo F, Foli A, et al. A patient with AL amyloidosis with negative free light chain results. Clin Chem Lab Med. (2016) 54:1035–7. doi: 10.1515/cclm-2015-0847
19. Garofalo M, Piccoli L, Romeo M, Barzago MM, Ravasio S, Foglierini M, et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat Commun. (2021) 12:3532. doi: 10.1038/s41467–021-23880–9
20. Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: A staging system for primary systemic amyloidosis. J Clin Oncol. (2004) 22:3751–7. doi: 10.1200/JCO.2004.03.029
21. Wechalekar AD, Schonland SO, Kastritis E, Gillmore JD, Dimopoulos MA, Lane T, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. (2013) 121:3420–7. doi: 10.1182/blood-2012-12-473066
22. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. (2012) 30:989–95. doi: 10.1200/JCO.2011.38.5724
23. Lilleness B, Ruberg FL, Mussinelli R, Doros G, Sanchorawala V. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood. (2019) 133:215–23. doi: 10.1182/blood-2018-06-858951
24. Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol Off J Am Soc Clin Oncol. (2012) 30:4541–9. doi: 10.1200/JCO.2011.37.7614
25. Palladini G, Schönland SO, Sanchorawala V, Kumar S, Wechalekar A, Hegenbart U, et al. Clarification on the definition of complete haematologic response in light-chain (AL) amyloidosis. Amyloid: Int J Exp Clin investigation: Off J Int Soc Amyloidosis. (2021) 28:1–2. doi: 10.1080/13506129.2020.1868810
26. Manwani R, Cohen O, Sharpley F, Mahmood S, Sachchithanantham S, Foard D, et al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood. (2019) 134:2271–80. doi: 10.1182/blood.2019000834
27. Muchtar E, Dispenzieri A, Leung N, Lacy MQ, Buadi FK, Dingli D, et al. Optimizing deep response assessment for AL amyloidosis using involved free light chain level at end of therapy: failure of the serum free light chain ratio. Leukemia. (2019) 33:527–31. doi: 10.1038/s41375-018-0258-y
28. Ravichandran S, Mahmood S, Wisniowski B, Sachchithanantham S, Popat R, Lachmann H, et al. A UK consensus algorithm for early treatment modification in newly diagnosed systemic light-chain amyloidosis. Br J Haematol. (2022) 198:328–32. doi: 10.1111/bjh.18216
29. Comenzo RL, Reece D, Palladini G, Seldin D, Sanchorawala V, Landau H, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. (2012) 26:2317–25. doi: 10.1038/leu.2012.100
30. Palladini G, Hegenbart U, Milani P, Kimmich C, Foli A, Ho AD, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood. (2014) 124:2325–32. doi: 10.1182/blood-2014-04-570010
31. Muchtar E, Wisniowski B, Palladini G, Milani P, Merlini G, Schönland S, et al. Graded renal response criteria for light chain (AL) amyloidosis. Blood. (2021) 138:2721–1. doi: 10.1182/blood-2021-149594
32. Muchtar E, Dispenzieri A, Wisniowski B, Palladini G, Milani P, Merlini G, et al. Graded cardiac response criteria for AL amyloidosis: the impact of depth of cardiac response on survival. Blood. (2021) 138:2720–0. doi: 10.1182/blood-2021-149222
33. Muchtar E, Dispenzieri A, Leung N, Lacy MQ, Buadi FK, Dingli D, et al. Depth of organ response in AL amyloidosis is associated with improved survival: grading the organ response criteria. Leukemia. (2018) 32:2240–9. doi: 10.1038/s41375-018-0060-x
34. Salinaro F, Meier-Ewert HK, Miller EJ, Pandey S, Sanchorawala V, Berk JL, et al. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. Eur Hear J Cardiovasc Imaging. (2017) 18:1057–64. doi: 10.1093/ehjci/jew298
35. Cohen OC, Ismael A, Pawarova B, Manwani R, Ravichandran S, Law S, et al. Longitudinal strain is an independent predictor of survival and response to therapy in patients with systemic AL amyloidosis. Eur Heart J. (2022) 43:333–41. doi: 10.1093/eurheartj/ehab507
36. Zumbo G, Barton SV, Thompson D, Sun M, Abdel-Gadir A, Treibel TA, et al. Extracellular volume with bolus-only technique in amyloidosis patients: Diagnostic accuracy, correlation with other clinical cardiac measures, and ability to track changes in amyloid load over time. J Magn Reson Imaging. (2018) 47:1677–84. doi: 10.1002/jmri.25907
37. Decker I, Goodman SA, Phillips SE, Lenihan DJ, Cornell RF. The six-minute walk test is a valuable measure of functional change following chemotherapy for AL (light-chain) cardiac amyloidosis. Br J haematology. (2017) 177:481–3. doi: 10.1111/bjh.2017.177.issue-3
38. Pulido V, Doros G, Berk JL, Sanchorawala V. The six-minute walk test in patients with AL amyloidosis: a single centre case series. Br J Haematol. (2017) 177:388–94. doi: 10.1111/bjh.2017.177.issue-3
39. Sidana S, Muchtar E, Sidiqi MH, Jevremovic D, Dispenzieri A, Gonsalves W, et al. Impact of minimal residual negativity using next generation flow cytometry on outcomes in light chain amyloidosis. Am J Hematol. (2020) 95:497–502. doi: 10.1002/ajh.25746
40. Kastritis E, Kostopoulos IV, Theodorakakou F, Fotiou D, Gavriatopoulou M, Migkou M, et al. Next generation flow cytometry for MRD detection in patients with AL amyloidosis. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2021) 28:19–23. doi: 10.1080/13506129.2020.1802713
41. Palladini G, Paiva B, Wechalekar A, Massa M, Milani P, Lasa M, et al. Minimal residual disease negativity by next-generation flow cytometry is associated with improved organ response in AL amyloidosis. Blood Cancer J. (2021) 11:34. doi: 10.1038/s41408–021-00428–0
42. Bomsztyk J, Ravichandran S, Giles HV, Berlanga O, Harding S, Khwaja J, et al. Use of matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS) free light chain assessment for the diagnosis and monitoring of systemic immunoglobulin light chain (AL) amyloidosis. Blood. (2022) 140:2351–3. doi: 10.1182/blood-2022–160018
43. Kastritis E, Kostopoulos IV, Terpos E, Paiva B, Fotiou D, Gavriatopoulou M, et al. Evaluation of minimal residual disease using next-generation flow cytometry in patients with AL amyloidosis. Blood Cancer J. (2018) 8:46. doi: 10.1038/s41408-018-0086-3
44. Muchtar E, Dispenzieri A, Jevremovic D, Dingli D, Buadi FK, Lacy MQ, et al. Survival impact of achieving minimal residual negativity by multi-parametric flow cytometry in AL amyloidosis. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2020) 27:13–6. doi: 10.1080/13506129.2019.1666709
45. Staron A, Burks EJ, Lee JC, Sarosiek S, Sloan JM, Sanchorawala V. Assessment of minimal residual disease using multiparametric flow cytometry in patients with AL amyloidosis. Blood Adv. (2020) 4:880–4. doi: 10.1182/bloodadvances.2019001331
46. Wu X, Zhou P, Wong SW, Rosenthal B, Chung A, Toskic D, et al. Using next generation sequencing to identify trackable clonotypic sequences for minimal residual disease testing in AL amyloidosis. Blood. (2023) 142:3398. doi: 10.1182/blood-2023–190355
47. Sarosiek S, Varga C, Jacob A, Fulciniti MT, Munshi N, Sanchorawala V. Detection of minimal residual disease by next generation sequencing in AL amyloidosis. Blood Cancer J. (2021) 11:117. doi: 10.1038/s41408–021-00511–6
48. Gibbons A, Ranjit Anderson N, Chakravorty L, Kwok F, Sidiqi H, Gibbs S, et al. Identifying early suboptimal hematological response in patients with AL amyloidosis treated with bortezomib-based chemotherapy. Blood. (2023) 142:2030. doi: 10.1182/blood-2023-179219
49. Fotiou D, Dimopoulos MA, Kastritis E. Systemic AL amyloidosis: current approaches to diagnosis and management. HemaSphere. (2020) 4:e454. doi: 10.1097/HS9.0000000000000454
50. Gertz MA, Cohen AD, Comenzo RL, Kastritis E, Landau HJ, Libby EN, et al. Birtamimab plus standard of care in light-chain amyloidosis: the phase 3 randomized placebo-controlled VITAL trial. Blood. (2023) 142:1208–18. doi: 10.1182/blood.2022019406
51. Comenzo RL, Vosburgh E, Simms RW, Bergethon P, Sarnacki D, Finn K, et al. Dose-intensive melphalan with blood stem cell support for the treatment of AL amyloidosis: one-year follow-up in five patients. Blood. (1996) 88:2801–6. doi: 10.1182/blood.V88.7.2801.bloodjournal8872801
52. Wechalekar AD, Sanchorawala V. Daratumumab in AL amyloidosis. Blood. (2022) 140:2317–22. doi: 10.1182/blood.2021014613
53. Sanchorawala V, Sarosiek S, Schulman A, Mistark M, Migre ME, Cruz R, et al. Safety, tolerability, and response rates of daratumumab in relapsed AL amyloidosis: results of a phase 2 study. Blood. (2020) 135:1541–7. doi: 10.1182/blood.2019004436
54. Roussel M, Merlini G, Chevret S, Arnulf B, Stoppa AM, Perrot A, et al. A prospective phase 2 trial of daratumumab in patients with previously treated systemic light-chain amyloidosis. Blood. (2020) 135:1531–40. doi: 10.1182/blood.2019004369
55. Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. (2021) 385:46–58. doi: 10.1056/NEJMoa2028631
56. Luo M, Zhu P, Nnane I, Xiong Y, Merlini G, Comenzo RL, et al. Population pharmacokinetics and exposure-response modeling of daratumumab subcutaneous administration in patients with light-chain amyloidosis. J Clin Pharmacol. (2022) 62:656–69. doi: 10.1002/jcph.1994
57. Staron A, Zheng L, Doros G, Connors LH, Mendelson LM, Joshi T, et al. Marked progress in AL amyloidosis survival: a 40-year longitudinal natural history study. Blood Cancer J. (2021) 11:139. doi: 10.1038/s41408-021-00529-w
58. Muchtar E, Sanchorawala V, Hassan H, Hegenbart U, Schonland S, Lee HC, et al. The impact of melphalan conditioning schedule on post-transplant outcomes in AL amyloidosis: A multicenter study with 1718 patients. Blood. (2023) 142:4784. doi: 10.1182/blood-2023-188115
59. Bellofiore C, Basset M, Benvenuti P, Nuvolone M, Foli A, Nanci M, et al. Daratumumab-based regimens versus cybord in newly diagnosed patients with AL amyloidosis and IIIb cardiac stage: A matched case-control study. Blood. (2023) 142:2028. doi: 10.1182/blood-2023–184681
60. Chakraborty R, Bhutani D, Mapara M, Reshef R, Maurer MS, Radhakrishnan J, et al. Reduced early mortality with daratumumab-based frontline therapy in systemic AL amyloidosis- experience from the Columbia amyloidosis multidisciplinary program. Blood. (2023) 142:4780. doi: 10.1182/blood-2023–187214
61. Oubari S, Hegenbart U, Schoder R, Steinhardt M, Papathanasiou M, Rassaf T, et al. Daratumumab in first-line treatment of patients with light chain amyloidosis and Mayo stage IIIb improves treatment response and overall survival. Haematologica. (2024) 109:220–30. doi: 10.3324/haematol.2023.283325
62. Abdallah N, Sidana S, Dispenzieri A, Lacy M, Buadi F, Hayman S, et al. Outcomes with early vs. deferred stem cell transplantation in light chain amyloidosis. Bone Marrow Transplant. (2020) 55:1297–304. doi: 10.1038/s41409-020-0964-8
63. Sharpley FA, Petrie A, Mahmood S, Sachchithanantham S, Lachmann HJ, Gillmore JD, et al. A 24-year experience of autologous stem cell transplantation for light chain amyloidosis patients in the United Kingdom. Br J Haematol. (2019) 187:642–52. doi: 10.1111/bjh.16143
64. Al Saleh AS, Sidiqi MH, Sidana S, Muchtar E, Dispenzieri A, Dingli D, et al. Impact of consolidation therapy post autologous stem cell transplant in patients with light chain amyloidosis. Am J Hematol. (2019) 94:1066–71. doi: 10.1002/ajh.25572
65. Rajasekariah H, McCaughan G, Ma CKK, Gorrie N, Milliken S, Ma D, et al. Orthotopic heart transplantation followed by autologous stem cell transplantation in patients with cardiac AL amyloidosis: results from a single centre prospective study. Blood. (2023) 142:3414. doi: 10.1182/blood-2023–184319
66. Gustine JN, Staron A, Szalat RE, Mendelson LM, Joshi T, Ruberg FL, et al. Predictors of hematologic response and survival with stem cell transplantation in AL amyloidosis: A 25-year longitudinal study. Am J Hematol. (2022) 97:1189–99. doi: 10.1002/ajh.26641
67. Bal S, Estrada-Merly N, Costa LJ, Qazilbash MH, Kumar S, D’Souza A. Outcomes of t(11;14) light chain (AL) amyloidosis after autologous stem cell transplantation: benchmark for new therapies. Blood Cancer J. (2023) 13:170. doi: 10.1038/s41408–023-00945–0
68. Warsame R, Kumar SK, Gertz MA, Lacy MQ, Buadi FK, Hayman SR, et al. Abnormal FISH in patients with immunoglobulin light chain amyloidosis is a risk factor for cardiac involvement and for death. Blood Cancer J. (2015) 5:e310. doi: 10.1038/bcj.2015.34
69. Landau H, Hassoun H, Rosenzweig MA, Maurer M, Liu J, Flombaum C, et al. Bortezomib and dexamethasone consolidation following risk-adapted melphalan and stem cell transplantation for patients with newly diagnosed light-chain amyloidosis. Leukemia. (2013) 27:823–8. doi: 10.1038/leu.2012.274
70. Ozga M, Zhao Q, Benson D, Elder P, Williams N, Bumma N, et al. AL amyloidosis: the effect of maintenance therapy on autologous stem cell transplantation outcomes. J Clin Med. (2020) 9. doi: 10.3390/jcm9113778
71. Cibeira MT, Oriol A, Lahuerta JJ, Mateos M-V, de la Rubia J, Hernández MT, et al. A phase II trial of lenalidomide, dexamethasone and cyclophosphamide for newly diagnosed patients with systemic immunoglobulin light chain amyloidosis. Br J Haematol. (2015) 170:804–13. doi: 10.1111/bjh.13500
72. Moreau P, Jaccard A, Benboubker L, Royer B, Leleu X, Bridoux F, et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed AL amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood. (2010) 116:4777–82. doi: 10.1182/blood-2010-07-294405
73. Kastritis E, Dialoupi I, Gavriatopoulou M, Roussou M, Kanellias N, Fotiou D, et al. Primary treatment of light-chain amyloidosis with bortezomib, lenalidomide, and dexamethasone. Blood Adv. (2019) 3:3002–9. doi: 10.1182/bloodadvances.2019000147
74. Palladini G, Milani P, Foli A, Obici L, Lavatelli F, Nuvolone M, et al. Oral melphalan and dexamethasone grants extended survival with minimal toxicity in AL amyloidosis: long-term results of a risk-adapted approach. Haematologica. (2014) 99:743–50. doi: 10.3324/haematol.2013.095463
75. Manwani R, Mahmood S, Sachchithanantham S, Lachmann HJ, Gillmore JD, Yong K, et al. Carfilzomib is an effective upfront treatment in AL amyloidosis patients with peripheral and autonomic neuropathy. Br J Haematol. (2019) 187:638–41. doi: 10.1111/bjh.16122
76. Xing Y, Yan J, Yu Z, Zhao J, Wang Y, Li X, et al. High-cutoff hemodialysis in multiple myeloma patients with acute kidney injury. Front Oncol. (2022) 12:1024133. doi: 10.3389/fonc.2022.1024133
77. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. (2020) 136:71–80. doi: 10.1182/blood.2019004460
78. Muchtar E, Dispenzieri A, Wisniowski B, Palladini G, Milani P, Merlini G, et al. Graded cardiac response criteria for patients with systemic light chain amyloidosis. J Clin Oncol Off J Am Soc Clin Oncol. (2023) 41:1393–403. doi: 10.1200/JCO.22.00643
79. Renz M, Torres R, Dolan PJ, Tam SJ, Tapia JR, Li L, et al. 2A4 binds soluble and insoluble light chain aggregates from AL amyloidosis patients and promotes clearance of amyloid deposits by phagocytosis (†). Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2016) 23:168–77. doi: 10.1080/13506129.2016.1205974
80. Wall JS, Kennel SJ, Williams A, Richey T, Stuckey A, Huang Y, et al. AL amyloid imaging and therapy with a monoclonal antibody to a cryptic epitope on amyloid fibrils. PloS One. (2012) 7:e52686. doi: 10.1371/journal.pone.0052686
81. Gertz MA, Landau H, Comenzo RL, Seldin D, Weiss B, Zonder J, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol Off J Am Soc Clin Oncol. (2016) 34:1097–103. doi: 10.1200/JCO.2015.63.6530
82. Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol. (2005) 79:319–28. doi: 10.1002/(ISSN)1096-8652
83. Palladini G, Merlini G. When should treatment of AL amyloidosis start at relapse? Early, to prevent organ progression. Blood Adv. (2019) 3:212–5. doi: 10.1182/bloodadvances.2018021253
84. Bomsztyk J, Ravichandran S, Giles HVD, Wright NJ, Berlanga O, Khwaja J, et al. Complete responses in AL amyloidosis are unequal - the impact of free light chain mass spectrometry in AL amyloidosis. Blood. (2024) 143(13):1259–68. doi: 10.1182/blood.2023022399
85. Palladini G, Barassi A, Klersy C, Pacciolla R, Milani P, Sarais G, et al. The combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in N-terminal natriuretic peptide type B (NT-proBNP) after chemotherapy best predicts survival in AL amyloidosis. Blood. (2010) 116:3426–30. doi: 10.1182/blood-2010-05-286567
86. Staron A, Mendelson LM, Joshi T, Sanchorawala V. Factors underlying impaired organ recovery despite a deep hematologic response in patients with systemic AL amyloidosis. Blood. (2023) 142:3410. doi: 10.1182/blood-2023-189956
87. Palladini G, Milani P, Foli A, Basset M, Russo F, Perlini S, et al. Presentation and outcome with second-line treatment in AL amyloidosis previously sensitive to nontransplant therapies. Blood. (2018) 131:525–32. doi: 10.1182/blood-2017-04-780544
88. Wechalekar AD, Cibeira MT, Gibbs SD, Jaccard A, Kumar S, Merlini G, et al. Guidelines for non-transplant chemotherapy for treatment of systemic AL amyloidosis: EHA-ISA working group. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2023) 30:3–17. doi: 10.1080/13506129.2022.2093635
89. Dispenzieri A, Lacy MQ, Zeldenrust SR, Hayman SR, Kumar SK, Geyer SM, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. (2007) 109:465–70. doi: 10.1182/blood-2006-07-032987
90. Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood. (2007) 110:3561–3. doi: 10.1182/blood-2007-07-099481
91. Basset M, Kimmich CR, Schreck N, Krzykalla J, Dittrich T, Veelken K, et al. Lenalidomide and dexamethasone in relapsed/refractory immunoglobulin light chain (AL) amyloidosis: results from a large cohort of patients with long follow-up. Br J Haematol. (2021) 195:230–43. doi: 10.1111/bjh.17685
92. Dispenzieri A, Buadi F, Laumann K, LaPlant B, Hayman SR, Kumar SK, et al. Activity of pomalidomide in patients with immunoglobulin light-chain amyloidosis. Blood. (2012) 119:5397–404. doi: 10.1182/blood-2012-02-413161
93. Sanchorawala V, Shelton AC, Lo S, Varga C, Sloan JM, Seldin DC. Pomalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 1 and 2 trial. Blood. (2016) 128:1059–62. doi: 10.1182/blood-2016-04-710822
94. Palladini G, Milani P, Foli A, Basset M, Russo F, Perlini S, et al. A phase 2 trial of pomalidomide and dexamethasone rescue treatment in patients with AL amyloidosis. Blood. (2017) 129:2120–3. doi: 10.1182/blood-2016-12-756528
95. Milani P, Sharpley F, Schönland SO, Basset M, Mahmood S, Nuvolone M, et al. Pomalidomide and dexamethasone grant rapid haematologic responses in patients with relapsed and refractory AL amyloidosis: a European retrospective series of 153 patients. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2020) 27:231–6. doi: 10.1080/13506129.2020.1767566
96. Jelinek T, Kryukova E, Kufova Z, Kryukov F, Hajek R. Proteasome inhibitors in AL amyloidosis: focus on mechanism of action and clinical activity. Hematol Oncol. (2017) 35:408–19. doi: 10.1002/hon.2351
97. Reece DE, Hegenbart U, Sanchorawala V, Merlini G, Palladini G, Bladé J, et al. Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase 1/2 study. Blood. (2011) 118:865–73. doi: 10.1182/blood-2011–02-334227
98. Reece DE, Hegenbart U, Sanchorawala V, Merlini G, Palladini G, Bladé J, et al. Long-term follow-up from a phase 1/2 study of single-agent bortezomib in relapsed systemic AL amyloidosis. Blood. (2014) 124:2498–506. doi: 10.1182/blood-2014-04-568329
99. Ravichandran S, Hall A, Jenner M, Garg M, Kishore B, Lachmann H, et al. A phase 1b dose-escalation study of carfilzomib in combination with thalidomide and dexamethasone in patients with relapsed/refractory systemic immunoglobulin light chain amyloidosis. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2023) 30:290–6. doi: 10.1080/13506129.2023.2169124
100. Cohen AD, Landau H, Scott EC, Liedtke M, Kaufman JL, Rosenzweig M, et al. Safety and efficacy of carfilzomib (CFZ) in previously-treated systemic light-chain (AL) amyloidosis. Blood. (2016) 128:645. doi: 10.1182/blood.V128.22.645.645
101. Dispenzieri A, Kastritis E, Wechalekar AD, Schönland SO, Kim K, Sanchorawala V, et al. A randomized phase 3 study of ixazomib-dexamethasone versus physician’s choice in relapsed or refractory AL amyloidosis. Leukemia. (2022) 36:225–35. doi: 10.1038/s41375-021-01317-y
102. Palladini G, Milani P, Malavasi F, Merlini G. Daratumumab in the treatment of light-chain (AL) amyloidosis. Cells. (2021) 10. doi: 10.3390/cells10030545
103. Kaufman GP, Schrier SL, Lafayette RA, Arai S, Witteles RM, Liedtke M. Daratumumab yields rapid and deep hematologic responses in patients with heavily pretreated AL amyloidosis. Blood. (2017) 130:900–2. doi: 10.1182/blood-2017-01-763599
104. Abeykoon JP, Zanwar S, Dispenzieri A, Gertz MA, Leung N, Kourelis T, et al. Daratumumab-based therapy in patients with heavily-pretreated AL amyloidosis. Leukemia. (2019) 33:531–6. doi: 10.1038/s41375-018-0262-2
105. Milani P, Fazio F, Basset M, Berno T, Larocca A, Foli A, et al. High rate of profound clonal and renal responses with daratumumab treatment in heavily pre-treated patients with light chain (AL) amyloidosis and high bone marrow plasma cell infiltrate. Am J Hematol. (2020) 95:900–5. doi: 10.1002/ajh.25828
106. Rosenbaum C, Liedtke M, Christos P, Kim H, Pogonowski K, Agudo N, et al. Daratumumab, pomalidomide and dexamethasone (DPd) in relapsed/refractory light chain amyloidosis previously exposed to daratumumab. Blood. (2023) 142:3402. doi: 10.1182/blood-2023–191133
107. Theodorakakou F, Fotiou D, Spiliopoulou V, Roussou M, Malandrakis P, Ntanasis-Stathopoulos I, et al. Outcomes of patients with light chain (AL) amyloidosis after failure of daratumumab-based therapy. Br J Haematol. (2023) 203:411–5. doi: 10.1111/bjh.19042
108. Bochtler T, Hegenbart U, Heiss C, Benner A, Moos M, Seckinger A, et al. Hyperdiploidy is less frequent in AL amyloidosis compared with monoclonal gammopathy of undetermined significance and inversely associated with translocation t(11;14). Blood. (2011) 117:3809–15. doi: 10.1182/blood-2010-02-268987
109. Fotiou D, Theodorakakou F, Gavriatopoulou M, Migkou M, Malandrakis P, Ntanasis-Stathopoulos I, et al. Prognostic impact of translocation t(11;14) and of other cytogenetic abnormalities in patients with AL amyloidosis in the era of contemporary therapies. Eur J Haematol. (2023) 111:271–8. doi: 10.1111/ejh.13993
110. Gertz MA, Lacy MQ, Dispenzieri A, Greipp PR, Litzow MR, Henderson KJ, et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood. (2005) 106:2837–40. doi: 10.1182/blood-2005–04-1411
111. Bochtler T, Hegenbart U, Kunz C, Granzow M, Benner A, Seckinger A, et al. Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J Clin Oncol Off J Am Soc Clin Oncol. (2015) 33:1371–8. doi: 10.1200/JCO.2014.57.4947
112. Muchtar E, Dispenzieri A, Kumar SK, Ketterling RP, Dingli D, Lacy MQ, et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia. (2017) 31:1562–9. doi: 10.1038/leu.2016.369
113. Premkumar VJ, Lentzsch S, Pan S, Bhutani D, Richter J, Jagannath S, et al. Venetoclax induces deep hematologic remissions in t(11;14) relapsed/refractory AL amyloidosis. Blood Cancer J. (2021) 11:10. doi: 10.1038/s41408-020-00397-w
114. Lebel E, Kastritis E, Palladini G, Milani P, Theodorakakou F, Aumann S, et al. Venetoclax in relapse/refractory AL amyloidosis-A multicenter international retrospective real-world study. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15061710
115. Roussel M, Pirotte M, Gounot R, Queru K, Rizzo O, Royer B, et al. B01 venetoclax targeted therapy in al amyloidosis patients: a retrospective analysis from the french amyloidosis network. HemaSphere. (2023) 7. doi: 10.1097/01.HS9.0000936096.74708.86
116. Orland M, Dima D, Ullah F, Awada H, Basali D, Faiman BM, et al. Outcomes of venetoclax-based therapy in patients with daratumumab-refractory t(11;14) positive light chain amyloidosis. Blood. (2023) 142:2031. doi: 10.1182/blood-2023–183030
117. Ailawadhi S, Chanan-Khan AA, Yannakou CK, Gibbs S, Khouri J, Chen Z, et al. First report on the effects of lisaftoclax (APG-2575) in combination with novel therapeutic regimens in patients with relapsed or refractory multiple myeloma (R/R MM) or immunoglobulin light-chain (Amyloid light-chain [AL]) amyloidosis. Blood. (2023) 142:2016. doi: 10.1182/blood-2023-188122
118. Khwaja J, Bomsztyk J, Bygrave C, Forbes A, Durairaj S, Fernandes S, et al. High responses rates with single agent belantamab mafodotin in relapsed systemic AL amyloidosis. Blood. (2023) 142:3408. doi: 10.1182/blood-2023-182042
119. Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. (2022) 387:495–505. doi: 10.1056/NEJMoa2203478
120. Stalker M, Garfall AL, Cohen AD, Vogl DT, Susanibar-Adaniya S, Djulbegovic M, et al. Safety and efficacy of teclistamab in patients with relapsed or refractory AL amyloidosis: A retrospective case series. Blood. (2023) 142:2035. doi: 10.1182/blood-2023–189277
121. Xu Y, Mao X, Que Y, Xu M, Li C, Almeida VDF, et al. The exploration of B cell maturation antigen expression in plasma cell dyscrasias beyond multiple myeloma. BMC Cancer. (2023) 23:123. doi: 10.1186/s12885–023-10591–1
122. Lebel E, Kfir-Erenfeld S, Asherie N, Grisariu S, Avni B, Elias S, et al. Feasibility of a novel academic anti-BCMA chimeric antigen receptor T-cell (CART) (HBI0101) for the treatment of relapsed and refractory AL amyloidosis. Blood. (2023) 142:538. doi: 10.1182/blood-2023-186450
123. Nicol M, Siguret V, Vergaro G, Aimo A, Emdin M, Dillinger JG, et al. Thromboembolism and bleeding in systemic amyloidosis: a review. ESC Hear Fail. (2022) 9:11–20. doi: 10.1002/ehf2.13701
124. Vilches S, Fontana M, Gonzalez-Lopez E, Mitrani L, Saturi G, Renju M, et al. Systemic embolism in amyloid transthyretin cardiomyopathy. Eur J Heart Fail. (2022) 24:1387–96. doi: 10.1002/ejhf.2566
125. Cappelli F, Tini G, Russo D, Emdin M, Del Franco A, Vergaro G, et al. Arterial thrombo-embolic events in cardiac amyloidosis: a look beyond atrial fibrillation. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. (2021) 28:12–8. doi: 10.1080/13506129.2020.1798922
126. Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. (2007) 116:2420–6. doi: 10.1161/CIRCULATIONAHA.107.697763
127. Martinez-Naharro A, Gonzalez-Lopez E, Corovic A, Mirelis JG, Baksi AJ, Moon JC, et al. High prevalence of intracardiac thrombi in cardiac amyloidosis. J Am Coll Cardiol. (2019) 73:1733–4. doi: 10.1016/j.jacc.2019.01.035
128. Larocca A, Cavallo F, Bringhen S, Di Raimondo F, Falanga A, Evangelista A, et al. Aspirin or enoxaparin thromboprophylaxis for patients with newly diagnosed multiple myeloma treated with lenalidomide. Blood. (2012) 119:933–9. doi: 10.1182/blood-2011-03-344333
129. Palumbo A, Cavo M, Bringhen S, Zamagni E, Romano A, Patriarca F, et al. Aspirin, warfarin, or enoxaparin thromboprophylaxis in patients with multiple myeloma treated with thalidomide: a phase III, open-label, randomized trial. J Clin Oncol Off J Am Soc Clin Oncol. (2011) 29:986–93. doi: 10.1200/JCO.2010.31.6844
130. Gould M, Zarrin-Khameh N, Sellin J. Small bowel amyloidosis. Curr Gastroenterol Rep. (2013) 15:350. doi: 10.1007/s11894-013-0350-4
131. Mumford AD, O’Donnell J, Gillmore JD, Manning RA, Hawkins PN, Laffan M. Bleeding symptoms and coagulation abnormalities in 337 patients with AL-amyloidosis. Br J Haematol. (2000) 110:454–60. doi: 10.1046/j.1365-2141.2000.02183.x
132. Kos CA, Ward JE, Malek K, Sanchorawala V, Wright DG, O’Hara C, et al. Association of acquired von Willebrand syndrome with AL amyloidosis. Am J Hematol. (2007) 82:363–7. doi: 10.1002/ajh.20829
133. Sundaram S, Rathod R. Gastric amyloidosis causing nonvariceal upper gastrointestinal bleeding. ACG Case Rep J. (2019) 6:3–4. doi: 10.14309/crj.0000000000000066
134. Sattianayagam PT, Gibbs SDJ, Pinney JH, Wechalekar AD, Lachmann HJ, Whelan CJ, et al. Solid organ transplantation in AL amyloidosis. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. (2010) 10:2124–31. doi: 10.1111/j.1600-6143.2010.03227.x
135. Kristen AV, Kreusser MM, Blum P, Schönland SO, Frankenstein L, Dösch AO, et al. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. J Hear Lung Transplant Off Publ Int Soc Hear Transplant. (2018) 37:611–8. doi: 10.1016/j.healun.2017.11.015
136. Davis MK, Kale P, Liedtke M, Schrier S, Arai S, Wheeler M, et al. Outcomes after heart transplantation for amyloid cardiomyopathy in the modern era. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. (2015) 15:650–8. doi: 10.1111/ajt.13025
137. Havasi A, Heybeli C, Leung N, Angel-Korman A, Sanchorawala V, Cohen O, et al. Outcomes of renal transplantation in patients with AL amyloidosis: an international collaboration through The International Kidney and Monoclonal Gammopathy Research Group. Blood Cancer J. (2022) 12:119. doi: 10.1038/s41408–022-00714–5
138. Brubaker AL, Urey MA, Taj R, Parekh JR, Berumen J, Kearns M, et al. Heart-liver-kidney transplantation for AL amyloidosis using normothermic recovery and storage from a donor following circulatory death: Short-term outcome in a first-in-world experience. Am J Transplant. (2023) 23:291–3. doi: 10.1016/j.ajt.2022.11.003
Keywords: amyloidosis, diagnosis, cardiac staging, response evaluation, treatment
Citation: Morè S, Manieri VM, Corvatta L, Morsia E, Poloni A and Offidani M (2024) AL amyloidosis: an overview on diagnosis, staging system, and treatment. Front. Hematol. 3:1378451. doi: 10.3389/frhem.2024.1378451
Received: 29 January 2024; Accepted: 23 April 2024;
Published: 08 May 2024.
Edited by:
Stefano Molica, Hull University Teaching Hospitals NHS Trust, United KingdomReviewed by:
Francesco Di Raimondo, University of Catania, ItalySamo Zver, University Medical Centre Ljubljana, Slovenia
Copyright © 2024 Morè, Manieri, Corvatta, Morsia, Poloni and Offidani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Offidani, TWFzc2ltby5vZmZpZGFuaUBvc3BlZGFsaXJpdW5pdGkubWFyY2hlLml0