 Jessica Nunes1,2
Jessica Nunes1,2 Dirk Loeffler1,2*
Dirk Loeffler1,2*- 1Department of Hematology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 2Department of Pathology and Laboratory Medicine, The University of Tennessee, Memphis, TN, United States
Hematopoietic stem cells (HSCs) can self-renew and differentiate for the entire life of an organism to produce new blood cells when needed. This process is regulated by asymmetric cell division (ACD), an evolutionarily conserved mechanism whereby cell fate determinants are unequally segregated into the daughter cells during division to instruct different cell fates. After many years of controversy, recent technical advances in microscopy, imaging, and bioinformatics make it now possible to visualize and quantify how factors segregate asymmetrically in dividing HSCs and lead to predictable changes in daughter cell fates many days later. While the molecular processes behind ACD in HSCs are still poorly understood, accumulating evidence suggests that lysosomes and other organelles, including mitochondria, autophagosomes, mitophagosomes, and recycling endosomes can segregate asymmetrically and act as cell fate determinants during divisions. Asymmetric segregation of lysosomes and mitochondria has been shown to predict mitochondrial activity, translation, and differentiation of HSC daughter cells and their offspring. This discovery and recent seminal findings show that lysosomes, once considered to be merely the trash bin of the cell, regulate many aspects of HSC biology and are crucial for the maintenance of quiescence and stem cell function. Here we provide a historical perspective and discuss the recent advances in our understanding of ACD and the role of lysosomes in HSC function. We discuss the limitations of past studies, talk about emerging concepts, and suggest critical next steps required to move the field forward.
1 Introduction
One of the biggest mysteries of life is the formation, maintenance, and regeneration of tissues. This process is fueled by the tremendous regenerative potential of very rare tissue-specific adult stem cells capable of both, (a) differentiation into mature cell types and (b) self-renewal to produce more stem cells required for long-term tissue maintenance (1). In the hematopoietic system, the regenerative potential of hematopoietic stem cells (HSCs) can be harnessed by transplanting donor HSCs to replace and/or supplement the recipient’s hematopoietic system (1). Transplanting HSCs is often the only curative therapy for many malignant and non-malignant hematological diseases (2) and is the foundation and forefront of modern regenerative medicine and gene therapy. A critical step of these life-saving therapies is the ex vivo culture of HSCs to edit their genome and/or expand their numbers before transplantation. HSCs make up less than 0.01% of bone marrow cells and are extremely rare (3). Their ex vivo expansion and maintenance are thus crucial for the successful and widespread application of any future regenerative therapy. However, despite recent progress (4), HSCs tend to die and/or differentiate in vitro and lose their regenerative properties quickly (5). Obtaining novel insights into the still poorly understood mechanisms of HSC fate decision-making is thus crucial to developing new and safe ex vivo HSC expansion and gene-editing protocols - a central aim of experimental hematology (6) and a prerequisite for advancing clinical applications tailored to specific diseases (1).
HSCs are multipotent and can give rise to all mature blood cells, while also producing new HSCs through self-renewal (1, 7). Their regenerative potential and the lifelong homeostasis of tissues rely on the ability of HSCs to make the ‘right’ decisions at the ‘right’ time to determine the future state of their offspring. These decisions include cell survival, proliferation, lineage choice, self-renewal, maturation, migration, activation, and senescence (8). Importantly, HSCs must adapt and adjust their fate decisions as different situations demand a different response (e.g. infection vs. bleeding) (9). These adaptations of HSC fates are thought to be orchestrated by cell-extrinsic and cell-intrinsic factors. Extrinsic factors include the cellular microenvironment (10) and secreted factors (11) and were recently reviewed in detail (12, 13). We thus focus here on the role of cell-intrinsic factors in HSC fate decision-making (Figure 1).
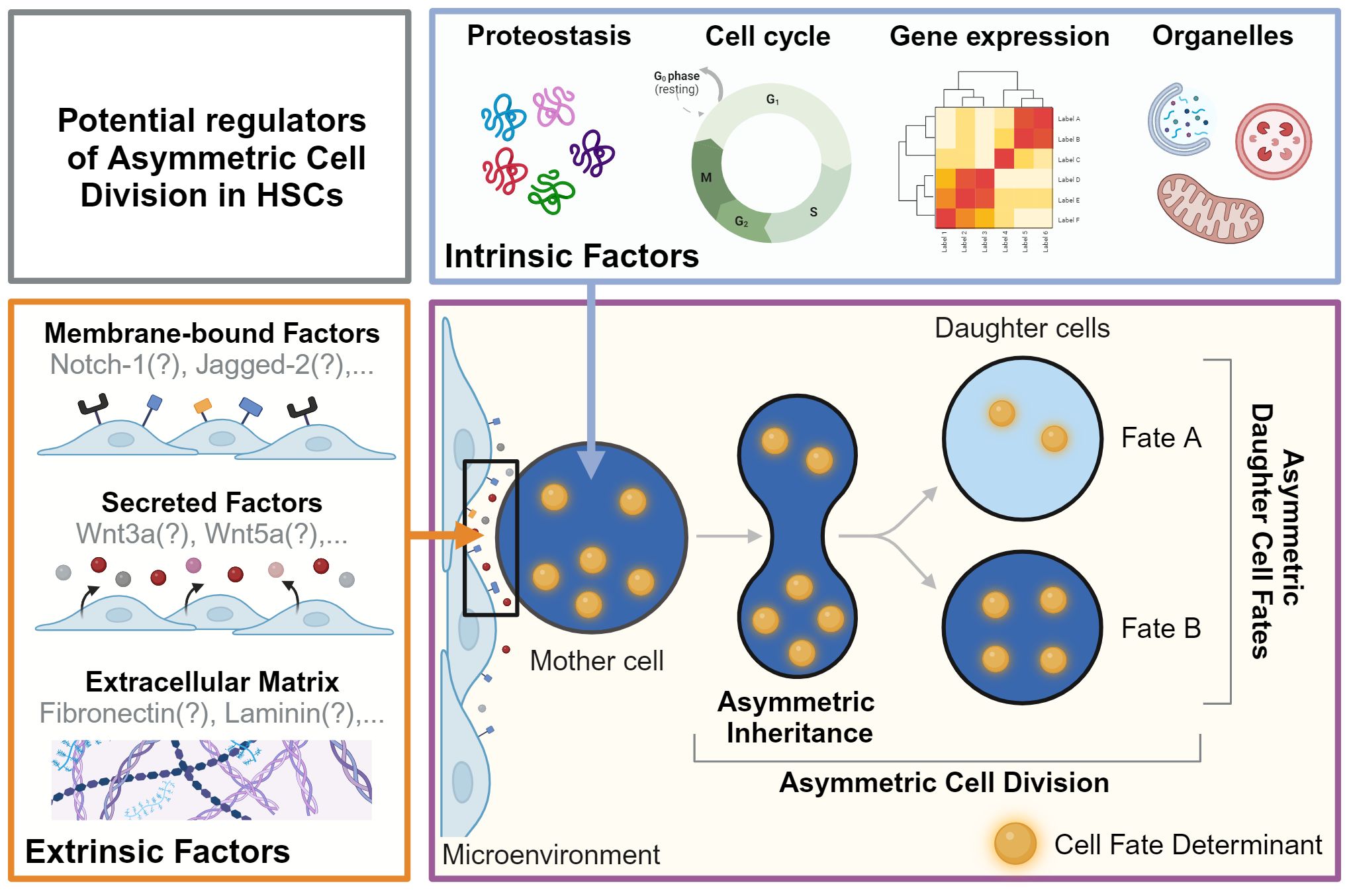
Figure 1 Potential regulators of asymmetric cell division in HSCs. Asymmetric Dell Division (ACD) is an evolutionarily conserved mechanism regulated by cell-intrinsic and -extrinsic factors. During ACD, cell fate determinants segregate asymmetrically to instruct changes in fate and behavior in daughter cells and their offspring. As the events during division and later cell fate acquisition can be many days apart, this process cannot be captured in a single snapshot. Live cell observations are thus a prerequisite to demonstrate functionally relevant ACD by linking asymmetric segregation to diverging daughter cell fates over time. The factors regulating ACD in HSC remain mostly unknown. Possible cell-intrinsic processes and regulators include proteostasis, gene expression, cell cycle regulators, and organelles such as lysosomes and mitochondria, which can act as cell fate determinants. Putative cell-extrinsic factors include membrane-bound factors, secreted factors, and the extracellular matrix in the microenvironment. Created with BioRender.com.
In the adult organism, most HSCs reside in the bone marrow and spend the majority of their lives outside the cell cycle in a reversible state of quiescence (14). Quiescent HSCs are at the apex of the hematopoietic hierarchy and their precise cell cycle regulation is essential to ensure the effective production of mature hematopoietic cells while preventing HSC exhaustion at the same time (15). In line with this view, HSC function correlates strongly with the depth of quiescence, the number of past divisions, and reduced cell cycle activity (16). Excessive stress and proliferation lead on the other hand to a loss of HSC function (17). To prevent cell cycle entry, quiescent HSCs express the cyclin-dependent kinase inhibitors p57 and p27 (18) and have low levels of the cyclin-dependent kinase 6 (CDK6) (16). Quiescent HSCs also have low mitochondrial activity, low reactive oxygen species (ROS) levels, rely on glycolysis and autophagy (19, 20), and have enlarged lysosomes, which were recently shown to be essential for the maintenance of quiescence (21). Despite being quiescent, most HSCs divide regularly and contribute to steady-state hematopoiesis over time (22, 23). When HSCs awake they exit the quiescent state and progress through the G1, S, and G2 phases of the cell cycle to divide (24). During this process, HSCs and/or their daughter cells choose from a variety of potential future cell fates, including self-renewal, differentiation, survival, cell death, proliferation, and quiescence.
How and when HSCs and/or their daughter cells decide to acquire these fates remains poorly understood. Many cell-intrinsic changes in metabolism, chromatin, gene expression, proteostasis, autophagy, and the cell cycle machinery have been shown to affect HSC fates (25–29). For instance, the repressive histone mark H3K27me3 is lost after G-CSF or M-CSF treatment of CD34+CD38- human hematopoietic progenitor cells during cytokine-induced lineage specification, thereby resulting in the recruitment of lineage-specifying transcription factors GATA-1 and PU.1 (30). Deletion of ubiquitin ligase Fbxw7 results in increased accumulation of its target c-MYC that drives cell cycle entry, differentiation, and exhaustion of HSCs (31). Knockdown of the autophagy genes Atg5 or Atg7 in mouse HSCs decreases their ability to regenerate and self-renew (32, 33). Abnormal activation of mitochondria results in the production of excessive metabolic byproducts and can disrupt HSC quiescence and function (34). A specific type of mitochondrial autophagy (mitophagy) which selectively clears damaged mitochondria was found to sustain self-renewal of HSCs via Nkx2-3, a transcription factor that is enriched in HSCs and can directly control transcription of the mitophagy regulator ULK1 which removes activated mitochondria (35). Although these studies show that certain genes and/or processes are involved in HSC fate regulation, when and how HSCs and their daughter cells decide to change fate often remains unknown.
So, how and when do HSCs make decisions? Evidence over the last two decades has shown that cell fate decisions can be regulated by stochastic processes such as transcriptional bursts or induced by cytokines and cell fate determinants acting at different times in the cell cycle. For instance, human embryonic stem cells (hESCs) in the G2/S/M phases of the cell cycle differentiate poorly, whereas hESCs in G1 are sensitive to differentiation cues (36). Whether such cell cycle-dependent mechanisms are also used to regulate cell fate decisions of HSCs is unclear (37). However, as recently demonstrated, HSC fate decisions can also occur during division through the asymmetric inheritance of cell fate determinants (38, 39). Using live-cell imaging, lysosomes and other organelles were found to segregate asymmetrically during HSC division and act as cell fate determinants by instructing different cell fates in HSC offspring (36, 38). Although this process, also known as asymmetric cell division (ACD), had long been known to act during the development of invertebrate organisms, it remained unclear whether it is also utilized by HSCs. However, after being controversial for decades, the ACD of HSCs is now taking center stage in stem cell biology. Here, we review the current knowledge about the regulation and consequence of cell fate determination in HSCs, with a special focus on ACD in HSCs.
2 Asymmetric cell division: early observations from invertebrates and definitions
Early studies on cell fate determination were pioneered by Charles O. Whitman in the 19th century. Whitman showed through direct observation of the leech embryo development that stereotypical, invariant cell divisions determine later cell fate during early cleavage stages (40). Later, in 1905, ACD was first shown by Edward Conklin, who observed that during divisions of the developing ascidian embryo, a yellow-colored portion of the cytoplasm is asymmetrically segregated (41). By following the segregating yellow cytoplasm over many cell divisions, Conklin showed that cells inheriting the deep yellow cytoplasm would later differentiate into muscle cells in the larva tail, while cells inheriting the light yellow material become the mesenchyme (41, 42). Although the mechanistic details remained unknown, Conklin was able to link mitotic segregation to later fates of differentiated cells and suggested that the ‘yellow cytoplasm’ might act as a cell fate determinant.
The concept of ACD has intrigued scientists for over a century, yet a molecular understanding of the underlying mechanisms has emerged only recently (43). Through studies of the developing embryos of invertebrate model organisms such as C. elegans and D. melanogaster, we now know of many evolutionary conserved regulators of ACD and have also learned that there are many ways cells use the ACD machinery. After Conklin’s seminal observation, it took almost a century to identify the first molecular cell fate determinant that is asymmetrically inherited during cell division. First observed in 1994 by Rhyu et al. during the division of the Sensory Organ Precursor (SOP) in Drosophila, this cell fate determinant is today known as NUMB (44). Through two rounds of asymmetric division, the SOP gives rise to the external sensory organ consisting of four cells, a neuron, a sheath cell, and two support cells. Because of its asymmetric segregation, NUMB can inhibit Notch signaling only in one SOP daughter cell, thereby instructing cell fate decisions. Importantly, deleting numb changes the cell fate of SOP descendants to produce only support cells, showing NUMB acts as a cell fate determinant (44). Later studies identified additional cell fate regulators in Drosophila including the atypical Protein Kinase C (aPKC), Partition Defective 6 (Par-6), and Bazooka (Baz), which form the Par complex, regulating apical polarity in larval brain neuroblasts (45). This complex directs phosphorylation and exclusion of NUMB from the apical cortex to establish the basal localization of cell fate determinants (45).
Work in C. elegans then showed that ACD can be broadly divided into four main stages (1): symmetry breaking (2), polarity establishment (3), segregation of cell fate determinants, and (4) spindle positioning (43). In the C. elegans embryo, symmetric breaking is induced by the fertilization and the entry of the sperm pronucleus, which leads to a local loss of actin and non-muscle myosin II-mediated cell surface contractions at the posterior pole (46). To establish polarity, the cortical actomyosin network then retracts until the contractile cortex covers the anterior and the non-contractile cortex covers the posterior half of the embryo (46). Cell fate determinants then segregate towards specific regions of the polarized mother cell and the spindle positions closer to the posterior cortex to generate a larger and a smaller daughter cell after division (46).
Based on these observations, ACD can be described as the unequal segregation of specific molecules into the daughter cells during division, that can act as developmental determinants causing the acquisition of different cell fates (47). Importantly, this means that the experimental proof for ACD, as in Conklin’s original experiment, requires linking asymmetric segregation of factors during division to predictable changes in the fate and/or behavior of daughter cells or their offspring (Figure 1). While evidence from invertebrate systems suggests that many factors, including organelles, RNA, and transcription factors can act as cell fate determinants (48), invertebrate models also show that cells can utilize evolutionary conserved polarity proteins in very different ways. For instance, in Drosophila neuroblasts (NB), in the presence of the adaptor protein Inscuteable, which binds members of the Par-6/Par-3 and the Pins/Gαi/Mud complexes, both complexes colocalize at the apical cortex (49). In contrast, in the SOP and the absence of Inscuteable, these complexes localize to opposite cortical domains during ACD (49). The differences between SOP and NB highlight that the precise molecular mechanisms of ACD can differ between cell types and tissues, and observations from invertebrates should not be directly extrapolated to mammalian cells.
3 Asymmetric cell division in HSCs: a decade-long controversy
The decision between self-renewal and differentiation has to be tightly regulated as the production of either too many or too few HSCs can lead to malignancies or tissue degeneration (50). Multiple mechanisms acting either on individual HSCs and/or the entire stem-cell population were suggested to explain how HSC numbers can be maintained for the life of an organism while at the same time creating differentiated offspring (51). Based on observations in invertebrate model organisms, ACD of HSCs was speculated as one possible mechanism (50). However, due to technical challenges, including extremely low HSC numbers, their slow cell cycle kinetics, their high motility, and low adhesion, neither divisions of highly purified HSCs, nor the asymmetric segregation of cell fate determinants during HSC divisions had been observed at the time.
Early evidence for ACD of HSCs came from in vitro and in vivo paired daughter cell assays after HSPC divisions, which showed that HSPC daughter cells can give rise to different-sized colonies and can differ in their ability to reconstitute the hematopoietic system after transplantation (52–54). Imaging living HSCs, isolated from transcriptional reporter mice, expressing GFP from the Hes1 or Tie2 locus later showed that reporter gene expression level of HSC daughters can diverge quickly within hours after division (Figures 2A, B). However, whether these changes were caused by events during division remained unclear, as the postmitotic changes in GFP expression were not linked to any asymmetric segregation during HSC divisions (55). Hence, alternative mechanisms of differential daughter cell fate acquisition not related to division could not be excluded.
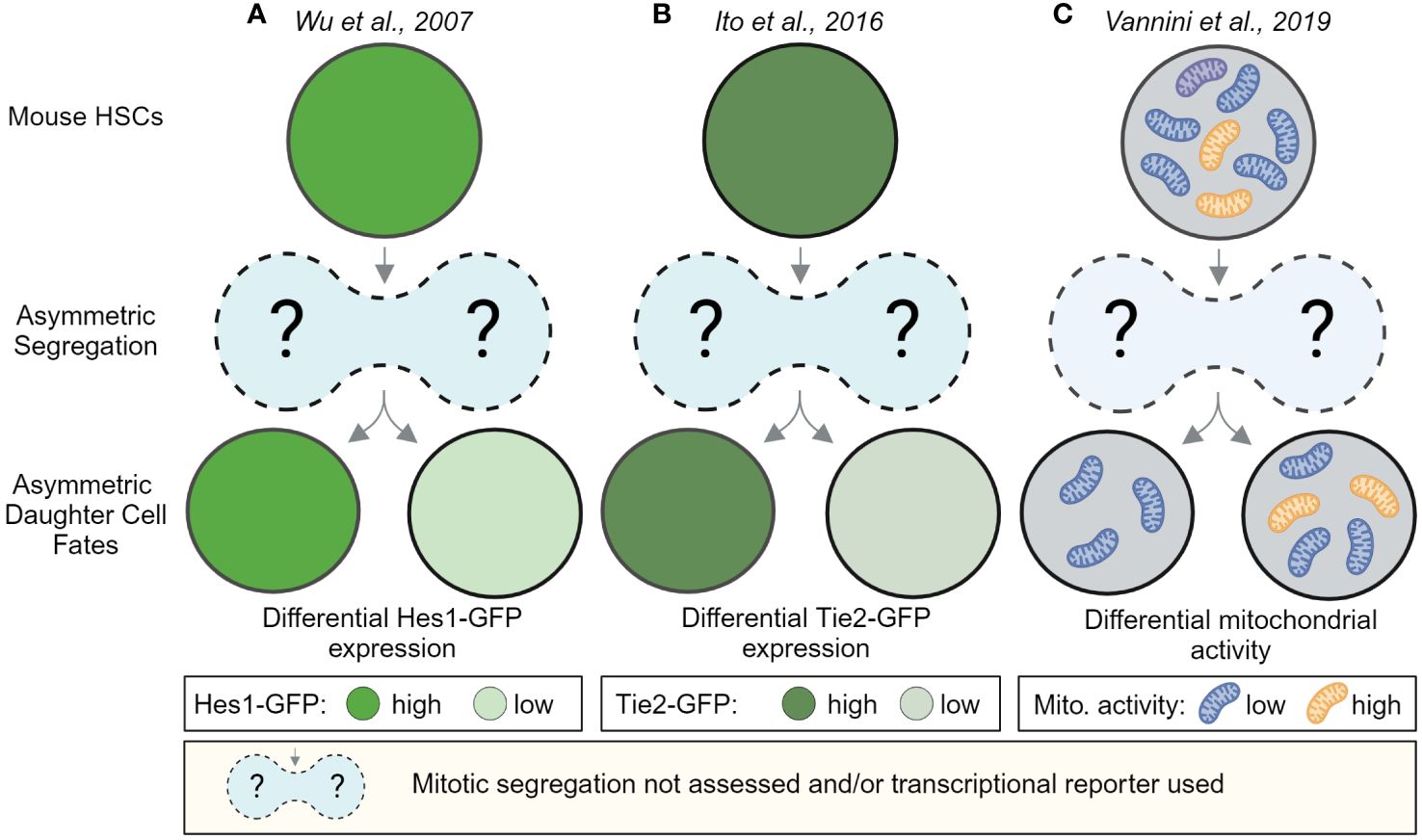
Figure 2 Postmitotic changes in marker expression provide evidence for asymmetric HSC daughter cell fates. HSC daughter cells can acquire asymmetric fates after division. In vitro, live-cell observations of HSCs showed that marker expression between HSC daughters can change over time. These studies provide evidence that HSC daughter cells can differ in the expression of the Notch target gene Hes1-GFP (A) the Angiopoietin receptor Tie2-GFP (B) and can differ in their mitochondrial activity as assessed by the fluorescent dye TMRM, an indicator for mitochondrial membrane potential (C). While, Wu et al. and Ito et al. used transcriptional reporters, which cannot be used to observe the asymmetric segregation of factors during mitosis, Vannini et al. analyzed TMRM levels hours after and not during divisions. These studies thus provide evidence for asymmetric daughter cell fates in HSCs but do not provide insights about events during HSC divisions. Created with BioRender.com.
Since the early 2000s, evidence for asymmetrically segregating factors in dividing hematopoietic cells has accumulated further supporting the idea that HSCs could use ACD (Figures 3A, B). However many of these studies were done in fixed cell lines and/or heterogeneous populations with very low HSC purities, and as recently revealed did not use reliable mitotic markers (56). It is thus possible that some of these studies analyzed postmitotic cells instead of cells during division. Consequently, only a few studies provided convincing evidence of asymmetric segregation during HSC divisions and the asymmetric segregation of many factors requires experimental validation using state-of-the-art tools that were often not available at the time. Importantly, providing convincing evidence for the asymmetric inheritance of cell fate determinants in HSCs is technically challenging as ACD requires (1) the identification of asymmetrically segregating factors during HSCs divisions that (2) lead to predictable changes in HSC daughter cell fate and/or behavior as in Conklin’s original observation (47, 57).
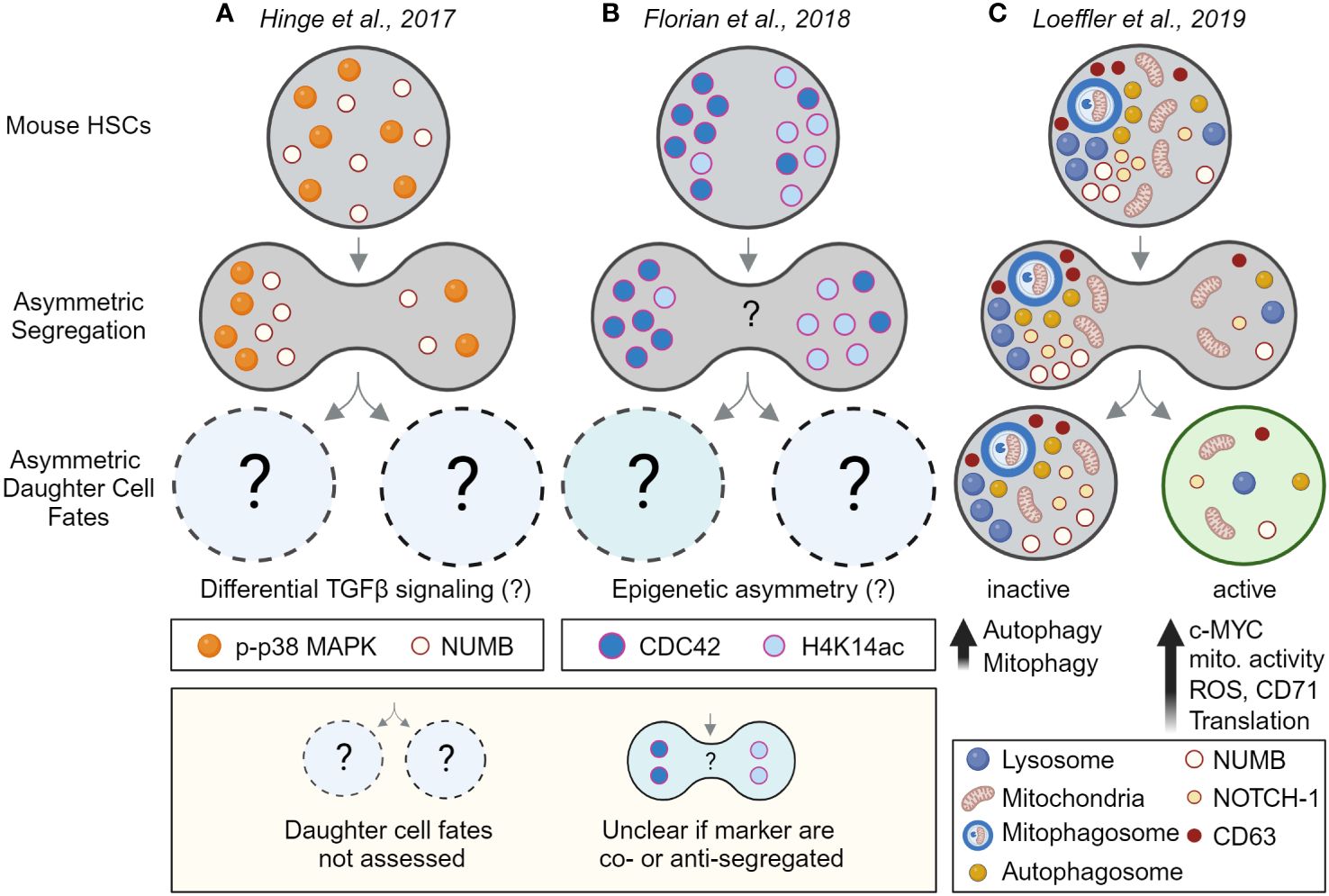
Figure 3 Evidence for asymmetric cell division in mouse HSCs by linking asymmetric segregation to later daughter cell fates. Evidence for (A, B) the asymmetric segregation of factors and (C) for asymmetric cell division in HSCs. Analyzing HSC divisions in fixed samples suggests (A) phospho-p38 MAP kinase (p-p38 MAPK) and NUMB, and (B) the RhoGTPase CDC42 and acetylated histone 4 (H4K14ac), segregate asymmetrically during HSC divisions. However, as the cells were fixed, future daughter cell fates could not be assessed. Although differential TGFβ signaling and epigenetic asymmetry between HSC daughter was suggested by other experiments (not shown), asymmetric segregation and daughter cell fate could not be directly linked to each other. That the observed asymmetric segregation of factors has functional consequences for HSC daughters could thus not be demonstrated directly. Also, the inheritance of CDC42 and H4K14ac was not assessed in the same experiment, it thus remains unclear if these markers segregate into the same or the opposite daughter cells. (C) Experimental proof that HSCs utilize ACD to regulate daughter cell fates. Long-term live-cell imaging and quantifications showed that lysosomes and other factors can segregate asymmetrically during HSC divisions and predict later changes in metabolic activity, translation, and differentiation in HSC daughters and their offspring. This study thus links for the first time asymmetric segregation of factors in dividing HSCs to later asymmetric daughter cell fates – thus demonstrating ACD of HSCs. Created with BioRender.com.
HSCs are extremely rare and less than 1% of cells are in division at any given time. Capturing highly purified dividing HSCs in fixed samples is thus both challenging and strongly biased as 99% of HSCs are excluded from the analysis. Live cell imaging on the other hand is the ideal tool for studying rare mitotic cells as 100% of HSC divisions are captured without bias. Choosing the right assay is thus critical for accurately studying HSC division dynamics and many classical in vitro and in vivo assays cannot be used to study ACD (see Table 1). Early attempts using live-cell imaging of more purified HSPCs identified AP2A2 as a positive regulator of HSC function and suggested asymmetric partitioning of AP2A2 in dividing mouse CD150+CD48-LSK HSCs (58). However, data acquisition and analysis of live-cell imaging experiments is challenging and requires specialized equipment, software, and know-how as cell viability and focus must be maintained for many days. At the same time, millions of acquired images need to be stored, transferred, and processed efficiently, to then segment, track, and analyze single HSCs over many cell generations (59, 60). Although Ting et al. were able to image living HSCs as they divide, 90% of HSCs with asymmetric AP2A2 died shortly after division, making it impossible to demonstrate that asymmetric mitotic segregation of AP2A2 determines HSC daughter cell fate acquisition (58). Whether HSCs use ACD thus could not be answered conclusively. Also, Zimdahl et al. used live-cell imaging to investigate whether NUMB is asymmetrically segregated in dividing hematopoietic cells. Overexpression of NUMB-CFP or NUMB-YFP in LSKs yielded contradicting results (61). While some cells showed asymmetric segregation of NUMB-CFP, no clear asymmetric segregation of NUMB-YFP could be observed during HSPC in vitro divisions (62). As it remained unclear which NUMB reporter faithfully reports the localization of endogenous NUMB, and Zimdahl et al. did not follow the HSC daughter cells after division, the putative unequal segregation of NUMB could not be linked to changes in later daughter cell fates. Whether HSCs use ACD thus remained unclear (47, 57).
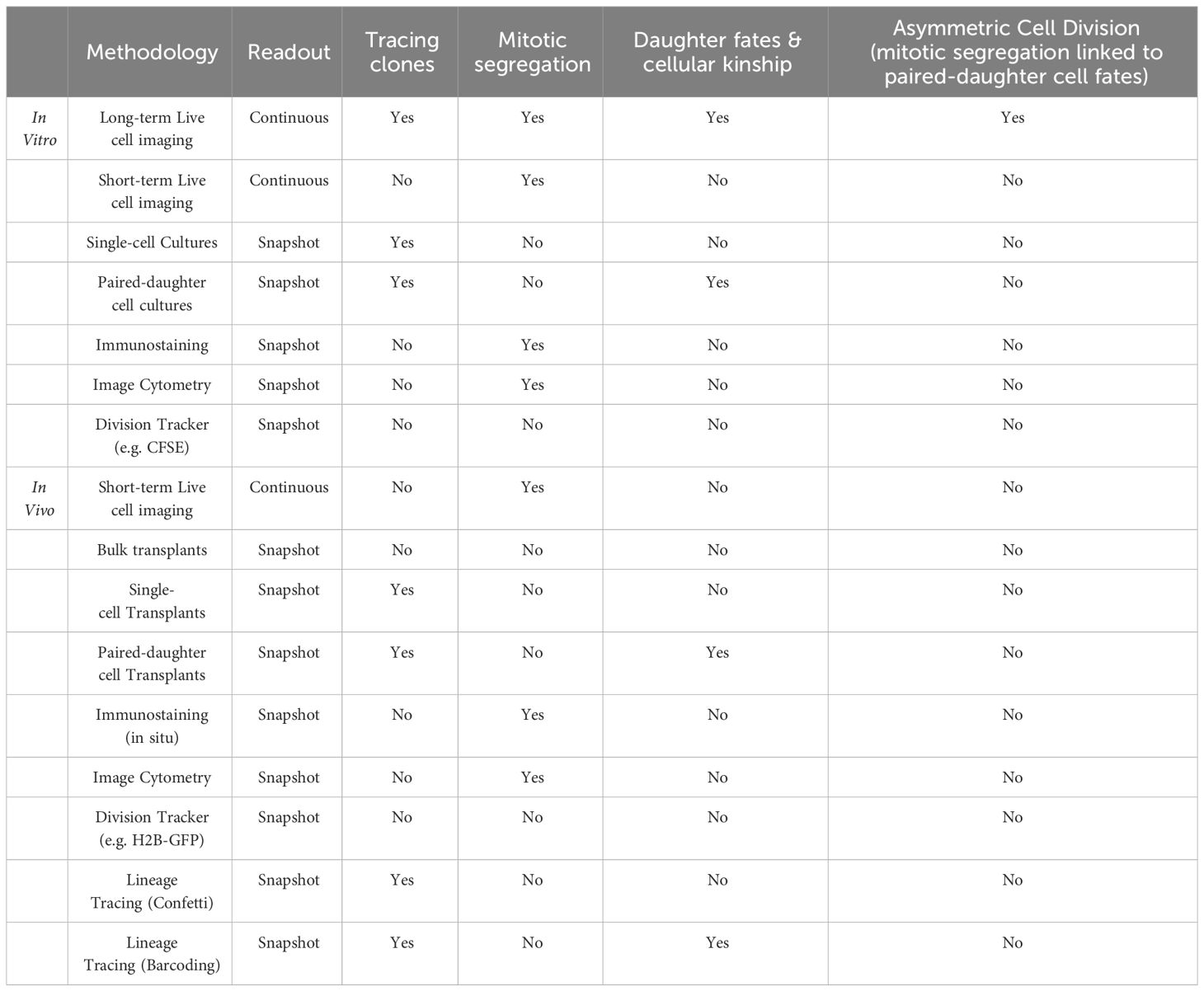
Table 1 Single-cell assays and their limitation to study asymmetric cell division.
While differential cell fate acquisition in paired-daughter cell assays and asymmetric mitotic segregation of factors suggested HSCs might use ACD, the deletion of multiple evolutionary conserved polarity proteins did not affect HSC function (63), arguing that HSCs might not use ACD after all. Neither the deletion of numb and numb-like in mouse HSPCs nor the deletion of Par-complex proteins aPKCζ or aPKCλ in LSK CD34−Flk2− HSCs affected the self-renewal and differentiation properties of HSCs (64, 65). Although it remains unknown why the loss of classical polarity proteins does not affect HSCs, it is possible that compensatory mechanisms, such as the recently discovered non-sense mediated decay (66), were triggered in these early genetic models and phenotypes masked.
Additional evidence against ACD in HSCs also came from Kiel et al., who tested the ‘Immortal strand hypothesis’ originally described by John Cairns in 1975. Cairns proposed that old and newly synthesized DNA strands might segregate asymmetrically during stem cell divisions - a mechanism that would reduce to chances of acquiring mutations in the stem cell pool. In his model, daughter cells fated to differentiate inherit newly synthesized DNA, while the older DNA strands are retained in daughters with stem cell properties (67). Kiel et al. injected BrdU into mice to label freshly synthesized DNA of HSCs in vivo. The analysis of BrdU label-retention at several time points after injection showed that HSCs did not segregate the newly synthesized DNA asymmetrically and thus did not retain older DNA strands as expected by Cairn’s model (67).
4 Direct evidence for asymmetric cell division in HSCs
Recent technical advances in microscopy, imaging, and bioinformatics now enable quantitative long-term live-cell imaging studies over many weeks and cell generations without affecting cell viability. Further, the development of sophisticated live cell imaging reporters including fluorescent proteins and cell-permeable fluorescent probes allows for the visualization and quantification of specific molecular events within dividing HSCs (see Table 2). Using novel software (60) and tissue culture coating to reduce the motility of hematopoietic suspension cells, the inheritance of factors during HSC divisions can now be quantified and linked to later changes in HSC daughter fates and behavior. Importantly, the quantification of asymmetric mitotic segregation and cell fate acquisition is done in the same experiment and relies on objective image-based single-cell dynamics quantifications and statistical analysis, instead of the manual scoring of symmetry vs. asymmetry by the human eye, as done in earlier studies. With advanced long-term single-cell imaging tools, and molecular profiling using scRNAseq, measuring how the asymmetric segregation of factors during HSC divisions is linked to later cell fate acquisition became possible. Using these tools, several recent studies provided direct experimental proof that HSCs can utilize ACD to regulate cell fate decisions (39, 71–73).
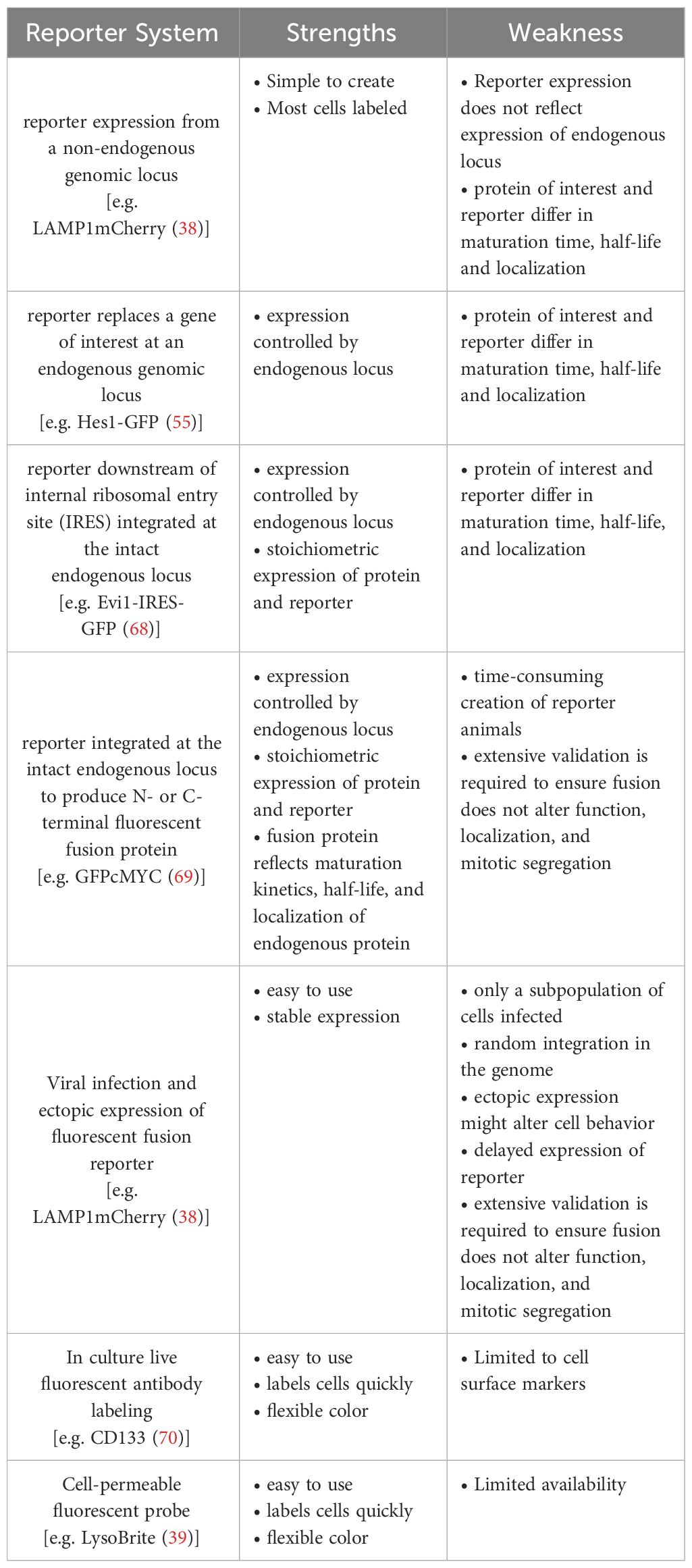
Table 2 Strength and weaknesses of continuous live cell imaging reporter.
Similar to Conklin’s original work a century ago, these studies showed that lysosomes and other organelles including autophagosomes, mitophagosomes, and recycling endosomes can segregate asymmetrically in dividing HSCs and predict the activity of their offspring after division (Figure 3C). Indicative of metabolic activation, HSC daughters receiving fewer lysosomes (LysoLow) upregulate the transcription factor c-MYC, increase mitochondrial membrane potential, produce more reactive oxygen species (ROS), upregulate the transferrin receptor CD71 and increase their overall translational activity. In contrast, HSC daughters receiving more lysosomes (LysoHigh) retain an overall lower metabolic state and show signs of increased autophagy as autophagosomes and mitophagosomes are asymmetrically co-inherited with lysosomes into LysoHigh daughters (38, 39), suggesting mitochondrial activation might be suppressed in LysoHigh daughters. Interestingly, while LysoHigh daughter cells also receive more CD63, NUMB, and NOTCH1, no clear evidence for asymmetric segregation of the evolutionarily conserved polarity proteins Inscuteable, PRKCζ, and PRKCI was found (65). Taken together, these findings demonstrate that HSCs use ACD to regulate daughter cell fate (Figure 3C) and suggest that asymmetric NUMB and lysosome inheritance might work together to modulate Notch signaling in HSC daughters to regulate cell fate decisions (38).
Long-term time-lapse imaging also showed recently that ACD is evolutionarily conserved and used by human HSCs (huHSCs). Highly purified CD49f+ cord blood-derived huHSCs were imaged for 2+ weeks and their offspring were tracked for four cell generations. As in mouse HSCs, lysosomes can segregate asymmetrically during huHSC divisions and predict later changes in marker expression in daughter cells (Figure 4C). In line with the idea that LysoHigh daughters retain stem cell properties, these daughters express higher levels of the huHSC marker CD49c, while LysoLow daughter cells give rise to offspring with high CD33 levels, indicative of myeloid differentiation (39). Importantly, the ability of huHSCs to differentiate into myeloid cells occurs almost exclusively in cells with few lysosomes, suggesting lysosomes may act as cell fate determinants and inhibit differentiation of LysoHigh huHSCs (39). While the mechanisms behind these observations remain poorly understood and need to be addressed in future studies, ACD of HSCs appears to be an evolutionary conserved mechanism from mice to humans.
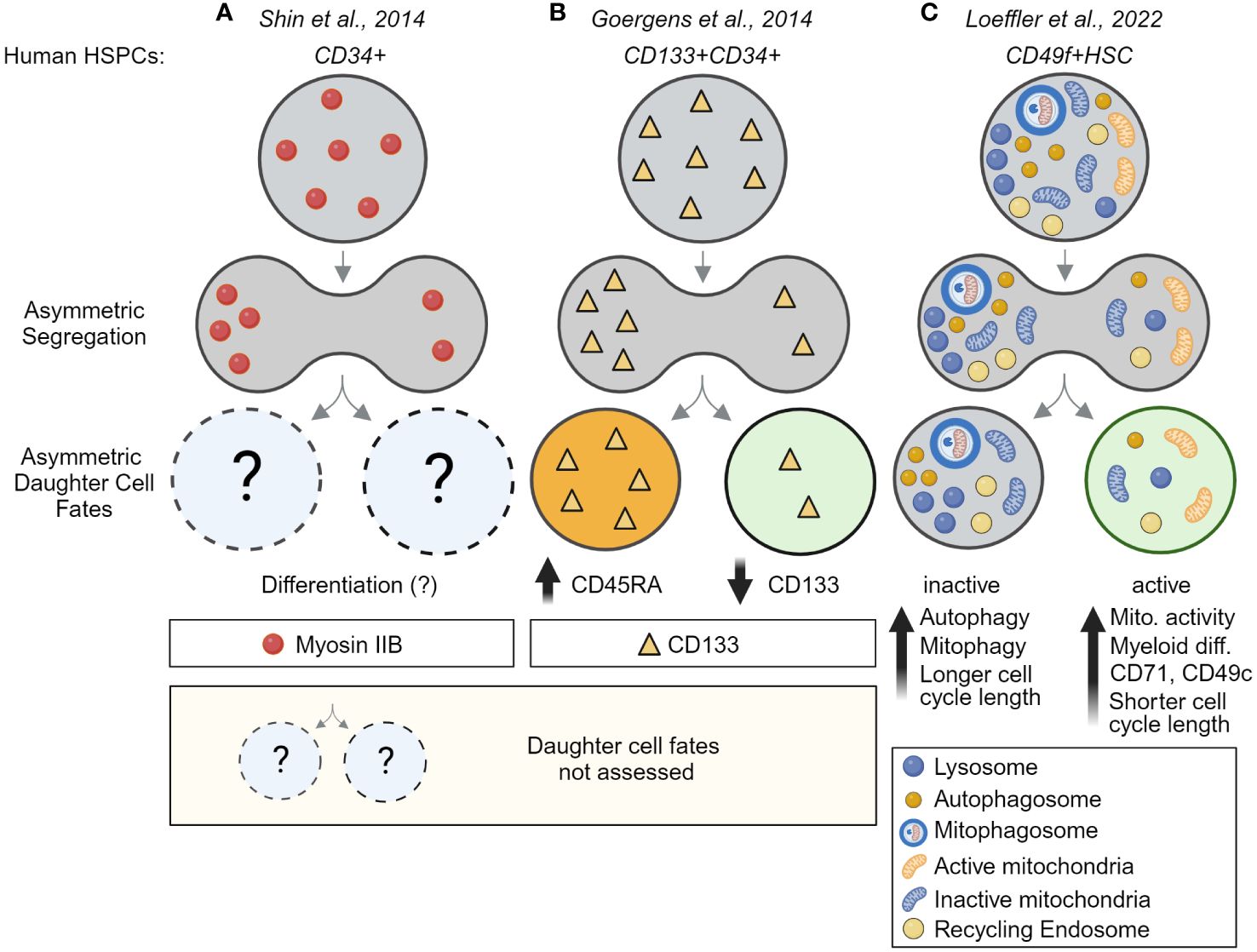
Figure 4 Asymmetric cell division is an evolutionarily conserved process – evidence for ACD in human HSCs. (A) Evidence for asymmetric segregation of Myosin IIB in dividing fixed CD34+ HSPCs. Because daughter cells were fixed, the asymmetric segregation could not be linked to future daughter cell fates. (B) Asymmetric inheritance of CD133 during the division of CD133+CD34+ HSPCs predicts CD45RA upregulation and links CD133 segregation to later differentiation and lineage choice. This study provides evidence for ACD in human multipotent progenitors but did not analyze human HSCs. (C) Direct demonstration of ACD in highly purified human HSCs. Long-term time-lapse imaging of HSC divisions their offspring over multiple cell generations showed lysosomes and other organelles, including autophagosomes, mitophagosomes, and recycling endosomes can segregate asymmetrically into the same HSC daughter cell, while active mitochondrial (TMRM) segregate into daughters receiving fewer lysosomes. Importantly, this asymmetric segregation of segregation predicts mitochondrial activity, myeloid differentiation, cell cycle length, and marker expression in HSC daughter and their offspring. This study thus provides the first direct evidence for the existence of asymmetric cell division in human HSCs. Created with BioRender.com.
To obtain further insights into the molecular programs driving cell fate divergence after ACD of HSCs, Wehling et al. developed trackSeq, a pipeline to image, track, and quantify HSC divisions in real time. Real-time analysis is critical to isolate and analyze the transcriptome of HSC daughters shortly after asymmetric division to understand the first steps in cell fate divergence (74). Using time-lapse imaging, single-cell picking, and scRNAseq, they were able to identify how gene expression programs diverge in closely related HSC daughter cells shortly after ACD. Because the kinship, mitotic segregation, and time after division were recorded, this approach dramatically reduced the measurement noise and improved the identification of candidate genes relevant for daughter cell fate divergence following ACD at the same time. Using the increased sensitivity, HSC daughters after ACD were found to activate different gene expression programs shortly after division, modulating translation, oxidative phosphorylation, cell cycle progression, and adhesion of HSC daughters. One of the top candidates, the Integrin β4 (ITGB4/CD104), was shown to be upregulated in LysoLow HSC daughters after ACD modulating the adhesion and motility of HSC daughters to LAMININ 511, an important component of the extracellular matrix in the bone marrow (74).
5 The evolving role of lysosomes - not just a bystander
Although paired-daughter cells were not analyzed directly, several recent studies using genetic models and lysosomal inhibitors support the idea that lysosomes are critical regulators of HSC function and might act as cell fate determinants (21, 75). These novel insights were surprising as lysosomes, after their discovery by Christian de Duve in the 1950s, were long considered static terminal organelles that are only required for cellular waste disposal and recycling and thus received little attention (76). However, this view changed dramatically, and it is clear today that lysosomes are critical for many cellular processes, including plasma membrane repair, regulating gene expression, metabolism, migration, and signaling (76). Lysosome numbers and composition respond to environmental cues and cellular needs, and lysosomes engage in functional interactions with other organelles via membrane contact sites (77, 78). Further, lysosomes are mobile and change in size, shape, and cellular localization (79).
As the terminal cellular compartment for degradation, lysosomes are central regulators of autophagy, the cellular process of recycling non-essential or damaged cellular material through lysosomal degradation (80). Autophagy is critical for HSC function as loss of ATG7 and ATG12 impair HSC repopulation activity and self-renewal (33, 81, 82). In contrast to autophagy, which relies on the step-wise formation, maturation of autophagosomes and their fusion with lysosomes, chaperone-mediated autophagy (CMA) involves the transportation of proteins directly into the lysosomal lumen through the lysosomal associated membrane protein 2a (LAMP2A) and the cytosolic chaperone HSC70 (80). CMA is required for HSC maintenance and function, as Lamp2a knock-out HSCs display impaired self-renewal (75). Furthermore, quiescent HSCs have higher basal CMA activity than HSCs undergoing activation, suggesting that lysosomes are critical for HSC function by maintaining quiescence. As such, quiescent HSCs have higher expression levels of lysosomal genes than activated HSCs with lower repopulation potential. However, a recent study showed that stem cell function and quiescence of activated HSCs can be restored by suppressing lysosomal activity with a V-ATPase inhibitor (21).
An important regulator of lysosomal function is the transcription factor EB (TFEB), a tumor suppressor that responds to stress signals and metabolic cues, like mitochondrial damage or nutrient starvation, via transcriptional activation of autophagy and lysosomal biogenesis genes (83, 84). TFEB is highly expressed in quiescent huHSCs, and MYC-induced repression of TFEB-associated lysosomal programs was shown to promote huHSC activation and anabolism (85). TFEB induces clearance of the transferrin receptor TfR1 from the membrane of huHSCs through endolysosomal degradation (85). Based on these observations Garcia-Prat et al. proposed that a TFEB-induced lysosomal program removes and degrades cell surface proteins to maintain huHSCs quiescence by making them more refractory to mitogenic signals (85).
Taken together, lysosomes regulate quiescence to protect HSCs from exhaustion (86). When needed, HSCs receive cues to exit their dormant state, become active, and adapt their metabolism to meet increased bioenergetic demands for cell growth and division (87). At the same time, cellular and metabolic activation need to be suppressed in a subset of HSCs so that they can re-enter quiescence and maintain the stem cell pool by self-renewal (87, 88). HSCs coordinate these processes using ACD and the asymmetric segregation of lysosomes that promotes quiescence. ACD might thus act as a protective mechanism that ensures one HSC daughter returns to a quiescent state.
6 Asymmetric inheritance of mitochondria in HSCs
Besides lysosomes, mitochondria can also segregate asymmetrically during divisions. Using photoactivatable green fluorescent protein (paGFP) tagged mitochondrial proteins to label “old” vs. “young” mitochondria, “old” mitochondria were originally found to partition asymmetrically in stemlike cells (SLCs) from immortalized human mammary epithelial cells (89). Interestingly, daughter cells inheriting fewer “old” mitochondria were more stemlike based on their ability to form mammospheres in culture (89). Similar to these findings, during HSC divisions, daughters receiving more lysosomes inherit more mitophagosomes, specialized autophagosomes responsible for degrading “old” and/or damaged mitochondria (38). However, in contrast to SLCs, HSC daughters inherit more mitophagosomes, and therefore “old” mitochondria are thought to retain stemness, suggesting HSCs inherit damaged cellular organelles and components while daughter cells destined for activation and differentiation receive fresh organelles (89). While the inheritance of damaged cellular constituents by HSCs might be at first glance counter-intuitive, similar patterns of segregation of “old” vs. “new” components have been observed in S. cerevisiae, where mother cells inherit damaged, lifespan-limiting material, and the “bud” is rejuvenated by receiving freshly produced highly functional organelles (48).
Using mito-EGFP mice to label all mitochondria and the fluorescent probe TMRM to quantify mitochondrial activity, Vannini et al. showed that the mitochondrial activity in HSC daughter cells can also differ after division (Figure 2C). Interestingly, this asymmetry in mitochondrial activity of HSC daughter cells increased in HSCs exposed to the NAD+-boosting agent nicotinamide riboside. Although only postmitotic events in paired daughter cells were measured, Vannini et al. speculated that “old” and active mitochondria segregate into opposite daughter cells during division (90). Whether differential mitochondrial activity in HSC daughter cells after division was a consequence of asymmetric segregation of active mitochondria or caused by other mechanisms remained unclear. Long-term imaging studies of dividing HSCs stained for both lysosomes and active mitochondria showed that differential mitochondrial activity in HSC daughters occurs after division, supporting the idea that changes in mitochondrial activity are induced after division (38). Because changes in mitochondrial activity in HSC daughters could be predicted based on asymmetric lysosomal segregation during HSCs, it is tempting to speculate that lower numbers of lysosomes permit and/or induce mitochondrial activity (38). Similar postmitotic changes in mitochondria were found using HSCs isolated from mito-Dendra2 mice (29). Instead of being asymmetrically segregated during division, the mito-Dendra2 expression increases in one HSC daughter approximately 50 minutes after division (29). However, as mito-Dendra2 does not reflect mitochondrial activity, it is not intuitively clear how to explain the sudden increase of the reporter in one daughter cell. As HSC daughter cells can diverge in their global translational activity after asymmetric lysosome segregation (38) (Figure 3C), the postmitotic increase of mito-Dendra2 in one HSC daughter might be explained by an increased production of the reporter in the daughter prone to activation.
Despite the conflicting evidence in mouse HSCs, recent live-cell imaging of dividing huHSCs shows that a small fraction of TMRM-positive active mitochondria are asymmetrically segregated during division (39) (Figure 4C). Importantly, although the fraction of asymmetrically segregating active mitochondria was small compared to lysosomes, the asymmetry was sufficient to predict later mitochondrial activity in huHSC daughter cells (39). Interestingly, lysosomes and active mitochondria segregate into different daughter cells during huHSC divisions (39), supporting the idea that the LysoLow daughter is prone to metabolic activation as shown in mouse HSCs. Contrary to earlier beliefs (91), it further demonstrates that the inheritance of organelles during HSCs division is a highly coordinated process (Figure 4C). Another important finding of this study is that the metabolic regulation of HSC daughter cells might be different between mice and humans (compare Figures 3C, 4C). However, the existence of any putative mechanisms specific to human HSCs will require further validation as the observed differences could simply reflect the differences between adult bone marrow-derived HSCs and huHSCs isolated from umbilical cord blood.
Taken together, the asymmetric segregation of activated mitochondria during mouse HSC divisions remains controversial. The available experimental data in mouse HSCs supports a model in which postmitotic changes in mitochondrial activity could be regulated by lysosomes. This model is supported by observations in other model systems, where mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics and mutations in lysosomal genes cause mitochondria dysfunction, as often observed in patients with Lysosomal Storage Disorders.
7 Evidence for asymmetric segregation of other organelles and markers
While recent live-cell imaging experiments have shown that lysosomes and mitochondria are asymmetrically segregated during HSC divisions and can be linked to later changes in HSC daughter cell fates, very little is known about the inheritance of other organelles and markers in HSCs. Myosin IIB (MIIB), the major non-muscle myosin II, was found to segregate asymmetrically in human HSPCs and suggested to regulate differentiation of CD34+ cells (Figure 4A), but mitotic segregation of MIIB was not linked to later cell fates. Autophagosomes, mitophagosomes, and recycling endosomes have also been found to asymmetrically co-segregate with lysosomes during HSC division. However, how this asymmetry relates to the mitotic segregation of centrosomes, endoplasmic reticulum and the maternal DNA strand that were reported to segregate asymmetrically in other model systems remains unclear (48). As the number of reported asymmetrically segregating factors increases, it appears that asymmetric inheritance is the norm rather than the exception. However, as the connection to later differential cell fate acquisition is challenging to analyze, it is often unclear whether these observed asymmetries influence cell fates, as we are only starting to reveal the underlying processes (48).
7.1 Role of the Rho GTPase CDC42
Rho GTPases are a family of small signaling G proteins known to regulate the cytoskeleton and cell polarity (92, 93). They exist in an inactive GDP-bound and an active GTP-bound form that was originally described to regulate cell polarity and ACD in yeast (94). CDC42 activity needs to be tightly regulated for appropriate HSC function as deficiency of CDC42 causes impaired adhesion, lodging, and retention of mouse HSCs leading to egress of HSCs from BM to distal organs and engraftment failure (95). In HSCs isolated from young mice, the Rho GTPase CDC42 was found to be polarized, while HSCs isolated from aged mice showed an apolar distribution of CDC42 (96, 97). In line with these observations, CDC42 segregates asymmetrically during divisions of young HSCs, while most divisions of aged HSCs are symmetric (97). Interestingly, both the loss in CDC42 polarity and asymmetric segregation can be reversed using the CDC42 inhibitor CASIN which also seems to restore the function of aged HSCs. While these observations suggest that CDC42 regulates HSC daughter cell fates, linking asymmetric CDC42 segregation to later cell fate acquisition has proven to be difficult. Extensive work on paired HSC daughter cells using scRNAseq, scATACseq, and single-cell transplants, suggested that HSC daughters differ strongly in their chromatin accessibility and function but not their transcriptome. Using mathematical modeling, data from different paired daughter cell assays were integrated to computationally match the frequencies of asymmetric daughter cell fates and maximize the correlation with asymmetric CDC42 segregation in fixed samples (Figure 3B). While this model suggests a possible link between CDC42 asymmetry and later paired-daughter cell fates, it is important to realize that the model can only assume that these two processes are linked (97). Direct experimental proof that CDC42 asymmetric segregation of CDC42 causes functional differences in HSC daughter cells is still missing and understanding the precise role of the Rho GTPases during HSCs division remains a key challenge for the field.
A recent study using paired daughter cell assays with in vivo transplantations and gene profiling experiments started to shed light on the regulation of Rho GTPases in HSCs. Using immunofluorescence staining of fixed cells they identified that the RhoGAP p190-B, a negative regulator of Rho GTPase signaling regulates asymmetric segregation of the phosphorylated p38 MAP kinase in HSC daughters (Figure 3A). They further showed that p190-B regulates TGF-β signaling activity in HSC daughters, a key pathway required for HSC quiescence (98). Interestingly, CD63 and YAP downstream of the Hippo pathways also modulate TGF-β signaling and were shown to regulate HSCs quiescence or to colocalize with polarized CDC42 (99), respectively. While CD63 can asymmetrically segregate during HSC divisions itself, CDC42 and YAP1 polarity depends on the polarity regulator Scribble (99). While further studies are required to understand how precisely CDC42, CD63, and YAP1 regulate TGF-β signaling in HSCs to establish and/or maintain quiescence, CDC42 also emerges as a key regulator of asymmetry in HSCs.
7.2 RNA binding proteins
RNA binding proteins (RBPs) regulate the localization and translation of mRNA and have emerged as important regulators of stem cell fate decisions (100). Several RBPs, including MSI2, SON, and SYNCRIP were shown to regulate HSC function, and their asymmetric segregation during HSC divisions was suggested (101, 102). While these studies establish that RBPs are important regulators for HSC fate determination, their role during division is less clear. Observations of asymmetrically segregating RBPs rely solely on the analysis of fixed cells treated with the tubulin depolymerization agent nocodazole. It is thus possible that RBP localization and inheritance changed by nocodazole as reported in other systems (56). Furthermore, as the asymmetric segregation of RBPs has not been linked to later HSC daughter fates, further validation, ideally using live-cell imaging and immunofluorescence without nocodazole and mitotic markers such as tubulin, will thus be required to firmly establish that RBPs act during HSC division.
7.3 Cells surface proteins
Although less well studied, cell surface markers were also suggested to segregate asymmetrically during HSC divisions, including CD53, CD63, CD71, CD62L and CD133 in human CD34+ HSPCs and Tie2, CD48, JAM-3 and Glut1 in mouse HSCs (103) (Figures 2B, 4B). While the asymmetric segregation of CD63 and CD71 was later shown to predict HSC daughter cell fates in dividing highly purified HSCs using live-cell imaging (38, 39), the asymmetric segregation of other cell surface markers is less clear and requires validation (see Table 3). As most surface markers were analyzed in paired-daughter cells hours after division, the observed changes in expression between sister cells may thus result from other mechanisms not related to division. Postmitotic changes in translational activity, recycling, and degradation, are well-established markers of cell fate divergence in HSC daughters. However, as these changes occur only after asymmetric lysosome segregation during division, they are downstream effects and do not instruct cell fate divergence themselves (38, 39). Additionally, surface markers are well known to get internalized through endocytosis and traffic through multiple organelles, including recycling endosomes and lysosomes for their degradation (38). Asymmetrically segregating degradation products of cell surface markers can thus be easily confused with functionally relevant asymmetric segregation. It will thus be important to show that the asymmetric segregation relies on markers located outside the lysosomal degradative machinery and is linked to downstream function.
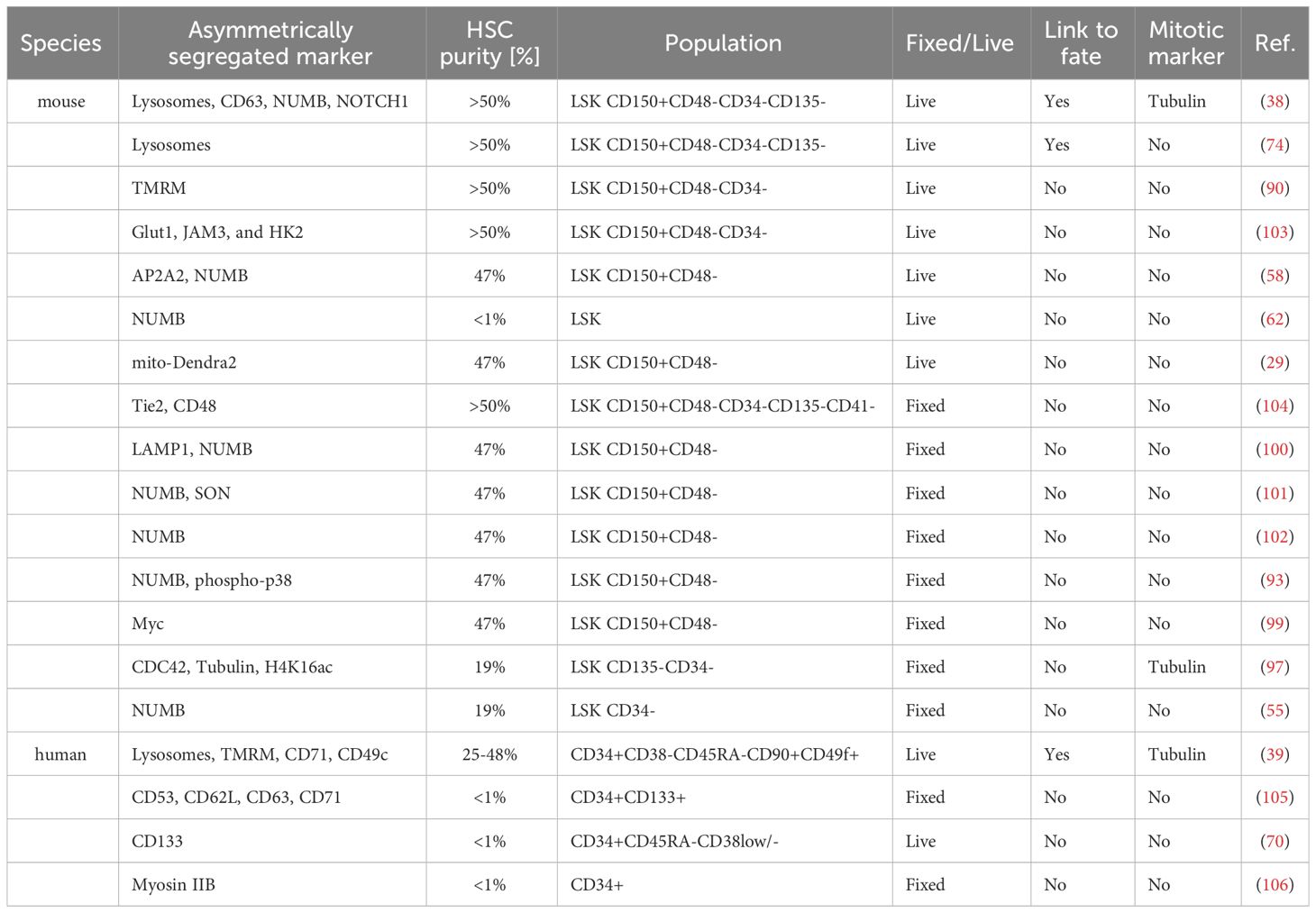
Table 3 Overview of studies evaluating asymmetric segregation of cellular components.
8 Discussion
HSCs need to carefully organize the division of their genetic material as well as other cellular components (107). While the inheritance of genetic material has long been recognized as a highly organized process, the mitotic segregation of organelles was initially thought to be stochastic (107). However, live-cell imaging studies of dividing mouse and human HSCs have recently shown that the inheritance of lysosomes and mitochondria are key regulators of HSC fate decisions and segregate in a coordinated manner (39, 107). Although the role of other organelles and factors during HSC division are less studied, accumulating evidence suggests that mitophagosomes, recycling endosomes, and CDC42 also segregate asymmetrically during HSC divisions. A key challenge for future studies will be to integrate these findings to create a comprehensive and cohesive picture of events during HSC divisions and how these events influence downstream cell fates of HSC daughters and their offspring.
The identification of the regulators and mechanisms of asymmetric organelle inheritance will be key moving forward. How are lysosomes asymmetrically partitioned during HSC divisions, and how do lysosomes instruct HSC daughter fates are central questions in the field. Lysosomes are known to move bidirectionally along microtubules during interphase, suggesting that similar mechanisms might be at play during HSC divisions (76). Retrograde movement from the cell periphery towards the perinuclear region is regulated via cytoplasmic dynein and its activator dynactin, while kinesins promote the anterograde movement from the perinuclear region towards the cell periphery (108–110). Many adaptor complexes are involved in mediating this coupling to dynein–dynactin or kinesins to regulate lysosomal positioning in response to different stimuli like cell starvation or growth factor stimulation (76). Evidence from Lis1-/- HSPCs suggests that NUMB segregation might be regulated by dynein-based transport mechanisms (62). As NUMB colocalizes closely with lysosomes in dividing HSCs, it is tempting to speculate that similar mechanisms regulate lysosome positioning via retrograde transport. Future studies devoted to understanding these complexes in HSCs will help us deduce how lysosomes are asymmetrically segregated during HSC development.
As HSCs reside in the bone marrow microenvironment, a better understanding of HSC-niche interactions could also provide novel insights into the extracellular cues regulating ACD in HSCs. While many niche factors regulating HSC function are known today, whether these factors also regulate ACD remains unclear as direct observations of HSC divisions in vivo are lacking. This would ideally be done using quantitative intravital microscopy at single-cell resolution capable of visualizing HSCs during division and tracking the HSC offspring for multiple cell generations. However, despite recent progress, in vivo, long-term live-imaging of HSCs is still impossible. The approximately one thousand HSCs in a mouse are scattered across the entire skeleton, are hidden in cubic centimeters of bone marrow, and divide on average every few weeks (111–113). Except for the calvaria, which harbors a thin sheet of bone marrow that can be imaged for short periods, most bones are inaccessible for in vivo live-cell imaging with single-cell resolution using current microscopes (114). The chances of finding a single HSC in vivo, capturing its divisions, and following its daughter cells until they divide again are not only extremely low but would at the same time require several weeks of continuous imaging of a large bone marrow volume and an unethical many week long immobilization of animals. In other words, live-cell imaging of dividing HSCs in vivo is a major technical barrier beyond today’s technical capabilities. For the foreseeable future, in vitro live-cell imaging will remain the only reliable way to quantify the ACD of HSCs. In combination with recently made progress in tissue clearing and 3-dimensional multicolor imaging of whole mouse femurs, dividing HSCs could be captured in situ in fixed samples. However, catching transient rare events such as divisions of extremely rare HSCs scattered across large bone marrow volumes and many bones will be challenging as even the most advanced light-sheet microscopes need to acquire, process, and analyze hundreds to thousands of terabytes of data to allow statistically meaningful quantitative analysis of highly purified HSC divisions.
In vivo, barcoding and fate mapping of HSCs could, in theory, be used to track HSC daughters long-term (115). For this purpose, Rodewald and colleagues developed a Polylox DNA cassette consisting of ten loxP sites integrated into the mouse genome to generate Cre recombinase-mediated unique cellular barcodes (115). Using this system, they were able to track the development of individual HSC clones for 9-11 months after birth and observed asymmetric expansion of large and small HSC clones with functional heterogeneity (115). However, studying ACD in vivo using this approach is currently not possible as HSC divisions cannot be directly observed, and the temporal resolution is limited. How intravital microscopy, barcoding, and fate mapping can be linked in a single experiment remains unclear.
Recently developed continuous long-term single-cell imaging tools thus remain the only way to link events during HSCs division to changes in cell fate and behavior many days or weeks later. While this approach does not recapitulate the in vivo microenvironment studying HSC behavior under controlled culture conditions, it will provide important insights into cell-intrinsic and cell-extrinsic mechanisms. These insights are important for ongoing biomedical efforts to maintain, expand, and/or manipulate HSCs ex vivo. As we delve deeper into understanding the intricate processes underlying ACD, it will also become important to understand the role of ACD in hematological malignancies. While multiple mechanisms for how ACD could contribute to leukemogenesis have been postulated, little experimental evidence is available today to back up these ideas (116, 117). As the technologies to study ACD reliably became only available recently, early studies proposing changes in asymmetry during divisions of leukemic cells could not functionally validate changes in daughter cell fates after division (55, 62, 118). It will thus be important to revisit some of the ideas proposed decades ago as we now possess the tools to quantify ACD and its impact on cell fate decisions. Future studies using live-cell imaging will need to determine how ACD changes during various stages of leukemia and whether manipulation of ACD can improve therapy.
Although single-cell tracking has enabled the first direct observations of ACD and other cell fate decisions over time, the technology is still in its infancy. The analysis of cellular genealogies remains challenging and requires interdisciplinary know-how in image analysis, computational methods, and statistics (8). The more widespread adaptation of these novel tools will be critical to improving our understanding of how HSCs utilize ACD to regulate healthy and malignant hematopoiesis.
Author contributions
JN: Visualization, Writing – original draft, Writing – review & editing. DL: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Alex’s Lemonade Stand Foundation (Grant GR-000003404) and the RUNX1 Research Program and the St. Jude Comprehensive Cancer Center Developmental Funds Grant (#PROJ-0003066 720202211).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee JY, Hong SH. Hematopoietic stem cells and their roles in tissue regeneration. Int J Stem Cells. (2020) 13:1–12. doi: 10.15283/ijsc19127
2. Riether C, Schurch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. (2015) 22:187–98. doi: 10.1038/cdd.2014.89
3. Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, Eaves CJ. Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc Natl Acad Sci U S A. (1990) 87:8736–40. doi: 10.1073/pnas.87.22.8736
4. Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature. (2019) 571:117–21. doi: 10.1038/s41586-019-1244-x
5. Rodling L, Schwedhelm I, Kraus S, Bieback K, Hansmann J, Lee-Thedieck C. 3D models of the hematopoietic stem cell niche under steady-state and active conditions. Sci Rep. (2017) 7:4625. doi: 10.1038/s41598-017-04808-0
6. Papa L, Djedaini M, Hoffman R. HSC expansion challenges the paradigm of unidirectional human hematopoiesis. Ann Ny Acad Sci. (2020) 1466:39–50. doi: 10.1111/nyas.14133
7. Till JE, McCulloch EA. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. 1961. Radiat Res. (2012) 178:AV3–7. doi: 10.1667/RRAV01.1
8. Loeffler D, Schroeder T. Understanding cell fate control by continuous single-cell quantification. Blood. (2019) 133:1406–14. doi: 10.1182/blood-2018-09-835397
9. Kelly LS, Darden DB, Fenner BP, Efron PA, Mohr AM. The hematopoietic stem/progenitor cell response to hemorrhage, injury, and sepsis: A review of pathophysiology. Shock. (2021) 56:30–41. doi: 10.1097/SHK.0000000000001699
10. Ritsma L, Ellenbroek SIJ, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, et al. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. (2014) 507:362–5. doi: 10.1038/nature12972
11. Roberts KJ, Kershner AM, Beachy PA. The stromal niche for epithelial stem cells: A template for regeneration and a brake on Malignancy. Cancer Cell. (2017) 32:404–10. doi: 10.1016/j.ccell.2017.08.007
12. Seshadri M, Qu CK. Microenvironmental regulation of hematopoietic stem cells and its implications in leukemogenesis. Curr Opin Hematol. (2016) 23:339–45. doi: 10.1097/MOH.0000000000000251
13. Zhang P, Zhang C, Li J, Han J, Liu X, Yang H. The physical microenvironment of hematopoietic stem cells and its emerging roles in engineering applications. Stem Cell Res Ther. (2019) 10:327. doi: 10.1186/s13287-019-1422-7
14. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. (2008) 135:1118–29. doi: 10.1016/j.cell.2008.10.048
15. Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. (2014) 141:4656–66. doi: 10.1242/dev.106575
16. Laurenti E, Frelin C, Xie S, Ferrari R, Dunant CF, Zandi S, et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. (2015) 16:302–13. doi: 10.1016/j.stem.2015.01.017
17. Bogeska R, Mikecin AM, Kaschutnig P, Fawaz M, Buchler-Schaff M, Le D, et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell. (2022) 29:1273–84 e8. doi: 10.1016/j.stem.2022.06.012
18. Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, et al. p57 and p27 Cooperate to Maintain Hematopoietic Stem Cell Quiescence through Interactions with Hsc70. Cell Stem Cell. (2011) 9:247–61. doi: 10.1016/j.stem.2011.07.003
19. Simsek T, Kocabas F, Zheng JK, DeBerardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. (2010) 7:380–90. doi: 10.1016/j.stem.2010.07.011
20. Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. (2011) 9:298–310. doi: 10.1016/j.stem.2011.09.010
21. Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, et al. Restraining lysosomal activity preserves hematopoietic stem cell quiescence and potency. Cell Stem Cell. (2020) 26:359–76 e7. doi: 10.1016/j.stem.2020.01.013
22. Weng C, Yu FL, Yang D, Poeschla M, Liggett LA, Jones MG, et al. Deciphering cell states and genealogies of human haematopoiesis. Nature. (2024) 627:389–98. doi: 10.1038/s41586-024-07066-z
23. Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, et al. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity. (2016) 45:597–609. doi: 10.1016/j.immuni.2016.08.007
24. Liu L, Michowski W, Kolodziejczyk A, Sicinski P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat Cell Biol. (2019) 21:1060–7. doi: 10.1038/s41556-019-0384-4
25. Ding Y, Liu Z, Liu F. Transcriptional and epigenetic control of hematopoietic stem cell fate decisions in vertebrates. Dev Biol. (2021) 475:156–64. doi: 10.1016/j.ydbio.2021.03.003
26. Dohla J, Kuuluvainen E, Gebert N, Amaral A, Englund JI, Gopalakrishnan S, et al. Metabolic determination of cell fate through selective inheritance of mitochondria. Nat Cell Biol. (2022) 24:148–54. doi: 10.1038/s41556-021-00837-0
27. Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. (2016) 17:183–93. doi: 10.1038/nrm.2016.8
28. Gao SW, Liu F. Novel insights into cell cycle regulation of cell fate determination. J Zhejiang Univ-Sc B. (2019) 20:467–75. doi: 10.1631/jzus.B1900197
29. Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, et al. Asymmetrically segregated mitochondria provide cellular memory of hematopoietic stem cell replicative history and drive HSC attrition. Cell Stem Cell. (2020) 26:420–30 e6. doi: 10.1016/j.stem.2020.01.016
30. Petruk S, Mariani SA, De Dominici M, Porazzi P, Minieri V, Cai JL, et al. Structure of nascent chromatin is essential for hematopoietic lineage specification. Cell Rep. (2017) 19:295–306. doi: 10.1016/j.celrep.2017.03.035
31. Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. (2008) 205:1395–408. doi: 10.1084/jem.20080277
32. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. (2011) 208:455–67. doi: 10.1084/jem.20101145
33. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. (2017) 543:205–+. doi: 10.1038/nature21388
34. Jin GX, Xu C, Zhang X, Long J, Rezaeian AH, Liu CF, et al. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat Immunol. (2018) 19:29–+. doi: 10.1038/s41590-017-0002-1
35. Hu MJ, Chen NC, Chen M, Chen F, Lu YK, Xu Y, et al. Transcription factor Nkx2-3 maintains the self-renewal of hematopoietic stem cells by regulating mitophagy. Leukemia. (2023) 37:1361–74. doi: 10.1038/s41375-023-01907-y
36. Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. (2013) 155:135–47. doi: 10.1016/j.cell.2013.08.031
37. Treichel S, Filippi MD. Linking cell cycle to hematopoietic stem cell fate decisions. Front Cell Dev Biol. (2023) 11:1231735. doi: 10.3389/fcell.2023.1231735
38. Loeffler D, Wehling A, Schneiter F, Zhang Y, Muller-Botticher N, Hoppe PS, et al. Publisher Correction: Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature. (2019) 573:E5. doi: 10.1038/s41586-019-1587-3
39. Loeffler D, Schneiter F, Wang W, Wehling A, Kull T, Lengerke C, et al. Asymmetric organelle inheritance predicts human blood stem cell fate. Blood. (2022) 139:2011–23. doi: 10.1182/blood.2020009778
40. Whitman CO. A contribution to the history of the germlayers in Clepsine. Boston: J Morphol. (1887). doi: 10.1002/jmor.1050010107
41. Conklin EG. The organization and cell-lineage of the ascidian egg. Journal of the Academy of Natural Sciences of Philadelphia (1905). doi: 10.5962/bhl.title.4801
42. Conklin EG. Mosaic development in ascidian eggs. J Exp Zoology. (1905) 2:145–223. doi: 10.1002/jez.1400020202
43. Gonczy P. Mechanisms of asymmetric cell division: flies and worms pave the way. Nat Rev Mol Cell Biol. (2008) 9:355–66. doi: 10.1038/nrm2388
44. Rhyu MS, Jan LY, Jan YN. Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell. (1994) 76:477–91. doi: 10.1016/0092-8674(94)90112-0
45. Goh LH, Zhou X, Lee MC, Lin SP, Wang HS, Luo Y, et al. Clueless regulates aPKC activity and promotes self-renewal cell fate in mutant larval brains. Dev Biol. (2013) 381:353–64. doi: 10.1016/j.ydbio.2013.06.031
46. Munro E, Nance J, Priess JR. Cortical flows powered by asymmetrical contraction transport PAR proteins to establish and maintain anterior-posterior polarity in the early C. elegans embryo. Dev Cell. (2004) 7:413–24. doi: 10.1016/j.devcel.2004.08.001
47. Horvitz HR, Herskowitz I. Mechanisms of asymmetric cell-division - 2 bs or not 2 bs, that is the question. Cell. (1992) 68:237–55. doi: 10.1016/0092-8674(92)90468-R
48. Sunchu B, Cabernard C. Principles and mechanisms of asymmetric cell division. Development. (2020) 147. doi: 10.1242/dev.167650
49. Neumüller RA, Knoblich JA. Dividing cellular asymmetry: asymmetric cell division and its implications for stem cells and cancer. Gene Dev. (2009) 23:2675–99. doi: 10.1101/gad.1850809
50. Schroeder T. Asymmetric cell division in normal and Malignant hematopoietic precursor cells. Cell Stem Cell. (2007) 1:479–81. doi: 10.1016/j.stem.2007.10.016
51. Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. (2006) 441:1068–74. doi: 10.1038/nature04956
52. Suda T, Suda J, Ogawa M. Disparate differentiation in mouse hemopoietic colonies derived from paired progenitors. Proc Natl Acad Sci U S A. (1984) 81:2520–4. doi: 10.1073/pnas.81.8.2520
53. Takano H, Ema H, Sudo K, Nakauchi H. Asymmetric division and lineage commitment at the level of hematopoietic stem cells: Inference from differentiation in daughter cell and granddaughter cell pairs. J Exp Med. (2004) 199:295–302. doi: 10.1084/jem.20030929
54. Ema H, Takano H, Sudo K, Nakauchi H. In vitro self-renewal division of hematopoietic stem cells. J Exp Med. (2000) 192:1281–8. doi: 10.1084/jem.192.9.1281
55. Wu M, Kwon HY, Rattis F, Blum J, Zhao C, Ashkenazi R, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. (2007) 1:541–54. doi: 10.1016/j.stem.2007.08.009
56. Loeffler D, Schneiter F, Schroeder T. Pitfalls and requirements in quantifying asymmetric mitotic segregation. Ann Ny Acad Sci. (2020) 1466:73–82. doi: 10.1111/nyas.14284
57. Pham K, Sacirbegovic F, Russell SM. Polarized cells, polarized views: asymmetric cell division in hematopoietic cells. Front Immunol. (2014) 5:1–14. doi: 10.3389/fimmu.2014.00026
58. Ting SB, Deneault E, Hope K, Cellot S, Chagraoui J, Mayotte N, et al. Asymmetric segregation and self-renewal of hematopoietic stem and progenitor cells with endocytic Ap2a2. Blood. (2012) 119:2510–22. doi: 10.1182/blood-2011-11-393272
59. Skylaki S, Hilsenbeck O, Schroeder T. Challenges in long-term imaging and quantification of single-cell dynamics. Nat Biotechnol. (2016) 34:1137–44. doi: 10.1038/nbt.3713
60. Hilsenbeck O, Schwarzfischer M, Skylaki S, Schauberger B, Hoppe PS, Loeffler D, et al. Software tools for single-cell tracking and quantification of cellular and molecular properties. Nat Biotechnol. (2016) 34:703–+. doi: 10.1038/nbt.3626
61. Toseland CP. Fluorescent labeling and modification of proteins. J Chem Biol. (2013) 6:85–95. doi: 10.1007/s12154-013-0094-5
62. Zimdahl B, Ito T, Blevins A, Bajaj J, Konuma T, Weeks J, et al. Lis1 regulates asymmetric division in hematopoietic stem cells and in leukemia. Nat Genet. (2014) 46:245–52. doi: 10.1038/ng.2889
63. Kandi R, Senger K, Grigoryan A, Soller K, Sakk V, Schuster T, et al. Cdc42-Borg4-Septin7 axis regulates HSC polarity and function. EMBO Rep. (2021) 22:e52931. doi: 10.15252/embr.202152931
64. Wilson A, Ardiet DL, Saner C, Vilain N, Beermann F, Aguet M, et al. Normal hemopoiesis and lymphopoiesis in the combined absence of numb and numblike. J Immunol. (2007) 178:6746–51. doi: 10.4049/jimmunol.178.11.6746
65. Sengupta A, Duran A, Ishikawa E, Florian MC, Dunn SK, Ficker AM, et al. Atypical protein kinase C (aPKCζ and aPKCλ) is dispensable for mammalian hematopoietic stem cell activity and blood formation (vol 108, pg 9957, 2011). P Natl Acad Sci USA. (2011) 108:12185–. doi: 10.1073/pnas.1103132108
66. Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Bio. (2015) 16:665–77. doi: 10.1038/nrm4063
67. Kiel MJ, He SH, Ashkenazi R, Gentry SN, Teta M, Kushner JA, et al. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. (2007) 449:238–U10. doi: 10.1038/nature06115
68. Kataoka K, Sato T, Yoshimi A, Goyama S, Tsuruta T, Kobayashi H, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. (2011) 208:2402–15. doi: 10.1084/jem.20110447
69. Lin WHW, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, et al. Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep. (2015) 13:2203–18. doi: 10.1016/j.celrep.2015.10.072
70. Gorgens A, Ludwig AK, Mollmann M, Krawczyk A, Durig J, Hanenberg H, et al. Multipotent hematopoietic progenitors divide asymmetrically to create progenitors of the lymphomyeloid and erythromyeloid lineages. Stem Cell Rep. (2014) 3:1058–72. doi: 10.1016/j.stemcr.2014.09.016
71. Yamamoto R, Wilkinson AC, Ooehara J, Lan X, Lai CY, Nakauchi Y, et al. Large-scale clonal analysis resolves aging of the mouse hematopoietic stem cell compartment. Cell Stem Cell. (2018) 22:600–7 e4. doi: 10.1016/j.stem.2018.03.013
72. Carrelha J, Meng Y, Kettyle LM, Luis TC, Norfo R, Alcolea V, et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. (2018) 554:106–11. doi: 10.1038/nature25455
73. Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood. (2016) 128:e20–31. doi: 10.1182/blood-2016-05-716480
74. Wehling A, Loeffler D, Zhang Y, Kull T, Donato C, Szczerba B, et al. Combining single-cell tracking and omics improves blood stem cell fate regulator identification. Blood. (2022) 140:1482–95. doi: 10.1182/blood.2022016880
75. Dong S, Wang Q, Kao YR, Diaz A, Tasset I, Kaushik S, et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature. (2021) 591:117–23. doi: 10.1038/s41586-020-03129-z
76. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. (2020) 21:101–18. doi: 10.1038/s41580-019-0185-4
77. Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. (2017) 356:1188–92. doi: 10.1126/science.aag2553
78. Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: The importance of ER membrane contact sites. Science. (2018) 361. doi: 10.1126/science.aan5835
79. Li P, Gu M, Xu H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem Sci. (2019) 44:110–24. doi: 10.1016/j.tibs.2018.10.006
80. Yim WWY, Mizushima N. Lysosome biology in autophagy. Cell Discovery. (2020) 6. doi: 10.1038/s41421-020-0141-7
81. Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. (2013) 494:323–7. doi: 10.1038/nature11895
82. Montazersaheb S, Ehsani A, Fathi E, Farahzadi R, Vietor I. An overview of autophagy in hematopoietic stem cell transplantation. Front Bioeng Biotech. (2022) 10. doi: 10.3389/fbioe.2022.849768
83. Yun S, Vincelette ND, Fernandez M, Yu XQ, Yang CY, Hitosugi T, et al. Tfeb links MYC signaling to epigenetic control of acute myeloid leukemia cell death and differentiation. Blood. (2020) 136:162–85. doi: 10.1182/blood-2020-137209
84. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. (2016) 129:2475–81. doi: 10.1242/jcs.146365
85. Garcia-Prat L, Kaufmann KB, Schneiter F, Voisin V, Murison A, Chen J, et al. TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell. (2021) 28:1838–50 e10. doi: 10.1016/j.stem.2021.07.003
86. Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. (2018) 553:418–26. doi: 10.1038/nature25022
87. Ito K, Bonora M, Ito K. Metabolism as master of hematopoietic stem cell fate. Int J Hematol. (2019) 109:18–27. doi: 10.1007/s12185-018-2534-z
88. Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature. (2016) 529:37–42. doi: 10.1038/nature16187
89. Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, et al. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. (2015) 348:340–3. doi: 10.1126/science.1260384
90. Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, et al. The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell. (2019) 24:405–+. doi: 10.1016/j.stem.2019.02.012
91. Bergeland T, Widerberg J, Bakke O, Nordeng TW. Mitotic partitioning of endosomes and lysosomes. Curr Biol. (2001) 11:644–51. doi: 10.1016/S0960-9822(01)00177-4
92. Florian MC, Dorr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. (2012) 10:520–30. doi: 10.1016/j.stem.2012.04.007
93. Hinge A, Xu J, Javier J, Mose E, Kumar S, Kapur R, et al. p190-B RhoGAP and intracellular cytokine signals balance hematopoietic stem and progenitor cell self-renewal and differentiation. Nat Commun. (2017) 8:14382. doi: 10.1038/ncomms14382
94. Etienne-Manneville S. Cdc42–the centre of polarity. J Cell Sci. (2004) 117:1291–300s. doi: 10.1242/jcs.01115
95. Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. (2007) 104:5091–6. doi: 10.1073/pnas.0610819104
96. Florian C, Dörr K, Scharffetter-Kochanek K, Zheng Y, Geiger H. CDC42 activity regulates hematopoietic stem cell aging and rejuvenation. Exp Dermatol. (2013) 22:E45–E. doi: 10.1016/j.stem.2012.04.007
97. Florian MC, Klose M, Sacma M, Jablanovic J, Knudson L, Nattamai KJ, et al. Aging alters the epigenetic asymmetry of HSC division. PloS Biol. (2018) 16:e2003389. doi: 10.1371/journal.pbio.2003389
98. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-β as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. (2009) 113:1250–6. doi: 10.1182/blood-2008-04-146480
99. Althoff MJ, Nayak RC, Hegde S, Wellendorf AM, Bohan B, Filippi MD, et al. Yap1-Scribble polarization is required for hematopoietic stem cell division and fate. Blood. (2020) 136:1824–36. doi: 10.1182/blood.2019004113
100. Herrejon Chavez F, Luo HZ, Cifani P, Pine A, Chu KL, Joshi S, et al. RNA binding protein SYNCRIP maintains proteostasis and self-renewal of hematopoietic stem and progenitor cells. Nat Commun. (2023) 14:2290. doi: 10.1038/s41467-023-38001-x
101. Luo H, Cortes-Lopez M, Tam CL, Xiao M, Wakiro I, Chu KL, et al. SON is an essential m(6)A target for hematopoietic stem cell fate. Cell Stem Cell. (2023) 30:1658–73 e10. doi: 10.1016/j.stem.2023.11.006
102. Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. (2010) 16:903–U101. doi: 10.1038/nm.2187
103. Girotra M, Trachsel V, Roch A, Lutolf MP. In vivo pre-instructed HSCs robustly execute asymmetric cell divisions in vitro. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21218225
104. Ito K, Carracedo A, Arai F, Ala U, Avigan D, Schafer Z, et al. A PML-PPAR-δ Pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance through the control of asymmetric division. Blood. (2012) 120:1350–8. doi: 10.1182/blood.V120.21.2327.2327
105. Beckmann J, Scheitza S, Wernet P, Fischer JC, Giebel B. Asymmetric cell division within the human hernatopoietic stem and progenitor cell compartment: identification of asymmetrically segregating proteins. Blood. (2007) 109:5494–501. doi: 10.1182/blood-2006-11-055921
106. Shin JW, Buxboim A, Spinler KR, Swift J, Christian DA, Hunter CA, et al. Contractile forces sustain and polarize hematopoiesis from stem and progenitor cells. Cell Stem Cell. (2014) 14:81–93. doi: 10.1016/j.stem.2013.10.009
107. Carlton JG, Jones H, Eggert US. Membrane and organelle dynamics during cell division. Nat Rev Mol Cell Biol. (2020) 21:151–66. doi: 10.1038/s41580-019-0208-1
108. Nakata T, Hirokawa N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane-transport. J Cell Biol. (1995) 131:1039–53. doi: 10.1083/jcb.131.4.1039
109. Matsushita M, Tanaka S, Nakamura N, Inoue H, Kanazawa H. A novel kinesin-like protein, KIF1Bβ3 is involved in the movement of lysosomes to the cell periphery in non-neuronal cells. Traffic. (2004) 5:140–51. doi: 10.1111/j.1600-0854.2003.00165.x
110. Guardia CM, Farías GG, Jia R, Pu J, Bonifacino JS. BORC functions upstream of kinesins 1 and 3 to coordinate regional movement of lysosomes along different microtubule tracks. Cell Rep. (2016) 17:1950–61. doi: 10.1016/j.celrep.2016.10.062
111. Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytomet. A. (2009) 75:14–24. doi: 10.1002/cyto.a.20674
112. Johnson C, Belluschi S, Laurenti E. Beyond “to divide or not to divide”: Kinetics matters in hematopoietic stem cells. Exp Hematol. (2020) 92:1–10 e2. doi: 10.1016/j.exphem.2020.11.003
113. Lucas D. Structural organization of the bone marrow and its role in hematopoiesis. Curr Opin Hematol. (2021) 28:36–42. doi: 10.1097/MOH.0000000000000621
114. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. (2015) 526:126–30. doi: 10.1038/nature15250
115. Pei WK, Feyerabend TB, Rössler J, Wang X, Postrach D, Busch K, et al. barcoding reveals haematopoietic stem cell fates realized. Nature. (2017) 548:456–+. doi: 10.1038/nature23653
116. Nimer SD. Myelodysplastic syndromes. Blood. (2008) 111:4841–51. doi: 10.1182/blood-2007-08-078139
117. Hawkins ED, Russell SM. Upsides and downsides to polarity and asymmetric cell division in leukemia. Oncogene. (2008) 27:7003–17. doi: 10.1038/onc.2008.350
Keywords: asymmetric cell division (ACD), hematopoietic stem cell (HSC), cell fate acquisition, lysosome, mitochondria, Cdc42, metabolism, quiescence
Citation: Nunes J and Loeffler D (2024) Asymmetric cell division of hematopoietic stem cells: recent advances, emerging concepts, and future perspectives. Front. Hematol. 3:1373554. doi: 10.3389/frhem.2024.1373554
Received: 19 January 2024; Accepted: 12 March 2024;
Published: 26 March 2024.
Edited by:
Marcus O. Muench, Vitalant Research Institute, United StatesReviewed by:
Jian Huang, Coriell Institute For Medical Research, United StatesTomer Itkin, Tel Aviv University, Israel
Copyright © 2024 Nunes and Loeffler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk Loeffler, ZGlyay5sb2VmZmxlckBzdGp1ZGUub3Jn