
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Hematol. , 25 January 2024
Sec. Blood Cancer
Volume 3 - 2024 | https://doi.org/10.3389/frhem.2024.1305741
This article is part of the Research Topic Artificial Intelligence in Hematology: Applications from Drug Design to Precision Medicine View all 4 articles
 Davide Crucitti1,2*
Davide Crucitti1,2* Carlos Pérez Míguez1
Carlos Pérez Míguez1 José Ángel Díaz Arias3
José Ángel Díaz Arias3 Diego Beltrán Fernandez Prada1
Diego Beltrán Fernandez Prada1 Adrián Mosquera Orgueira1,3
Adrián Mosquera Orgueira1,3Developing new drugs is a complex and formidable challenge, intensified by rapidly evolving global health needs. De novo drug design is a promising strategy to accelerate and refine this process. The recent introduction of Generative Artificial Intelligence (AI) algorithms has brought new attention to the field and catalyzed a paradigm shift, allowing rapid and semi-automatic design and optimization of drug-like molecules. This review explores the impact of de novo drug design, highlighting both traditional methodologies and the recently introduced generative algorithms, as well as the promising development of Active Learning (AL). It places special emphasis on their application in oncological drug development, where the need for novel therapeutic agents is urgent. The potential integration of these AI technologies with established computational and experimental methods heralds a new era in the rapid development of innovative drugs. Despite the promising developments and notable successes, these technologies are not without limitations, which require careful consideration and further advancement. This review, intended for professionals across related disciplines, provides a comprehensive introduction to AI-driven de novo drug design of small organic molecules. It aims to offer a clear understanding of the current state and future prospects of these innovative techniques in drug discovery.
Modern medicine’s progress is tightly linked to drug design innovations. Developing new drugs is critical for global health, but the process is costly and time-consuming, often taking several years and costing over a billion dollars (1, 2). Improvements in the drug design phase can greatly reduce these expenses and make it more accessible.
Traditionally, drug design was mainly experimental. However, in the 1990s, computational techniques like de novo molecular design began to emerge (2, 3). De novo design is a set of computational methods that can be used to design a compound without using a previously known one as a starting point. This approach aims to automate the creation of new chemical structures tailored to specific molecular characteristics. It leverages knowledge from existing, effective molecules to design novel ones with unique structural features. In drug design, de novo methods focus on generating molecules with unique drug-like qualities, differentiating them from current treatments. This approach presents a multifaceted challenge as it seeks to design molecules that satisfy various pharmaceutical criteria, including biological activity, target selectivity, and optimal ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles. Despite their innovative nature, these methods faced challenges such as the difficulty of synthesizing the molecules they proposed and the need for specialized computational skills, limiting their broad application in the field of drug discovery (4).
A significant change occurred in 2017 with the introduction of generative AI in de novo design (5, 6). This breakthrough revitalized interest in the field and inspired solutions to previous limitations. These AI algorithms, utilizing vast data on bioactivity, toxicity, and protein structures, have streamlined the process of identifying and refining drug candidates. The emergence of various models, each employing distinct AI architectures, has led to a rapid proliferation of innovative methods. This expansion has significantly enhanced the role of these technologies in the realm of drug discovery (7, 8).
Pharmaceutical companies are now integrating these algorithms into their drug design processes, often in collaboration with AI firms (9). Drugs developed using these methods, like DSP-1181, EXS21546, and DSP-0038, have reached clinical trials (10), demonstrating the effectiveness of generative algorithms in producing viable therapeutic agents. While these compounds primarily target well-researched biological targets and do not innovate structural or binding properties, they validate the utility of generative algorithms in producing effective therapeutic agents.
Generative drug design has demonstrated its maturity and effectiveness. These methods stand to substantially change the pharmaceutical industry, even without considering additional advancements. They enable the rapid generation of alternative treatments once a new drug target is discovered, thereby significantly enhancing their role in both drug discovery and development. They also automate aspects of drug optimization, streamlining the development process.
Despite the rapid progress of these methods, it’s vital to critically assess their limitations. Most of their validation is based on computational benchmarks, which may not fully address real-world challenges (11). Integrating these algorithms into the design-make-test-analyze (DMTA) cycle is essential for a comprehensive efficacy assessment. This integration not only validates their effectiveness but also identifies areas for improvement, fostering wider adoption and refined performance.
This review introduces various de novo algorithms and their role in drug discovery, aimed at professionals in related fields. It covers the advantages and challenges of AI in drug discovery, detailing how these technologies integrate with existing processes. Topics include computational strategies for molecule selection, experimental validation of compounds’ efficacy, and limitations like synthetic feasibility and chemical space exploration Figure 1. Evaluation metrics for these algorithms are also discussed. It also discusses limitations such as synthetic feasibility and chemical space exploration, as well as metrics to evaluate algorithms. Finally, the review includes case studies highlighting the impact of these technologies in real-world drug development, specifically in oncology and hematology.
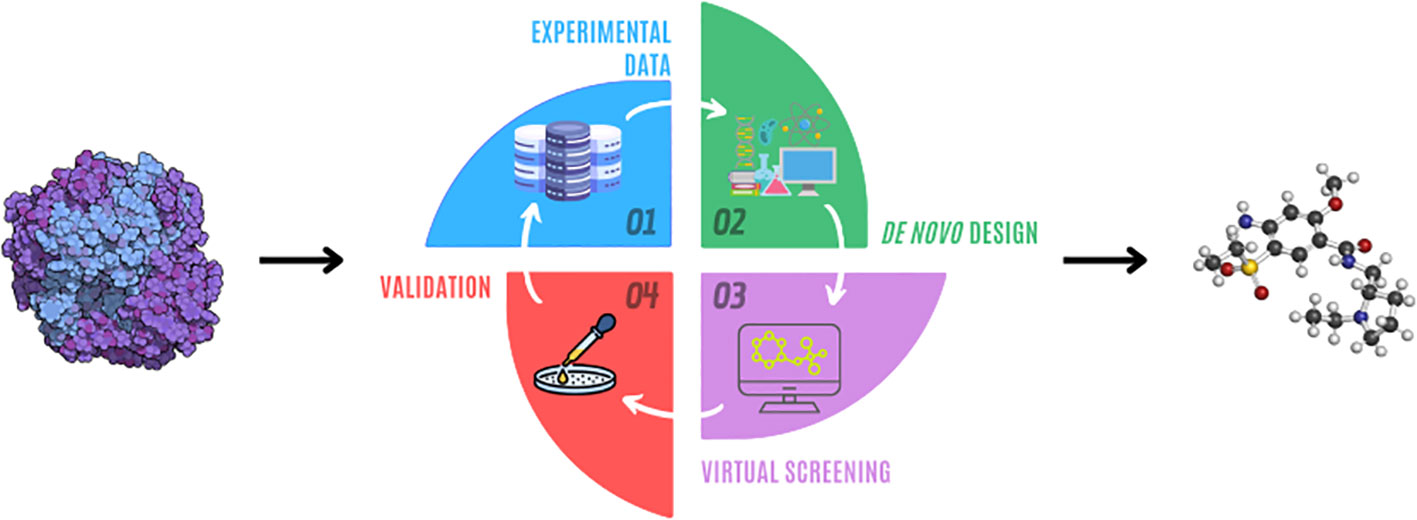
Figure 1 Drug design process.
Drug development is a complex, iterative, and multi-stage process that aims to convert a biological hypothesis into a clinically effective drug (4, 12). This iterative phase utilizes a feedback-oriented approach. It begins with designing molecules based on existing data, followed by computational analysis for selection and ranking. The chosen candidates are then synthesized and tested experimentally, including evaluations on isolated targets, cell cultures, and animal models.
The early stage of drug discovery involves identifying and validating biological targets that can be influenced by potential drugs to change disease progression. These targets vary, including proteins, mutated genes, specific nucleic acid sequences, or components of pathogens (13). Validation is crucial to confirm the therapeutic relevance of these targets. To this end, an array of molecular techniques are used for gene and protein-level verification, while cell-based assays further substantiate the biological significance of the targets in a more complex environment. The introduction of AI in this phase has accelerated the processing of large datasets, speeding up target validation.
After validating drug targets, the subsequent phase in drug discovery is identifying “hits,” molecules that affect the activity of the target. This phase emphasizes exploring a diverse range of molecular structures, as some may show initial activity but prove challenging to optimize. High-throughput screening (HTS) has been a traditional method, testing vast compound libraries against the target. However, HTS is costly and inefficient, often screening over 50,000 compounds with low hit rates and high costs per compound (4). The rise of de novo drug design offers a more efficient alternative for hit identification, reducing the reliance on extensive experimental validation. These AI algorithms utilize existing biomedical data to optimize the experimental design and more precisely explore the vast chemical space, which contains an estimated to drug-like molecules (14). A notable limitation of conventional algorithms is their tendency to explore wide chemical regions without considering the synthesizability of proposed molecules.
After identifying potential drug molecules, the next stage in drug discovery is transforming these “hits” into “lead” compounds, aiming to improve their effectiveness, specificity, and overall suitability as drugs. The main strategy involves altering the core structure, or scaffold, of the molecule and adjusting its substituents. This approach provides insights into how specific changes affect interactions with the target and synthesizing a series of molecules with a common scaffold is easier than a set of entirely diverse compounds.
AI has become a valuable tool in this phase. AI algorithms do more than just analyze the effects of different changes; they actively suggest modifications to enhance the potential of lead candidates. AI can incrementally improve a known hit molecule or, based on its training, generate entirely new molecules likely to be effective in the same category.
The final stage involves refining lead compounds to ensure their effectiveness, safety, and compliance with clinical standards. This stage requires meticulous structural modifications, aiming to prepare drug candidates for clinical trials. AI significantly aids this process by forecasting a drug’s behavior and proposing structural changes, thus accelerating the development of promising candidates.
Refining drug candidates is a collaborative effort combining a chemist’s expertise, laboratory testing, computational modeling, and AI insights. The ability of AI to predict key aspects, such as a the interaction of a drug with its target and its physical and chemical properties, is particularly valuable. De novo AI provides chemists with structural modification suggestions, facilitating exploration and expediting the optimization process. Consequently, AI enables chemists to work more effectively, accelerating the journey towards viable drug candidates.
The landscape of drug discovery is a complex and dynamic field that necessitates strategies for identifying and optimizing molecules. Ranging from the broad exploration of chemical space to nuanced structural modifications, these strategies present unique advantages and challenges. These are exemplified in Figure 2. This section outlines key strategies commonly used in the field.
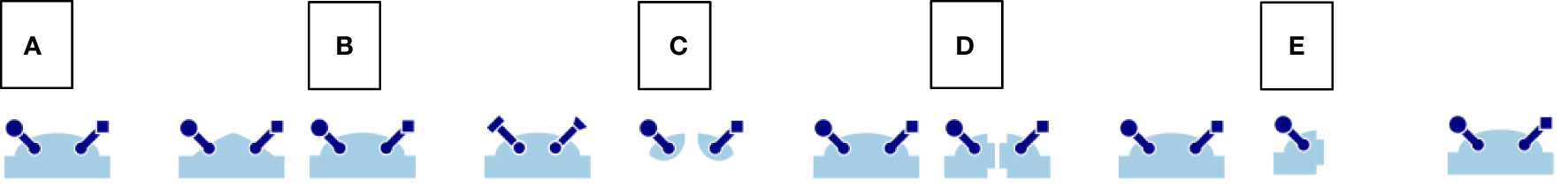
Figure 2 Drug design strategies. (A) — Scaffold hopping, (B) — Scaffold decoration, (C) — Fragment Linking, (D) — Fragment Merging, (E) — Fragment Growing.
Chemical space sampling involves selecting a diverse subset of molecules from a vast array, using computational tools to maximize discovery potential while balancing the synthesizability of these molecules (4, 15–19).
Scaffold hopping is used in hit-to-lead and optimization phases to find novel lead molecules by modifying the core structure of a molecules, the scaffold, while maintaining similar activity. This strategy has evolved with AI, enabling the generation of alternative scaffolds and suggesting optimal substituents for target interactions (20–23). Although this process results in structurally distinct compounds, their binding mechanisms to the target typically remain similar, positioning scaffold hopping more as an optimization step than an exploratory one.
In scaffold decoration, functional groups are added to the scaffold to enhance interaction with the target, thereby improving efficacy or selectivity. This approach is pivotal in the later stages of drug development, allowing for the fine-tuning of molecular properties and evaluating the influence of each substituent over the activity (24, 25).
Distinct from other methods, this approach begins with small molecules, named fragments, which have shown target-binding affinity (26–29). These fragments are then elaborated upon using various techniques. Fragment Linking involves joining two or more fragments that interact with different sites on the target to create a single molecule with improved binding affinity. Fragment Merging combines overlapping fragments into a unified molecular structure. Lastly, Fragment Growing expands the fragment by adding parts to enhance its binding affinity to the target.
The fusion of AI with computational chemistry has led to the development of de novo design methods, which have found applications in areas ranging from drug discovery to materials science (30). These innovative techniques are focused on synthesizing a variety of new, diverse, and efficient chemical compounds. In contrast to traditional AI applications that mainly predict molecular properties or interactions with biological targets, these new methods are centered on the creation of novel molecules. Their objective is to explore and map extensive areas of the chemical space, going beyond the confines of existing drug-like molecule databases.
De novo molecular design primarily employs two strategies. The first, “Holistic Generation,” involves creating an entire molecule from scratch, which is particularly useful for initial discovery phases and for exploring chemical spaces broadly. The second strategy, “Iterative Generation,” builds molecules in a step-by-step manner, which is more suited for refining molecules or tailoring them for specific purposes. A selection of de novo design methodologies which have been developed over time can be found in Table 1.
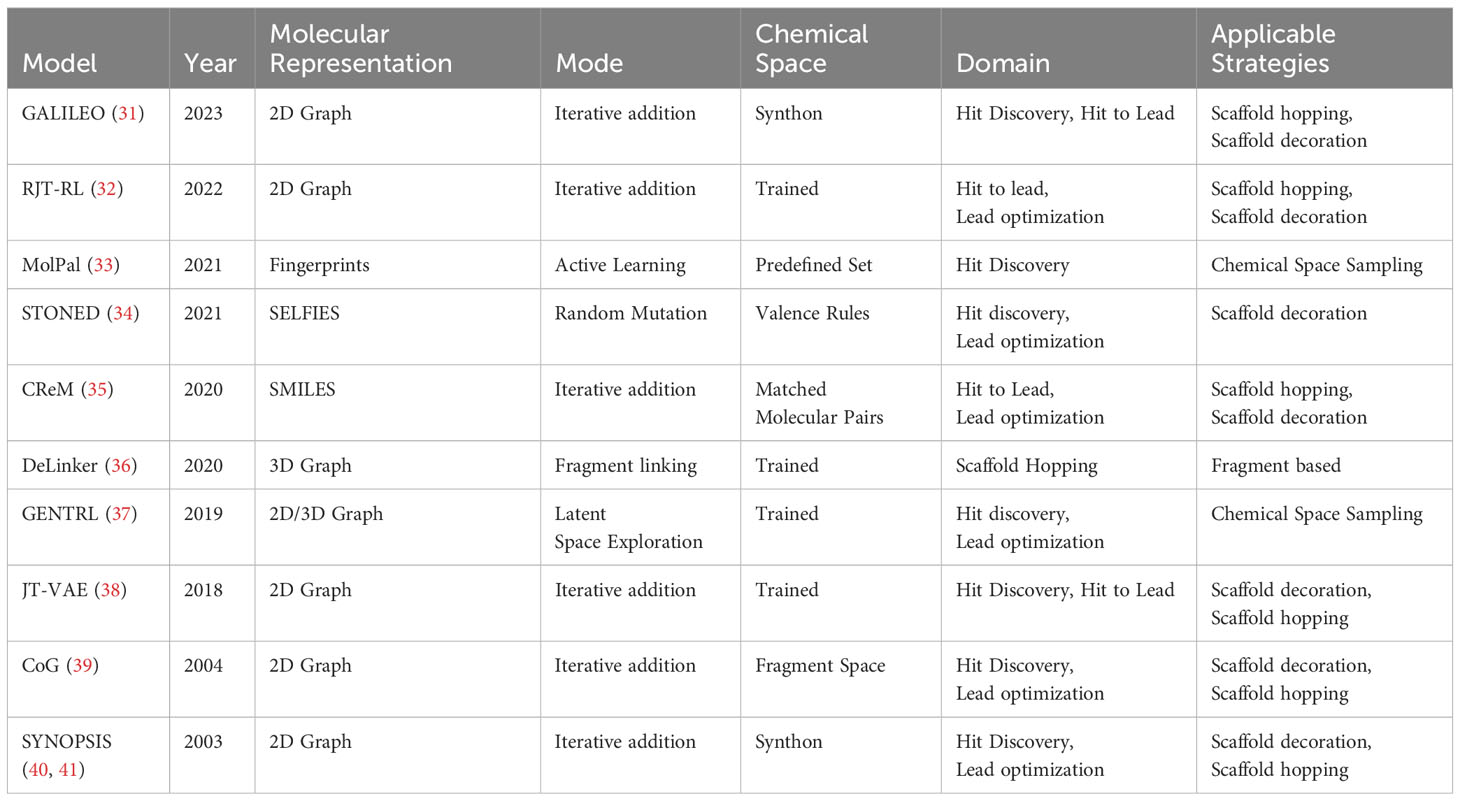
Table 1 A list of relevant de novo methodologies which have been developed over time.
Molecules, being tangible entities that cannot be directly manipulated through computations, need methods to represent their properties and characteristics. These range from quantum mechanics-based electron distribution models to basic chemical formulas. The representations are communicated through various encodings, such as the structural formula, which visually outlines molecular topologies. Encodings for molecular structure generation are typically view atoms as indivisible units connected by covalent bonds.
String-based encodings like the Wiswesser Line Notation (42) and SMILES (43) represent molecular topology of organic molecules through character sequences. However, limitations in SMILES led to the development of DeepSMILES (44), ensuring syntactic correctness, and SELFIES (45), ensuring both syntactical validity and chemical space adherence.
Graph-based encodings depict molecules as nodes (atoms) linked by edges (bonds) (46), with 2D versions showing connections and 3D versions adding spatial information. While 3D encodings offer more detail, they also add complexity in algorithm training.
Feature-based encodings use molecular descriptors (47) or fingerprints (48) to detail molecules based on properties or experimental data, useful for comparison and property prediction but less effective for structure creation.
AI can define encodings by converting molecular structures into a continuous numerical space, which can be decoded back (8). Trained on extensive molecular datasets, they capture essential features and allow for the exploration of new molecular structures. Challenges in learning encodings include accurately representing chemical space and ensuring relevance to drug development, as molecules close in latent space might have divergent biological and chemical properties.
Access to extensive data is crucial to train algorithms. Key to this is the availability of public databases. Prominent among these are ChEMBL (49), PubChem (50), BindingDB (51), and DrugBank (52), which offer essential bioactivity data for drug discovery. ChemSpider (53) aggregates information on molecular properties and available compounds. CompTox (54) is a resource for toxicity and environmental hazards. GDB17 (55) lists organic compounds up to 17 atoms, while QM9 (56) provides a subset with quantum mechanically determined molecular conformations. ZINC (57) catalogs commercially available compounds, and Enamine’s REAL database lists synthesizable molecules.
The Protein Data Bank (PDB) (58) provides structural details of proteins and nucleic acids, crucial for understanding molecular interactions. Uniprot (59) offers protein sequences and functional annotations, which help target selection and validation. The Therapeutic Target Database (TTD) (60) focuses on well-studied therapeutic targets.
The field of de novo drug design has been significantly advanced by generative algorithms, each offering unique advantages and applications. A training dataset of molecular structures is essential for these techniques. These use a set of known molecules to train a generative algorithm which is then used to generate novel molecular structures. Key AI methods are summarized below, with a detailed discussion available in the comprehensive review by Martinelli (7).
● Recurrent Neural Networks (RNNs): Predominant in generative drug design, RNNs excel in generating new molecular structures by identifying patterns in training data. Notable developments include the first model by Olivecrona et al. (61) and subsequent advancements like DrugEx (62, 63), which emphasizes multi-objective optimization, including toxicity considerations.
● Latent Space Exploration: Latent space exploration methodologies are another common generative drug design technique. Among these, VAEs have garnered considerable attention, and were the first architecture specifically applied to drug generation in 2017 (5). Subsequent studies have reinforced their utility and applicability (7, 8). VAEs function through a dual neural network architecture, comprised of an encoder that maps molecular structures into a latent space, and a decoder that reverses this transformation. Models have been designed to accommodate both 2D and 3D molecular representations (64). The PASITHEA model introduced “deep dreaming” to molecular design (65). This approach employs a deep neural network, trained to predict specific molecular properties. By reversing the network and inputting desired property values, one can generate molecules optimized for those properties, an improvement over the more abstract latent space of VAEs.
● Generative Adversarial Networks (GANs): This architecture involves two competing neural networks: a generator and a discriminator. The generator’s primary objective is to create molecular structures that the discriminator cannot distinguish from genuine molecules. The first example in generative drug design was ORGANIC (66) and other ones were rapidly developed, such as ATNC (67) and MolGAN (68) which improved the rate of valid molecules generated. These methodologies have been applied especially in the field of molecular optimization (69–71).
● Transformer-based Models: Drawing inspiration from natural language models like BERT, transformers view molecules as sequences of tokens. These models typically employ a string based molecular representation. A notable example is ChemBERTa (72). Transformers have also shown utility in suggesting structural modifications during the optimization phases of drug design (73, 74).
Rule-based techniques, formulated before trained generative algorithms, provide a structured approach to exploring the chemical space. Combinatorial chemistry principles are used to construct a well-defined chemical space and a set of traversal rules for these techniques. Combinatorial chemistry involves the creation of molecules by combining molecular substructures or atoms based on human-defined rules. These methodologies differ according to the rules employed in the combinatorial process and the exploratory algorithms used to select molecules to analyze from these potentially extensive sets.
The chemical space can be defined in several ways:
1. Valence Rules: This approach focuses on combinations of atoms adhering to valence rules. An example is GDB-17 (55), which lists all organic molecules with up to 17 atoms of C, N, O, and S.
2. Fragment Spaces: Techniques like BRICS (75) and RECAP (76) employ sets of small molecular fragments that are linked together through a set of manually defined rules.
3. Synthon Method: Proposed in 2007 (77), it uses generalized reactions on available reagents to enumerate synthetically accessible molecules. Notable methodologies include DOGS (78), Synth-On (79), and pre-compiled commercial libraries (80, 81).
4. Definition of Substituents: This method involves defining a set of substituents that will be applied to a molecular scaffold, similar to traditional scaffold decoration approaches.
5. Matched Molecular Pairs Analysis: First proposed in 2005 (82), it utilizes a catalog of structural transformations known to enhance molecular properties. It is useful in lead optimization (35, 83) and enhancing ADMET properties (84, 85).
The main strategies employed to explore the defined chemical space can be grouped in two categories:
1. Random Generation/Mutation: This approach often uses molecular string representations to either create entirely new molecules or iteratively change existing ones. An example is the STONED algorithm (34).
2. Evolutionary Molecular Modification: These methods begin with a molecule and applies atomic or fragment modifications using evolutionary algorithms. It is particularly effective for scaffold hopping or decoration. Examples include GB-GA (86) and EvoMol (87) which use valence rules. CoG (39) uses fragment spaces, while SYNOPSIS (40, 41) and GALILEO (31) are based on synthon spaces.
Active Learning (AL) is a novel introduction to the field of de Novo Drug Design for the exploration of chemical space. AL operates on the principle that models can achieve higher accuracy with fewer data by intelligently selecting their training samples. Central to AL is the concept of uncertainty. AL addresses epistemic uncertainty, related to the lack of knowledge in certain areas of the modeled space by the model (88). For instance, an unfamiliar molecular structure can increase uncertainty in predictions due to insufficient knowledge in that area. AL reduces uncertainty by adding new data to the training set.
AL has traditionally been used to improve model predictions, but is now also used to explore the chemical space. It identifies and investigates molecules linked with high uncertainty, assumed to be those presenting less-explored molecular structures. By doing so, AL enables more efficient exploration with fewer experiments. These approaches employ AI techniques like Bayesian machine learning algorithms and Ensemble learning to estimate uncertainty. The process follows an iterative process, it identifies molecules with high uncertainty, tests them and adds the results to the training set. This results in the refinement of the model and the expansion of the training set, which is effectively the set of screened compounds. Predictions from the model are generally discarded, as the method is primarily used to explore and not predict. Unlike the methods previously outlined, this methodology is purely explorative and does not generate novel structures, the structures for this exploration can come from different sources, such as databases, combinatorial chemistry, or generative algorithms. Strategies that include molecules predicted to have desirable properties can be used to combine the exploration and optimization steps. In fact, it has been shown that purely exploratory approaches effectively explore the chemical space but do not identify many active compounds (89). A schematic representation of the process using AL is exemplified in Figure 3.
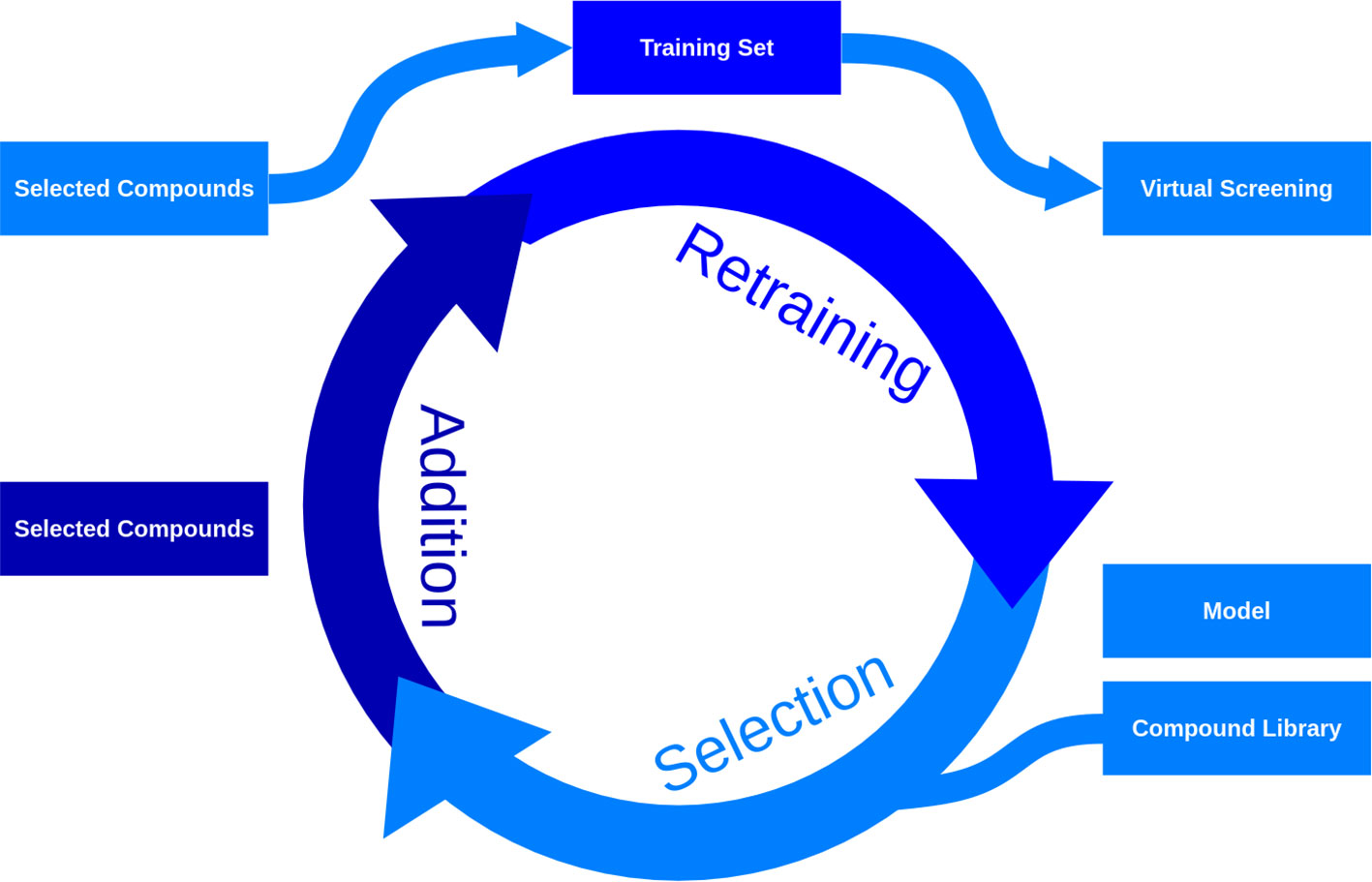
Figure 3 Iterative selection process through active learning.
However, AL requires regular generation of new data, making it more suited for virtual rather than experimental screening. While these strategies do not improve the accuracy of the prediction, they optimize the quantity of tested compounds; making it possible to screen larger libraries. Despite this, it has been applied in the context of automated experimental testing (90).
The application of AL to drug discovery has been explored since the early 2000s, but most of these studies were retrospective in nature (91). The focus has only recently shifted towards identifying and evaluating specific AL techniques that could be effectively integrated into the drug discovery process (33), but it is still an emerging field with slow and limited adoption (7, 89). Three experimental high-throughput screening results datasets were used to demonstrate the potential of AL, showing a sixfold increase in Enrichment Factor over traditional virtual screening methods (89).
Virtual screening is a key technique in drug design used to assess and prioritize molecular candidates. It uses principles from chemistry, statistics, and empirical data to predict how molecules will behave in terms of activity, toxicity, and suitability as drugs.
Since it’s expensive and complex to synthesize and test every molecule created by AI, virtual screening is essential. It helps focus on the most promising molecules, making drug discovery more efficient. These methods also estimate molecular properties for machine learning training when experimental data is scarce. However, they can’t replace actual laboratory tests. When generative algorithms are trained exclusively on computed data, their performance is governed by the accuracy of the underlying theoretical models. It is advisable to use experimental data to train algorithms and to use computational techniques to filter and prioritize whenever feasible.
In the early stages of drug discovery, virtual screening is crucial for identifying diverse and unique molecules rather than just focusing on similar, high-affinity compounds.
Rule-based filters serve as rapid evaluation systems to categorize molecules based on predetermined criteria related to properties like bioactivity or synthetic feasibility. These systems typically use two-dimensional molecular structures to pinpoint promising candidates and weed out potential “false positives”—molecules that initially appear viable but fail in later testing stages. Tools like Lipinski’s Rule of 5 (92) assess oral bioavailability, while the QED metric (93) estimates therapeutic potential. Structural filters, such as PAINS (94) and BRENK (95), identify molecules that could interfere with bioassays or prove unsuitable as lead candidates. Additionally, instruments like the Synthetic Accessibility Score (96) and automated retrosynthetic analysis (97–99) aid in eliminating hard-to-synthesize molecules. However, it’s important to recognize that while these methods are broadly useful, they are primarily tailored for known drugs and targets. This specialization can limit their applicability in assessing novel structures (100).
Ligand-based virtual screening, employed in drug discovery when a target’s structure is unknown, it is computationally simple and identifies ligands with similar binding characteristics. Its effectiveness hinges on the availability of known ligands interacting with the target, a limitation when such data is scarce. In de novo design, focusing on structurally similar compounds is less preferred and as such these methodologies are often not appropriate.
Key techniques in this approach include Quantitative Structure-Activity Relationship (QSAR), which correlates molecular structure with biological activity, aiding in predicting the behavior of new molecules in terms of activity and toxicity (62, 101, 102). Pharmacophore Modeling identifies essential molecular features for biological activity, using known ligands to create models for finding new molecules with similar characteristics (31, 103, 104). Structural Similarity assesses the resemblance between molecules, facilitating the discovery of new compounds similar to active ones, or exploring diverse molecular structures for further study (17, 48, 105).
Structure-based virtual screening is a technique in drug discovery that uses the 3D structures of both the drug molecule and its target. It can use either experimentally determined structures or accurate computational models of the target, made through methods like homology modeling or AI tools like AlphaFold (106, 107). This approach can be useful even without data on binding ligands.
Molecular Docking is commonly used in structure-based virtual screening to predict binding modes (108). This knowledge aids in evaluating the interaction’s quality and provides comparative insights based on other known ligands (109–111). Molecular simulations, typically conducted through Molecular Dynamics or Monte Carlo techniques, aim to predict the structural, thermodynamic, and kinetic attributes of the molecule-target interaction (112). These simulations can be computationally demanding, making them most effective when focused on a refined list of candidates. One common application is the estimation of the Free Energy of binding through methods like Alchemical Free Energy, Thermodynamic integration or Free Energy Perturbation (113).
Drug development involves several experimental assays to test a drug’s effectiveness, selectivity, and safety. These tests, essential at every stage of development, fall into three main categories: biochemical, biophysical, and cell-based (114, 115).
Biochemical Assays: Used early in drug development, these tests measure how a drug interacts with its target at a molecular level by measuring its effects on function, often using indicators like fluorescent signals. Common types include FRET and ALPHAscreen. They are cost-effective and suitable for large-scale screening, but need follow-up tests for confirmation.
Biophysical Assays: These assays focus on the direct physical interaction between a drug and its target rather than functional outcomes. Techniques like X-ray crystallography and NMR are used here. They are more resource-intensive but provide detailed information about the interaction, helping to refine drug candidates (115).
Cell-Based Assays: Conducted throughout drug development, these tests assess a drug’s effect within living cells. They are crucial for understanding a drug’s overall impact, including its ability to enter cells and potential side effects. Techniques like cell viability and reporter gene assays are commonly used.
After these laboratory assays, promising drug candidates are then tested in animal models to further assess their efficacy and safety before moving on to human clinical trials.
Evaluating the efficacy of generative models is essential to select the most suitable model for distinct tasks and to pave the way for innovations that outpace current constraints. A variety of metrics have been devised to serve this purpose.
Assessing how thoroughly a generative model explores the chemical space is central to its evaluation, with various metrics offering different perspectives on this crucial aspect. The Internal Diversity Metric gauges the structural diversity within generated compound collections, whereas the concept of richness focuses on counting the number of unique compounds produced (116). The Fréchet ChemNet Distance (117) provides numerical insights into the alignment of generated molecules with a target distribution, serving as an indicator of potential biases. Complementing these is the Coverage Score (118), which quantifies the model’s ability to sample molecules from larger datasets. The #Circles Metric (119) takes a more comprehensive approach by examining the overlap in structural diversity between two chemical sets and assessing the impact of introducing new molecules to the sampling range. For a more detailed exploration of these and other methodologies, the work by Xie et al. (119) serves as a valuable resource.
Beyond metrics, benchmarking suites like GuacaMol (116) and MOSES (120) provide holistic evaluations by employing a range of these metrics across different tasks, which include both sampling of the chemical space and fine-tuning of specific physicochemical properties. Ciepliński et al. (121, 122) have developed a framework that evaluates models based on their effectiveness in molecular docking simulations with protein targets, going beyond simple physicochemical assessments which are often insufficient when optimizing a drug structure. This approach has revealed limitations in current models, including the generation of improbable molecules and a tendency to underperform compared to the top molecules in public databases. Gao and Coley (97) have introduced an approach to assess the real-world synthesizability of generated molecular structures employing retrosynthetic analysis tools.
In 2018, Merk et al. (123) set a landmark by using Generative AI to design and then synthesize and experimentally validate inhibitors for the nuclear receptors RXRα/β/γ and PPARα/γ/δ. They utilized a deep RNN, trained on a vast dataset of over 540,000 SMILES of bioactive compounds. This was fine-tuned using 25 fatty acid mimetics known for their agonistic activity. The system generated 1,000 molecular structures, with 93% being chemically valid and 90% being unique. Out of 49 computationally high-scoring compounds, five were synthesized, and four demonstrated promising bioactivities. However, these novel molecules resembled the training set compounds, highlighting the need for further innovation in the generation of diverse structures.
DDR1, a kinase implicated in cancer and Inflammatory bowel disease, is a promising therapeutic target. Zhavoronkov et al. (37) demonstrated the power of deep learning in a ‘hit-to-lead’ optimization campaign, identifying a DDR1 inhibitor in 46 days. By leveraging an existing DDR1 inhibitory molecular scaffold, they generated 30,000 structures adjusting the substituent groups of this scaffold to enhance its pharmacological efficacy which were then computationally screened. From this, six were synthesized based on their synthetic feasibility and target profile. Two showcased potent inhibitory and pharmacokinetic properties.
In 2022, Xiaoqin et al. (24) also utilized deep learning to design selective inhibitors for DDR1 using a generative scaffold decorator. Refining a scaffold active against FGFR but also interacting with DDR1, they generated over 19,000 molecular structures. After filtering and computational simulations two compounds were synthesized. Both demonstrated significant anti-inflammatory activity in animal tests, with one emerging as the most selective DDR1 inhibitor to date. These studies underscore the potential of generative AI in accelerating ‘hit-to-lead’ drug discovery campaigns.
The application of de novo techniques in oncological drug design has primarily focused on kinase targets, largely due to the extensive research and abundant data availability (124). As AI methodologies advance, there’s a growing expectation that these tools will be applied to lesser-studied targets. While these studies have yielded successful results, the molecules generated do not show significant structural or binding mode differences compared to known ligands. Consequently, de novo techniques have shown more success in drug optimization rather than in the discovery of entirely novel hits.
In 2021, Yu et al. (22) undertook a scaffold hopping campaign to identify novel structures for JAK1 inhibition. To enhance this process, they used a neural network with Graph-Based Variational Autoencoders, training the model on scaffolds derived from drug-like compounds and fine-tuning it with the structure and bioactivity of known kinase inhibitors. The objective was to hop through different scaffolds while retaining similar side chains, as these two were encoded separately into the model. With a known JAK1 inhibitor as the starting scaffold, they generated 30,000 molecular structures. These underwent a screening process considering physicochemical properties, structural alerts, and their alignment with established inhibitors. A QSAR model and molecular docking were used to prioritize 25 molecules. 7 of these were synthesized, all of which demonstrated JAK1 inhibitory potential in experimental trials. While the inhibitory activity of the identified molecules is significant, they did not conduct kinase screening tests to assess selectivity, and in consequence it is difficult to appreciate the real value of the compounds.
In 2022, Jang et al. focused on FLT3 (125), a critical kinase in hematopoiesis. When mutated in acute myeloid leukemia (AML), it is often associated with adverse outcomes. Their approach involved enhancing the FLT3 selectivity of a previously active molecule against breast cancer cells, which was predicted to interact with FLT3. A deep learning generative model was trained on drug-like SMILES structures and fine-tuned with known FLT3 binders. The model generated over 10,000 structures. These were filtered, and the resulting molecules were ranked based on their binding affinities derived from Alchemical Free Energy calculations. The most promising compound was synthesized and tested in cellular cultures and on the protein. It showed affinity for the mutated FLT3 variant and inhibited FLT3-mutated AML cell proliferation. The study, however, has limitations: it leaned heavily on molecular docking simulations without experimental assays for kinase selectivity, and did not test on healthy cell lines, leaving potential toxicity unexplored. Additionally, the molecules produced strongly resembled known FLT3 inhibitors, questioning the novelty of molecules designed through this approach. The findings are promising, but comprehensive experimental validation is required.
In their 2023 study, Zhu et al. engaged in a ‘hit-to-lead’ campaign targeting SIK2 (25). They utilized a proprietary AI system, with an existing inhibitor as the reference, to produce molecules with varied substituents. Each molecule, out of the 5,000 candidates, was docked onto the protein structure modelled by AlphaFold and evaluated on structural and protein interaction criteria. They were then grouped into 56 clusters based on structural similarities and hydrogen bond interactions. From each cluster, two molecules were synthesized and underwent in vitro and in vivo testing. One molecule stood out for its potent inhibitory activity and ideal ADMET properties.
In this article, we have explored a range of Artificial Intelligence (AI) methodologies applied in drug design. Currently, the field is evolving, with these methodologies primarily applied in experimental settings. To date, the results, while promising in accelerating and standardizing drug optimization for known targets, have not substantially led to the creation of novel de novo structures with distinct inhibition patterns from existing inhibitors. The aspiration to simplify and automate drug design for new targets or novel targeting patterns remains largely unfulfilled. Despite the rapid development of these methodologies, there is still a significant journey ahead before we can identify a set of methodologies robust enough to be widely adopted as standard practices. This journey will necessitate extensive experimental validation of both current and future techniques. The initial positive results, however, do suggest a potential for broader adoption in the future. Trained generative algorithms have notably altered the landscape of de novo drug design. Initially received with skepticism, focusing more on theoretical potential than practical application, these techniques have gradually gained recognition. This is particularly evident with some AI-designed drugs progressing to clinical trials. Yet, their success has largely been confined to well-characterized protein targets, indicating a notable limitation in their ability to explore new or under-researched biological targets.
While the potential of AI to drive de novo molecular design is significant, this field faces several challenges. AI methodologies have proven successful in targeting well-studied proteins, yet advancements in the design of molecules significantly different from known ligands remain limited. Available methods typically optimize molecules within established chemical spaces, raising questions about their applicability to genuine de novo drug design. The challenge is determining whether the current methodologies need refinement, or if the approach itself is unsuitable for this type of problem.
A key area of focus has been trained generative algorithms, which have brought new life to de novo drug design. Their ability to explore chemical spaces beyond their training set structures is still a concern (122). Efforts are ongoing to expand the range of molecular structures these algorithms can explore (7, 126). Yet, it remains challenging to assess their effectiveness in spanning chemical space. There are metrics for comparing molecule sets, but no methodology currently enables the enumeration of the chemical space accessible to generative algorithms. Such a methodology would be crucial for comparing the diversity and breadth of chemical spaces generated by these algorithms with those from combinatorial chemistry and existing chemical databases.
Data availability is also a challenge for trained generative algorithms. The bias towards well-explored molecular structures and targets in currently available datasets can limit their ability to generate diverse and innovative molecular designs for new targets (11).
Another significant limitation is the uncertainty regarding the quantity of molecules that need to be sampled from these algorithms to find a promising hit. These algorithms are expected to guide experimental design, but there are no comprehensive benchmarks of their effectiveness. Their limited application in large-scale hit discovery programs makes it difficult to assess their suitability for extensive de novo drug discovery and the resources needed. These methods have the potential to streamline the hit discovery process and reduce the number of necessary tests. However, this efficiency must be balanced against the higher synthetic efforts required for molecules generated through these algorithms, as opposed to those identified via high-throughput screening from commercial compound libraries. While advancements in synthesizability have been made, the necessity for custom synthesis will always persist. It is important to determine the extent to which these algorithms can reduce testing needs. Virtual screening can play a significant role in filtering compounds. This process predominantly depends on structure-based methods, as ligand-based methods are less effective outside well-characterized chemical spaces. The drawback of this reliance is the increased demand for computational resources, which exceeds that of traditional virtual screening methods. This raises questions about the circumstances under which these novel AI methodologies may offer advantages over established techniques. Determining the right balance between computational effort and the efficiency of hit discovery remains a challenge.
The synthesizability of molecules generated by AI algorithms remains a challenging issue (4, 7, 97, 124). Despite this, significant progress has been made towards addressing this problem. New algorithms that can generate molecules more amenable to synthesis and improved methods for evaluating synthesizability, are expected to mitigate this limitation in the near future.
Rule-based systems offer an advantage over trained generative techniques in that they do not require training data and allow the enumeration of the explorable chemical space. Despite their longstanding presence, these systems have been primarily used to optimize hits or leads, with limited application in full de novo design. The success of these methods depends on the formulation of effective rules for generation. These rules must not only ensure synthesizability, but also ensure the creation of a diverse chemical spaces. Recent progress in this area involves generating synthon spaces from a set of generic reactions, potentially leading to more easily synthesizable molecules. The limited availability of publicly available reactions sets poses limitations, and it is unclear how much these methods improve synthesizability over other approaches.
Active Learning (AL) shows promise in optimizing virtual screenings, but integrating experimental data with computational results is a problem yet to be resolved. A methodology allowing such integration would enable a more targeted search process.
Another focus area is multi-objective optimization, which is crucial for developing effective and safe drugs. The ligands that bind effectively to the target must also have desirable physicochemical properties, selectivity, and an acceptable ADMET profile. Advances are being made in this field, using multi-objective optimization techniques from other disciplines, raising hopes for a solution in the near future.
Finally, the widespread application of these methodologies is hampered by their limited diffusion and ease of use. De novo design requires an integrated approach combining computational science, chemistry, and biology. Many models lack this interdisciplinary aspect and are therefore computationally sophisticated but less practical from a chemical or biological standpoint. This gap restricts their utility, especially for experimental chemists who require user-friendly tools. There are some commercial software with intuitive interfaces, but their use is still limited by the need for extensive computational knowledge and high costs (127, 128).
Methodologies for de novo drug design utilizing AI have been developed, yet their application in large-scale campaigns remains limited. The effectiveness of these methods compared to traditional approaches is still an open question. Therefore, a critical development area involves devising metrics to compare these AI methodologies against conventional methods and using de novo design in larger drug development campaigns.
Advancements in broadening the chemical space accessible to trained generative algorithms are being done (126). Future progress will be key in assessing whether these algorithms can create novel molecules distinct from their training data. This will clarify if such limitations are inherent to these methods or if they can be surpassed for more innovative molecular design. The capability of these algorithms to generate novel structures will significantly indicate their flexibility and influence in drug discovery.
Active Learning (AL) emerges as a potent tool in AI-based de novo drug design. Primarily used in compound screening, AL’s potential extends to optimizing identified hits—an area ripe for exploration. These methods could potentially replace evolutionary algorithms in rule-based systems, emphasizing the generation of unique or underexplored molecules. Currently, ensemble methods dominate AL, but integrating Bayesian models, renowned for their efficacy (88), could significantly enhance the exploration of chemical spaces.
Transitioning to the next phase of development, hybrid systems that merge exploratory and trained methodologies present a promising path for comprehensive chemical space exploration. Rule-based systems efficiently probe molecular structures akin to identified hits. In contrast, trained generative algorithms have proven effective in refining structures within explored chemical spaces. A cohesive de novo drug generation pipeline might first employ AL for rapid hit discovery, followed by Rule-Based systems for initial optimization. Subsequently, trained generative algorithms could refine these hits further, offering a layered approach composed of broad initial scans followed by focused lead tuning. This integrated strategy could substantially advance de novo drug design capabilities.
Furthermore, retrosynthetic analysis tools are advancing, particularly in addressing synthesizability challenges. Improvements in these tools may lessen the emphasis on generating only synthetically feasible molecules, paving the way for more creative and expansive molecular design.
Artificial Intelligence (AI) holds potential to significantly impact de novo drug design, yet it’s important to approach its current state and future prospects with a balanced view. The challenges outlined in this review are critical, and addressing them is key to harnessing AI’s full potential in this field. While we anticipate improvements in the efficiency and effectiveness of drug development through AI, it’s important to recognize that these advancements will be gradual and contingent upon overcoming significant hurdles.
AI methodologies have introduced noteworthy changes in drug design, including improvements in algorithmic efficiency and the exploration of new chemical spaces. However, it’s crucial to note that these advancements are still in their nascent stages. The transformative impact of AI on drug design is promising, but it is not without its complexities and limitations.
Our discussion has covered a range of AI methodologies, exploring their integration into the drug discovery process and highlighting both their strengths and weaknesses. We have provided examples of AI applications in drug design, demonstrating the rapid development in this area. However, it’s evident that most AI algorithms have found more success in optimizing molecular structures than in groundbreaking de novo design.
Looking to the future, the role of AI in drug design appears promising, but its trajectory is not without uncertainty. Developments in algorithmic sophistication and application breadth are anticipated, yet they will likely face challenges in surpassing the current limitations. As the field evolves, it is hoped that AI will not only supplement but also enhance traditional drug design methodologies. However, this evolution will require a careful and considered approach, ensuring that new technologies are robust, reliable, and truly beneficial in the quest for novel therapeutic solutions.
DC: Writing – original draft, Writing – review & editing. CP: Visualization, Writing – review & editing. JA: Writing – review & editing. DF: Writing – review & editing. AM: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the grant 'JI-2022-Astrazenca-2 Germline Predisposition to Myelodysplastic Syndromes in Adults: Expanding Diagnostic Precision and Evidence of Pathogenicity.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Economics (2016) 47:20–33. doi: 10.1016/j.jhealeco.2016.01.012
2. Clark DE ed. Evolutionary algorithms in molecular design. 1st. Weinheim, Federal Republic of Germany: Wiley (2000). doi: 10.1002/9783527613168
3. Moon JB, Howe WJ. 3D database searching and de novo construction methods in molecular design. Tetrahedron Comput Method (1990) 3:697–711.- doi: 10.1016/0898-5529(90)90168-8
4. Lipinski CA. Overview of hit to lead: the medicinal chemist’s role from HTS retest to lead optimization hand off. In: Hayward MM, Bikker JA, Ellingboe JW, Freeman-Cook KD, Gilbert AM, Harrison RK, et al, editors. Lead-seeking approaches. Berlin, Heidelberg: Springer Berlin Heidelberg (2009). p. 1–24. doi: 10.1007/7355_2009_4
5. Gómez-Bombarelli R, Wei JN, Duvenaud D, Hernández-Lobato JM, Sánchez-Lengeling B, Sheberla D, et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci (2018) 4:268–76. doi: 10.1021/acscentsci.7b00572
6. Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci (2018) 4:120–31. doi: 10.1021/acscentsci.7b00512
7. Martinelli DD. Generative machine learning for de novo drug discovery: A systematic review. Comput Biol Med (2022) 145:105403. doi: 10.1016/j.compbiomed.2022.105403
8. Wigh DS, Goodman JM, Lapkin AA. A review of molecular representation in the age of machine learning. WIREs Comput Mol Sci (2022):e1603. doi: 10.1002/wcms.1603
9. Mak K-K, Balijepalli MK, Pichika MR. Success stories of AI in drug discovery - where do things stand? Expert Opin. Drug Discov (2022) 17:79–92. doi: 10.1080/17460441.2022.1985108
10. drug discovery AI. assessing the first AI-designed drug candidates to go into human clinical trials | CAS (2022). Available at: https://www.cas.org/resources/cas-insights/drug-discovery/ai-designed-drug-candidates (Accessed September 8, 2023).
11. Volkamer A, Riniker S, Nittinger E, Lanini J, Grisoni F, Evertsson E, et al. Machine learning for small molecule drug discovery in academia and industry. Artif Intell Life Sci (2023) 3:100056. doi: 10.1016/j.ailsci.2022.100056
12. Hughes J, Rees S, Kalindjian S, Philpott K. Principles of early drug discovery. Br J Pharmacol (2011) 162:1239–49. doi: 10.1111/j.1476-5381.2010.01127.x
13. Patrick GL. An introduction to medicinal chemistry. Fifth. Oxford: Oxford University Press (2013). 789 p.
14. Polishchuk PG, Madzhidov TI, Varnek A. Estimation of the size of drug-like chemical space based on GDB-17 data. J Comput Aided Mol Des (2013) 27:675–9. doi: 10.1007/s10822-013-9672-4
15. Boehm M, Wu T-Y, Claussen H, Lemmen C. Similarity searching and scaffold hopping in synthetically accessible combinatorial chemistry spaces. J Med Chem (2008) 51:2468–80. doi: 10.1021/jm0707727
16. Bajusz D, Rácz A, Héberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Cheminform (2015) 7:20. doi: 10.1186/s13321-015-0069-3
17. Garcia-Hernandez C, Fernández A, Serratosa F. Ligand-based virtual screening using graph edit distance as molecular similarity measure. J Chem Inf Model (2019) 59:1410–21. doi: 10.1021/acs.jcim.8b00820
18. Bender A, Mussa HY, Glen RC, Reiling S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J Chem Inf Comput Sci (2004) 44:1708–18. doi: 10.1021/ci0498719
19. Raymond JW, Blankley CJ, Willett P. Comparison of chemical clustering methods using graph- and fingerprint-based similarity measures. J Mol Graphics Model (2003) 21:421–33. doi: 10.1016/S1093-3263(02)00188-2
20. Schüller A, Suhartono M, Fechner U, Tanrikulu Y, Breitung S, Scheffer U, et al. The concept of template-based de novo design from drug-derived molecular fragments and its application to TAR RNA. J Comput Aided Mol Des (2008) 22:59–68. doi: 10.1007/s10822-007-9157-4
21. Grisoni F, Merk D, Consonni V, Hiss JA, Tagliabue SG, Todeschini R, et al. Scaffold hopping from natural products to synthetic mimetics by holistic molecular similarity. Commun Chem (2018) 1:44. doi: 10.1038/s42004-018-0043-x
22. Yu Y, Xu T, Li J, Qiu Y, Rong Y, Gong Z, et al. A novel scalarized scaffold hopping algorithm with graph-based variational autoencoder for discovery of JAK1 inhibitors. ACS Omega (2021) 6:22945–54. doi: 10.1021/acsomega.1c03613
23. Krueger B, Dietrich A, Baringhaus K-H, Schneider G. Scaffold-hopping potential of fragment-based de novo design: the chances and limits of variation. CCHTS (2009) 12:383–96. doi: 10.2174/138620709788167971
24. Tan X, Li C, Yang R, Zhao S, Li F, Li X, et al. Discovery of pyrazolo[3,4-d]pyridazinone derivatives as selective DDR1 inhibitors via deep learning based design, synthesis, and biological evaluation. J Med Chem (2022) 65:103–19. doi: 10.1021/acs.jmedchem.1c01205
25. Zhu W, Liu X, Li Q, Gao F, Liu T, Chen X, et al. Discovery of novel and selective SIK2 inhibitors by the application of AlphaFold structures and generative models. Bioorganic Medicinal Chem (2023) 91:117414. doi: 10.1016/j.bmc.2023.117414
26. Erlanson DA. Introduction to fragment-based drug discovery. In: Davies TG, Hyvönen M, editors. Fragment-based drug discovery and X-ray crystallography. Berlin, Heidelberg: Springer Berlin Heidelberg (2011). p. 1–32. doi: 10.1007/128_2011_180
27. Turner LD, Trinh CH, Hubball RA, Orritt KM, Lin C-C, Burns JE, et al. From fragment to lead: de novo design and development toward a selective FGFR2 inhibitor. J Med Chem (2022) 65:1481–504. doi: 10.1021/acs.jmedchem.1c01163
28. Penner P, Martiny V, Bellmann L, Flachsenberg F, Gastreich M, Theret I, et al. FastGrow: on-the-fly growing and its application to DYRK1A. J Comput Aided Mol Des (2022) 36:639–51. doi: 10.1007/s10822-022-00469-y
29. Wills S, Sanchez-Garcia R, Dudgeon T, Roughley SD, Merritt A, Hubbard RE, et al. Fragment merging using a graph database samples different catalogue space than similarity search. J Chem Inf Model (2023) 63:3423–37. doi: 10.1021/acs.jcim.3c00276
30. Liu Y, Yang Z, Yu Z, Liu Z, Liu D, Lin H, et al. Generative artificial intelligence and its applications in materials science: Current situation and future perspectives. J Materiomics (2023) 9:798–816. doi: 10.1016/j.jmat.2023.05.001
31. Meyenburg C, Dolfus U, Briem H, Rarey M. Galileo: Three-dimensional searching in large combinatorial fragment spaces on the example of pharmacophores. J Comput Aided Mol Des (2022) 37:1–16. doi: 10.1007/s10822-022-00485-y
32. Ishitani R, Kataoka T, Rikimaru K. Molecular design method using a reversible tree representation of chemical compounds and deep reinforcement learning. J Chem Inf Model (2022) 62:4032–48. doi: 10.1021/acs.jcim.2c00366
33. Graff DE, Shakhnovich EI, Coley CW. Accelerating high-throughput virtual screening through molecular pool-based active learning. Chem Sci (2021) 12:7866–81. doi: 10.1039/d0sc06805e
34. Nigam A, Pollice R, Krenn M, Gomes G dos P, Aspuru-Guzik A. Beyond generative models: superfast traversal, optimization, novelty, exploration and discovery (STONED) algorithm for molecules using SELFIES. Chem Sci (2021) 12:7079–90. doi: 10.1039/d1sc00231g
35. Polishchuk P. CReM: chemically reasonable mutations framework for structure generation. J Cheminformatics (2020) 12:28. doi: 10.1186/s13321-020-00431-w
36. Imrie F, Bradley AR, van der Schaar M, Deane CM. Deep generative models for 3D linker design. J Chem Inf Model (2020) 60:1983–95. doi: 10.1021/acs.jcim.9b01120
37. Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol (2019) 37:1038–40. doi: 10.1038/s41587-019-0224-x
38. Jin W, Barzilay R, Jaakkola T. Junction tree variational autoencoder for molecular graph generation. [preprint]. (2019). doi: 10.48550/arXiv.1802.04364.
39. Brown N, McKay B, Gilardoni F, Gasteiger J. A graph-based genetic algorithm and its application to the multiobjective evolution of median molecules. J Chem Inf Comput Sci (2004) 44:1079–87. doi: 10.1021/ci034290p
40. Vinkers HM, de Jonge MR, Daeyaert FFD, Heeres J, Koymans LMH, van Lenthe JH, et al. SYNOPSIS: SYNthesize and OPtimize system in silico. J Med Chem (2003) 46:2765–73. doi: 10.1021/jm030809x
41. Daeyaert F, Deem MW. A pareto algorithm for efficient de novo design of multi-functional molecules. Mol Inf (2017) 36:1600044. doi: 10.1002/minf.201600044
42. Wiswesser WJ. 107 years of line-formula notations (1861-1968). J Chem Doc (1968) 8:146–50. doi: 10.1021/c160030a007
43. Weininger D. SMILES, a chemical language and information system. 1. Introduction to Method. encoding rules. J Chem Inf Comput Sci (1988) 28:31–6. doi: 10.1021/ci00057a005
44. O’Boyle N, Dalke A. DeepSMILES: an adaptation of SMILES for use in machine-learning of chemical structures. [preprint]. (2018), Chemistry. doi: 10.26434/chemrxiv.7097960.v1.
45. Krenn M, Häse F, Nigam A, Friederich P, Aspuru-Guzik A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach Learn: Sci Technol (2020) 1:045024. doi: 10.1088/2632-2153/aba947
46. Kimber TB, Chen Y, Volkamer A. Deep learning in virtual screening: recent applications and developments. IJMS (2021) 22:4435. doi: 10.3390/ijms22094435
47. Todeschini R, Consonni V. Handbook of molecular descriptors. 1st ed. Weinheim, Federal Republic of Germany: Wiley. (2000). doi: 10.1002/9783527613106
48. Cereto-Massagué A, Ojeda MJ, Valls C, Mulero M, Garcia-Vallvé S, Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods (2015) 71:58–63. doi: 10.1016/j.ymeth.2014.08.005
49. Mendez D, Gaulton A, Bento AP, Chambers J, De Veij M, Félix E, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res (2019) 47:D930–40. doi: 10.1093/nar/gky1075
50. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2023 update. Nucleic Acids Res (2023) 51:D1373–80. doi: 10.1093/nar/gkac956
51. Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res (2016) 44:D1045–53. doi: 10.1093/nar/gkv1072
52. Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res (2006) 34:D668–672. doi: 10.1093/nar/gkj067
53. Pence HE, Williams A. ChemSpider: an online chemical information resource. J Chem Educ (2010) 87:1123–4. doi: 10.1021/ed100697w
54. Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, Baker NC, et al. The CompTox Chemistry Dashboard: a community data resource for environmental chemistry. J Cheminform (2017) 9:61. doi: 10.1186/s13321-017-0247-6
55. Ruddigkeit L, van Deursen R, Blum LC, Reymond J-L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J Chem Inf Model (2012) 52:2864–75. doi: 10.1021/ci300415d
56. Ramakrishnan R, Dral PO, Rupp M, von Lilienfeld OA. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data (2014) 1:140022. doi: 10.1038/sdata.2014.22
57. Tingle BI, Tang KG, Castanon M, Gutierrez JJ, Khurelbaatar M, Dandarchuluun C, et al. ZINC-22─A free multi-billion-scale database of tangible compounds for ligand discovery. J Chem Inf Model (2023). doi: 10.26434/chemrxiv-2022-82czl
59. The UniProt Consortium, Bateman A, Martin M-J, Orchard S, Magrane M, Ahmad S, et al. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res (2023) 51:D523–31. doi: 10.1093/nar/gkac1052
60. Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, et al. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res (2024) 52(D1):D1465–D1477. doi: 10.1093/nar/gkad751
61. Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de-novo design through deep reinforcement learning. J Cheminform (2017) 9:48. doi: 10.1186/s13321-017-0235-x
62. Šícho M, Luukkonen S, Van Den Maagdenberg HW, Schoenmaker L, Béquignon OJM, Van Westen GJP. DrugEx: deep learning models and tools for exploration of drug-like chemical space. J Chem Inf Model (2023) 63:3629–3636. doi: 10.1021/acs.jcim.3c00434
63. Liu X, Ye K, Van Vlijmen HWT, Emmerich MTM, IJzerman AP, Van Westen GJP. DrugEx v2: de novo design of drug molecules by Pareto-based multi-objective reinforcement learning in polypharmacology. J Cheminform (2021) 13:85. doi: 10.1186/s13321-021-00561-9
64. Drotár P, Jamasb AR, Day B, Cangea C, Liò P. Structure-aware generation of drug-like molecules. [preprint]. (2021). doi: 10.48550/arXiv.2111.04107
65. Shen C, Krenn M, Eppel S, Aspuru-Guzik A. Deep molecular dreaming: inverse machine learning for de-novo molecular design and interpretability with surjective representations. Mach Learn: Sci Technol (2021) 2:11. doi: 10.1088/2632-2153/ac09d6
66. Sanchez-Lengeling B, Outeiral C, Guimaraes GL, Aspuru-Guzik A. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). [preprint]. (2017), Chemistry. doi: 10.26434/chemrxiv.5309668.v3
67. Putin E, Asadulaev A, Vanhaelen Q, Ivanenkov Y, Aladinskaya AV, Aliper A, et al. Adversarial threshold neural computer for molecular de novo design. Mol. Pharmaceutics (2018) 15:4386–97. doi: 10.1021/acs.molpharmaceut.7b01137
68. De Cao N, Kipf T. MolGAN: An implicit generative model for small molecular graphs. (2018). doi: 10.48550/arXiv.1805.11973
69. Maziarka Ł, Pocha A, Kaczmarczyk J, Rataj K, Danel T, Warchoł M. Mol-CycleGAN: a generative model for molecular optimization. J Cheminform (2020) 12:2. doi: 10.1186/s13321-019-0404-1
70. Jacobs I, Maragoudakis M. De novo drug design using artificial intelligence applied on SARS-CoV-2 viral proteins ASYNT-GAN. BioChem (2021) 1:36–48. doi: 10.3390/biochem1010004
71. Bai Q, Tan S, Xu T, Liu H, Huang J, Yao X. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Briefings Bioinf (2021) 22:bbaa161. doi: 10.1093/bib/bbaa161
72. Ahmad W, Simon E, Chithrananda S, Grand G, Ramsundar B. ChemBERTa-2: towards chemical foundation models (2022). Available at: http://arxiv.org/abs/2209.01712 (Accessed September 16, 2023).
73. He J, You H, Sandström E, Nittinger E, Bjerrum EJ, Tyrchan C, et al. Molecular optimization by capturing chemist’s intuition using deep neural networks. J Cheminform (2021) 13:26. doi: 10.1186/s13321-021-00497-0
74. Tysinger EP, Rai BK, Sinitskiy AV. Can we quickly learn to “Translate” Bioactive molecules with transformer models? J Chem Inf Model (2023) 63:1734–44. doi: 10.1021/acs.jcim.2c01618
75. Degen J, Wegscheid-Gerlach C, Zaliani A, Rarey M. On the art of compiling and using “Drug-like” Chemical fragment spaces. ChemMedChem (2008) 3:1503–7. doi: 10.1002/cmdc.200800178
76. Lewell XQ, Judd DB, Watson SP, Hann MM. RECAP retrosynthetic combinatorial analysis procedure: A powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J Chem Inf Comput Sci (1998) 38:511–22. doi: 10.1021/ci970429i
77. Cramer RD, Soltanshahi F, Jilek R, Campbell B. AllChem: generating and searching 1020 synthetically accessible structures. J Comput Aided Mol Des (2007) 21:341–50. doi: 10.1007/s10822-006-9093-8
78. Hartenfeller M, Zettl H, Walter M, Rupp M, Reisen F, Proschak E, et al. DOGS: Reaction-Driven de novo Design of Bioactive Compounds. PloS Comput Biol (2012) 8:e1002380. doi: 10.1371/journal.pcbi.1002380
79. Zabolotna Y, Volochnyuk DM, Ryabukhin SV, Gavrylenko K, Horvath D, Klimchuk O, et al. SynthI: A new open-source tool for synthon-based library design. J Chem Inf Model (2022) 62:2151–63. doi: 10.1021/acs.jcim.1c00754
80. Hoffmann T, Gastreich M. The next level in chemical space navigation: going far beyond enumerable compound libraries. Drug Discovery Today (2019) 24:1148–56. doi: 10.1016/j.drudis.2019.02.013
81. Alnammi M, Liu S, Ericksen SS, Ananiev GE, Voter AF, Guo S, et al. Evaluating scalable supervised learning for synthesize-on-demand chemical libraries. J Chem Inf Model (2023) 63(17):5513–28. doi: 10.1021/acs.jcim.3c00912
82. Leach AG, Jones HD, Cosgrove DA, Kenny PW, Ruston L, MacFaul P, et al. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure. J Med Chem (2006) 49:6672–82. doi: 10.1021/jm0605233
83. Yang Z, Shi S, Fu L, Lu A, Hou T, Cao D. Matched molecular pair analysis in drug discovery: methods and recent applications. J Med Chem (2023) 66:4361–77. doi: 10.1021/acs.jmedchem.2c01787
84. Cucurull-Sanchez L. Successful identification of key chemical structure modifications that lead to improved ADME profiles. J Comput Aided Mol Des (2010) 24:449–58. doi: 10.1007/s10822-010-9361-5
85. Dossetter AG, Douglas A, O’Donnell C. A matched molecular pair analysis of in vitro human microsomal metabolic stability measurements for heterocyclic replacements of di-substituted benzene containing compounds – identification of those isosteres more likely to have beneficial effects. Med Chem Commun (2012) 3:1164. doi: 10.1039/c2md20155k
86. Jensen JH. A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space. Chem Sci (2019) 10:3567–72. doi: 10.1039/c8sc05372c
87. Leguy J, Cauchy T, Glavatskikh M, Duval B, Da Mota B. EvoMol: a flexible and interpretable evolutionary algorithm for unbiased de novo molecular generation. J Cheminform (2020) 12:55. doi: 10.1186/s13321-020-00458-z
88. Yu J, Wang D, Zheng M. Uncertainty quantification: Can we trust artificial intelligence in drug discovery? iScience (2022) 25:104814. doi: 10.1016/j.isci.2022.104814
89. Van Tilborg D, Grisoni F. Traversing chemical space with active deep learning. [preprint] Chem (2023). doi: 10.26434/chemrxiv-2023-wgl32
90. Reker D. Practical considerations for active machine learning in drug discovery. Drug Discovery Today: Technol (2019) 32-33:73–9. doi: 10.1016/j.ddtec.2020.06.001
91. Reker D, Schneider G. Active-learning strategies in computer-assisted drug discovery. Drug Discovery Today (2015) 20:458–65. doi: 10.1016/j.drudis.2014.12.004
92. Lipinski CA. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev (2016) 101:34–41. doi: 10.1016/j.addr.2016.04.029
93. Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem (2012) 4:90–8. doi: 10.1038/nchem.1243
94. Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem (2010) 53:2719–40. doi: 10.1021/jm901137j
95. Brenk R, Schipani A, James D, Krasowski A, Gilbert I, Frearson J, et al. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem (2008) 3:435–44. doi: 10.1002/cmdc.200700139
96. Ertl P, Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform (2009) 1:8. doi: 10.1186/1758-2946-1-8
97. Gao W, Coley CW. The synthesizability of molecules proposed by generative models. J Chem Inf Model (2020) 60:5714–23. doi: 10.1021/acs.jcim.0c00174
98. Coley CW, Thomas DA, Lummiss JAM, Jaworski JN, Breen CP, Schultz V, et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science (2019) 365:eaax1566. doi: 10.1126/science.aax1566
99. Genheden S, Thakkar A, Chadimová V, Reymond J-L, Engkvist O, Bjerrum E. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J Cheminform (2020) 12:70. doi: 10.1186/s13321-020-00472-1
100. Yang Z-Y, Yang Z-J, He J-H, Lu A-P, Liu S, Hou T-J, et al. Benchmarking the mechanisms of frequent hitters: limitation of PAINS alerts. Drug Discovery Today (2021) 26:1353–8. doi: 10.1016/j.drudis.2021.02.003
101. Toropov AA, Toropova AP. QSPR/QSAR: state-of-art, weirdness, the future. Molecules (2020) 25:1292. doi: 10.3390/molecules25061292
102. Ballabio D, Grisoni F, Consonni V, Todeschini R. Integrated QSAR models to predict acute oral systemic toxicity. Mol. Inf (2019) 38:1800124. doi: 10.1002/minf.201800124
103. Palmeira A, Rodrigues F, Sousa E, Pinto M, Vasconcelos MH, Fernandes MX. New uses for old drugs: pharmacophore-based screening for the discovery of P-glycoprotein inhibitors: pharmacophore-based screening for the discovery of P-glycoprotein inhibitors. Chem. Biol. Drug Design (2011) 78:57–72. doi: 10.1111/j.1747-0285.2011.01089.x
104. Mousa LA, Hatmal MM, Taha M. Exploiting activity cliffs for building pharmacophore models and comparison with other pharmacophore generation methods: sphingosine kinase 1 as case study. J Comput Aided Mol Des (2022) 36:39–62. doi: 10.1007/s10822-021-00435-0
105. Riniker S, Landrum GA. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J Cheminform (2013) 5:26. doi: 10.1186/1758-2946-5-26
106. Kuhlman B, Bradley P. Advances in protein structure prediction and design. Nat Rev Mol Cell Biol (2019) 20:681–97. doi: 10.1038/s41580-019-0163-x
107. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
108. Caballero J. The latest automated docking technologies for novel drug discovery. Expert Opin. Drug Discov (2021) 16:625–45. doi: 10.1080/17460441.2021.1858793
109. Desaphy J, Raimbaud E, Ducrot P, Rognan D. Encoding protein–ligand interaction patterns in fingerprints and graphs. J Chem Inf Model (2013) 53:623–37. doi: 10.1021/ci300566n
110. Renner S, Derksen S, Radestock S, Mörchen F. Maximum common binding modes (MCBM): Consensus docking scoring using multiple ligand information and interaction fingerprints. J Chem Inf Model (2008) 48:319–32. doi: 10.1021/ci7003626
111. Yasuo N, Sekijima M. Improved method of structure-based virtual screening via interaction-energy-based learning. J Chem Inf Model (2019) 59:1050–61. doi: 10.1021/acs.jcim.8b00673
112. Braun E, Gilmer J, Mayes HB, Mobley DL, Monroe JI, Prasad S, et al. Best practices for foundations in molecular simulations [Article v1.0]. Living J. Comput. Mol. Sci (2019) 1:5957–7. doi: 10.33011/livecoms.1.1.5957
113. Wade AD, Bhati AP, Wan S, Coveney PV. Alchemical free energy estimators and molecular dynamics engines: accuracy, precision, and reproducibility. J Chem Theory Comput (2022) 18:3972–87. doi: 10.1021/acs.jctc.2c00114
114. Blay V, Tolani B, Ho SP, Arkin MR. High-Throughput Screening: today’s biochemical and cell-based approaches. Drug Discovery Today (2020) 25:1807–21. doi: 10.1016/j.drudis.2020.07.024
115. Renaud J-P, Chung C, Danielson UH, Egner U, Hennig M, Hubbard RE, et al. Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discov (2016) 15:679–98. doi: 10.1038/nrd.2016.123
116. Brown N, Fiscato M, Segler MHS, Vaucher AC. GuacaMol: benchmarking models for de novo molecular design. J Chem Inf Model (2019) 59:1096–108. doi: 10.1021/acs.jcim.8b00839
117. Preuer K, Renz P, Unterthiner T, Hochreiter S, Klambauer G. Fréchet chemNet distance: A metric for generative models for molecules in drug discovery. J Chem Inf Model (2018) 58:1736–41. doi: 10.1021/acs.jcim.8b00234
118. Woodward DJ, Bradley AR, van Hoorn WP. Coverage score: A model agnostic method to efficiently explore chemical space. J Chem Inf Model (2022) 62(12):4391–402. doi: 10.1021/acs.jcim.2c00258
119. Xie Y, Xu Z, Ma J, Mei Q. How much space has been explored? Measuring the chemical space covered by databases and machine-generated molecules. [preprint]. (2023). doi: 10.48550/arXiv.2112.12542.
120. Polykovskiy D, Zhebrak A, Sanchez-Lengeling B, Golovanov S, Tatanov O, Belyaev S, et al. Molecular sets (MOSES): A benchmarking platform for molecular generation models. Front Pharmacol (2020) 0. doi: 10.3389/fphar.2020.565644
121. Cieplinski T, Danel T, Podlewska S, Jastrzebski S. We Should at Least Be Able to Design Molecules That Dock Well (2021). Available at: http://arxiv.org/abs/2006.16955 (Accessed July 8, 2022).
122. Ciepliński T, Danel T, Podlewska S, Jastrzbski S. Generative models should at least be able to design molecules that dock well: A new benchmark. J Chem Inf Model (2023) 63(11):3238–47. doi: 10.1021/acs.jcim.2c01355
123. Merk D, Friedrich L, Grisoni F, Schneider G. De novo design of bioactive small molecules by artificial intelligence. Mol. Inf (2018) 37:1700153. doi: 10.1002/minf.201700153
124. Stanley M, Segler M. Fake it until you make it? Generative de novo design and virtual screening of synthesizable molecules. Curr Opin Struct Biol (2023) 82:102658. doi: 10.1016/j.sbi.2023.102658
125. Jang SH, Sivakumar D, Mudedla SK, Choi J, Lee S, Jeon M, et al. PCW-A1001, AI-assisted de novo design approach to design a selective inhibitor for FLT-3(D835Y) in acute myeloid leukemia. Front Mol Biosci (2022) 9:1072028. doi: 10.3389/fmolb.2022.1072028
126. Lee S, Jo J, Hwang SJ. Exploring chemical space with score-based out-of-distribution generation. [preprint]. (2022). doi: 10.48550/arXiv.2206.07632
127. Ivanenkov YA, Polykovskiy D, Bezrukov D, Zagribelnyy B, Aladinskiy V, Kamya P, et al. Chemistry42: an AI-driven platform for molecular design and optimization. J Chem Inf Model (2023) 63:695–701. doi: 10.1021/acs.jcim.2c01191
Keywords: de novo, drug design, artificial intelligence, cheminformatics, machine learning
Citation: Crucitti D, Pérez Míguez C, Díaz Arias JÁ, Fernandez Prada DB and Mosquera Orgueira A (2024) De novo drug design through artificial intelligence: an introduction. Front. Hematol. 3:1305741. doi: 10.3389/frhem.2024.1305741
Received: 02 October 2023; Accepted: 05 January 2024;
Published: 25 January 2024.
Edited by:
Shuaiying Cui, Boston University, United StatesReviewed by:
Dragos Horvath, UMR7140 Chimie de la Matière Complexe, FranceCopyright © 2024 Crucitti, Pérez Míguez, Díaz Arias, Fernandez Prada and Mosquera Orgueira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Crucitti, ZGF2aWRlLmNydWNpdHRpQHNlcmdhcy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.