 Michael O’Dwyer
Michael O’Dwyer Siobhan Glavey3
Siobhan Glavey3 Roisin McAvera
Roisin McAvera Alessandro Natoni
Alessandro Natoni- 1Department of Hematology, School of Medicine, University of Galway, Galway, Ireland
- 2Lambe Institute for Translational Research, School of Medicine, College of Medicine Nursing and Health Sciences, University of Galway, Galway, Ireland
- 3Multiple Myeloma Research Group, Department of Pathology, Royal College of Surgeons in Ireland, Department of Pathology, Beaumont Hospital, Dublin, Ireland
- 4Hematology, Department of Translational and Precision Medicine, Sapienza University, Rome, Italy
- 5Discipline of Pharmacology and Therapeutics, School of Medicine, College of Medicine, Nursing and Health Sciences, University of Galway, Galway, Ireland
There is growing recognition of the importance of sialylation as a critical post translational modification in cancer. In this article we review the role of increased cell surface sialylation (hypersialylation) in Multiple Myeloma as it relates to cellular trafficking and immune evasion. Knowledge of the specific effects of sialic acid on cell trafficking machinery and modulation of immune cell interactions will identify opportunities for therapeutic interventions. The available evidence indicates that hypersialylation facilitates disease progression and negatively impacts on response to treatment and overall survival. Further research is required to fully elucidate the mechanisms through which hypersialylation influences disease biology and therapy resistance with the ultimate goal of developing new treatment approaches to improve the outcomes of patients with Multiple Myeloma.
1 Introduction to multiple myeloma and sialylation
Multiple Myeloma (MM) is a clonal malignancy of antibody producing plasma cells, involving the bone marrow (BM) and associated with clinical features of anemia, bone destruction, renal impairment and hypercalcemia. Despite advances in the treatment of MM the majority of patients will eventually develop resistance and die from their disease, with significantly shorter survival in patients with high-risk genetic features (1). The BM microenvironment has a major influence on the pathogenesis of MM, in which malignant plasma cells have bidirectional interactions with the BM cellular and non-cellular elements. The multiple and complex interactions between MM cells and various BM cell types, contribute to neoplastic plasma cell growth, proliferation, and drug resistance (2). A frequently overlooked mediator of cell-to-cell interactions and communication in the tumor microenvironment (TME) is glycosylation. All cells are covered by a dense coating of glycans, which post-translationally modify multiple proteins or lipids, significantly impacting their function (3). Glycosylation of extracellular structures plays a critical role in cell-to-cell interactions and is known to influence cellular trafficking, signaling and the immune response. Sialic acid residues, which decorate the end of glycan (sugar) structures on the cell surface play a key role in many of these processes (4). Glycosylation is not a template driven process and glycans on proteins or lipids are produced by combined action of a number of different enzymes. In the case of sialic acid-coated glycans (sialoglycans), the final transfer of sialic acids to the underlying glycan chain is catalyzed by an enzymatic family of sialyltransferases (Table 1), and takes place in the Golgi apparatus (5). Glycan binding proteins are known as lectins and the best-known family of sialoglycan binding lectins are the Selectins. E, P and L-Selectin, expressed predominantly on endothelial cells, platelets and lymphocytes, play an important role in cellular trafficking and platelet adhesion. Well known Selectin ligands, include P-selectin glycoprotein ligand-1 (PSGL-1), CD43 and CD44, which when appropriately post translationally modified by sialic acid and fucose (to generate sialyl Lewis X/sLeX) can bind to E and P-Selectin (6). The other main family of sialoglycan binding lectins are the Siglecs (4). The Siglecs are sialic acid binding immunoglobulin like lectins. This is a large family of receptors, which are expressed by immune cells. While some are activating receptors, containing an immunoreceptor tyrosine-based activation (ITAM) motif in their intracytoplasmic tail, most Siglecs are inhibitory receptors, with an intracellular immunoreceptor tyrosine-based inhibitory (ITIM) motif, analogous to PD-1 and CD47. It is now well recognized that increased sialylation (hypersialylation) correlates with advanced stage and inferior prognosis in many forms of cancer. Indeed, by exploiting sialic binding receptors, such as the Selectins and Siglecs, hypersialylation promotes cancer cell survival by enabling dissemination, enhancing survival and by modulating immune cell responses (7).
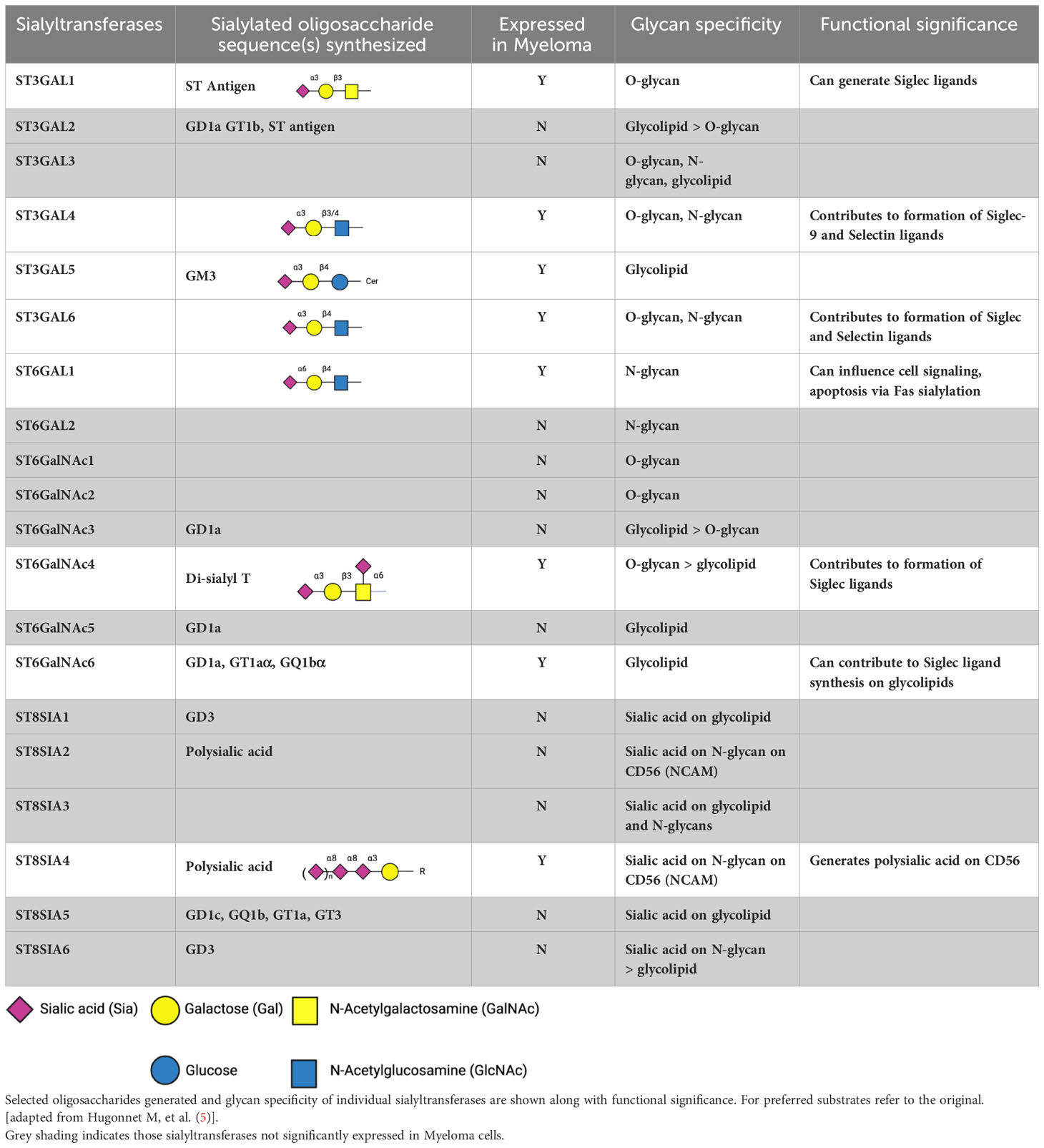
Table 1 Human sialyltransferases, focusing on those more highly expressed in Multiple Myeloma, based on an expression cut off > 10 transcripts per million (nTPM) in cell lines in Human Protein Atlas and also detected in > 1% cells by single cell RNA seq in bmbrowser.org.
2 Role of sialoglycans in cellular trafficking and adhesion
Many glycosyltransferases have important roles in regulation of leukocyte trafficking, through regulation of the expression of selectin ligands. These include α 1,3 fucosyltransferases and α 2,3 sialyltransferases (8). These glycosylation dependent mechanisms of immune cell trafficking are also functional in hematological malignancies. We first identified the potential importance of hypersialylation in disease progression of MM, when we discovered an important role for the sialyltransferase ST3GAL6 in the biology of MM. In the UK MRC Myeloma IX trial overexpression of ST3GAL6 was an independent prognostic marker of poor overall survival (OS) (9). We identified an important role for ST3GAL6 in cellular trafficking and drug resistance and found that it played a major role in generation of E-selectin ligands, influencing trafficking in and out of the bone marrow niche (10). This suggests that there may be benefit in blocking the interaction of E-selectin and its ligands to overcome drug resistance. Indeed, the success of this strategy has been demonstrated in Acute Myeloid Leukemia (AML) with the E-selectin inhibitor uproleselan (GMI-1271) having entered clinical trials with encouraging results (11). A study was also conducted to assess the efficacy of uproleselan in preventing gastrointestinal toxicity in patients receiving high dose melphalan as conditioning for stem cell transplantation. The rationale for this study was that the expression of E-selectin is increased at sites of inflammation, resulting in increased extravasation of inflammatory leukocytes into inflamed tissue associated with chemotherapy toxicity. Blockade of E-selectin could therefore reduce this inflammation (11). While this study has been completed, results have not yet been reported (NCT04682405). We also found that sialylation influenced cell adhesion not only by mediating Selectin binding but also α4 integrin mediated binding to VCAM1 and MadCAM. In an in-vivo model we found that pretreatment with a sialyltransferase inhibitor improved survival of SCID Beige mice injected with MM.1S cells expressing sLeX (12). In effect, using cell sorting with the HECA452 antibody we were able to generate a subclone of MM.1S cells which were enriched for the expression of E-selectin ligands. While low dose bortezomib was largely ineffective in this model, the combination with sialyltransferase inhibitor treatment was synergistic. Thus, we concluded that de-sialylation of MM cells likely overcame cell adhesion mediated drug resistance (CAM-DR) in vivo. Conceivably, de-sialylation could be relevant for cell adhesion mediated immune resistance as well. In more recent work we found that MM cells can bind platelets in a selectin dependent manner, which can be blocked using a P-selectin antibody and is more pronounced with disease progression (13). Platelet cloaking is a potential mechanism of immune evasion as it can lead to inhibition of NK mediated killing. In summary, there are several different ways in which sialoglycans can influence cellular adhesion and drug resistance. Figure 1 demonstrates potential interactions of sialoglycans on MM cells with E and P Selectin.
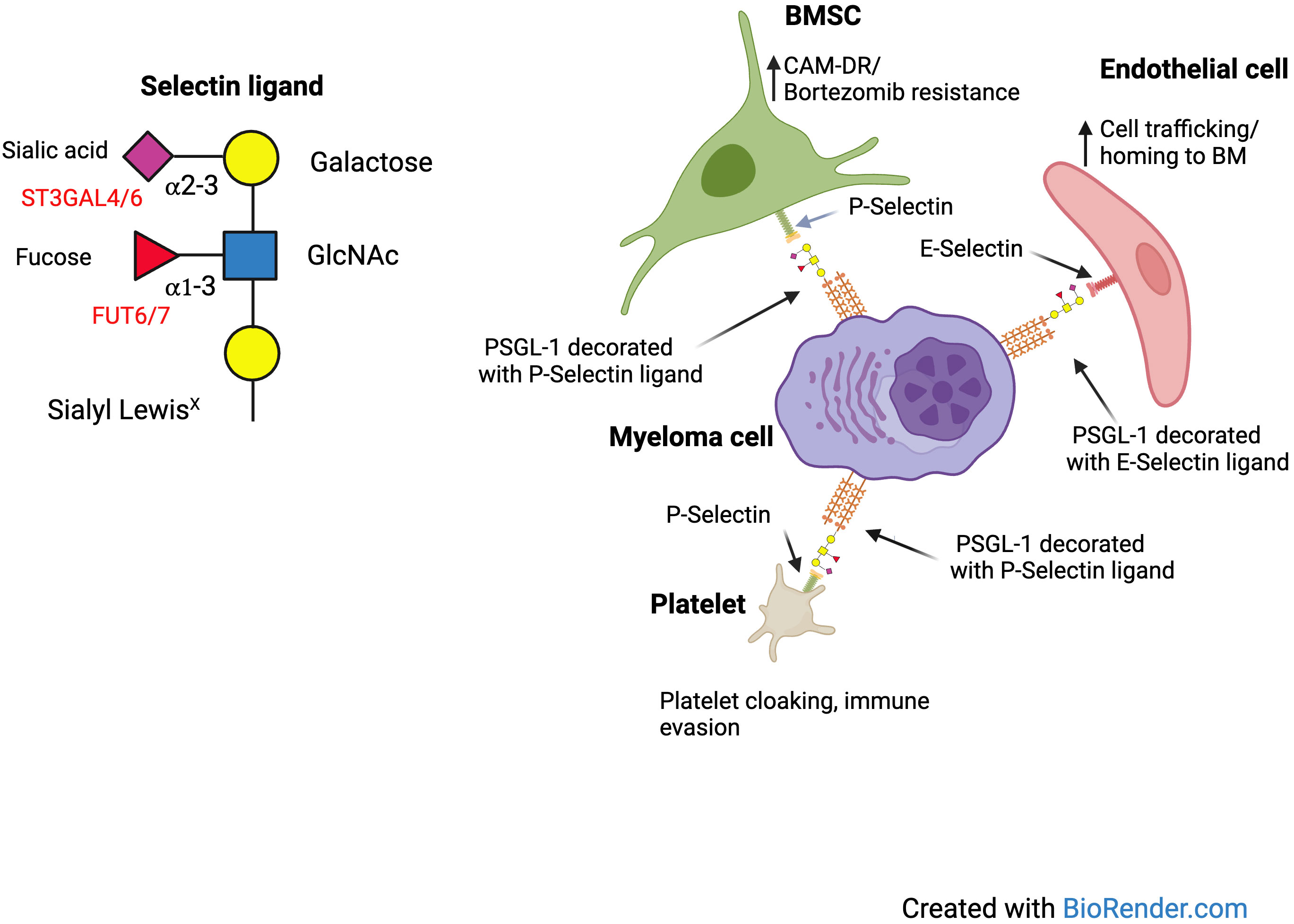
Figure 1 Interactions of selectins and their ligands on MM cells.
3 Siglec ligands and immune evasion in MM
Following the discovery of the Siglec family of receptors, it became clear that sialoglycans behave as self-associated molecular patterns (SAMPs), which are recognized by inhibitory Siglec receptors to suppress autoimmune responses. It is now well accepted that this axis could also be exploited by tumor cells to mediate cancer immune evasion (14). Tumor cells and stromal cells in the TME can induce the expression of Siglec receptors on NK cells, T cells and macrophages. There is now an extensive literature linking hypersialylation of tumor cells with immune evasion in many different cancers. These interactions can induce macrophage polarization, inhibit dendritic activation, leading to reduced antigen dependent T cell responsiveness, as well as inhibit direct activation of T cells and induce exhaustion. Presentation of sialylated antigen by dendritic cells has also been reported to induce T cell tolerance with expansion of antigen specific regulatory T cells (Tregs) (15). Reprogramming of tumor stroma by tumor cells can also lead to enhanced sialylation and induction of Siglec ligands, which induce T cell exhaustion (16). Among the first insights in this regard, were reports that interaction of sialoglycan SAMPs on tumor cells with Siglecs on NK cells could lead to inhibition of NK cell function (17, 18). This prompted us to explore whether MM cells expressed sialoglycans capable of acting as Siglec ligands and whether the expression of these ligands influenced the cytotoxicity of NK cells (19, 20). We found that MM cell lines, as well as primary samples from patients with MM, strongly expressed ligands for Siglec-7 and Siglec-9. Siglec ligands were also present on plasma cells from individuals with MGUS. While the Siglec-7 receptor was equally expressed on NK cells from healthy donors and MM patients, we found that expression, based on the mean fluorescent intensity (MFI), was significantly higher on NK cells from MM patients (p = 0.0043). When the surface of MM cells was desialylated using a sialidase enzyme (neuraminidase), co-culture with NK cells led to significantly enhanced killing under different conditions (naïve, IL-2 activated and expanded NK cells). Using an alternate approach, blockage of sialic acid incorporation on glycoproteins in the Golgi, using STI, we again observed enhanced killing by NK cells following co-culture and also ADCC in the presence of Daratumumab. Interestingly, pre-treatment of MM cells led to enhanced detection of CD38 by flow cytometry, suggesting that sialylation of CD38 could potentially mask this important therapeutic epitope in some cells. We did not observe this to be the case for BCMA, though previous work suggested BCMA could be internalized following sialylation (21). From this work, we concluded that sialylation of MM cells could protect from NK cell mediated cytotoxicity and reasoned that this was at least in part due to interactions between Siglec-7 and its ligands. Indeed, knock out (KO) of Siglec-7 in NK cells led to significant enhancement of killing of MM cells in co-culture, supporting this conclusion.
Given an apparent important role for Siglec-7 ligands in immune evasion, we then sought to determine the identity of potential glycoproteins that could function as Siglec-7 ligands. To elucidate the identity of glycoproteins that could function as Siglec-7 ligands in MM, mass spectrometry was carried out on proteins bound to Siglec-7 Fc chimera-magnetic bead complexes after incubation with cell lysates from MM cell lines untreated or treated with neuraminidase. PSGL-1 emerged as the primary Siglec-7 ligand expressed in 3/3 cell lines. Furthermore, CD43 was also identified as a Siglec-7L expressed in one cell line, consistent with previous work showing that CD43 is a high affinity ligand for Siglec-7 (22). These results identified PSGL-1 as a new, broadly expressed Siglec-7 ligand in MM. We have also confirmed PSGL-1 to be the main E-selectin ligand on MM cells, expressing sLeX (23). PSGL-1 along with CD43 recently emerged in a genome wide CRISPR screen as being associated with resistance to NK cells (24). PSGL-1 is of particular interest in MM. In the Human Protein Atlas, proteomic analysis shows the highest level of PSGL-1 in MM patients and MM cell lines expressed the highest levels of mRNA for PSGL-1 (25). The expression of PSGL-1 has previously been shown to increase with disease progression in MM (26, 27).
Siglec-7 predominantly binds sialylated O-glycans. In addition to PSGL-1 and CD43, O-linked glycoproteins such as CD34, CD44, CD45, and MUC-1 have the potential to function as Siglec-7 ligands provided they possess the appropriate post translational glycan modifications (28). On the other hand, Siglec-9 ligands appear to be predominantly expressed on N-linked glycans, though sialylated MUC-1 has been reported to be a Siglec-9 ligand. It is important to recognize that sialylated glycolipids (gangliosides), such as GD2 and GD3 expressing disialyl T antigens can also function as ligands for both Siglec-7 and Siglec-9 (29). GD2 for example, was recently demonstrated to be a Siglec-7 ligand (30). In addition to functioning as a Siglec-7 ligand, it has also been reported that PSGL-1 can bind Siglec-5 (31, 32). Siglec- 5 was identified as a new inhibitory T cell immune checkpoint molecule and studies suggest that blockade of Siglec-5 could serve as a new strategy to enhance anti-tumor T cell functions. Cancer cell lines bind Siglec-5 indicating the presence of Siglec-5 ligands and it is conceivable that MM cells may express ligands for Siglec-5, given the strong expression of PSGL-1, though this remains to be explored. Other potential Siglec receptors that could be important in MM include Siglec-10, which plays an important role in regulating the phagocytic function of macrophages (33). The best recognized ligand of Siglec-10 is CD24. CD24 has been reported to be associated with a favorable prognosis in MM but this preceded the use of CD38 monoclonal antibodies, for which macrophages are important effector cells (34). A re-evaluation of the role of CD24 in the era of CD38 antibodies is warranted. Siglec-15 has recently emerged as a new target for cancer immunotherapy (35). Interestingly, Siglec-15 is expressed on osteoclasts, and engagement of Siglec-15, which contains an ITAM motif, leads to differentiation and function of osteoclasts, which are central feature to myeloma bone disease. The function of Siglec-15 seems to be sialic acid dependent since treatment with sialidase inhibited osteoclast development (36). Finally, treatment of 5T2MM mice with a Siglec-15 inhibitor led to prevention of bone loss (37).
4 Sialyltransferases involved in Siglec-7 ligand synthesis and potential role in myeloma
The mere expression of PSGL-1 or CD43 does not necessarily imply the expression of Siglec ligands. The expression of Siglec ligands depends on the presence of appropriate post translational modifications, which are mediated by glycosyltransferases. The generation of Siglec ligands is dependent on the combined activity of a number of glycosyltransferases, including sialyltransferases which deposit a disialic structure on a carbohydrate backbone, typically on O-linked glycans. Gene knockout (KO) and knock-in (KI) studies suggest an important role for specific sialyltransferases in the generation of Siglec-7 ligands (38). The α2,3 sialyltransferase ST3GAL1 or ST3GAL2 can generate sialyl T (ST) antigen on an O-glycan, which via further sialylation via ST6GALNAC1/2/3/4 can add an α2,6 linked sialic to a core N-Galactosamine to generate a disialyl T (dST) antigen, capable of binding to Siglec-7. ST3GAL1, ST6GALNAC4 and ST6GALNAC1 have all been reported to be involved in generation of Siglec-7 ligands in cancer cells (22, 39, 40). As noted above, glycolipids can also function as Siglec-7 ligands. ST3GAL1 and ST6GALNAC4 are both highly expressed in a subset of MM patients. The Human Protein Atlas proteomic analysis shows that of all cancers ST3GAL1 is most highly expressed in the serum of MM patients and MM cell lines have the highest mRNA expression of all cancer cell lines screened. This is similarly the case for ST6GALNAC4 (24). While a causal link remains to be proven, ST6GALNAC4 has recently been shown to be induced by MYC, an oncogene strongly associated with disease progression in MM patients (41). Publically available single cell RNA seq data (www.bmbrowser.org) shows the presence of transcripts for a number of sialyltransferases, including ST3GAL1 and ST6GALNAC4 transcripts in MM cells from patients (42). We have also seen strong cytoplasmic staining for ST3GAL1 in plasma cells of MM patients on immunohistochemistry, especially in those with t(4,14) (unpublished data). High levels of ST3GAL1 have been associated with inferior PFS +/- OS in a number of studies. In the UKMRC Myeloma IX study, expression was independent of ISS but elevated levels of ST3GAL1 were associated with inferior PFS (p = 0.0147) with a trend for poor OS (p=0.0695). Interestingly, in this study, increased ST3GAL1 expression was associated with hypodiploidy (p=1.2 e-05, 1q gain (p=0.01) and t(4,14) (0.009) (43, 44). Meanwhile, within the COMPASS dataset elevated ST3GAL1 expression was associated with both inferior progression free survival (PFS) (p=0.0123) and OS (p=0.0039) (45) (Figure 2A). Once again in this study we saw significantly higher expression of ST3GAL1 in patients with t(4,14) (p=0.006). We also observed that elevated levels of expression of ST3GAL4, which may be involved in either Selectin or Siglec-9 ligand synthesis, were associated with inferior PFS and OS (Figure 2B).
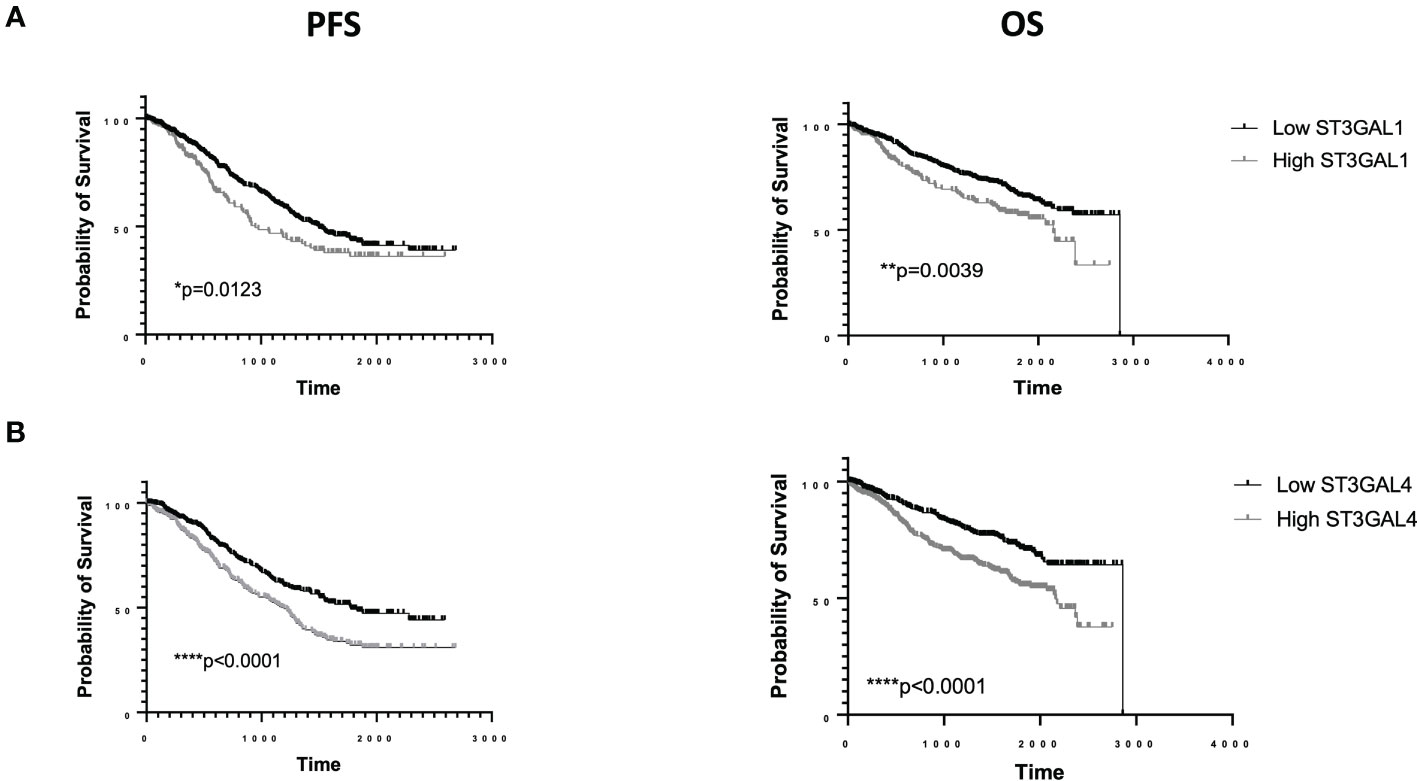
Figure 2 RNA sequencing data from the COMMPASS dataset. Elevated levels of ST3GAL1 transcripts are associated with inferior PFS and OS (A). Elevated levels of ST3GAL4 transcripts are associated with inferior PFS and OS (B).
Supporting this hypothesis, ST3GAL1 was recently identified as the main contributor to Siglec-7 ligand synthesis in pancreatic cancer and was associated with driving tumor associated macrophage differentiation (38). There is indeed abundant literature linking expression of Siglec ligands with macrophage polarization in cancer (39, 46, 47). This has yet to be addressed in MM. Conceivably, Siglec-7 and Siglec-9 ligands expressed by MM cells facilitate the accumulation of M2 polarized macrophages within the BM, leading to immune suppression and reduced response to immunotherapies, such as CD38 monoclonal antibodies. Preliminary analysis of BM samples from patients with MM and compared to normal controls suggests a significantly higher percentage of Siglec-7 positive monocytes/macrophages as well as much higher intensity of expression as determined by MFI (unpublished data). Data from the bone marrow browser confirm that all relevant immune subsets in MM patients, including NK cells and monocytes express Siglec-7 receptors (41).
Using single cell RNA sequencing (scRNA seq) analysis, Francesco Maura and colleagues recently demonstrated that factors associated with sustained measurable residual disease (MRD) negativity in patients receiving intensive quadruplet induction with Carfilzomib, lenalidomide and dexamethasone in combination with Daratumumab (KRD-Dara) included a sufficient number of functional CD16 positive NK cells at baseline and evidence of increased monocyte activation and expansion following Daratumumab exposure (48). We have already shown that hypersialylation and expression of Siglec ligands can inhibit Daratumumab induced ADCC. Our preliminary observations suggest the possibility that interaction between Siglec ligands on MM cells and Siglecs on monocyte/macrophages could result in failure to achieve the necessary activation and expansion of monocytes required for optimal efficacy of Daratumumab. Clonal T cell expansion is also important for sustained responses in Dara treated patients. Hypersialylation and expression of Siglec ligands also has the potential to negatively affect T cell responses as well as negatively affect the function of antigen presenting dendritic cells. Apart from potential negative effects on both innate and adaptive cellular immune responses, hypersialylation has the potential to inhibit complement dependent cytotoxicity. A recent study reports that sialylation of the complement inhibitory protein CD55 by ST3GAL1 facilitated immune evasion in breast cancer (49). O-linked desialylation of CD55 by ST3GAL1 silencing resulted in increased C3 deposition and complement mediated lysis of breast cancer cells and enhanced sensitivity to ADCC. This could be relevant to MM as Daratumumab resistance is accompanied by increased expression of complement inhibitory proteins, including CD55 (50).
5 Sialoglycans and T cell redirected therapies
Given the emerging importance of T cell redirected therapies such as CAR-T and bispecific antibodies (BsAbs) in the treatment of MM, consideration of the potential impact of sialylation is worth considering. In other cancers there is growing evidence implicating Siglec ligands in immune evasion of T cells (51–53). Increased expression of Siglec receptors has been documented on intratumoral T cells, co-expressed with other exhaustion markers e.g. PD-1. Importantly, desialylation leads to enhanced T cell activation and cytotoxicity. Sialylation of the Fas receptor could also inhibit apoptosis induction by Fas-L expressing T cells (54). Sialic acid-containing glycans on the surface of both T cells and antigen presenting cells (APCs) are alternative ligands of CD28 that compete with binding to CD80 on the APC, resulting in attenuated co-stimulation (55). Tumor cell glycosylation can negatively impact the function of CAR-T cells and strategies to reduce tumor cell glycosylation can enhance the efficacy of CAR-T cells. It has been proposed that this could be due to interference of glycans with formation of a proper immunological synapse. Apart from the potential benefit of reducing expression of Siglec ligands it is also known that N-glycosylation of PD-L1 is critical to its function. Therefore, it is possible that disruption of tumor glycosylation could lead to reduced expression of functional inhibitory ligands. In one study treatment of tumor cells with the glucose/mannose analogue 2-deoxy-D-glucose (2DG) to disrupt the N-glycan coating on the cell surface resulted in enhanced CAR-T cell activity in different xenograft mouse models of pancreatic adenocarcinoma (56). It is quite likely that interactions of sialylated glycans with Siglecs exposed on activated CAR-T cells could contribute to CAR-T cell inhibition. Indeed, a high throughput CRISPR screen implicated key enzymes involved in Siglec ligand biosynthesis (GNE and GALE) in resistance to BCMA CAR-T cells (57). The top hit GNE is a rate limiting enzyme involved in sialic acid biosynthesis (58). GALE is required for glycoprotein and glycolipid biosynthesis and GALE KO cells have greatly reduced sialylation (59). Increased activity of sialidase secreting CAR-T cells in the setting of solid tumors has been reported (60). Another study utilizing a genome wide CRISPR activation screen identified increased expression of CD43, recently described as a Siglec-7 ligand, as being associated with resistance to a CD20/CD3 bispecific antibody. Importantly, resistance due to CD43 was dependent on its sialylation (61). A recent report showed that sialic-acid removal was synergistic with a HER2 targeting bispecific T cell engager treatment in vitro (62).
In the context of MM, factors currently associated with resistance to T cell redirected therapies include loss of antigen/target epitope, loss of MHC I, low diversity of pre-therapy TCR repertoire, hyper-expanded CD8 clones with an exhausted phenotype, bone marrow stromal cell (BMSC) mediated immune resistance and increased numbers of immune suppressive cells within the BM microenvironment (63, 64) BMSCs may impair the activity of T cell-redirecting bispecific antibodies against tumor cells by suppressing T-cell activation, forming a physical barrier that hampers T-cell penetration and by inducing cell adhesion mediated resistance in tumor cells (65–67). Understanding precisely how BMSCs hinder immune function is currently an area of intense research.
6 A potential role of sialoglycans and inflammatory stromal cells in myeloma
It is becoming increasingly clear that an inflammatory BM microenvironment is associated with resistance to T cell redirected therapy in patients with MM. The BM microenvironment is composed of a number of discrete cell types, many of which are known to play an immune suppressive role and promote disease progression in MM. These include myeloid derived suppressor cells (MDSCs), Tregs and of course mesenchymal stromal cells (MSCs)/BMSCs. However, while the tumor promoting role of the immune suppressive BM microenvironment was well recognized in MM, until recently a detailed characterization of the individual cell types was lacking. Following a detailed analysis of matched MM and control bone marrows, both of CD138 positive and CD138 negative fractions by scRNA seq, Tom Cupedo and colleagues have recently shown the presence of a distinctive inflammatory signature in BM stromal cells in patients that is absent in BM stromal cells of normal controls (68). This inflammatory signature is driven by TNF-α and IL-1β signaling, as a result of chronic inflammation in the bone marrow, which may be amplified by neutrophils. Intriguingly, this inflammatory stromal signature persists following treatment, even in patients who achieve MRD negativity with induction therapy. The authors speculated whether this MM specific inflamed stroma could play a role in MM pathobiology and contribute to disease persistence and relapse. It has previously been reported that MSCs can induce resistance to CAR-T cells (67). BM-cancer associated fibroblasts (CAFs) were found to suppress CAR-T cells through both contact-dependent and cytokine-mediated effects. They also noted that when BCMA-CAR-T cells were stimulated and cocultured with BM-CAFs, the surface expression of inhibitory receptors such as PD-1 was significantly upregulated on CAR-T cells, whereas BM-CAFs simultaneously overexpressed inhibitory ligands such as PD-L1. Contact dependent intrinsic resistance was previously reported (66). Adhesion of MM cells to stroma led to induction of anti-apoptotic molecules, which protected MM cells from CAR mediated cytotoxicity. Since integrin mediated attachment to stroma in MM is dependent on sialylation of α4 integrins, there is the potential this could be blocked using desialylation approaches (12).
We recently reported that stromal cells with an inflammatory signature have an immune suppressive phenotype, which is, at least in part, sialic acid dependent (16). In murine models of both MM and colorectal cancer we found that MSCs grown in tumor conditioned media induce T cell exhaustion in a sialic acid dependent manner. Tumor conditioning led to upregulation of sialyltransferases, increases in cell surface sialic acids and expression of Siglec-E ligands. Siglec-E is the murine homolog of Siglec-7 and 9. When co-cultured with activated T cells these tumor conditioned MSCs blocked T cell proliferation and induced T cell exhaustion. Treatment of MSCs with a sialyltransferase inhibitor reduced sialylation of MSCs and restored T cell proliferation along with increases in CD25 expression and Granzyme B content. Similarly, evaluating biopsies from patients with colorectal cancer, we found that the secretome of tumor cells induced a similar phenotype in human MSCs/CAFs with induction of ligands for Siglec-7 and Siglec-9. Intratumoral T cells had an exhausted phenotype with increased expression of Siglec-7 and Siglec-9 along with increased expression of PD-1. When co-cultures were performed with intratumoral T cells and conditioned MSCs/CAFs, T cell activation and proliferation could be restored following STI treatment of MSCs/CAFs with a decline in expression of Siglec receptors and other exhaustion markers such as PD-1. Analysis of Tom Cupedo’s scRNA seq data generated from inflamed stromal cells of MM patients indicates close similarity to the pattern of sialyltransferases induced in our model. We believe that ongoing inflammation in the bone marrow, could be a major driver of immune suppression in a sialic dependent manner.
From the above, it is clear that the interaction of Siglec ligands on MM cells has the potential for multiple different consequences, facilitating disease progression in MM (Figure 3).
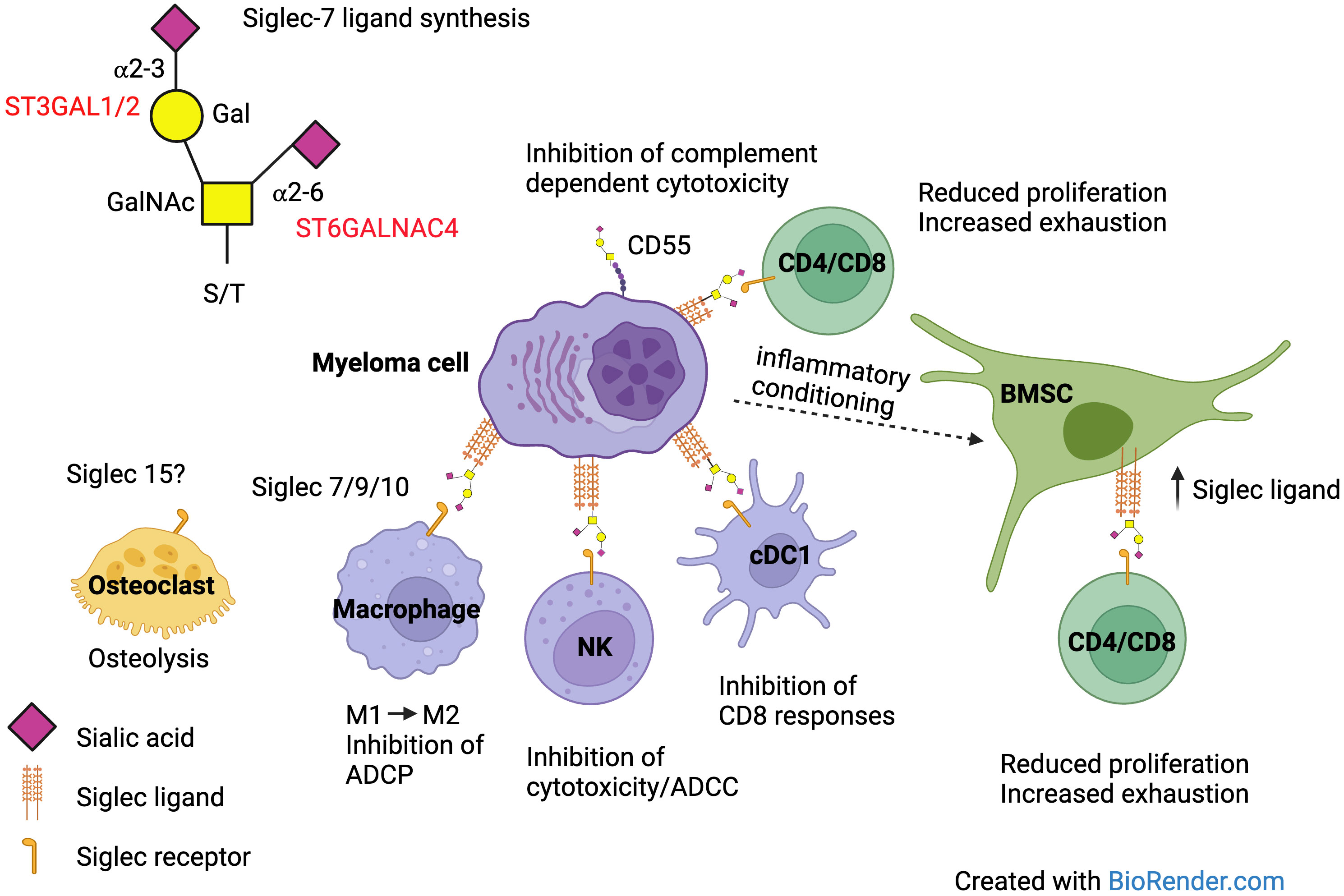
Figure 3 Siglec ligand expression on MM cells can affect the function of multiple different cells within the BM microenvironment via interaction with Siglec receptors. Antibody dependent cellular phagocytosis (ADCP), antibody dependent cellular cytotoxicity (ADCC).
7 Therapeutic targeting of the sialoglycan-Siglec axis
Could this sialoglycan-Siglec axis be therapeutically targeted? Based on an original concept developed by Carolyn Bertozzi, sialidase conjugated antibodies are being developed as a means of desialylating tumor cells at the cell surface (69, 70). In preclinical models of HER2 positive breast cancer, the delivery of sialidase HER2 conjugated antibodies proved more active than HER2 antibody treatment alone, with the combination with PD-1 blockade adding synergism. This approach is now being tested in the clinic with non-targeted antibodies conjugated with sialidase in solid tumors (NCT05259696) and promising initial clinical data were recently presented (71). In the context of MM sialidase conjugated antibodies could be tested in combination with CD38 monoclonal antibodies or even bispecific antibodies, should further preclinical data support this.
An alternative approach is sialyltransferase inhibition. Shortly after the sialyltransferase inhibitor 3FaxNeu5Ac was first described we started to evaluate the activity of this drug in MM. This drug, originally developed by Paulson and colleagues, was the first cell permeable sialyltransferase inhibitor (72). In its peracetylated form it readily diffuses across the cell membrane, is deacetylated by cellular esterases and subsequently form CMP-3Fax-Neu5Ac, which acts as a poor substrate for sialyltransferases by virtue of the electron withdrawing effects of the fluorine substituent. As a result, CMP-3Fax-Neu5Ac builds up to high levels inside the cell, impairing the action of all sialyltransferases, both in vitro and in vivo (6). Our initial interest was to assess the ability of this drug to inhibit cellular trafficking and overcome cell adhesion mediated drug resistance (CAM-DR) in MM models, both in-vitro and in-vivo, as noted above, before moving on to study its effect on immune modulation. In initial in-vivo studies, with systemic administration of free drug, we confirmed previous observations that at higher doses the drug had potential for nephrotoxicity (12, 73). Bull and colleagues reported that intratumoral injections with the same sialyltransferase inhibitor blocked tumor sialic acid expression in vivo and suppressed tumor growth in multiple tumor models (74). Sialic acid blockade had a major impact on the immune cell composition of the tumor, enhancing tumor-infiltrating NK cell and CD8+ T-cell numbers while reducing regulatory T-cell and myeloid regulatory cell numbers. Sialic acid blockade enhanced cytotoxic CD8+ T-cell-mediated killing of tumor cells in part by facilitating antigen-specific T-cell-tumor cell clustering. Sialic acid blockade also synergized with adoptive transfer of tumor-specific CD8+ T cells in vivo and enhanced CpG immune adjuvant therapy by increasing dendritic cell activation and subsequent CD8+ T-cell responses. Their data emphasize the crucial role of sialic acids in tumor immune evasion and provided proof of concept that sialic acid blockade creates an immune-permissive TME for CD8+ T-cell-mediated tumor immunity, either as single treatment or in combination with other immune-based intervention strategies. Local intratumoral delivery is however not practical for treatment of disseminated cancers. Perhaps this issue can be overcome by targeted delivery to the BM using nanoparticles encapsulating the inhibitor (75, 76). Tumor targeting could also be enhanced by the functionalization of nanoparticles with monoclonal antibodies, though it may also be necessary to also target the stromal compartment for optimal efficacy (16).
Finally, an alternative novel approach would be to specifically target MM specific mucins, such as PSGL-1, by conjugating the secreted protease of C1 esterase inhibitor (StcE) to a nanobody targeting a MM specific antigen, such as BCMA. StcE is a bacterial protease from Escherichia coli and cleaves mucin domains by recognizing a discrete peptide- and glycan-based motif. Carolyn Bertozzi and colleagues have shown StcE to be highly effective in eliminating mucins, such as PSGL-1 and CD43 from the cell surface and abolishing Siglec-7 binding (28). Her group recently developed an anti HER2 conjugate, termed αHER2-eStcE, which exhibited high on-target activity as quantified by mixed cell assays in a variety of cell types expressing different mucin proteins (77). In cell lines, functional mixed cell assays replicated the biophysical and immunological effects of treatment with the wild-type StcE enzyme and showed high on-target selectivity. In an orthotopic mouse model of breast cancer, previously shown to be driven by sialic acid-mediated immunological silencing, αHER2-eStcE treatment decreased tumor burden and increased survival.
8 Conclusions
Emerging evidence implicates hypersialylation in the biology of MM, suggesting hypersialylation could play an important role in cellular trafficking, drug resistance and resistance to immune therapies, through modulation of the tumor microenvironment. Future research is required to extend these findings and establish the true importance of sialylation in in cell-cell communication, immune cell infiltration and effective anti-tumor immunity in the tumor microenvironment. disease progression. Understanding the impact of sialylation in the TME and how it affects MM disease progression is likely to uncover new therapeutic targets. Hopefully, this will lead to new innovative approaches to improve the outcome of MM patients, particularly in those with high-risk MM.
Author contributions
MO’D: Conceptualization, Data curation, Formal analysis, Writing – original draft, Writing – review & editing. SG: Data curation, Formal analysis, Writing – original draft, Writing – review & editing. RM: Data curation, Formal analysis, Writing – original draft, Writing – review & editing. AN: Writing – original draft, Writing – review & editing. AR: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AR is supported by a Science Foundation Ireland (SFI) Frontiers for the Future Award (19/FFP/6446), Starting Investigator Award (15/SIRG/3456), and Galway University Foundation.
Acknowledgments
Multiple Myeloma Research Foundation for access to data from COMMPASS study. Figures were created using Biorender.
Conflict of interest
MO’D is a founder of ONK Therapeutics, a company developing engineered NK cell therapies and a member of its Board of Directors. The subject matter of the review does not directly relate to the activities of ONK Therapeutics. He is also co-inventor on three related patents US20210186999A1, US20220389112 and US2017032727899A1 and AR is a co-inventor on patent US20210186999A1, which relate to therapeutic strategies employing inhibitors of sialylation. These patents are assigned to the University of Galway and are not currently being commercially exploited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Duane C, O'Dwyer M, Glavey S. Adoptive immunotherapy and high-risk myeloma. Cancers (Basel) (2023) 15:2633. doi: 10.3390/cancers15092633
2. Samur MK, Szalat R, Munshi NC. Single-cell profiling in multiple myeloma: insights, problems, and promises. Blood (2023) 142:313–24. doi: 10.1182/blood.2022017145
3. Glavey SV, Huynh D, Reagan MR, Manier S, Moschetta M, Kawano Y, et al. The cancer glycome: carbohydrates as mediators of metastasis. Blood Rev (2015) 29:269–79. doi: 10.1016/j.blre.2015.01.003
4. Smith BAH, Bertozzi CR. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat Rev Drug Discov (2021) 20:217–43. doi: 10.1038/s41573-020-00093-1
5. Hugonnet M, Singh P, Haas Q, von Gunten S. The distinct roles of sialyltransferases in cancer biology and onco-immunology. Front Immunol (2021) 12:799861. doi: 10.3389/fimmu.2021.799861
6. Natoni A, Macauley MS, O'Dwyer ME. Targeting selectins and their ligands in cancer. Front Oncol (2016) 6:93. doi: 10.3389/fonc.2016.00093
7. Rodrigues E, Macauley MS. Hypersialylation in cancer: modulation of inflammation and therapeutic opportunities. Cancers (Basel) (2018) 10:207. doi: 10.3390/cancers10060207
8. Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunol. Rev (2009) 230:97–113. doi: 10.1111/j.1600-065X.2009.00795.x
9. Glavey SV, Manier S, Natoni A, Sacco A, Moschetta M, Reagan MR, et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood (2014) 124:1765–76. doi: 10.1182/blood-2014-03-560862
10. Natoni A, Smith TAG, Keane N, McEllistrim C, Connolly C, Jha A, et al. E-selectin ligands recognised by HECA452 induce drug resistance in myeloma, which is overcome by the E-selectin antagonist, GMI-1271. Leukemia (2017) 31:2642–51. doi: 10.1038/leu.2017.123a
11. DeAngelo DJ, Jonas BA, Liesveld JL, Bixby DL, Advani AS, Marlton P, et al. Phase 1/2 study of uproleselan added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia. Blood (2022) 139:1135–46. doi: 10.1182/blood.2021010721
12. Natoni A, Farrell ML, Harris S, Falank C, Kirkham-McCarthy L, Macauley MS, et al. Sialyltransferase inhibition leads to inhibition of tumor cell interactions with E-selectin, VCAM1, and MADCAM1, and improves survival in a human multiple myeloma mouse model. Haematologica (2020) 105:457–67. doi: 10.3324/haematol.2018.212266
13. Natoni A, Cerreto M, De Propris MS, Petrucci MT, Fazio F, Intoppa S, et al. Sialofucosylation enables platelet binding to myeloma cells via P-selectin and suppresses NK cell-mediated cytotoxicity. Cancers (Basel) (2023) 15:2154. doi: 10.3390/cancers15072154
14. Adams OJ, Stanczak MA, von Gunten S, Läubli H. Targeting sialic acid-Siglec interactions to reverse immune suppression in cancer. Glycobiology (2018) 28:640–7. doi: 10.1093/glycob/cwx108
15. Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LA, Schetters ST, Engels S, et al. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci USA (2016) 113(12):3329–34. doi: 10.1073/pnas.1507706113
16. Egan H, Treacy O, Lynch K, Leonard NA, O'Malley G, Reidy E, et al. Targeting stromal cell sialylation reverses T cell-mediated immunosuppression in the tumor microenvironment. Cell Rep (2023) 42:112475. doi: 10.1016/j.celrep.2023.112475
17. Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol (2014) 10:69–75. doi: 10.1038/nchembio.1388
18. Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest (2014) 124:1810–20. doi: 10.1172/JCI65899
19. Daly J, Carlsten M, O'Dwyer M. Sugar free: novel immunotherapeutic approaches targeting siglecs and sialic acids to enhance natural killer cell cytotoxicity against cancer. Front Immunol (2019) 10:1047. doi: 10.3389/fimmu.2019.01047
20. Daly J, Sarkar S, Natoni A, Stark JC, Riley NM, Bertozzi CR, et al. Targeting hypersialylation in multiple myeloma represents a novel approach to enhance NK cell-mediated tumor responses. Blood Adv (2022) 6:3352–66. doi: 10.1182/bloodadvances.2021006805
21. Huang HW, Chen CH, Lin CH, Wong CH, Lin KI. B-cell maturation antigen is modified by a single N-glycan chain that modulates ligand binding and surface retention. Proc Natl Acad Sci USA (2013) 110:10928–33. doi: 10.1073/pnas.1309417110
22. Wisnovsky S, Möckl L, Malaker SA, Pedram K, Hess GT, Riley NM, et al. Genome-wide CRISPR screens reveal a specific ligand for the glycan-binding immune checkpoint receptor Siglec-7. Proc Natl Acad Sci USA (2021) 118:e2015024118. doi: 10.1073/pnas.2015024118
23. O'Dwyer M, Kirkham-McCarthy L, Cerreto M, Foà R, Natoni A. PSGL-1 decorated with sialyl Lewisa/xpromotes high affinity binding of myeloma cells to P-selectin but is dispensable for E-selectin engagement. Sci Rep (2024) 14:1756. doi: 10.1038/s41598-024-52212-2
24. Dufva O, Gandolfi S, Huuhtanen J, Dashevsky O, Duàn H, Saeed K, et al. Single-cell functional genomics reveals determinants of sensitivity and resistance to natural killer cells in blood cancers. Immunity (2023) 56:2816–2835.e13. doi: 10.1016/j.immuni.2023.11.008
25. Available online at: https://www.proteinatlas.org.
26. Azab AK, Quang P, Azab F, Pitsillides C, Thompson B, Chonghaile T, et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood (2012) 119:1468–78. doi: 10.1182/blood-2011-07-368050
27. Ferguson ID, Patiño-Escobar B, Tuomivaara ST, Lin YT, Nix MA, Leung KK, et al. The surfaceome of multiple myeloma cells suggests potential immunotherapeutic strategies and protein markers of drug resistance. Nat Commun (2022) 13:4121. doi: 10.1038/s41467-022-31810-6
28. Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, et al. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci USA (2019) 116:7278–87. doi: 10.1073/pnas.1813020116
29. Schnaar RL. Gangliosides as siglec ligands. Glycoconj J (2023) 40:159–67. doi: 10.1007/s10719-023-10101-2
30. Theruvath J, Menard M, Smith BAH, Linde MH, Coles GL, Dalton GN, et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat Med (2022) 28:333–44. doi: 10.1038/s41591-021-01625-x
31. Vuchkovska A, Glanville DG, Scurti GM, Nishimura MI, White P, Ulijasz AT, et al. Siglec-5 is an inhibitory immune checkpoint molecule for human T cells. Immunology (2022) 166:238–48. doi: 10.1111/imm.13470
32. Pepin M, Mezouar S, Pegon J, Muczynski V, Adam F, Bianchini EP, et al. Soluble Siglec-5 associates to PSGL-1 and displays anti-inflammatory activity. Sci Rep (2016) 6:37953. doi: 10.1038/srep37953
33. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
34. Gross Even-Zohar N, Pick M, Hofstetter L, Shaulov A, Nachmias B, Lebel E, et al. CD24 is a prognostic marker for multiple myeloma progression and survival. J Clin Med (2022) 11:2913. doi: 10.3390/jcm11102913
35. Sun J, Lu Q, Sanmamed MF, Wang J. Siglec-15 as an emerging target for next-generation cancer immunotherapy. Clin Cancer Res (2021) 27:680–8. doi: 10.1158/1078-0432.CCR-19-2925
36. Ishida-Kitagawa N, Tanaka K, Bao X, Kimura T, Miura T, Kitaoka Y, et al. Siglec-15 protein regulates formation of functional osteoclasts in concert with DNAX-activating protein of 12 kDa (DAP12). J Biol Chem (2012) 287:17493–502. doi: 10.1074/jbc.M111.324194
37. Tremblay G, Moraitis A, Vanderkerken K, Filion M. Inhibition of Siglec-15 to prevent bone loss in the 5T2MM model of multiple myeloma. J Clin Oncol (2015) 33:e19534. doi: 10.1200/jco.2015.33.15_suppl.e19534
38. Büll C, Nason R, Sun L, Van Coillie J, Madriz Sørensen D, Moons SJ, et al. Probing the binding specificities of human Siglecs by cell-based glycan arrays. Proc Natl Acad Sci USA (2021) 118:e2026102118. doi: 10.1073/pnas.2026102118
39. Rodriguez E, Boelaars K, Brown K, Eveline Li RJ, Kruijssen L, Bruijns SCM, et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat Commun (2021) 12:1270. doi: 10.1038/s41467-021-21550-4
40. Chang LY, Liang SY, Lu SC, Tseng HC, Tsai HY, Tang CJ, et al. Molecular basis and role of siglec-7 ligand expression on chronic lymphocytic leukemia B cells. Front Immunol (2022) 13:840388. doi: 10.3389/fimmu.2022.840388
41. Smith BAH, Deutzmann A, Correa KM, Delaveris CS, Dhanasekaran R, Dove CG, et al. MYC-driven synthesis of Siglec ligands is a glycoimmune checkpoint. Proc Natl Acad Sci USA (2023) 120:e2215376120. doi: 10.1073/pnas.2215376120
42. Available online at: www.bmbrowser.org.
43. Morgan GJ, Davies FE, Gregory WM, Bell SE, Szubert AJ, Navarro Coy N, et al. Cyclophosphamide, thalidomide, and dexamethasone as induction therapy for newly diagnosed multiple myeloma patients destined for autologous stem-cell transplantation: MRC Myeloma IX randomized trial results. Haematologica (2012) 97:442–50. doi: 10.3324/haematol.2011.043372
44. Glavey S, Wu P, Murillo LS, Loughrey C, Roccaro AM, Morgan GJ, et al. Low expression of the FUCA1 gene is an adverse prognostic factor in myeloma and combined with high sialyltransferase gene expression identifies patients at increased risk of early disease progression and death. Blood (2013) 122:1864. doi: 10.1182/blood.V122.21.1864.1864
45. Available online at: https://themmrf.org/finding-a-cure/personalized-treatment-approaches/.
46. Bärenwaldt A, Läubli H. The sialoglycan-Siglec glyco-immune checkpoint - a target for improving innate and adaptive anti-cancer immunity. Expert Opin Ther Targets (2019) 23:839–53. doi: 10.1080/14728222.2019.1667977
47. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol (2016) 17:1273–81. doi: 10.1038/ni.3552
48. Maura F, Boyle EM, Coffey D, Maclachlan K, Gagler D, Diamond B, et al. Genomic and immune signatures predict clinical outcome in newly diagnosed multiple myeloma treated with immunotherapy regimens. Nat Cancer (2023) 4(12):1660–74. doi: 10.1038/s43018-023-00657-1
49. Lin WD, Fan TC, Hung JT, Yeo HL, Wang SH, Kuo CW, et al. Sialylation of CD55 by ST3GAL1 facilitates immune evasion in cancer. Cancer Immunol Res (2021) 9:113–22. doi: 10.1158/2326-6066.CIR-20-0203
50. Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood (2016) 128:959–70. doi: 10.1182/blood-2016-03-703439
51. Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von Gunten S, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest (2018) 128:4912–23. doi: 10.1172/JCI120612
52. Haas Q, Boligan KF, Jandus C, Schneider C, Simillion C, Stanczak MA, et al. Siglec-9 regulates an effector memory CD8+ T-cell subset that congregates in the melanoma tumor microenvironment. Cancer Immunol Res (2019) 7:707–18. doi: 10.1158/2326-6066.CIR-18-0505
53. Haas Q, Markov N, Muerner L, Rubino V, Benjak A, Haubitz M, et al. Siglec-7 represents a glyco-immune checkpoint for non-exhausted effector memory CD8+ T cells with high functional and metabolic capacities. Front Immunol (2022) 13:996746. doi: 10.3389/fimmu.2022.996746
54. Swindall AF, Bellis SL. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem (2011) 286:22982–90. doi: 10.1074/jbc.M110.211375
55. Edgar LJ, Thompson AJ, Vartabedian VF, Kikuchi C, Woehl JL, Teijaro JR, et al. Sialic acid ligands of CD28 suppress costimulation of T cells. ACS Cent Sci (2021) 7:1508–15. doi: 10.1021/acscentsci.1c00525
56. Greco B, Malacarne V, De Girardi F, Scotti GM, Manfredi F, Angelino E, et al. Disrupting N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid Malignancies. Sci Transl Med (2022) 14:eabg3072. doi: 10.1126/scitranslmed.abg3072
57. Ramkumar P, Abarientos AB, Tian R, Seyler M, Leong JT, Chen M, et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv (2020) 4:2899–911. doi: 10.1182/bloodadvances.2019001346
58. Hinderlich S, Weidemann W, Yardeni T, Horstkorte R, Huizing M. UDP-glcNAc 2-epimerase/manNAc kinase (GNE): A master regulator of sialic acid synthesis. Top Curr Chem (2015) 366:97–137. doi: 10.1007/128_2013_464
59. Broussard A, Florwick A, Desbiens C, Nischan N, Robertson C, Guan Z, et al. Human UDP-galactose 4'-epimerase (GALE) is required for cell-surface glycome structure and function. J Biol Chem (2020) 295:1225–39. doi: 10.1074/jbc.RA119.009271
60. Durgin JS, Thokala R, Johnson L, Song E, Leferovich J, Bhoj V, et al. Enhancing CAR T function with the engineered secretion of C. perfringens neuraminidase. Mol Ther (2022) 30:1201–14. doi: 10.1016/j.ymthe.2021.11.014
61. Decker CE, Young T, Pasnikowski E, Chiu J, Song H, Wei Y, et al. Genome-scale CRISPR activation screen uncovers tumor-intrinsic modulators of CD3 bispecific antibody efficacy. Sci Rep (2019) 9:20068. doi: 10.1038/s41598-019-56670-x
62. Szijj PA, Gray MA, Ribi MK, Bahou C, Nogueira JCF, Bertozzi CR, et al. Chemical generation of checkpoint inhibitory T cell engagers for the treatment of cancer. Nat Chem (2023) 15(11):1636–47. doi: 10.1038/s41557-023-01280-4
63. Lee H, Ahn S, Maity R, Leblay N, Ziccheddu B, Truger M, Chojnacka M, et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med (2023) 29:2295–306. doi: 10.1038/s41591-023-02491-5
64. Friedrich MJ, Neri P, Kehl N, Michel J, Steiger S, Kilian M, et al. The pre-existing T cell landscape determines the response to bispecific T cell engagers in multiple myeloma patients. Cancer Cell (2023) 41:711–725.e6. doi: 10.1016/j.ccell.2023.02.008
65. Verkleij CPM, Broekmans MEC, van Duin M, Frerichs KA, Kuiper R, de Jonge AV, et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv (2021) 5:2196–215. doi: 10.1182/bloodadvances.2020003805
66. Holthof LC, van der Schans JJ, Katsarou A, Poels R, Gelderloos AT, Drent E, et al. Bone marrow mesenchymal stromal cells can render multiple myeloma cells resistant to cytotoxic machinery of CAR T cells through inhibition of apoptosis. Clin Cancer Res. (2021) 27:3793–803. doi: 10.1158/1078-0432.CCR-20-2188
67. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood (2022) 139:3708–21. doi: 10.1182/blood.2021012811
68. de Jong MME, Kellermayer Z, Papazian N, Tahri S, Hofste Op Bruinink D, Hoogenboezem R, et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat Immunol (2021) 22:769–80. doi: 10.1038/s41590-021-00931-3
69. Xiao H, Woods EC, Vukojicic P, Bertozzi CR. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci USA (2016) 113:10304–9. doi: 10.1073/pnas.1608069113
70. Gray MA, Stanczak MA, Mantuano NR, Xiao H, Pijnenborg JFA, Malaker SA, et al. Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat Chem Biol. (2020) 16:1376–84. doi: 10.1038/s41589-020-0622-x
71. Available online at: https://clinicaltrials.gov/ct2/show/NCT05259696.
72. Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, et al. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat Chem Biol (2012) 8:661–8. doi: 10.1038/nchembio.999
73. Macauley MS, Arlian BM, Rillahan CD, Pang PC, Bortell N, Marcondes MC, et al. Systemic blockade of sialylation in mice with a global inhibitor of sialyltransferases. J Biol Chem. (2014) 289:35149–58. doi: 10.1074/jbc.M114.606517
74. Büll C, Boltje TJ, Balneger N, Weischer SM, Wassink M, van Gemst JJ, et al. Sialic acid blockade suppresses tumor growth by enhancing T-cell-mediated tumor immunity. Cancer Res (2018) 78:3574–88. doi: 10.1158/0008-5472.CAN-17-3376
75. Natoni A, Bohara R, Pandit A, O'Dwyer M. Targeted approaches to inhibit sialylation of multiple myeloma in the bone marrow microenvironment. Front Bioeng Biotechnol (2019) 7:252. doi: 10.3389/fbioe.2019.00252
76. Büll C, Boltje TJ, van Dinther EA, Peters T, de Graaf AM, Leusen JH, et al. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano (2015) 9:733–45. doi: 10.1021/nn5061964
Keywords: multiple myeloma, glycosylation, sialic acid, selectin, Siglec, immune evasion, immunotherapy, cellular trafficking
Citation: O’Dwyer M, Glavey S, McAvera R, Natoni A and Ryan AE (2024) Hypersialylation and multiple myeloma. Front. Hematol. 3:1360149. doi: 10.3389/frhem.2024.1360149
Received: 22 December 2023; Accepted: 01 February 2024;
Published: 19 February 2024.
Edited by:
Tao Zhang, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Yongwei Zheng, Guangzhou Bio-Gene Technology Co., Ltd., ChinaGovindarajan Thangavelu, University of Minnesota Twin Cities, United States
Copyright © 2024 O’Dwyer, Glavey, McAvera, Natoni and Ryan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael O’Dwyer, bWljaGFlbC5vZHd5ZXJAdW5pdmVyc2l0eW9mZ2Fsd2F5Lmll