 Laura Pelagatti1†
Laura Pelagatti1† Giulia Pozzi2†
Giulia Pozzi2† Samuele Cortellazzi2
Samuele Cortellazzi2 Cristina Mancini3Eugenia Martella3Luca Pagliaro1,4Mariateresa Giaimo1,4
Cristina Mancini3Eugenia Martella3Luca Pagliaro1,4Mariateresa Giaimo1,4 Giovanni Roti1,4Marco Vitale5
Giovanni Roti1,4Marco Vitale5 Cecilia Carubbi2*
Cecilia Carubbi2* Elena Masselli1,2*
Elena Masselli1,2*- 1Hematology and Bone Marrow Transplant (BMT) Unit, Parma University Hospital (AOU-PR), Parma, Italy
- 2Anatomy Unit, Department of Medicine and Surgery (DiMeC), University of Parma, Parma, Italy
- 3Pathology Unit, Parma University Hospital (AOU-PR), Parma, Italy
- 4Translational Hematology and Chemogenomics (THEC), Department of Medicine and Surgery (DiMeC), University of Parma, Parma, Italy
- 5Faculty of Medicine and Surgery, University Vita-Salute San Raffaele, Milan, Italy
Introduction: Polycythemia vera (PV) is a JAK2-mutated myeloproliferative neoplasm (MPN) characterized by clonal erythrocytosis and an intrinsic risk of transformation into acute myeloid leukemia (AML), known as blast-phase (BP) disease, a condition typified by dismal prognosis. In PV, the evolution to BP generally occurs through an overt fibrotic progression, represented by the post-PV myelofibrotic (MF) stage. However, direct leukemic transformation from PV may also occur in up to ~50% of patients. In this study, we sought to shed light on the morphological, clinical, and molecular features that may differentiate BP arising from a direct transition from the PV stage (post-PV-BP) from those evolving through a diagnosis of post-PV myelofibrosis (post-PV-MF-BP).
Methods and results: We retrospectively analyzed a cohort of post-PV-BP (n=5) and post-PV-MF-BP (n=5). We found that BP arising from PV directly displayed significantly lower leukocyte count (median 2.93 × 109/L, range: 2.30–39.40 vs. median 41.05 × 109/L, range: 5.46–58.01; P=0.03), and spleen diameter (14.0 cm, range: 11.5–20.0 vs. 25.5 cm, range: 18–26; P=0.03) as compared to those experiencing an overt fibrotic stage. The most striking differences emerged from bone marrow (BM) morphological analysis: all post-PV-BP were characterized by significantly higher cellularity (median 70%, range: 60%–98% vs. 28%, range: 2%–41%, P=0.0245), lower degree of fibrosis (fibrosis grade 1 vs. fibrosis grade 3 in all cases, P=0.008) and dysplastic features involving all three lineages, most prominently the erythroid and megakaryocytic compartment. Next-generation sequencing (NGS) analysis revealed that post-PV-BP cases were enriched in mutations located in genes involved in DNA methylation such as DNMT3A, IDH1/2, and TET2 (45% vs. 15%, P=0.038).
Discussion: With all the limits of the small number of patients for each cohort, our data suggest that BPs that arise directly from PV present a peculiar phenotype, consistent with the molecular signature of the disease, typified by mutations of genes occurring with a high frequency in Myelodysplastic Syndromes (MDS) and MDS/MPN. Further studies in larger cohorts are warranted to translate these observations into robust evidence that may advise therapeutic choices.
1 Introduction
Polycythemia vera (PV) is a JAK2-mutated myeloproliferative neoplasm (MPN) typified by clonal erythrocytosis. Along with primary myelofibrosis (PMF) and essential thrombocythemia (ET), PV belongs to the classic Philadelphia-negative MPN. PV can lead to significant morbidity and increased mortality, with patients experiencing a reduced quality of life, a high rate of vascular events, and the potential intrinsic risk of disease evolution. Mutations in JAK2 Exon 12-15 encompass the vast majority (97%–98%) of PV mutational landscape (1). In addition, over 50% of patients harbor non-driver mutations in specific myeloid genes with the most frequent being TET2 (18%), ASXL1 (15%), and SH2B3 (3%) (2).
Approximately 15% of patients with PV experience a fibrotic progression of their disease over time, termed post-PV myelofibrosis (post-PV-MF) (2) and about 3% evolve, over a period of 10 years, into accelerated (MPN-AP) and blast phase (MPN-BP) disease (3), operationally defined by 10% to 19% or ≥ 20% myeloid blasts in the peripheral blood or bone marrow, respectively (4). However, despite this formal classification, MPN patients with 5% or more circulating/marrow blasts can be on a spectrum that leads to MPN-BP, with similar poor survival rates (5).
Clinical and molecular risk factors for leukemic evolution have been identified by the Mayo-Florence study conducted on 410 patients with MPN-BP, with 97 patients whose condition progressed from an antecedent PV, either directly (46 patients) or through a post-PV-MF stage (51 patients). Hematological risk factors are leukocytosis ≥11 × 109/uL (Florence cohort) or ≥15 × 109/uL (Mayo cohort), older age (> 67 years, both cohorts), abnormal karyotype (Mayo cohort), presence of IDH2 and RUNX1 mutations (Mayo cohort), and SRSF2 mutations (Florence cohort) (3).
On these bases, investigators generated a mutation-enhanced international prognostic system for chronic phase disease (MIPSS-PV), which included presence of adverse mutations (SRSF2) (two points); abnormal karyotype (one point); age >67 years (three points); and leukocyte count ≥15 × 109/uL (two points) (6). Additionally, it is well established that previous treatment with radiophosphorous, chlorambucil, or pipobroman increases the risk of leukemic transformation (3).
The evolution to AP and BP is an almost invariably fatal event in the course of PV (and MPN in general), with a median overall survival (OS) of 13 months for AP and 3–5 months for BP, due to disease aggressiveness and limited therapeutic options, especially for transplant ineligible patients (4). The lack of standardized treatment(s) mirrors our still fragmented understanding of the molecular mechanisms underlying the progression of an MPN to AP/BP. It is currently acknowledged that the acquisition of additional somatic mutations and epigenetic alterations in hematopoietic progenitor and stem cells play a pivotal role in leukemic transformation, accounting for the selection and expansion of the malignant clone on one side, and on the other side, the generation of a tumor supportive proinflammatory microenvironment. Two main models of leukemic transformation have been proposed: (i) the acquisition of additional mutations by the JAK2/CALR/MPL-positive clone (7) and (ii) the emergence and expansion of a leukemic clone distinct from the clone bearing an activating driver mutation, eventually overcoming the MPN JAK2/CALR/MPL-mutated clone (7). In the first setting, the most common genetic alteration is represented by TP53 mutations, detected in approximately 15% of patients with JAK2V617F (showing cooperative activity) and 25% of patients with CALR mutations. In the second scenario, a decrease in variant allele frequency (VAF) or loss of pre-existing driver mutation is usually observed (4). In addition to these two scenarios, another possible mechanism has been suggested, focused on the timing of occurrence of mutations involving chromatin modifiers, spliceosome complex components, DNA methylation modifiers, tumor suppressors, and transcriptional regulators. These mutations are described as coexisting mutations since their presence within a clone can precede the acquisition of the driver mutation or can occur subsequently. Grabek et al. (8) suggested that specific coexisting mutations in the same driver-mutated clone may favor direct leukemic transformation while others may drive progression to myelofibrosis which in turn may evolve into AP/BP.
Independent from the molecular pathway leading to MPN-AP/BP, it is well established that the mutational profile and overall prognosis of acute myeloid leukemia (AML) resulting from an antecedent MPN remains distinct from that in de novo AML (2, 4, 9). Indeed, mutations in FLT3 and NPM1 are rarely seen in MPN-BP, while, in addition to driver mutations, frequently mutated genes are ASXL1 (30%), TET2 (25%), SRSF2 (22%), RUNX1 (20%), and TP53 (17%). Moreover, 25% and 46% of patients, harbor mutations in 3 and ≥4 genes, respectively. TP53 is the only individual mutation to correlate with shorter OS (9).
In PV, the evolution to MPN-AP/BP generally occurs through a fibrotic progression, represented by the secondary (post-PV) MF stage, whose diagnosis is codified by the IWG-MRT criteria (10). However, direct leukemic transformation from a florid disease (PV) to AP/BP may also occur. Data from the Mayo-AGIMM study indicate that cases of post-PV AP/BP without a documented MF phase range from 37% (AGIMM cohort) to 53% (Mayo cohort). Similar rates were found for post-ET BP (58% and 55% in the two cohorts, respectively) (3).
So far, studies have mainly focused on the comparison of the clinical and biological characteristics of MPN-AP/BP vs. de novo AML. Here, we analyzed the morphological, clinical, and molecular features of patients with post-PV AP/BP arising from a direct progression from PV to leukemic phase (overall, termed post-PV-BP), and compared them with those transitioning through a documented overt fibrotic stage (termed post-PV-MF-BP), documenting that post-PV-BP is characterized by unique features, distinct from post-PV-MF-BP.
2 Methods
The current study is a single-center descriptive retrospective review of 10 consecutive cases of AP/BP evolving from an antecedent PV, either directly, without a documented overt fibrotic progression (termed post-PV-BP, five patients, with two AP and three BP), or through a documented MF stage, i.e., diagnosis of post-PV-MF (termed post-PV-MF-BP, five patients, all BP), followed at the Hematology and BMT Unit of Parma University Hospital.
Demographic, clinical, and molecular data at the time of PV diagnosis and leukemic transformation were collected in a dedicated database after patient de-identification by univocal alphanumeric codes (Ethical Committee approval: protocol 6923, 17/02/2020). Diagnosis of PV, post-PV-MF, and AP/BP was determined according to the WHO/IWG-MRT criteria (10, 11).
Cytogenetic analysis was performed according to the International System for Human Cytogenetic Nomenclature criteria. High-risk karyotype included monosomal karyotype and single or multiple cytogenetic abnormalities of −7, inv(3)(q21.3q26.2)/t(3,3)(q21.3;q26.2), i(17)(q10)/-17/abn(17p) (3). NGS analysis for target myeloid genes was performed by Myeloid Solutions Panel (SOPHiA Genetics, Saint Sulpice, Switzerland). Alignment, base calling, and variant annotation were performed with SOPHiA DDM software and investigating available databases (such as NCBI, COSMIC, GNOMAD, CLINVAR, and ExAC). Pathogenic relevant mutations (category A and B) were characterized by a functional analysis through Hidden Markov Models (FATHMM) score >0.90, while genetic variants that have been flagged as an SNP in the Catalogue of Somatic Mutations In Cancer (COSMIC) database and/or reported as of undetermined significance/benign/likely benign in ClinVar database belonged to category C or D and had a low FATHMM score (<0.5).
Bone marrow biopsies were fixed in neutral buffered formalin, decalcified with ethylenediamine tetra-acetic acid and paraffin embedded for routine diagnostic purposes. Paraffin sections were used for hematoxylin-eosin staining and for reticulin staining. BM age-adjusted cellularity was evaluated according to the European Myelofibrosis Network (EUMNET) consensus (12), and the grading of BM fibrosis was assessed semi-quantitatively following the European consensus guidelines (12). BM sections were also immunostained by using antibodies against CD34 (clone QBend/10), CD61 (clone2f2), CD71 (clone MRQ-48), and myeloperoxidase (polyclonal antibody) (Ventana-Roche, Basilea, Switzerland). Immunostaining was carried out with polymeric system Ultraview 3,3′-diaminobenzidine DAB IHC Detection Kit (Ventana-Roche, Basilea, Switzerland), on a BenchMark ULTRA IHC/ISH instrument (Roche, Basilea, Switzerland), in accordance with the manufacturer’s protocols. Images were captured and analyzed by an Olympus DP22 digital camera (Ventana-Roche, Basilea, Switzerland).
Immunophenotyping was performed using BD FACSLyric™ Flow Cytometer (BD™ Biosciences, Franklin Lakes, NJ, USA). Peripheral blood samples (3–5 mL in EDTA tubes) were processed with lyse and wash protocol. After labeling with antiCD45-APC-H7, antiCD34-PerCP-Cy5.5, antiCD33-PE, antiCD71-APC, and CD117-PE-Cy7 (all from BD™ Biosciences), the different cell populations were identified according to a sequential and cumulative gating strategy, adapted from the ISHAGE guidelines for stem and progenitor cell analysis by flow cytometry (13), combining specific immunostaining with the analysis of lateral dispersion, or side scatter (SSC), and frontal dispersion, or forward scatter (FSC).
For statistical analysis, numerical variables were summarized by their median and range, and categorical variables by count and relative frequency (percentage). Differences in the distribution of continuous variables were calculated by Mann–Whitney/Kruskal–Wallis tests, while categorical variable comparisons were established by χ2/Fisher’s exact test. A P value <0.05 was considered statistically significant. Analysis was performed with dedicated software (Prism 5, GraphPad Software Inc., San Diego, CA, USA).
3 Results
3.1 Demographic, clinical, and laboratory characteristics at the time of PV diagnosis and leukemic transformation
In both cohorts (post-PV-BP and post-PV-MF-BP), 40% of patients were male (2 out of 5). Median follow-up from PV diagnosis was 83 months (range: 15–190) for post-PV-BP and 118 months (range: 28–301) for post-PV-MF-BP (P=0.6, ns), with a mortality rate of 80% (4 out of 5) and 100% (5 out of 5), respectively. Concerning the two APs, post-PV-AP2 died soon after the diagnosis for infectious complications, while post-PV-AP5 was eligible for intensive chemotherapy and underwent allogenic bone marrow transplant (alloHSCT) 10 months after disease progression.
The clinical and laboratory features of PV stage for post-PV-BP and post-PV-MF are summarized in Table 1. Median age at the time of PV diagnosis was similar in the two groups (67 years, range: 49–84 for post-PV-BP and 63 years, range: 42–70 for post-PV-MF-BP). All patients were JAK2V617F mutated, while no mutations of JAK2 exon 12 were found in either group. No differences in hematologic parameters (CBC counts, LDH levels, spleen longitudinal diameter, and history of major thrombotic events) were observed at the time of PV stage in the two groups. BM biopsy revealed a scattered/loose network of reticulin with no/few intersections (fibrosis grade 0/0–1) in all post-PV-BP and in the vast majority (4 out of 5, 80%) of post-PV-MF-BP, with only one patient (1 out of 5, 20%, post-PV-MF-BP2) presenting a fibrosis grade 1. Despite fibrosis grade ≥ 1 at the time of PV diagnosis being associated with reduced myelofibrosis-free survival (14, 15), time to progression to MF of this patient was 96 months, as compared to a median 69 months of the other post-PV-MF cases (range: 11–203 months).
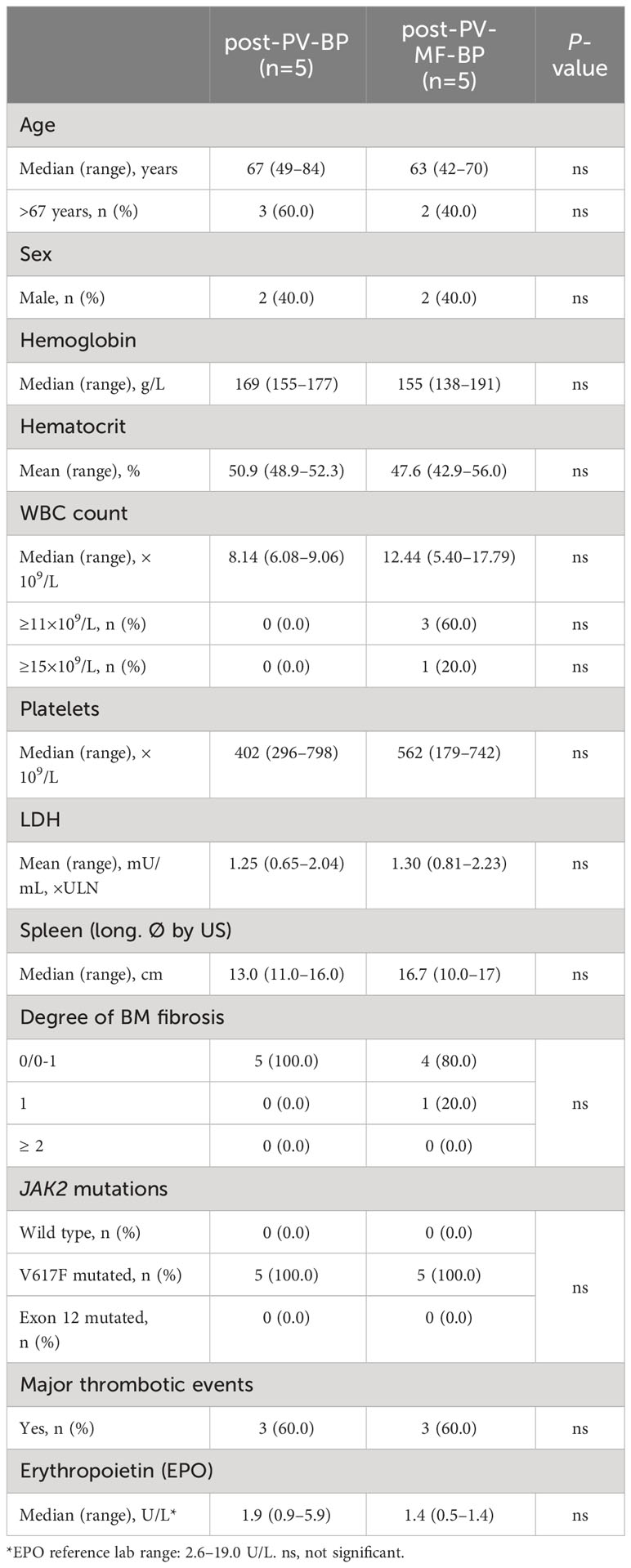
Table 1 Demographic, clinical, and laboratory features at the time of PV diagnosis in post-PV-BP and post-PV-MF-BP.
Clinical and laboratory features at the time of evolution to BP are summarized in Table 2. Also at this stage, median age at diagnosis was similar in the two groups (76 years, range: 56–85 for post-PV-BP and 69 years, range: 66–79 for post-PV-MF-BP). Median time to progression from initial PV diagnosis was comparable as well: 7 years (range: 1–14) for post-PV-BP and 8 years (range: 2–24) for post-PV-MF-BP.
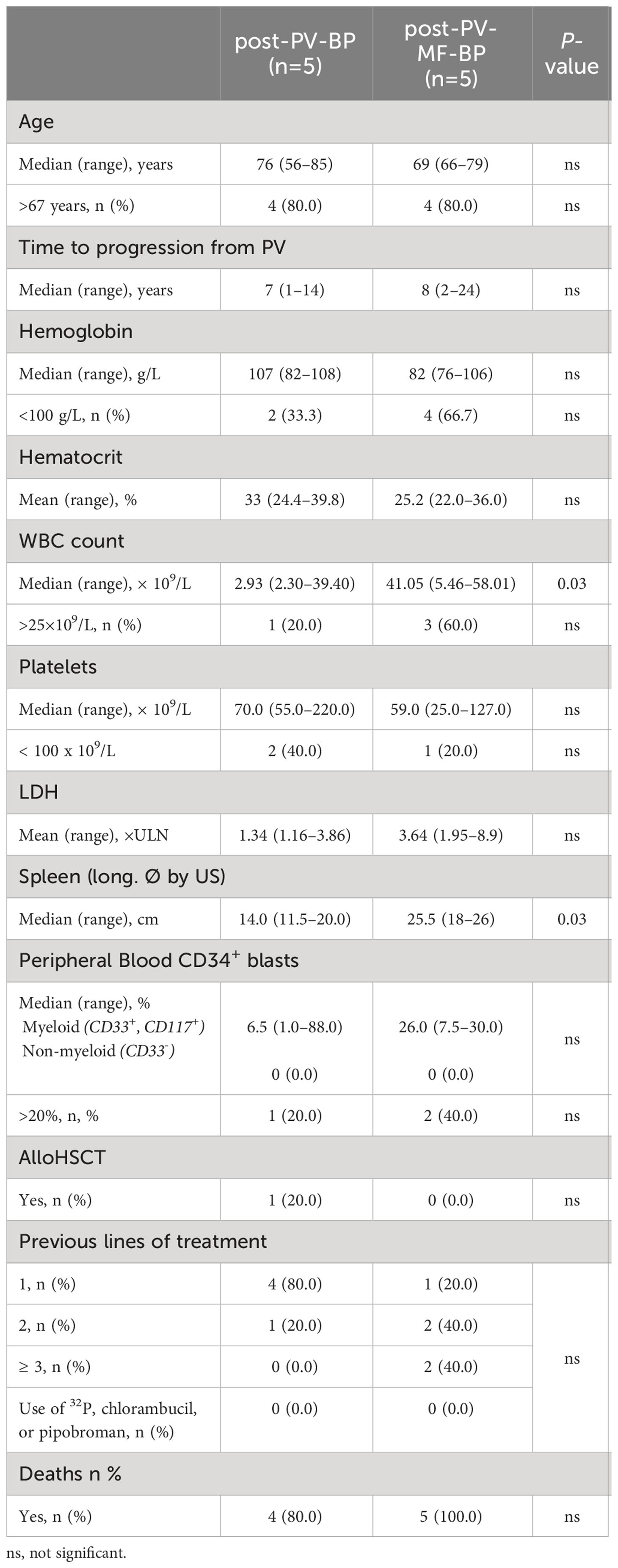
Table 2 Demographic, clinical, and laboratory features at the time of leukemic evolution in post-PV-BP and post-PV-MF-BP.
At the time of BP transformation, post-PV-MF-BP patients presented with a significantly higher WBC count (median 41.05 × 109/L, range: 5.46–58.01) as compared to post-PV-BP patients (median 2.93 × 109/L, range: 2.30–39.40, P=0.03) and a significantly larger splenomegaly (median longitudinal diameter by US: 25.5 cm, range: 18–26 vs. 14.0, range: 11.5–20.0, P=0.03). No differences in terms of hemoglobin and hematocrit levels, platelet counts, LDH levels, and percentage of peripheral blood myeloid blasts (CD34+, CD117+, CD33+) were found. Lines of cytoreductive treatment prior to BP are summarized in Table 2: the majority (4 out of 5, 80%) of post-PV-BP underwent only one line of treatment during PV stage, represented by hydroxyurea, while, as expected, post-PV-MF-BP mostly experienced multiple lines of treatment (≥2 in 4 out of 5, 80%), represented by hydroxyurea (all patients), interferon (1 out of 5, 20%), busulfan (1 out of 5, 20%) during PV stage, and the JAK1/2-inhibitor ruxolitinib during the MF stage (3 out of 5, 60%). Of note, history of treatment did not include radiophosphorous, chlorambucil, or pipobroman for both categories.
No differences in OS after diagnosis of leukemic transformation were detected in the two groups (13 months in post-PV-BP vs. 5 months in post-PV-MF-BP, P=0.390).
3.2 Bone marrow features at the time of leukemic transformation
Bone marrow morphological features and immunophenotype and cytogenetic data during AP/BP for post-PV-BP and post-PV-MF-BP are reported in Table 3.
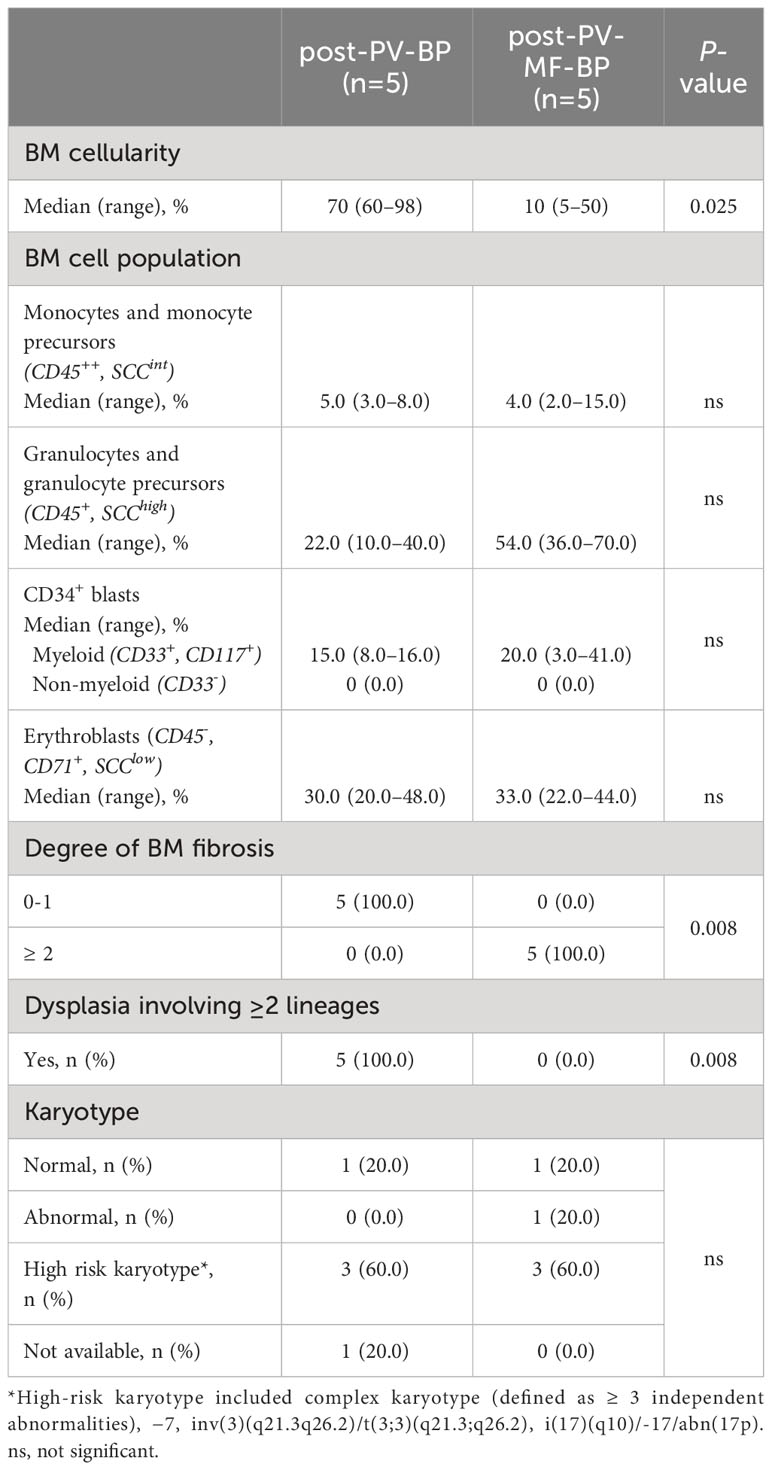
Table 3 Bone marrow morphological and cytogenetic features at the time of leukemic evolution in post-PV-BP and post-PV-MF-BP.
Post-PV-MF-BP are characterized by a significantly lower cellularity as compared to post-PV-BP (28%, range: 2%–41% vs. 70%, range: 60%–98%, P=0.0245; Table 3; Figures 1A–D), with marked reduction of all three hematopoietic lineages (megakaryocytic, erythroid, and myeloid). This is highlighted in Figures 2B, D, F with CD61-, CD71-, and MPO-immunostaining, respectively.
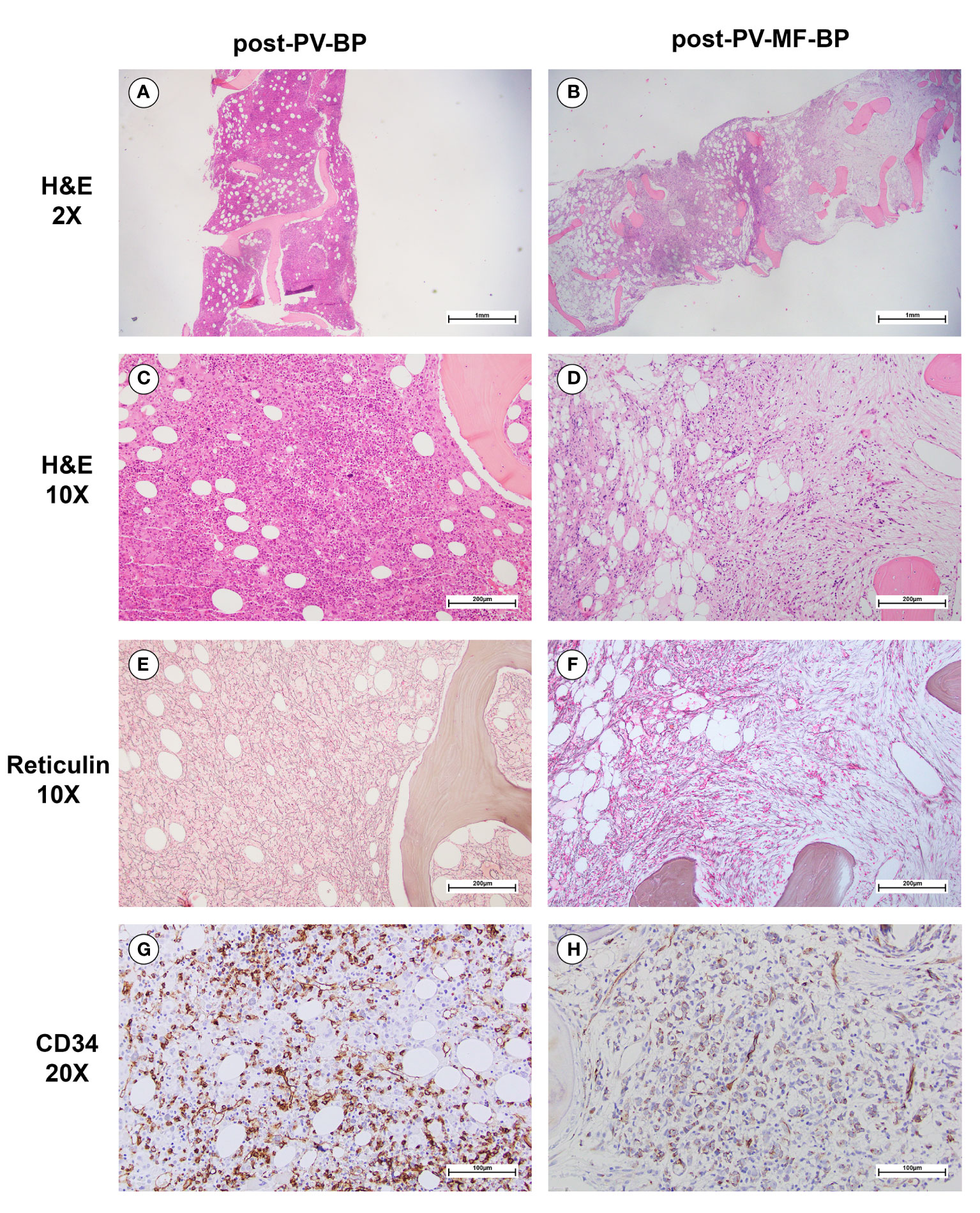
Figure 1 Morphological bone marrow features of a representative post-PV-BP and post-PV-MF-BP. (A–D) H&E staining showing increased BM cellularity in post-PV-BP (A, C); and dramatically reduced BM cellularity, with cellular streaming in post-PV-MF-BP (B, D) (panels A and B: original magnification 2x, scale bar: 1mm; panels C and D: original magnification 10x, scale bar: 200μm). (E) Reticulin staining of post-PV-BP BM biopsy, showing a loose network of reticulin fibers with few intersections (fibrosis grade 0–1) (original magnification 10x, scale bar: 200 μm). (F) Reticulin staining of post-PV-MF-BP BM biopsy, showing a diffuse and dense network of reticulin fibers with extensive intersections (fibrosis grade 3) (original magnification 10x, scale bar: 200 μm). (G, H) CD34-immunostaining of post-PV-BP (G) and post-PV-MF-BP BM biopsy (H) (original magnification 20x, scale bar: 100 μm) showing similar infiltrates of immature myeloid cells.
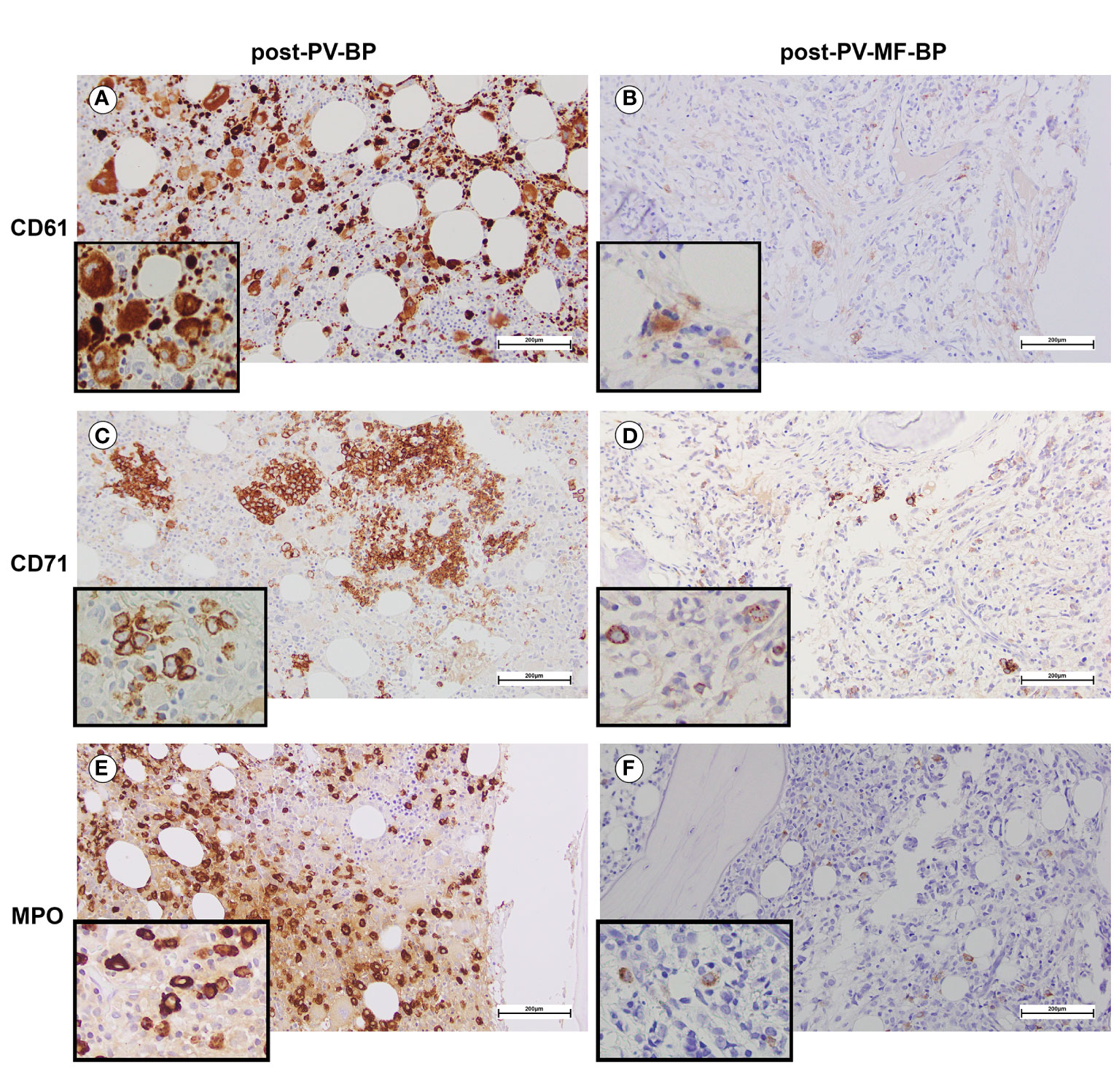
Figure 2 BM immunostaining assessing erythroid, megakaryocytic, and myeloid lineages in a representative post-PV-BP and post-PV-MF-BP. (A, B) CD61-immunostaining of post-PV-BP (A) and post-PV-MF-BP BM biopsy (B). Megakaryocyte hyperplasia with dysplastic features (tight clusters of hypolobulated/mono-nucleated megakaryocytes, insert) is detected in post-PV-BP (A) vs. megakaryocyte hypoplasia with morphological atypia (hypolobulation, insert) in post-PV-MF-BP (B) (original magnification 20×, scale bar: 200 µm; insert 40×). (C, D) CD71-immunostaining of a post-PV-BP (C) and a post-PV-MF-BP BM biopsy (D) showing a marked reduction of the erythroid compartment in post-PV-MF-BP (D) vs. scattered clusters of residual erythropoiesis with megaloblasts (insert) in post-PV-BP (C) (original magnification 20×, scale bar: 200 µm; insert 40×). (E, F) Myeloperoxidase immunostaining of a post-PV-BP (E) and a post-PV-MF-BP (F) showing hyperplastic (post-PV-BP) vs. reduced (post-PV-MF-BP) myelopoiesis (original magnification 20×, scale bar: 200 µm; insert ×40).
By contrast, post-PV-BP displayed clusters of residual dysplastic erythropoiesis (Figure 2C), megakaryocytic and myeloid hyperplasia with altered topographical dislocation and peculiar dysplastic features (Figures 2A, E), further documented by May-Grunwald Giemsa staining of bone marrow aspirates described in detail below (Figure 3).
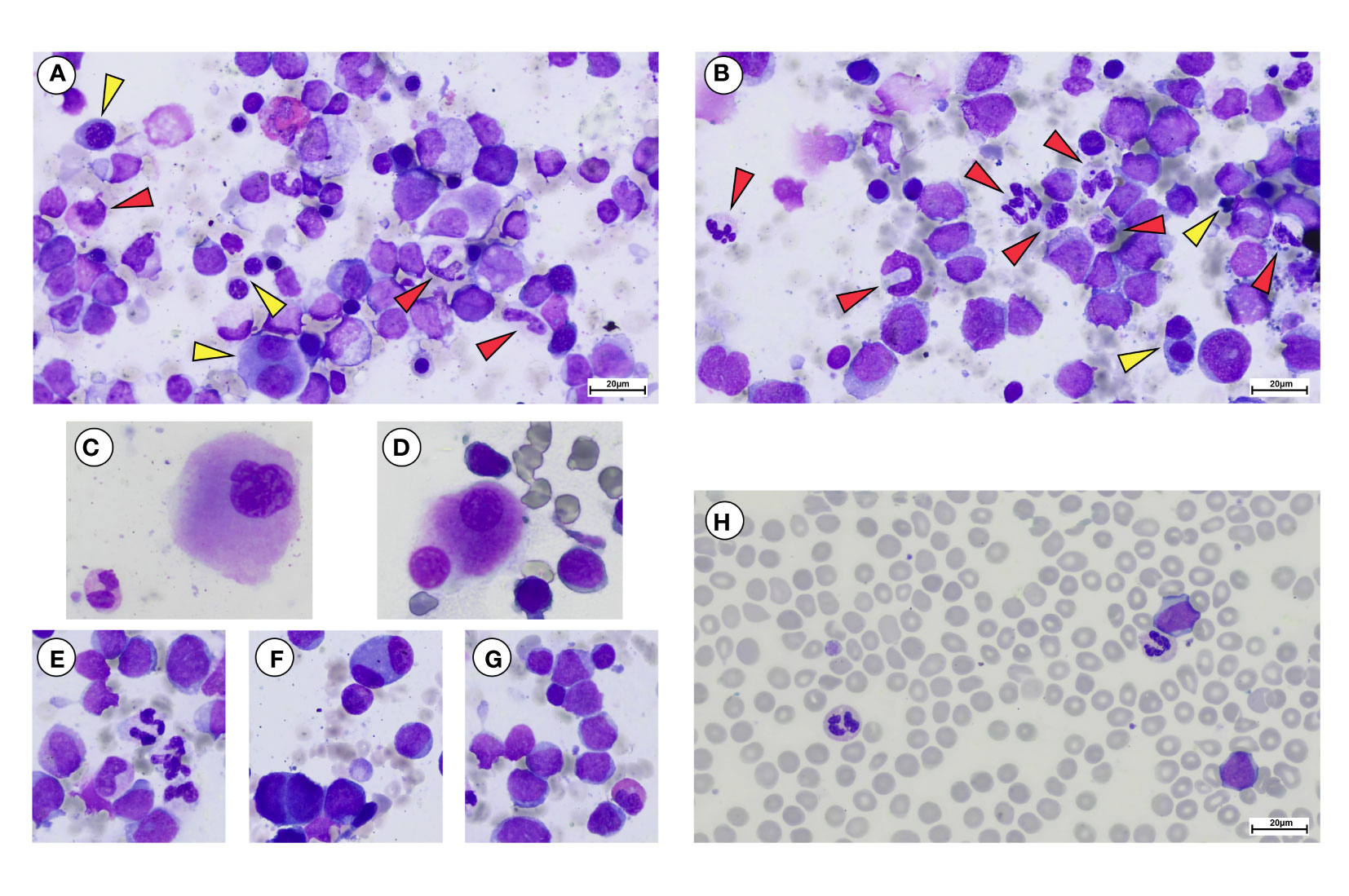
Figure 3 May Grunwald-Giemsa staining of bone marrow aspirate and peripheral blood smear of a representative post-PV-BP. (A–G) Overview of erythroid myeloid and megakaryocytic lineages showing dysplastic features in bone marrow aspirate. (A, B) Erythroid lineage (yellow arrows) showing dysplastic features including presence of chromatin bridges, nuclear/cytoplasmatic maturation asynchrony, bi-nucleated, and giant erythroblasts with nuclear shape abnormalities. (A, B) Myeloid lineage (red arrows) showing dysplastic features including nuclear segmentation defects/hypersegmentation, chromatin clumping, hypo-granular cytoplasm, and cytoplasmic vacuoles. (C, D) Hypolobulated dysplastic megakaryocytes. (E) Dysgranulopoiesis, with myeloid precursors and neutrophils with hypo/agranular cytoplasm, multiple vacuoles causing nuclear dislocation and nuclear segmentation defects. (F) Bi-nucleated erythroblasts. (G) Myeloid blasts with a thin rim of agranular cytoplasm. A dysplastic hyposegmented eosinophil can also be observed. (H) Peripheral blood smear showing myeloid blasts, recapitulating the same characteristic of the BM counterpart. Dysplastic vacuolated neutrophils and a giant platelet can be observed as well (original magnification 40×, scale bar: 20 µm).
In post-PV-MF-BP, indeed, bone marrow architecture is altered by extensive deposition of reticulin fibers, arranged in coarse bundles with numerous intersections, and consequent cellular streaming, compatible with BM fibrosis grade 3 in all subjects, in contrast to post-PV-BP, which, instead, displayed only a mild fibrosis (grade 1 in all cases, P=0.008, Table 3; Figures 1E, F), in some cases already present at the time of PV diagnosis (Table 1).
Despite the absolute reduction in overall BM cellularity, BM aspirate immunophenotyping (Table 3) revealed a similar % of myeloid (CD34+, CD117+, and CD33+) blasts, consistent with the CD34 immunostaining of bone marrow biopsies (Figures 1G, H). Flow cytometric analysis of BM cell populations documented a similar % of monocytes/monocyte precursors (CD45++, SCCint), granulocyte/granulocyte precursors (CD45+, SCChigh), and nucleated erythroid cells (CD45-, CD71+, and SCClow).
Finally, in both groups, majority of patients (3 out of 5, 60%) presented a high-risk karyotype.
One of the most striking aspects was that all patients of the post-PV-BP group demonstrated dysplastic features involving all three lineages, while no sign of dysplasia was detectable in the post-PV-MF-BP cohort (P=0.008). May-Grunwald-Giemsa staining of BM aspirates from a representative post-PV-BP case (Figures 3A–G) recapitulates these findings. Erythroid dysplasia is clearly documented by the presence of bi-nucleated giant erythroblasts, chromatin bridges, nuclear/cytoplasm maturation asynchrony, and severe nuclear shape abnormalities (yellow arrows in panels A–B, and panel F). Myeloid dysplastic features included neutrophil and granulocyte precursors with nuclear segmentation defects/hyper-segmentation, chromatin clumping, agranular/hypo-granular cytoplasm, and cytoplasmatic vacuoles leading to nuclear dislocation (red arrows in panels A–B and panel E). Hypolobulated/micro-megakaryocytes are as shown in panels C and D, respectively.
Panels G and H document BM and PB myeloid blast morphology, respectively, typified by a thin rim of agranular cytoplasm. In panel H, PB dysplastic neutrophils are also detected.
3.3 Molecular features at the time of leukemic transformation
Table 4 summarizes the distribution of pathogenic relevant mutations [category A and B, Functional Analysis through Hidden Markov Models (FATHMM) score >0.90] in the two cohorts (post-PV-BP and post-PV-MF-BP). Nucleotide change, amino-acid substitution, type of mutation (e.g., missense and frameshift), and variant allele frequency (VAF %) are reported for each category A and category B mutation in Supplementary Table 1.
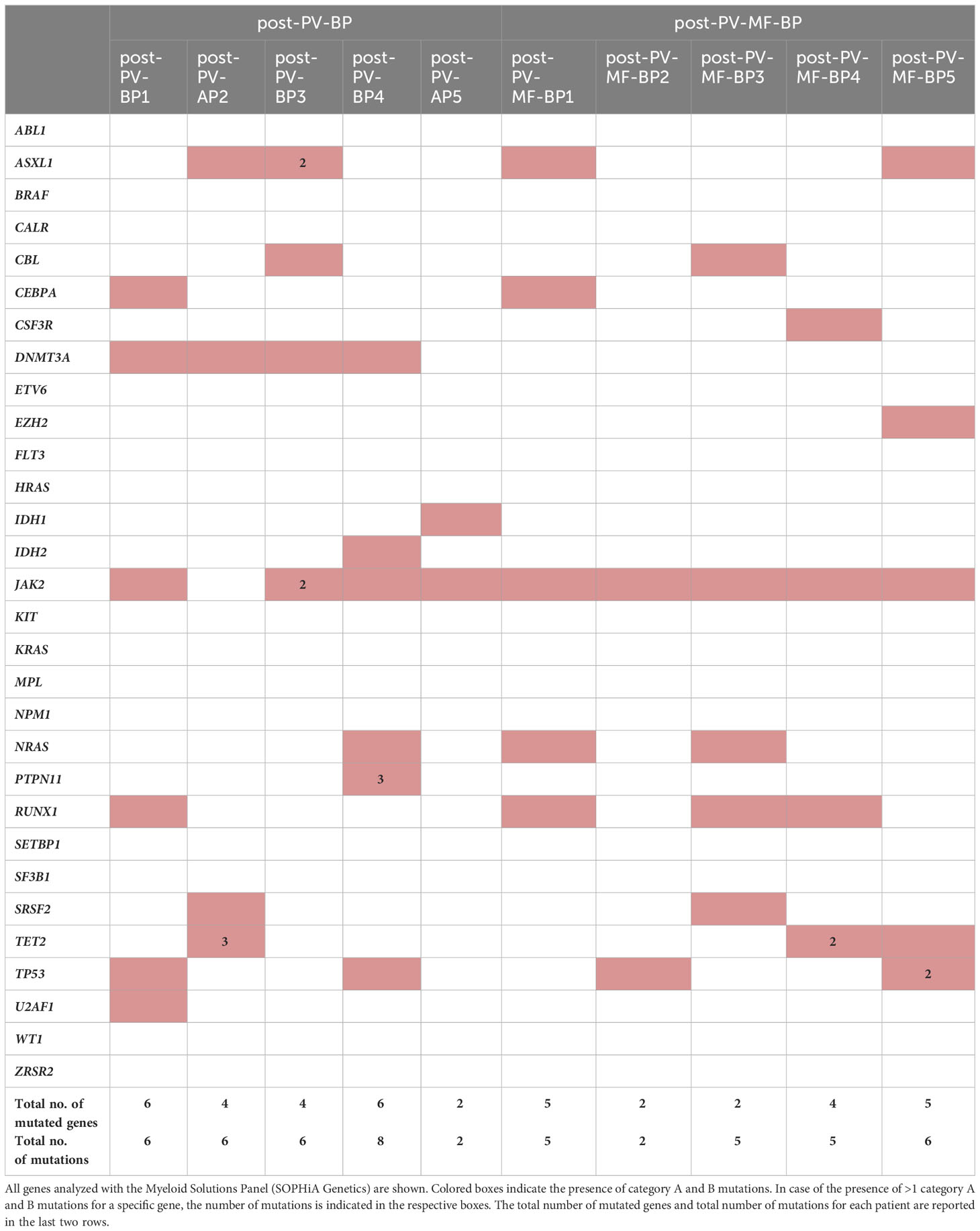
Table 4 Distribution of pathogenetically relevant (category A and B) mutations in cohort 1 (post-PV-BP1-5) and in cohort 2 (post-PV-MF-BP1-5).
Most commonly mutated genes in the overall AP/BP population (post-PV-BP + post-PV-MF-BP) were JAK2V617F, as expected, mutated in 9 out of 10 patients (90%); followed by ASXL1, DNMT3A, RUNX1, and TP53 (4 out of 10, 40%); and TET2 and NRAS (3 out of 10, 30%). Two patients presented SRSF2 mutation (2 out of 10, 20%), one (1 out of 10, 10%) IDH1, and one (1 out of 10, 10%) IDH2 mutation. All patients carried at least two different mutated genes (median number of mutated genes: 5, range: 2–6), with half of the patients carrying multiple mutations of the same gene, mainly TET2. Overall, the median number of mutations was 6 (range: 2–8). In line with the current literature, none of the patients presented FLT3 and/or NPM1 mutations, more frequent in de-novo AML (4). Of note, post-PV-AP2 lost JAK2V617F mutation at the time of leukemic evolution, while post-PV-BP3 also displayed a non-canonical, germline missense JAK2 mutation [c.3188G>A, p.(Arg1063His), rs77375493], characterized by a G to A substitution at position 3188 listed as probably pathogenetic (category B, score 0.94) in the COSMIC database. Also known as JAK2R1063H, this mutation is currently emerging as a functionally relevant mutation in myeloid proliferation, likely cooperating with JAK2V617F to favor leukemic transformation (16). Of note, post-PV-BP3 was the only patient for whom NGS data of the PV phase were available and allowed us to compare the mutational status of the chronic vs. blast phase (Supplementary Table 3). As expected, JAK2R1063H was already present at the time of PV diagnosis (VAF 54%), together with JAK2V617F (VAF 11.8%). Notably, DNMT3A mutation was also detected already at this stage. During leukemic evolution, we registered an increased in the VAF of both mutations (75% for JAK2R1063H and 49% for JAK2V617F) and the acquisition of two different ASXL1 mutations and CBL mutation, likely driving clonal evolution.
Comparison of pathogenic relevant mutations of myeloid genes in the two groups showed that DNMT3A mutations were present in majority of post-PV-BP (4 out of 5, 80%) and completely absent in the post-PV-MF-BP (P=0.048, Table 4). DNMT3A, together with IDH1/2 and TET2, segregated along the DNA methylation pathway (5). Notably, all post-PV-BP presented at least one mutation in these genes (with one case, PV-BP2, harboring four different mutations; three in TET2; and one in DNMT3A). Overall, the frequency of mutations in genes involved in the DNA methylation pathway (DNMT3A, IDH1/2, and TET2) is significantly higher in the post-PV-BP cohort as compared to post-PV-MF-BP (45% vs. 15%, P=0.038).
We also analyzed the frequency of mutations that have been flagged as an SNP in COSMIC database and/or reported as of undetermined significance/benign/likely benign in ClinVar database. All these mutations belong to category C or D and have a low score (<0.5) according to FATHMM and are reported in Supplementary Table 2. No significant enrichment of category C/D mutations was found by comparing post-PV-BP and post-PV-MF-BP. The analysis of category C/D mutations in ClinVar database allowed us to pinpoint recurrent germline variants harbored by all patients (both post-PV-BP and post-PV-MF-BP) located in the JAK2 (rs2230722 and rs2230724), HRAS (rs12628), ASXL1 (rs6058694), and TP53 (rs2454206) genes that have been variably implicated in increased cancer susceptibility (17, 18).
4 Discussion
The natural history of PV encompasses the evolution into post-PV-MF, usually occurring as a late event with reported 10-, 15-, and 20-year incidences of 27.4%, 39.9%, and 61.1%, respectively (19), and the transformation into AP/BP, with incidence estimates of 3%–7% (20). Despite the progress in our understanding of the genetic basis of leukemogenesis (as acknowledged by the 2022 WHO classification), post-PV-AP/BP still lacks a comprehensive characterization and falls into the AML category defined by differentiation (21). Thus, disease-specific clinical, laboratory, and molecular markers are eagerly awaited, to improve diagnosis and define personalized therapeutic approaches.
Depending on the study cohorts, one-third up to approximately one-half of leukemic evolutions occur directly from the PV stage, i.e., without a documented overt fibrotic phase. In this study, we targeted the differential morphological, clinical, and molecular features of AP/BP with a direct onset from the PV stage and those of AP/BP arising after the diagnosis of post-PV-MF. To this end, we performed a single-center retrospective descriptive study on 10 post-PV acute myeloid leukemias: 5 post-PV-BP (with two AP), and 5 post-PV-MF-BP.
The two cohorts displayed similar hematologic parameters at the time of PV diagnosis. By contrast, at the time of leukemic evolution, AP/BP directly arising from PV displayed significantly lower leukocyte counts and spleen diameters as compared to those passing through an overt fibrotic phase, likely due to the compensatory extramedullary hematopoiesis that typifies overt fibrotic stages. The most striking differences emerged from BM morphological analysis: all post-PV-BP were characterized by significantly higher cellularity, lower degree of fibrosis, and dysplastic features involving all three lineages, most prominently the erythroid and megakaryocytic compartments. Of note, these aspects were attributable to the intrinsic characteristics of the disease and not to the iatrogenic effects of previous treatment(s), since all 10 patients underwent hydroxyurea for comparable periods (post-PV-BP: median 76 months, range: 16–144 vs. post-PV-MF-BP: 67 months, range: 7–102, P=0.463).
Recent studies have provided valuable insights into the genomic and molecular alterations driving the transition to blast phase in MPNs (7, 8, 17). Mutations in genes such as TP53, ASXL1, IDH1/2, and RUNX1 have been identified as key drivers of BP-MPN, contributing to increased genomic instability, resistance to therapy, and clonal evolution (8). Understanding these molecular events is crucial for identifying novel therapeutic targets and designing personalized treatment approaches. NGS analysis of our two cohorts shows that post-PV-BP is significantly enriched in DNMT3A mutations, and overall, in mutations of genes that segregate along the DNA methylation pathway (i.e., TET2 and IDH1/2).
DNMT3A and TET2 are the most frequently identified variants among patients with clonal hematopoiesis of indetermined potential (CHIP) and provide selective advantage that foster clonal expansion and myeloid skewing. Both genes are more frequently mutated in MDS and MDS/MPN as compared to PV (22) and both increase in frequency in secondary AML (either post-MDS or post-MPN) (23). However, while DNMT3A mutations are generally present in early-stage disease, TET2 mutation can occur either before or after the acquisition of JAK2V617F mutation, leading to a biclonal disease or to a competitive advantage driving clonal dominance (24). In our cohort, we were able to retrieve NGS data at the time of PV diagnosis for one patient (post-PV-BP3), demonstrating that DNMT3A and JAK2 mutations were already present in the chronic stage. Based on the accumulating evidence on the molecular hits during the history of these disorders, we can speculate that the acquisition of DNMT3A mutation might occur as an early genetic event that might play a role in the dysplastic phenotype that typifies post-PV-BP. Indeed, clonality studies are necessary to answer this question and to assess whether the pre-leukemic clones in the two examined patient groups are independent or not from the clones driving chronic phase PV or post-PV-MF.
Our analysis also detected a non-canonical mutation of JAK2, namely, JAK2R1063H. A growing body of literature is highlighting the potential clinical significance of this germline variant in the MPN setting. Indeed, JAK2R1063H weakly hyperactivates JAK2/STAT5 signaling as compared to the acquired somatic V617F, but likely cooperates with another germline variant, JAK2E846D, in hereditary erythrocytosis (25) and with JAK2V617F itself in MPNs, favoring a more aggressive disease phenotype with significantly higher neutrophil counts and enrichment in thrombotic events (26). A potential pro-thrombotic role of this variant is further supported by a whole-exome sequencing study conducted in 22 young ischemic stroke patients with familial clustering of stroke, in which JAK2R1063H was detected in a proband with embolic stroke of undetermined source and prothrombotic status (27). Finally, in a subsequent next-generation sequencing study on 2154 MPN, MPN-BP, and AML patients, JAK2R1063H was predicted to have a functional consequence on JAK2 by three distinct predictive algorithms and was associated with an increased risk of leukemic transformation when combined with canonical JAK2 mutation (16).
NGS analysis also allowed information retrieval on genetic variations that have been flagged as an SNP and/or reported as of undetermined significance/benign/likely benign. Although no differences were found among post-PV-BP and post-PV-MF-BP, our analysis shed light onto germline variants of potential interest in MPN-BP, such as JAK2 (rs2230722 and rs2230724), HRAS (rs12628), ASXL1 (rs6058694), and TP53 (rs2454206).
HRAS rs12628, originally described by Taparowsky et al. (28), was shown to be associated with increased risk of oral carcinoma, colon, gastric and bladder cancer, and cutaneous melanoma (18, 29–31). This genetic variant has also been associated with syndromic congenital disorders, such as Noonan syndrome, Noonan-related syndrome, and Costello Syndrome (32). To the best of our knowledge, no data are currently available on the contribution of this SNP to hematologic malignancies. JAK2 rs2230722, located in exon 6, and JAK2 rs2230724, present in exon 19 have already been described in MPNs (17), with the latter being associated with the progression to BP, especially in individuals older than 45 years old (33) and therefore suggested as a potential genetic marker of leukemic progression in MPNs. ASXL1 rs6058694 has been reported in AML (34) and in a case report of triple-negative ET unresponsive to therapy (35). Additionally, TP53 rs2454206 has been extensively investigated as a germline predisposition factor for cancer. Despite conflicting data, this SNP may increase the risk of colorectal cancer (36), chronic lymphocytic leukemia (37), and acute myeloid leukemia with adverse molecular and cytogenetic risk (38).
With all the limits of the small number of patients studied and the descriptive intent, this is the first report formally describing recurrent morphological, clinical, and hematological stigmata that differentiate BPs that arise directly from PV from those occurring after post-PV-MF. This peculiar phenotype is consistent with the molecular signature of the disease, typified by mutations of genes involved in the DNA methylation pathways, occurring with a high frequency in MDS and MDS/MPN, leading us to speculate that BP occurring directly from the “florid” phase might be characterized by a distinct physiopathological mechanism.
Further studies in larger cohorts are warranted to translate these observations into robust evidence that may guide diagnosis and advise targeted therapeutic choices, like, for instance, hypomethylating agents.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Area Vasta Emilia Nord Ethical Commitee (prot. 6923, 17/02/2020). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
LPe: Investigation, Writing – original draft, Data curation. GP: Data curation, Investigation, Methodology, Writing – original draft. SC: Methodology, Investigation, Writing – original draft. CM: Methodology, Writing – review & editing. EuM: Methodology, Writing – review & editing. LPa: Writing – review & editing, Investigation. MG: Writing – review & editing, Investigation. GR: Writing – review & editing, Supervision. MV: Conceptualization, Supervision, Writing – review & editing. CC: Conceptualization, Data curation, Writing – original draft, Supervision, Writing – review & editing. ElM: Conceptualization, Data curation, Funding acquisition, Writing – original draft, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Fondi Locali per la Ricerca 2023, Quota Prodotti di Ricerca, Parma University to ElM and CC.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2024.1356561/full#supplementary-material
References
1. Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. (2007) 356:459–68. doi: 10.1056/NEJMoa065202
2. Tefferi A, Barbui T. Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. (2023) 98:1465–87. doi: 10.1002/ajh.27002
3. Tefferi A, Mudireddy M, Mannelli F, Begna KH, Patnaik MM, Hanson CA, et al. Blast phase myeloproliferative neoplasm: Mayo-AGIMM study of 410 patients from two separate cohorts. Leukemia. (2018) 32:1200–10. doi: 10.1038/s41375-018-0019-y
4. Ajufo HO, Waksal JA, Mascarenhas JO, Rampal RK. Treating accelerated and blast phase myeloproliferative neoplasms: progress and challenges. Ther Adv Hematol. (2023) 14:20406207231177282. doi: 10.1177/20406207231177282
5. Patel AA, Odenike O. SOHO state of the art updates and next questions | Accelerated phase of MPN: what it is and what to do about it. Clin Lymphoma Myeloma Leuk. (2023) 23:303–9. doi: 10.1016/j.clml.2023.01.011
6. Tefferi A, Guglielmelli P, Lasho TL, Coltro G, Finke CM, Loscocco GG, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. (2020) 189:291–302. doi: 10.1111/bjh.16380
7. Mannelli F. Acute myeloid leukemia evolving from myeloproliferative neoplasms: many sides of a challenging disease. J Clin Med. (2021) 10:436. doi: 10.3390/jcm10030436
8. Grabek J, Straube J, Bywater M, Lane SW. MPN: the molecular drivers of disease initiation, progression and transformation and their effect on treatment. Cells. (2020) 9:1901. doi: 10.3390/cells9081901
9. McNamara CJ, Panzarella T, Kennedy JA, Arruda A, Claudio JO, Daher-Reyes G, et al. The mutational landscape of accelerated- and blast-phase myeloproliferative neoplasms impacts patient outcomes. Blood Adv. (2018) 2:2658–71. doi: 10.1182/bloodadvances.2018021469
10. Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. (2013) 122:1395–8. doi: 10.1182/blood-2013-03-488098
11. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 128:462–3. doi: 10.1182/blood-2016-06-721662
12. Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. (2005) 90:1128–32.
13. Sutherland DR, Anderson L, Keeney M, Nayar R, Chin-Yee I. The ISHAGE guidelines for CD34+ cell determination by flow cytometry. International Society of Hematotherapy and Graft Engineering. J Hematother. (1996) 5:213–26. doi: 10.1089/scd.1.1996.5.213
14. Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Randi ML, et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood. (2012) 119:2239–41. doi: 10.1182/blood-2011-11-393819
15. Barraco D, Cerquozzi S, Hanson CA, Ketterling RP, Pardanani A, Gangat N, et al. Prognostic impact of bone marrow fibrosis in polycythemia vera: validation of the IWG-MRT study and additional observations. Blood Cancer J. (2017) 7:e538. doi: 10.1038/bcj.2017.17
16. Benton CB, Boddu PC, DiNardo CD, Bose P, Wang F, Assi R, et al. Janus kinase 2 variants associated with the transformation of myeloproliferative neoplasms into acute myeloid leukemia. Cancer. (2019) 125:1855–66. doi: 10.1002/cncr.31986
17. Torres DG, Barbosa Alves EV, Araújo de Sousa M, Laranjeira WH, Paes J, Alves E, et al. Molecular landscape of the JAK2 gene in chronic myeloproliferative neoplasm patients from the state of Amazonas, Brazil. BioMed Rep. (2023) 19:98. doi: 10.3892/br.2023.1680
18. Tomei S, Adams S, Uccellini L, Bedognetti D, De Giorgi V, Erdenebileg N, et al. Association between HRAS rs12628 and rs112587690 polymorphisms with the risk of melanoma in the North American population. Med Oncol. (2012) 29:3456–61. doi: 10.1007/s12032-012-0255-3
19. Bai J, Ai L, Zhang L, Yang FC, Zhou Y, Xue Y. Incidence and risk factors for myelofibrotic transformation among 272 Chinese patients with JAK2-mutated polycythemia vera. Am J Hematol. (2015) 90:1116–21. doi: 10.1002/ajh.24191
20. Tefferi A, Alkhateeb H, Gangat N. Blast phase myeloproliferative neoplasm: contemporary review and 2024 treatment algorithm. Blood Cancer J. (2023) 13:108. doi: 10.1038/s41408-023-00878-8
21. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
22. Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. (2011) 25:1217–9. doi: 10.1038/leu.2011.77
23. Fried I, Bodner C, Pichler MM, Lind K, Beham-Schmid C, Quehenberger F, et al. Frequency, onset and clinical impact of somatic DNMT3A mutations in therapy-related and secondary acute myeloid leukemia. Haematologica. (2012) 97:246–50. doi: 10.3324/haematol.2011.051581
24. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. (2017) 129:667–79. doi: 10.1182/blood-2016-10-695940
25. Kapralova K, Horvathova M, Pecquet C, Fialova Kucerova J, Pospisilova D, Leroy E, et al. Cooperation of germ line JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia. Blood. (2016) 128:1418–23. doi: 10.1182/blood-2016-02-698951
26. Mambet C, Babosova O, Defour JP, Leroy E, Necula L, Stanca O, et al. Cooccurring JAK2 V617F and R1063H mutations increase JAK2 signaling and neutrophilia in myeloproliferative neoplasms. Blood. (2018) 132:2695–9. doi: 10.1182/blood-2018-04-843060
27. Ilinca A, Martinez-Majander N, Samuelsson S, Piccinelli P, Truvé K, Cole J, et al. Whole-exome sequencing in 22 young ischemic stroke patients with familial clustering of stroke. Stroke. (2020) 51:1056–63. doi: 10.1161/STROKEAHA.119.027474
28. Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature. (1982) 300:762–5. doi: 10.1038/300762a0
29. Johne A, Roots I, Brockmöller J. A single nucleotide polymorphism in the human H-ras proto-oncogene determines the risk of urinary bladder cancer. Cancer Epidemiol Biomarkers Prev. (2003) 12:68–70.
30. Catela Ivkovic T, Loncar B, Spaventi R, Kapitanovic S. Association of H-ras polymorphisms and susceptibility to sporadic colon cancer. Int J Oncol. (2009) 35:1169–73. doi: 10.3892/ijo_00000433
31. Pandith AA, Shah ZA, Khan NP, Baba KM, Wani MS, Siddiqi MA. HRAS T81C polymorphism modulates risk of urinary bladder cancer and predicts advanced tumors in ethnic Kashmiri population. Urol Oncol. (2013) 31:487–92. doi: 10.1016/j.urolonc.2011.03.004
32. Sol-Church K, Stabley DL, Nicholson L, Gonzalez IL, Gripp KW. Paternal bias in parental origin of HRAS mutations in Costello syndrome. Hum Mutat. (2006) 27:736–41. doi: 10.1002/humu.20381
33. Zhong Y, Wu J, Ma R, Cao H, Wang Z, Ding J, et al. Association of Janus kinase 2 (JAK2) polymorphisms with acute leukemia susceptibility. Int J Lab Hematol. (2012) 34:248–53. doi: 10.1111/j.1751-553X.2011.01386.x
34. Pellegrino M, Sciambi A, Treusch S, Durruthy-Durruthy R, Gokhale K, Jacob J, et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. (2018) 28:1345–52. doi: 10.1101/gr.232272.117
35. Zaidi U, Shahid S, Fatima N, Ahmed S, Sufaida G, Nadeem M, et al. Genomic profile of a patient with triple negative essential thrombocythemia, unresponsive to therapy: A case report and literature review. J Adv Res. (2017) 8:375–8. doi: 10.1016/j.jare.2017.04.001
36. Elshazli RM, Toraih EA, Elgaml A, Kandil E, Fawzy MS. Genetic polymorphisms of TP53 (rs1042522) and MDM2 (rs2279744) and colorectal cancer risk: An updated meta-analysis based on 59 case-control studies. Gene. (2020) 734:144391. doi: 10.1016/j.gene.2020.144391
37. Ounalli A, Moumni I, Mechaal A, Chakroun A, Barmat M, Rhim REE, et al. TP53 Gene 72 Arg/Pro (rs1042522) single nucleotide polymorphism increases the risk and the severity of chronic lymphocytic leukemia. Front Oncol. (2023) 13:1272876. doi: 10.3389/fonc.2023.1272876
38. Tripon F, Iancu M, Trifa A, Crauciuc GA, Boglis A, Balla B, et al. Association Analysis of TP53 rs1042522, MDM2 rs2279744, rs3730485, MDM4 rs4245739 Variants and Acute Myeloid Leukemia Susceptibility, Risk Stratification Scores, and Clinical Features: An Exploratory Study. J Clin Med. (2020) 9:1672. doi: 10.3390/jcm9061672
Keywords: polycythemia vera, blast-phase, accelerated phase, bone marrow morphology, mutational profile
Citation: Pelagatti L, Pozzi G, Cortellazzi S, Mancini C, Martella E, Pagliaro L, Giaimo M, Roti G, Vitale M, Carubbi C and Masselli E (2024) Morphological, clinical, and molecular profiling of post-polycythemia vera accelerated/blast phase occurring with and without antecedent secondary myelofibrosis. Front. Hematol. 3:1356561. doi: 10.3389/frhem.2024.1356561
Received: 15 December 2023; Accepted: 13 March 2024;
Published: 18 April 2024.
Edited by:
Anna Rita Migliaccio, Campus Bio-Medico University, ItalyReviewed by:
Ioanna Triviai, Max Planck Institute for Molecular Genetics, GermanyTayler Van Denakker, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2024 Pelagatti, Pozzi, Cortellazzi, Mancini, Martella, Pagliaro, Giaimo, Roti, Vitale, Carubbi and Masselli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Carubbi, Y2VjaWxpYS5jYXJ1YmJpQHVuaXByLml0; Elena Masselli, ZWxlbmEubWFzc2VsbGlAdW5pcHIuaXQ=
†These authors have contributed equally to this work