 Kunapa Iam-arunthai1Tawatchai Suwanban1Pravinwan Thungthong1
Kunapa Iam-arunthai1Tawatchai Suwanban1Pravinwan Thungthong1 Supat Chamnanchanunt1,2*Suthat Fucharoen3
Supat Chamnanchanunt1,2*Suthat Fucharoen3- 1Division of Hematology, Department of Medicine, Rajavithi Hospital, College of Medicine, Rangsit University, Bangkok, Thailand
- 2Department of Clinical Tropical Medicine, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand
- 3Thalassemia Research Center, Institute of Molecular Biosciences, Mahidol University, Nakhon Pathom, Thailand
Background: Thalassemia is a genetic hemoglobinopathy in which a defective globin chain can cause transfusion-dependent anemia and other complications. As genotype interactions lead to variations in the clinical course among patients with thalassemia, clinical factors may help predict survival in the types of thalassemia complicated by gene interactions.
Aim: This study aimed to determine the clinical factors associated with survival in patients with thalassemia. We retrospectively reviewed the medical records of patients with thalassemia older than 15 years between 2002 and 2020 that were available at the Rajavithi Hospital. Data on the clinical parameters, laboratory tests, treatments, and survival status were collected and analyzed.
Results: Of the 478 thalassemia patients included, 68.8% were women, and the mean age was 41 ± 17 years. The most common type of thalassemia was β-thalassemia (53.3%). Male sex, low body mass index, the thalassemia type, comorbidities, low hemoglobin level, high ferritin level, and regular blood transfusion were significantly associated with short-term survival. However, only the thalassemia type (β-thalassemia, p = 0.048) and the co-inheritance of the α- and β-thalassemia genotypes (p = 0.03) were independently associated with death. The overall survival rates among patients with α-thalassemia, β-thalassemia, and co-inheritance of the α- and β-thalassemia genotypes were 98.1%, 90.6%, and 75.0%, respectively. The death rate was 6.3%, and the most common cause of death was infection.
Conclusion: The thalassemia genotype was a predictive factor of survival, and co-inheritance of the α- and β-thalassemia genotypes results in a shorter-term survival compared with other types, especially transfusion-dependent thalassemia. These results can be applied in clinical settings to predict and possibly extend the life expectancy of patients with thalassemia.
1 Introduction
Thalassemia is a genetic hemoglobinopathy and is considered to be among the global public health issues (1–4). Mutations in the globin gene result in abnormal globin chain synthesis, ultimately leading to chronic hemolytic anemia. Thalassemia has alpha (α) and beta (β) forms, and the disease severity is categorized based on the transfusion dependency and complications. Moreover, it can be classified based on its clinical severity and the degree of anemia. The standard phenotype-based classification that is widely applied is transfusion-dependent thalassemia (TDT) or non-transfusion-dependent thalassemia (NTDT). Furthermore, while thalassemia major manifests as severe anemia in the neonatal period, some patients with thalassemia intermedia only have asymptomatic anemia (5). The severity of thalassemia is determined by the number of mutated genes—four in α-thalassemia and two in β-thalassemia (6). In Thailand, while approximately 40% of the Thai population carries either α- or β-thalassemia (7, 8), only 1% of patients with thalassemia have TDT (7).
Patients with TDT suffer from chronic anemia, which leads to short life expectancy. Furthermore, although life expectancy can be improved by regular blood transfusions, multiple side effects occur both during (allergic reaction) or after transfusion (such as iron overload or hemochromatosis). In hemochromatosis, iron deposits in the visceral organs (the myocardium, liver, and endocrine glands) lead to organ dysfunction (e.g., congestive heart failure, cirrhosis, and endocrinopathies associated with diabetes mellitus) (9). Thus, novel treatment strategies for thalassemia have been developed to balance the necessary blood transfusion and iron accumulation to increase the survival rate (7, 8). Numerous studies have demonstrated that an optimization of the blood transfusions/iron chelation protocols induces a high survival rate that is close to the life expectancy of the general population (8–12). However, the survival rate of thalassemia is also dependent on several other factors, including disease severity, adequate blood transfusion, blood quality, and preventive iron deposition. The prevalence of cardiopulmonary and bone deformities remains high among western thalassemia patients with high ferritin levels, whereas among eastern TDT patients, infection is the main cause of complications (8, 12–15). It has been hypothesized that different clinical outcomes are the results of the interaction between the α- and β-thalassemia genotypes, which can compensate to yield a normal lifespan in patients with minimal complications (8, 16). The co-inheritance of the α- with β-thalassemia genotypes can affect genotype-based categorization, similar to that observed in other hemoglobinopathy combinations, such as α-thalassemia and sickle cell anemia-sickle hemoglobin (HbS) (17). However, only a few studies have investigated the survival and predictive factors that can influence the outcomes during the clinical course of the α- and β-thalassemia genotypes. Thus, the present study aimed to identify the clinical factors that determine the survival rate in patients with α- and β-thalassemia and in those with co-inheritance of the α- and β-thalassemia genotypes.
2 Method
2.1 Study setting
This retrospective study reviewed the medical records of adult patients (aged >18 years) with thalassemia at the Rajavithi Hospital, Bangkok, Thailand, between 2002 and 2020. The eligibility criteria were thalassemia diagnosed using a high-performance liquid column chromatography (HPLC)-based hemoglobin analysis for β-thalassemia and the gap-PCR method for α-thalassemia (18). The primer sequences used were suitable for diagnosing 5 α0-thalassemia deletions and 2 α+-thalassemia deletions. In addition, for α-thalassemia, molecular diagnosis of α0-thalassemia (category Southeast Asian or Thai deletion) and Hemoglobin Constant Spring (HbCS) was performed in patients with hemoglobin H (HbH) disease. Based on the risk of death (19), the sample size was calculated as at least 300 patients, ensuring that the study achieved 95% confidence interval and 5% significance level the risk of death hazard ratio equal to 2.8 in a prior study.
2.2 Recruitment and data collection method
The information collected included demographic characteristics and diagnosis based on hemoglobin analysis, first diagnosis with follow-up period, body mass index (BMI) [calculated as BMI = Wt/(Ht)2, where weight (Wt) was in kilograms and height (Ht) was in meters], comorbidities, treatment-related thalassemia, and cause of death. Comorbidities during the follow-up period were categorized according to the guidelines provided in the OPTIMAL CARE study (14). Thalassemia patients were divided into two groups: TDT, which consisted of patients requiring regular blood transfusions every 2–6 weeks for survival, and NTDT, which consisted of patients who may or may not require an occasional transfusion (12, 20). Data analysis was conducted to determine the clinical characteristics, treatments, causes of death, and the survival rates.
2.3 Ethical consideration
The study was approved by and carried out in accordance with the Ethics Committee of the Rajavithi Hospital, no. 050/2562 (Rajavithi Hospital). The Ethics Committee of the Rajavithi Hospital has granted a waiver for informed consent. All patient data were de-coded, and written informed consent was not required as this was a retrospective study.
2.4 Data management and statistical analysis
Categorical data were expressed as number and percentages. Continuous parameters were demonstrated as the mean with standard deviation if the parameters were normally distributed and as median if the parameters were not normally distributed. The Cox proportional hazards model was utilized to evaluate the relationship between risk factors and survival time, whereas the Kaplan–Meier method was used for survival analysis. Data were analyzed using SPSS Statistics version 22.0 (license 22.0; IBM, Armonk, NY, USA). A p-value less than 0.05 was considered statistically significant.
3 Results
3.1 Patient characteristics
The data of 478 patients with thalassemia who matched our inclusion criteria were included in this study. Of these patients, 329 (68.8%) were women, the mean age was 41 ± 17 years (range, 16–91 years), and the median BMI was 19.9 kg/m2 (range, 12.8–40.4 kg/m2). The distribution of the thalassemia types was as follows: 51.9% had β-thalassemia/HbE, 29.7% had HbH, 8.4% had combined HbH CS disease, 4.6% had HbAE Bart’s disease, 1.6% had homozygous HbCS, 1.5% had Hb EF Bart’s disease and homozygous β-thalassemia, and 0.8% had Hb AE with CS disease. Men predominantly had β-thalassemia (21.1%), while women constituted the majority of those with α-thalassemia (35.4%). Individuals with combined α- and β-thalassemia were primarily women (6 out of 8). The baseline clinical characteristics of all patients are presented in Table 1. The pre-transfusion ferritin level was 1,052 ng/mL (range, 17–41,677 ng/mL), and 20.3% of the patients had ferritin levels >2,500 ng/mL. The last ferritin level was lower by 875 ng/mL (range, 34–25,034 ng/mL), whereas 18.6% of the patients had ferritin levels >2,500 ng/mL. The pre-transfusion hemoglobin level was 7.3 g/dL (range, 1.8–12 g/dL), and the last hemoglobin level was 7.6 g/dL (range, 2.2–14.9 g/dL). Hypertension (33.7%) was the most common comorbid condition. Most of the patients (79.3%) had NTDT, almost half of all patients (45.4%) required iron chelator therapy for iron overload, and 19% of the patients underwent splenectomy. The mean age of the patients in the death group was 39.2 ± 2.7 years, and the time to developing complications did not differ statistically between the death and survival groups. Complications occurred in 19.2% of patients with thalassemia, which included liver disease, endocrinopathies, and cardiac disease. All patients were followed up in the hospital for at least 10 years (median follow-up of 16.1 years; range, 9.8–16.1 years).
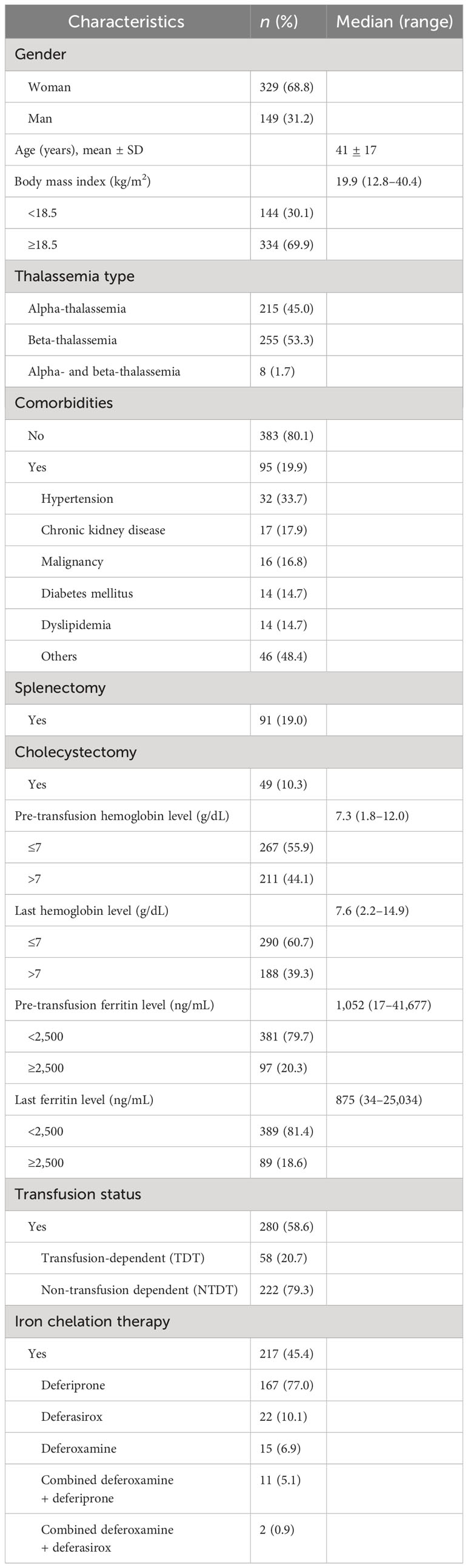
Table 1 Characteristics of 478 thalassemia patients.
3.2 Risk factors for death
Factors were classified using incidence death rate per 1,000 person-years and are presented in Table 2. The following factors were statistically significant in the model male sex (p = 0.02), BMI <18.5 kg/m2 (p = 0.013), co-inheritance of the α- and β-thalassemia genotypes (p < 0.001), comorbidities (p = 0.02), pre-transfusion hemoglobin ≤7 g/dL (p = 0.01), last hemoglobin ≤7 g/dL (p = 0.001), pre-transfusion ferritin level ≥2,500 ng/mL (p = 0.004), last ferritin level ≥2,500 ng/mL (p < 0.001), and requirement of blood transfusion (p = 0.013). The univariate analyses identified the following variables as associated with death: gender [male: hazard ratio (HR) = 2.28, 95%CI = 1.11–4.66, p = 0.024]; BMI <18.5 kg/m2 (HR = 2.40, 95%CI = 1.17–4.91, p = 0.017); thalassemia type (β-thalassemia: HR = 5.31, 95%CI = 1.84–15.29, p = 0.002; co-inheritance of the α- and β-thalassemia genotypes: HR = 14.45, 95%CI = 2.65–78.90, p = 0.002); blood transfusion (HR = 2.93, 95%CI = 1.20–7.18, p = 0.018); and pre-transfusion ferritin level >2,500 ng/mL (HR = 2.79, 95%CI = 1.34–5.79, p = 0.006) (Table 3). Subsequently, the multivariate logistic regression analyses revealed that the thalassemia type was independently associated with increased risk of death (β-thalassemia type: adjusted HR = 3.21, 95%CI = 1.01–10.21, p = 0.048; co-inheritance of the α- and β-thalassemia genotypes: adjusted HR = 13.17, 95%CI = 2.40–72.44, p = 0.003).
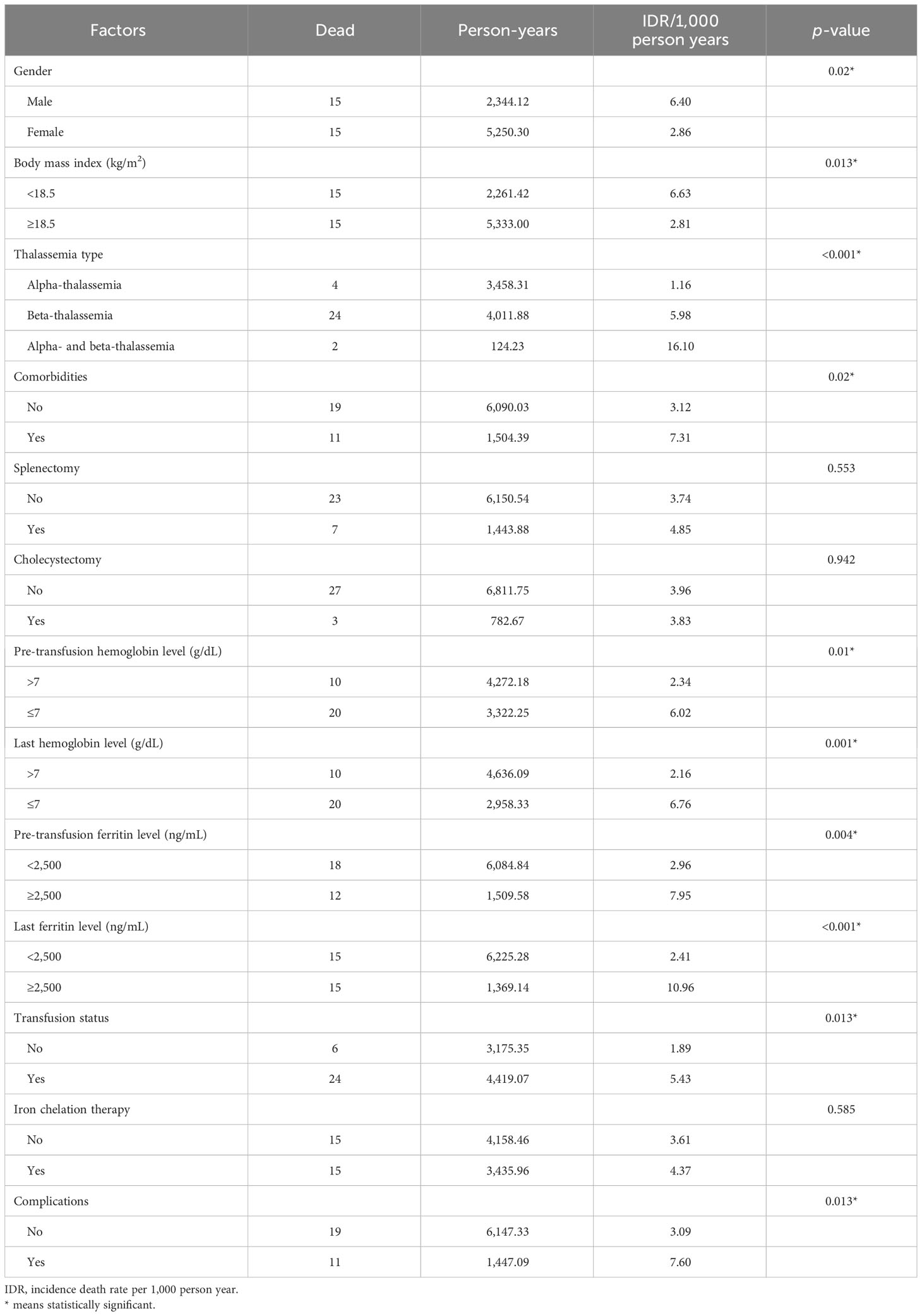
Table 2 Incidence death rate per 1,000 person-years classified by factors.
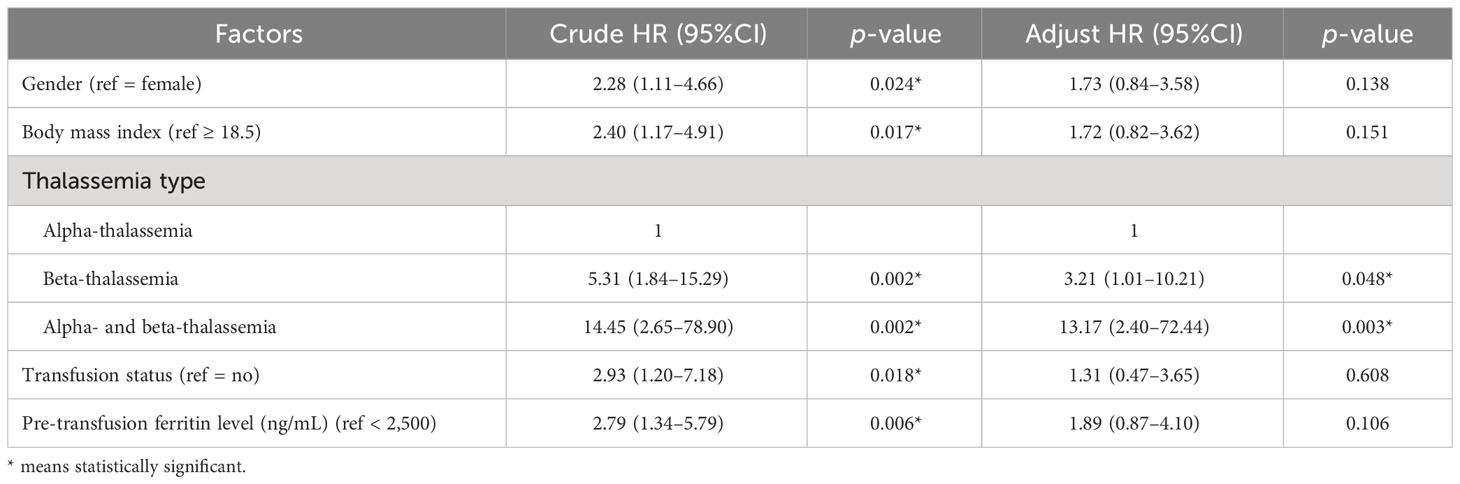
Table 3 Cox proportional hazards model for the risk factors associated with death.
After treatment, the overall survival rate among patients with thalassemia was 93.7%, and the survival rates of patients with α-thalassemia, β-thalassemia, and co-inheritance of the α- and β-thalassemia genotypes were 98.1%, 90.6%, and 75.0%, respectively (Figure 1). A comparison of the overall survival rates classified by thalassemia genotype and phenotype indicated that the survival rate among α-thalassemia-TDT patients was 100%, whereas that for the NTDT group was 98.5%. In β-thalassemia, the survival rate among TDT patients was 92.3%, which was higher than that of NTDT patients (88.3%). The overall survival rates of patients with co-inheritance of the α- and β-thalassemia genotypes were 66.7% in the TDT group and 50.0% in the NTDT group (Figure 2). We recorded 30 deaths (15 men and 15 women) during the follow-up period, with the most common cause of death being infection (50.0%), followed by heart failure (23.3%), and then hepatocellular carcinoma, upper gastrointestinal bleeding, and cancers (6.7% in each). Among the death-related infection cases, five patients died from gut-associated organisms (Vibrio and Escherichia coli), followed by four with Pseudomonas septicemia, and one each with Klebsiella spp. and Acinetobacter infection. Two patients died with tuberculosis and one from advanced AIDS infection. The remaining cases had unidentified organisms. Patients with cancer had gynecologic malignancies (n = 4), hepatocellular carcinoma (n = 3), lung cancer (n = 2), breast cancer (n = 2), thyroid cancer (n = 1), prostate cancer (n = 1), cholangiocarcinoma (n = 1), leukemia (n = 1), and lymphoma (n = 1). Intracerebral hemorrhage and severe metabolic acidosis each accounted for 3.3% of all deaths (Table 4). Thalassemia survival patients were shown in Table 5.
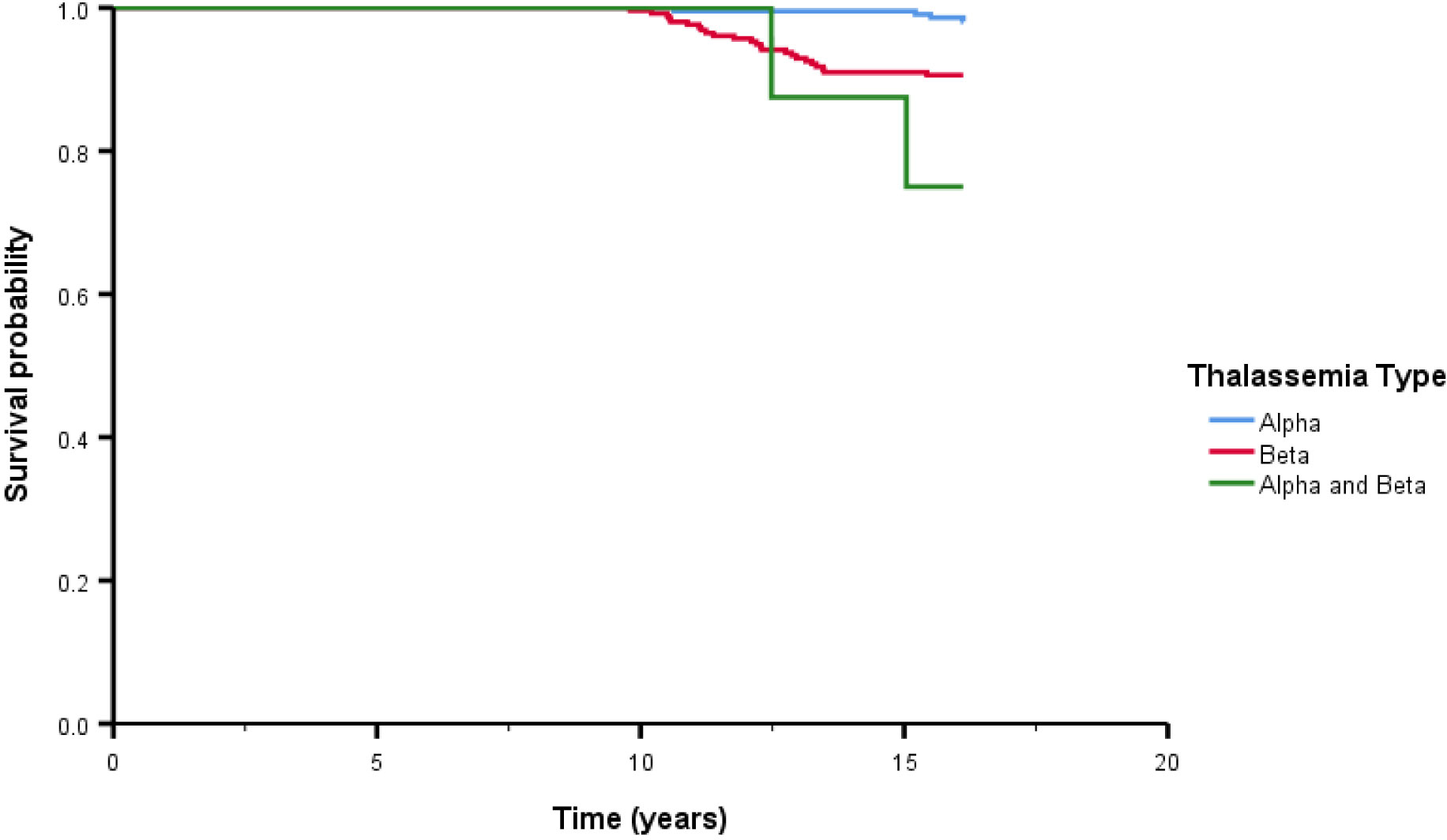
Figure 1 The overall survival after treatment among thalassemia patients classified by thalassemia type. Alpha means alpha thalassemia genotype, Beta means beta thalassemia genotype.
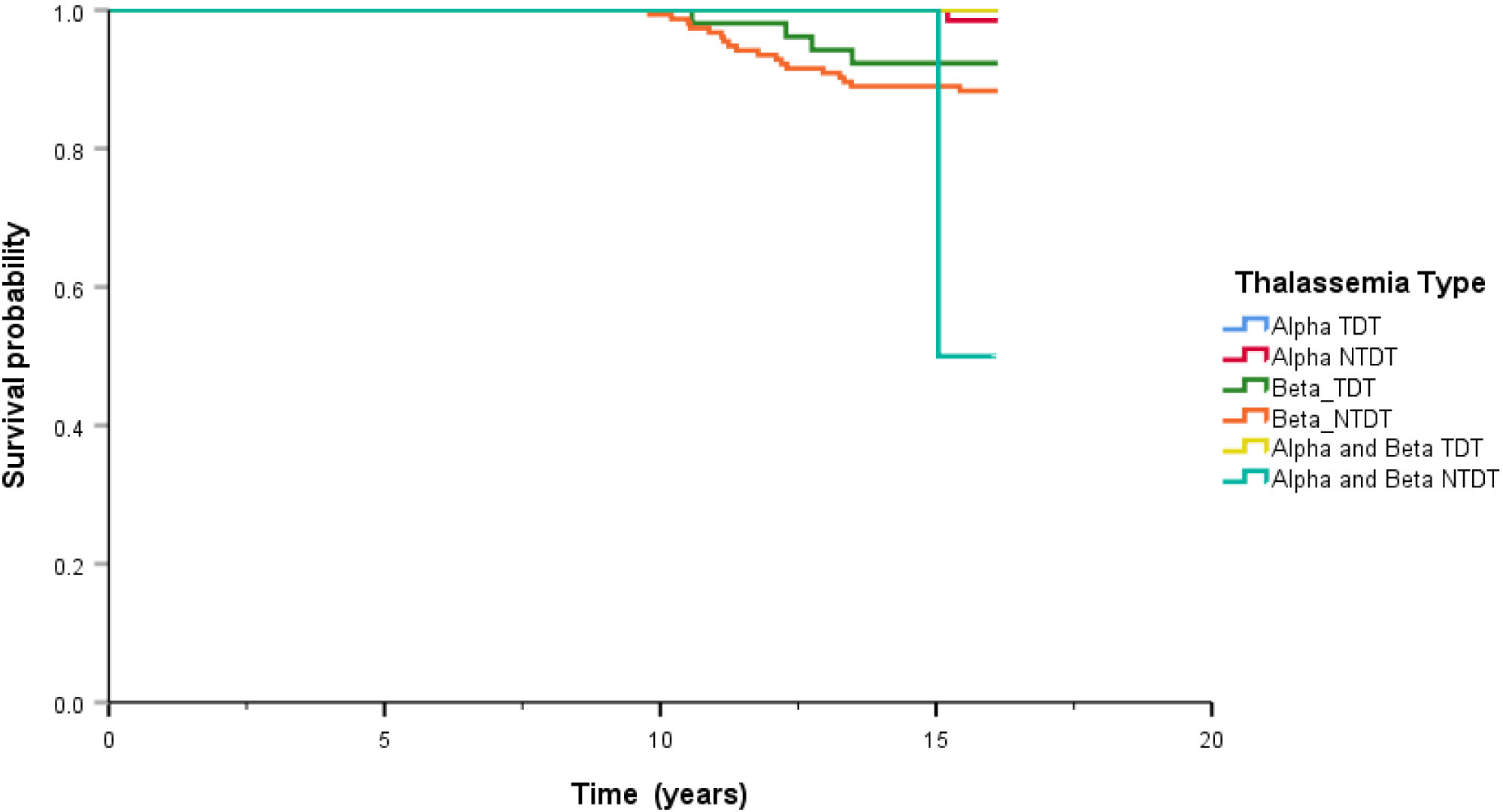
Figure 2 Comparison of overall survival classified by genotype of thalassemia. Alpha means alpha thalassemia genotype, Beta means beta thalassemia genotype, TDT means transfusion dependence thalassemia, and NTDT means non-transfusion dependence thalassemia.
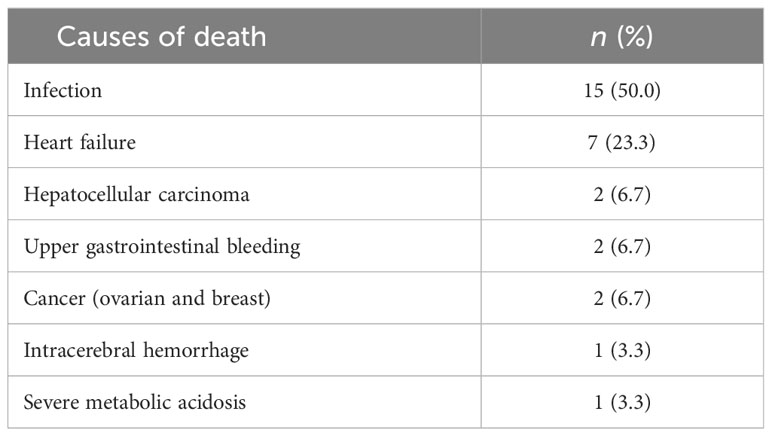
Table 4 Causes of death among 30 thalassemia patients.
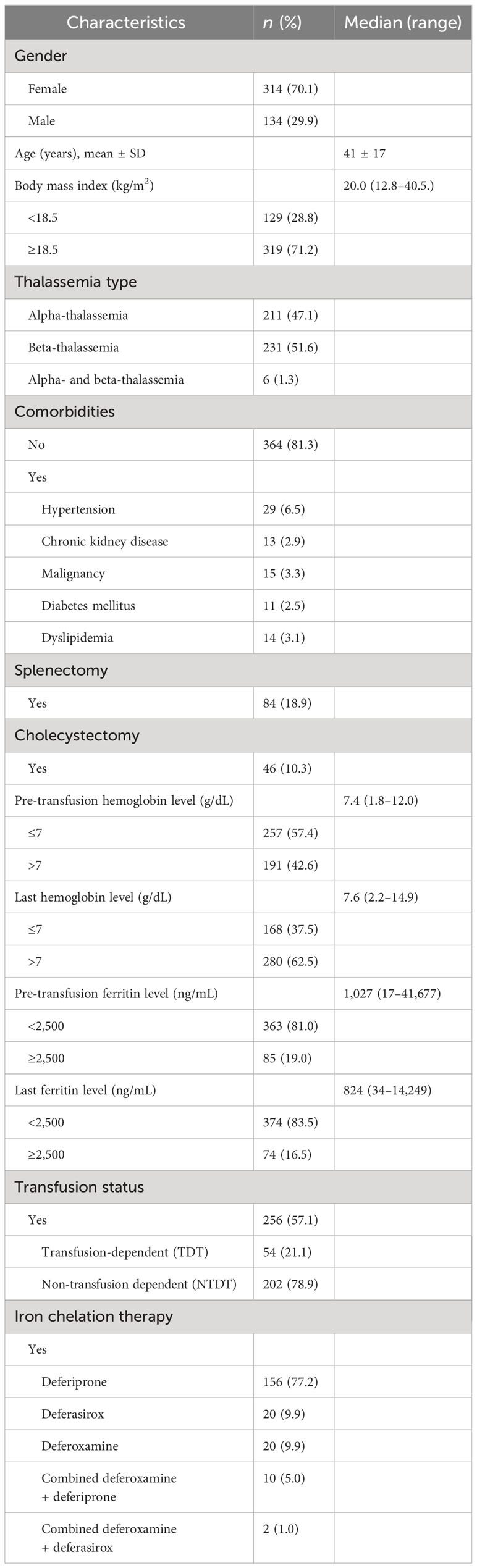
Table 5 Characteristics of 448 thalassemia survival patients.
4 Discussion
This retrospective cohort study aimed to identify the clinical predictors of death and survival rates in Thai patients with thalassemia and to compare the results of this study with those of other studies (8, 9, 14). It is interesting to note that our cohort comprised predominately older women (aged >40 years), similar to another study (14), and that only 19% of the patients underwent splenectomy. This indicates that the availability of blood transfusion has reduced the need for splenectomy. Severe anemia was observed in about half of the cohort, both initially and during follow-up, a finding similar to that of other Thai studies (8, 9). A balance between blood transfusion and iron depletion has been proposed to increase the survival rates among patients with thalassemia (12, 21).
We demonstrated seven clinical factors that were significantly associated with death rate, namely, male sex, low BMI, thalassemia type, comorbidities, low hemoglobin level, high ferritin level, and requirement of regular blood transfusion. These findings are in agreement with those of multiple studies of clinical factors associated with survival rates in thalassemia (22–25). The greater risk due to male sex may be explained by the fact that thalassemia in women might be associated with lower oxidative stress, which in turn affects the cardiac function and incidence of infections (24). This effect is similar to that observed in hemochromatosis, which may also require regular blood transfusion and is associated with greater incidence of cardiac dysfunction and infection. In addition, several studies have demonstrated that high ferritin levels (>2,500 ng/mL) are associated with short-term survival in patients with thalassemia (13, 26). A low BMI typically reflects nutritional deficiencies among patients with thalassemia, and malnutrition can lead to anemia that requires regular blood transfusion (22). Thus, malnutrition is associated with TDT and is thereby related to iron overload, cardiac dysfunction, and increased infection rate. However, it should be noted that most published studies have been conducted on patients with thalassemia major, and it is possible that the type of thalassemia is significantly associated with complications and death. We demonstrated that thalassemia major (β-thalassemia) had a more severe clinical course than α-thalassemia. As blood transfusions are needed to treat anemia, high ferritin levels can be commonly observed among TDT patients (8, 12, 15). This observation potentially reflects the association between transfusion dependence and more severe thalassemia conditions. The difference in the survival rates among various studies may be attributed to sample size disparities or the methods used for calculating the survival rate, which are based on the thalassemia major or thalassemia intermedia types. Similarly, the higher overall survival rate observed in our cohort may be attributed to the thalassemia type, which has also been reported by other studies (27, 28). Patients with thalassemia major are at a higher risk of mortality than those with other types of thalassemia, and the underlying etiology is the need for regular blood transfusions and consequent iron accumulation, leading to organ dysfunction (29). Other factors that may influence the survival of patients with thalassemia include time to diagnosis, availability of healthcare services, regular monitoring, proper treatment, and early iron chelation therapy. Contrary to the results presented here, a study from Great Britain found that 50% of patients with thalassemia intermedia died at a young age (<35 years) (6). Limitations in the study design and data analysis may have influenced the non-significant finding for age of complication onset.
There were hypotheses of the deletion of the α-globin gene with the β-thalassemia genotype affecting the hyperunstable globin variant, leading to easy hemolytic condition (30–32). The pathophysiology of co-inheritance is not clear to explain this phenomenon related to the survival rate. Interestingly, our data showed support that the co-inheritance of the α- and β-thalassemia genotypes with TDT reduced the survival rate. This combined hemoglobinopathy (α- and β-thalassemia) is contradictory to that of another study in patients with co-inheritance of α-thalassemia and other hemoglobinopathy types (HbS), especially with respect to improving the survival rate (17). Some studies have mentioned other clinical factors that might be related to the prognosis of α-thalassemia (10, 33, 34). Similarly to β-thalassemia leading to anemia that needs regular blood transfusions, complications might develop earlier among patients with co-inheritance of the α- and β-thalassemia types. It has been known that co-inheritance of the α- and β-thalassemia genotypes is relatively common in Southeast Asia (35). Blood transfusion with concordant iron chelation therapy was prescribed for the patients in our study. Iron overload due to long-term blood transfusion among patients with thalassemia is the major cause of complications, such as cardiac dysfunction, and 92 patients experienced complications (e.g., liver dysfunction, endocrinopathies, and cardiac disease). In our cohort, more than half were categorized as TDT, and iron chelation therapy, given twice in a 3-month interval, was advised when high serum ferritin levels were >1,000 ng/mL (14). While a few patients used deferasirox owing to its low cost, most patients (77%) received deferiprone and did not develop cardiac problems. This can be explained by the cardioprotective effect of deferiprone, which exhibits greater iron mobilization. This increases the heart signal and improves heart function in patients with iron overload (36). Surprisingly, we found a high incidence of infection (50% of the study population). This observation is different from the results of a previous study (27). In another study, it was demonstrated that thalassemia patients with iron overload predominantly suffered from cardiac complications, including heart failure and arrhythmia (29). We hypothesized that the high ferritin levels in our cohort have rendered the patients susceptible to bacterial infection, with the underlying mechanism involving impaired phagocytic function of macrophages and neutrophils, alternated T lymphocytes, lower numbers and function of natural killer cells, increased numbers and function of B lymphocytes, impaired secretion of immunoglobulin, and suppression of the complement system function (11). In support of this hypothesis, Teawtrakul et al. have demonstrated that a high ferritin level is a significant risk factor to bacterial infection in patients with TDT (15). The strength of this study lies in our observational study supporting the hypothesis of co-inherited thalassemia patients having low survival rates. The limitations of this study include its retrospective design and the fact that the cohort was recruited from a tertiary care center with a small sample size for subgroup analysis. Although we demonstrated poor survival among patients with co-inheritance of the α- and β-thalassemia genotypes and TDT, future studies on a larger population are required to validate the results of the present study. A case–control study may be needed to make conclusions on clinical parameters for use in the global medical fraternity, including exploring potential differences in the efficacy and compliance between the various iron chelators used in clinical practice.
5 Conclusion
The overall survival rate among patients with thalassemia was 93.7%, and it was higher in those with α-thalassemia-TDT. However, the co-inheritance of the α- and β-thalassemia genotypes with TDT led to poor survival, and it was found that the most common cause of death was infection. Our results suggest the usefulness of clinical parameters in estimating the survival rates among patients with thalassemia.
Data availability statement
Available data and materials upon request, please directly contact correspondence author for data request according to the regulation of the Ethical Committees. Requests to access the datasets should be directed toU3VwYXQuY2hhQG1haGlkb2wuYWMudGg=.
Ethics statement
The studies involving humans were approved by The Ethics Committee of the Rajavithi Hospital-Number 050/2562 (Rajavithi Hospital). The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because all patient data were decoded and this was a retrospective study, written informed consent was not required.
Author contributions
KI-a: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. SC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TS: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Writing – review & editing. PT: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Writing – review & editing. SF: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by a research grant from Rajavithi Hospital, the Multicenter Research Grant from the Thai Society of Hematology, Specific League Funds from Mahidol University, and partially supported by the ICTM grant of the Faculty of Tropical Medicine. The funders have no role in the process of designing, processing, and writing this manuscript.
Acknowledgments
The authors thank Lertnapa Lertum, Supasri Keawpai, and Chanjira Sae-Lim for supporting thalassemia patients. Wannakorn Homsuwan, Associate Professor Dusit Sujirarat for rechecking and confirming data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia: molecular biology and clinical medicine. Hemoglobin (1997) 21:299–319. doi: 10.3109/03630269709000664
2. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ (2001) 79:704–12.
3. Weatherall DJ, Clegg JB. Thalassaemia: classification, genetics and relationship to other inherited disorders of haemoglobin. Thalassaemia Syndromes (2001), 121–32. doi: 10.1002/9780470696705.ch3
4. Premawardhena A, Fisher CA, Olivieri NF, De Silva S, Arambepola M, Perera W, et al. Haemoglobin E beta thalassaemia in Sri Lanka. Lancet (2005) 366:1467–70. doi: 10.1016/S0140-6736(05)67396-5
5. Haddad A, Tyan P, Radwan A, Mallat N, Taher A. beta-thalassemia intermedia: A bird’s-eye view. Turk J Haematol (2014) 31:5–16. doi: 10.4274/Tjh.2014.0032
6. Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet (2000) 355:2051–2. doi: 10.1016/S0140-6736(00)02357-6
7. Panich V, Pornpatkul M, Sriroongrueng W. The problem of thalassemia in Thailand. Southeast Asian J Trop Med Public Health (1992) 23 Suppl 2:1–6.
8. Winichakoon P, Tantiworawit A, Rattanathammethee T, Hantrakool S, Chai-Adisaksopha C, Rattarittamrong E, et al. Prevalence and risk factors for complications in patients with nontransfusion dependent alpha- and beta-thalassemia. Anemia (2015), 793025. doi: 10.1155/2015/793025
9. Ekwattanakit S, Siritanaratkul N, Viprakasit V. A prospective analysis for prevalence of complications in Thai nontransfusion-dependent Hb E/beta-thalassemia and alpha-thalassemia (Hb H disease). Am J Hematol (2018) 93:623–9. doi: 10.1002/ajh.25046
10. Viprakasit V. Alpha thalassemia syndromes: from clinical and molecular diagnosis to bedside management. EHA Hematol. Educ. Program (2013) 7:329–38.
11. Teawtrakul N, Jetsrisuparb A, Sirijerachai C, Chansung K, Wanitpongpun C. Severe bacterial infections in patients with non-transfusion-dependent thalassemia: prevalence and clinical risk factors. Int J Infect Dis (2015) 39:53–6. doi: 10.1016/j.ijid.2015.09.001
12. Chuncharunee S, Teawtrakul N, Siritanaratkul N, Chueamuangphan N. Review of disease-related complications and management in adult patients with thalassemia: A multi-center study in Thailand. PloS One (2019) 14:e0214148. doi: 10.1371/journal.pone.0214148
13. Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica (2004) 89:1187–93.
14. Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood (2010) 115:1886–92. doi: 10.1182/blood-2009-09-243154
15. Teawtrakul N, Jetsrisuparb A, Pongudom S, Sirijerachai C, Chansung K, Wanitpongpun C, et al. Epidemiologic study of major complications in adolescent and adult patients with thalassemia in Northeastern Thailand: the E-SAAN study phase I. Hematology (2018) 23:55–60. doi: 10.1080/10245332.2017.1358845
16. Kan YW, Nathan DG. Mild thalassemia: the result of interactions of alpha and beta thalassemia genes. J Clin Invest (1970) 49:635–42. doi: 10.1172/JCI106274
17. Rumaney MB, Bitoungui VJN, Vorster AA, Ramesar R, Kengne AP, Ngogang J, et al. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PloS One (2014) 9:e100516. doi: 10.1371/journal.pone.0100516
18. Kho SL, Chua KH, George E, Tan JA. A novel gap-PCR with high resolution melting analysis for the detection of alpha-thalassaemia Southeast Asian and Filipino beta degrees -thalassaemia deletion. Sci Rep (2015) 5:13937. doi: 10.1038/srep13937
19. Vitrano A, Calvaruso G, Lai E, Colletta G, Quota A, Gerardi C, et al. Survival comparability between thalassemia major versus thalassemia intermedia. Blood (2015) 126:2141–1. doi: 10.1182/blood.V126.23.2141.2141
20. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood (2010) 115:4331–6. doi: 10.1182/blood-2010-01-251348
21. Betts M, Flight PA, Paramore LC, Tian L, Milenkovic D, Sheth S. Systematic literature review of the burden of disease and treatment for transfusion-dependent beta-thalassemia. Clin Ther (2020) 42:322–337.e322. doi: 10.1016/j.clinthera.2019.12.003
22. Goldberg EK, Neogi S, Lal A, Higa A, Fung E. Nutritional deficiencies are common in patients with transfusion-dependent thalassemia and associated with iron overload. J Food Nutr Res (Newark) (2018) 6:674–81. doi: 10.12691/jfnr-6-10-9
23. Al-Hafidh M, Younis MS, Al-Taee KF. Survival rate and mortality causes in patients with β-thalassemia major in Nineveh Governorate, Iraq. PJMHS (2020) 14:1274–7.
24. Daar S, Al Khabori M, Al Rahbi S, Hassan M, El Tigani A, Pennell DJ. Cardiac T2* MR in patients with thalassemia major: a 10-year long-term follow-up. Ann Hematol (2020) 99:2009–17. doi: 10.1007/s00277-020-04117-z
25. Wanchaitanawong W, Tantiworawit A, Piriyakhuntorn P, Rattanathammethee T, Hantrakool S, Chai-Adisaksopha C, et al. The association between pre-transfusion hemoglobin levels and thalassemia complications. Hematology (2021) 26:1–8. doi: 10.1080/16078454.2020.1856513
26. Ansari-Moghaddam A, Adineh HA, Zareban I, Mohammadi M, Maghsoodlu M. The survival rate of patients with beta-thalassemia major and intermedia and its trends in recent years in Iran. Epidemiol Health (2018) 40:e2018048. doi: 10.4178/epih.e2018048
27. Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, et al. Survival and causes of death in thalassaemia major. Lancet (1989) 2:27–30. doi: 10.1016/S0140-6736(89)90264-X
28. Telfer P, Coen PG, Christou S, Hadjigavriel M, Kolnakou A, Pangalou E, et al. Survival of medically treated thalassemia patients in Cyprus. Trends and risk factors over the period 1980-2004. Haematologica (2006) 91:1187–92.
29. Latifi SM, Zandian K. Survival analysis of β-thalassemia major patients in Khouzestan province referring to Shafa hospital. Sci Med J (AJUMS) (2010) 9:83–92.
30. Bowie LJ, Reddy PL, Beck KR. Alpha thalassemia and its impact on other clinical conditions. Clin Lab Med (1997) 17:97–108. doi: 10.1016/s0272-2712(18)30234-8
31. Singer ST, Kim HY, Olivieri NF, Kwiatkowski JL, Coates TD, Carson S, et al. Hemoglobin H-constant spring in North America: an alpha thalassemia with frequent complications. Am J Hematol (2009) 84:759–61. doi: 10.1002/ajh.21523
32. Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med (2011) 13:83–8. doi: 10.1097/GIM.0b013e3181fcb468
33. Kan YW. Molecular pathology of alpha-thalassemia. Ann N Y Acad Sci (1985) 445:28–36. doi: 10.1111/j.1749-6632.1985.tb17172.x
34. Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev (2012) 26 Suppl 1:S31–34. doi: 10.1016/S0268-960X(12)70010-3
35. Li J, Xie XM, Liao C, Li DZ. Co-inheritance of alpha-thalassaemia and beta-thalassaemia in a prenatal screening population in mainland China. J Med Screen (2014) 21:167–71. doi: 10.1177/0969141314548203
Keywords: alpha-thalassemia, beta-thalassemia, causes of death, survival rate, risk factor
Citation: Iam-arunthai K, Suwanban T, Thungthong P, Chamnanchanunt S and Fucharoen S (2024) Predicting factors of survival rates among alpha- and beta-thalassemia patients: a retrospective 10-year data analysis. Front. Hematol. 3:1339026. doi: 10.3389/frhem.2024.1339026
Received: 15 November 2023; Accepted: 19 January 2024;
Published: 05 February 2024.
Edited by:
Immacolata Tartaglione, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Gil Cunha De Santis, Hemocentro Foundation of Ribeirão Preto, BrazilOluwabukola Gbotosho, University of Cincinnati, United States
Copyright © 2024 Iam-arunthai, Suwanban, Thungthong, Chamnanchanunt and Fucharoen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Supat Chamnanchanunt, c3VwYXQuY2hhQG1haGlkb2wuYWMudGg=