 Eleni Besiridou1
Eleni Besiridou1 Osheen Bhatt1Andrea Yurukova1
Osheen Bhatt1Andrea Yurukova1 Anshupa Sahu2
Anshupa Sahu2 Sarah Kinkley2
Sarah Kinkley2 Victoria Fischer1
Victoria Fischer1 Ioanna Triviai1,2*
Ioanna Triviai1,2*- 1Stem Cell Biology Research Group, Department for Stem Cell Transplantation, University Medical Center Hamburg – Eppendorf, Hamburg, Germany
- 2Department of Computational Biology, Max Planck Institute for Molecular Genetics, Berlin, Germany
In this study, we report an unprecedented case of graft failure following hematopoietic stem cell transplantation (HSCT) in a patient with a myeloproliferative neoplasm (MPN) who had achieved bone marrow (BM) remission. We outline the intricate cellular components of splenic hematopoiesis, which are derived from both the recipient and the donor, underpinning the persistence of fibrosis. Our investigation characterizes the distinct cellular populations that play a pivotal role in graft exhaustion and introduces an unusual hypothesis regarding the perpetuation of a malignant niche in extramedullary sites within the context of HSCT.
Introduction
Extramedullary hematopoiesis (EMH) is widely considered to be the main contributor to the development of splenomegaly in myeloproliferative neoplasms (MPN). The analysis of spleens from MPN patients revealed that EMH is characterized by the presence of large clusters of atypical megakaryocytes, which are accompanied by increased numbers of erythroid and myeloid cells, leading to the effacement of the red pulp (1). Regardless of their mutational profile, which is often reported to differ from that of the BM, there is a direct correlation between splenic aberrant megakaryocyte proliferation and the development of fibrosis in the spleen (1). The fibrotic tissue in the spleens of MPN patients was observed to contain larger numbers of smooth muscle alpha-actin positive (αSMA+) myofibroblasts and reticulocytes, which are the primary contributors to collagen deposition (2), than that of the spleens of healthy participants (3).
HSCT represents the only curative approach for MPN by eliminating the neoplastic clones and resolving fibrosis. A successful HSCT results in a complete histological response in the BM and the normalization of the spleen size. HSCT has been shown to effectively decrease or eradicate EMH in the spleen, as evidenced by the clearance of the megakaryocyte clusters. Although the number of αSMA+ cells remains unchanged after HSCT, there is a gradual subsidence of splenic fibrosis (3).
In this study, we present an intriguing case of deteriorating hypersplenism despite achieving complete disease remission in the BM after HSCT. Our objective was to elucidate the spleen enlargement by investigating splenic function as a repository of residual disease clones and a niche for donor cells and also to identify the potential cellular interactions related to hypersplenism and graft failure.
Case description
We report the case of a 65-year-old male patient diagnosed with primary myelofibrosis (PMF)-related secondary acute myeloid leukemia (sAML). At the age of 29 years, the patient suffered a lung arterial embolism after an accident. The peripheral blood (PB) count showed thrombocythemia of 1,299/nL, and the consecutive BM biopsy revealed an essential thrombocythemia phenotype. In 2013, the patient presented with constitutional symptoms, splenomegaly, and anemia. The diagnosis was revised to primary myelofibrosis (PMF) with a CALR-K385fs* mutation and a high-risk Dynamic International Prognostic Scoring System (DIPSS) score. The initiated treatment with ruxolitinib followed by momelotinib and histone-deacetylase inhibitors did not show a sufficient effect in terms of the reduction of splenomegaly and the symptom response. In 2017, the patient developed a post-PMF sAML and was referred to the University Medical Center Hamburg-Eppendorf, where he underwent a mismatched unrelated donor allogeneic HSCT (MMUD alloHSCT) on 30 June 2017. At the time of the HSCT, the patient was highly dependent on transfusions (i.e., they were receiving eight units of packed red blood cells per month for 1 year), and their spleen was palpable at eight fingers below the ribcage. The leukocyte engraftment was detected on day 12 post-transplantation. Disease-complete remission (CR) was achieved at 30 days, with sustained confirmation 6 months post-HSCT. After engraftment, the patient was diagnosed with acute graft-versus-host disease (GvHD) of the skin and gut and later with a cytomegalovirus (CMV) infection, and was treated with valganciclovir and foscarnet. Under the antiviral treatment, he developed pancytopenia and, again, became transfusion-dependent. In September 2017, the patient’s spleen was palpable at the level of the umbilicus. In December 2017, they were diagnosed with chronic GvHD of the gastrointestinal tract. Progressive bicytopenia (erythrocytic and megakaryocytic lineages) due to hypersplenism was observed with high transfusion dependency and consecutive siderosis of internal organs. A splenectomy, performed on 30 June 2018, led to a rapid recovery of the blood platelet count, with it reaching 100/nL–150/nL within 1 week. A donor blood chimerism of 99.9% was documented on 15 November 2018, with a normalized blood profile reported on 2 January 2019 (considered the date of maximal response). Unfortunately, the patient died on 20 February 2019 because of an infection due to the immunosuppression for chronic GvHD (disseminated herpes zoster). A schematic diagram of the patient’s history is depicted in Figure 1A.
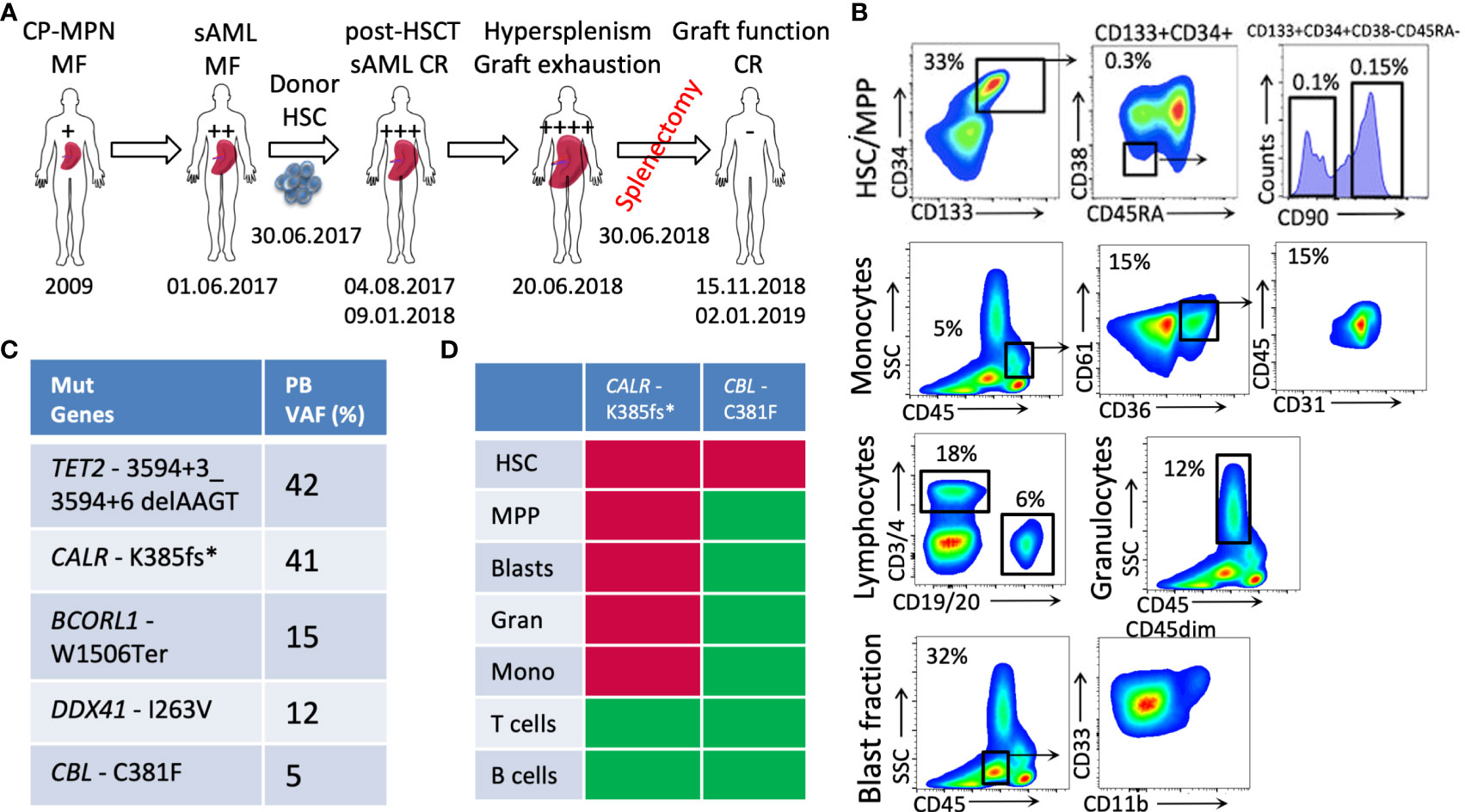
Figure 1 Patient case presentation and disease description. (A) A schematic diagram of the patient’s medical history; (B) the phenotypic characterization of PB cells (HSCs: CD133+CD34+CD90+; blasts: CD45dimCD33+ and/or CD11b+; monocytes: CD45+CD61+CD36+CD31+, lymphocytes, and granulocytes) at the sAML stage before HSCT; (C) the genetic lesions determined in the patient’s PB at the stage of sAML using targeted exon sequencing; (D) the genotyping of PB fractions—red: mutated; green: non-mutated (CP-MPN, chronic phase MPN; sAML, secondary AML; HSCT, hematopoietic stem cell transplantation; CR, complete remission; HSCs, hematopoietic stem cells; PB, peripheral blood; VAF, variant allele frequency).
Diagnostic assessment
We performed an extensive analysis of the patient’s PB before (at the sAML stage) and in conjunction with the splenic tissue after HSCT (at graft exhaustion) (Figures 1B–D, 2). The exon-targeted sequencing of PB DNA for 54 myeloid malignancy-related genes to reveal the mutational profile of circulating cells was performed as previously published (4). A germline single-nucleotide polymorphism (SNP) in the TET2 gene was also detected in the patient at the time of the HSCT, which was then used for chimerism analysis (Sanger sequencing). In addition, common SNP variations were detected in the donor (the EIF2S2 gene, through PCR), as shown in Figure 2E.
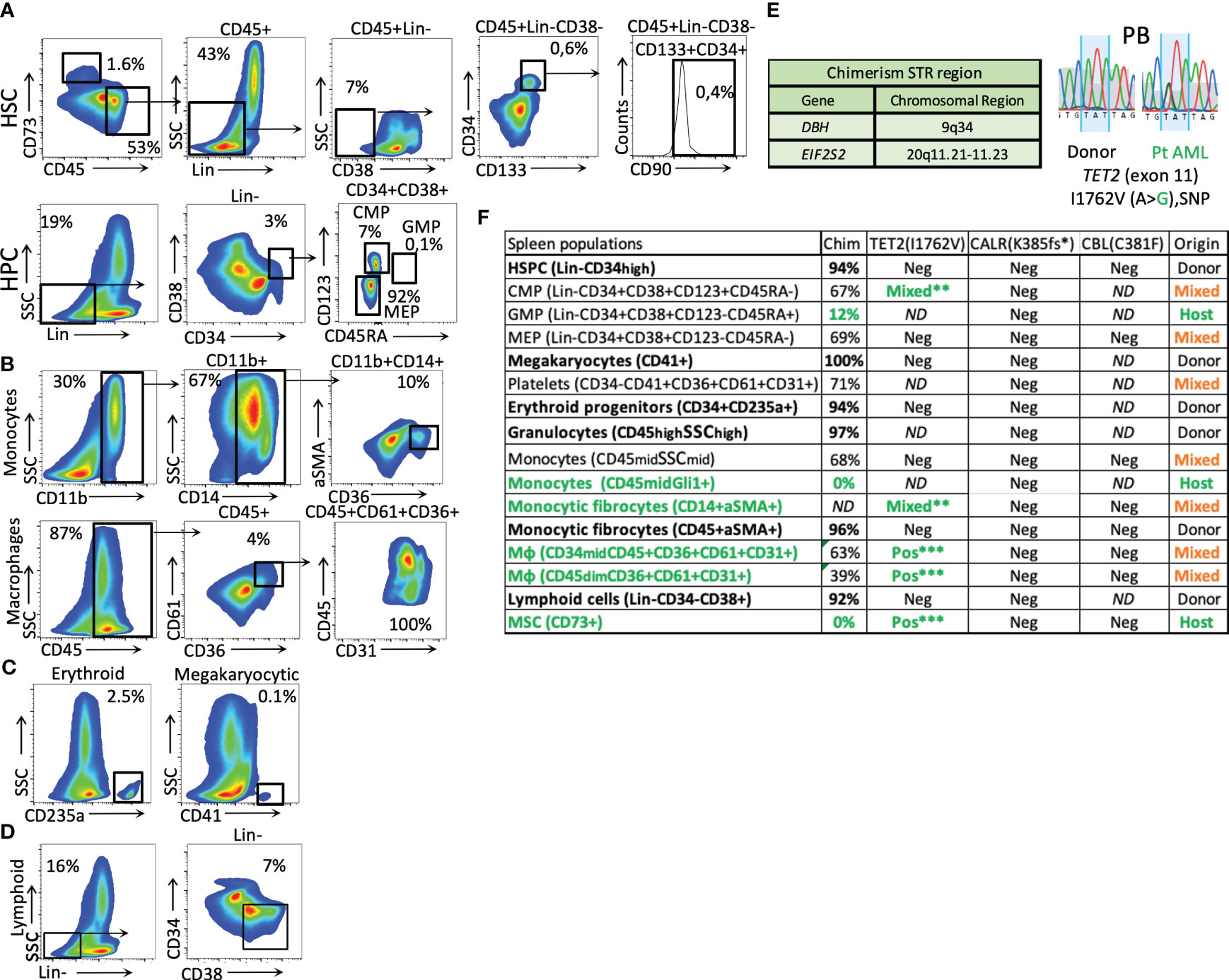
Figure 2 Phenotypical splenic cell characterization and genotyping after HSCT. (A) splenic HSCs (Lin-CD45+CD73−CD38−CD133+CD34+CD90+) and mesenchymal origin cells (CD73+) (the percentage related to all events). (B) Monocytic and macrophage fractions. (C) Erythroid and megakaryocytic precursors (from the CD34+ fraction). (D) Lymphoid cells (Lin-CD34−CD38+). (E) STR regions were used for chimerism analysis and patient TET2 gene-specific germline SNPs were used to identify host-origin hematopoietic cells in the splenic tissue. (F) Table of cell fraction origin in the patient’s spleen, as defined by chimerism and SNP detection methods. The green color represents host-derived cells, the bold font represents the donor origin, and the orange color represents the mixed chimerism populations. The analysis of mutated CARL and CBL genes is included. (The stars mark the intensity of the patient germline SNP detection signal by Sanger sequencing. (**, mixed alleles, ***heterozygous, 50% VAF); Mφ, macrophages.
A large portion of the acquired splenic tissue from the splenectomy operation was used for fibrosis staining, while another portion was subjected to tissue dissociation/cell sorting for further analyses. The hematopoietic and mesenchymal origin cells were phenotypically characterized using fluorescence-activated cell sorting (FACS) and sorted to determine their mutational burden, chimerism status, and global gene expression. Both the PB- and splenic-sorted populations were genotyped for the genetic lesions identified at the sAML stage using Sanger sequencing. The splenic cell fractions were analyzed for their donor/host origin by chimerism, as described above. The transcriptional profiles of the sorted fractions were obtained using whole-cytoplasmic-RNA sequencing.
A comparative analysis of the transcriptomic data pertaining to the BM and mobilized blood (mob blood) hematopoietic stem cells (HSCs) from the control healthy donors (four donors and seven donors, respectively) (Figures 3A, B) was performed using the Bioconductor R package DESeq2 (5). Pathway enrichment analysis for differentially expressed genes (adjusted p-value < 0.05) was performed using the R package pathfindR (6). The fibrosis-related cells were analyzed for fibrotic genes using quantitative PCR (qPCR) and their expression relative to each other (sorted splenic fractions), and a control culture of BM-derived expanded mesenchymal stromal cells (MSCs) stimulated towards fibrogenesis was compared (Figures 4C–E).
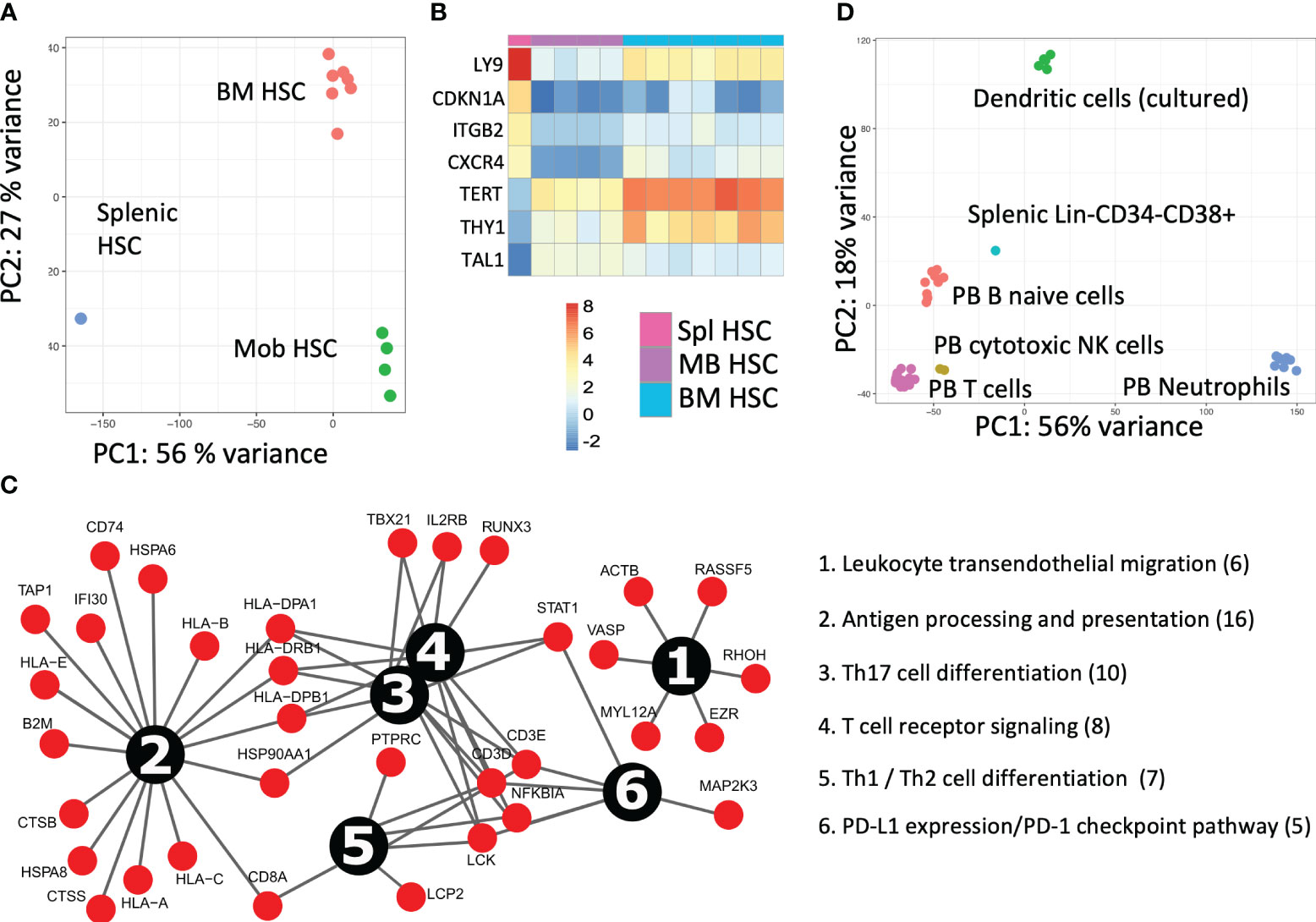
Figure 3 Splenic cell fraction transcriptomic characterization after HSCT (A) The PCA analysis for the transcriptomic comparison of patient splenic HSCs with their bone marrow (BM) (n = 7) and mobilized blood (mob blood) (n = 4) of the control donors (own data). (B) Expression analysis of primary deregulated genes and (C) pathway analysis of upregulated genes in splenic HSCs. (D) The transcriptomic analysis of the dominant lymphoid donor origin population in the spleen (Lin-CD34−CD38+). The PCA comparative analysis of the splenic lymphoid graft population to PB B-naive cells (n = 9), T cells (n = 14), cytotoxic NK cells (n = 2), neutrophils (n = 10), and dendritic cells (n = 6) from the control donors (Blueprint Consortium data).
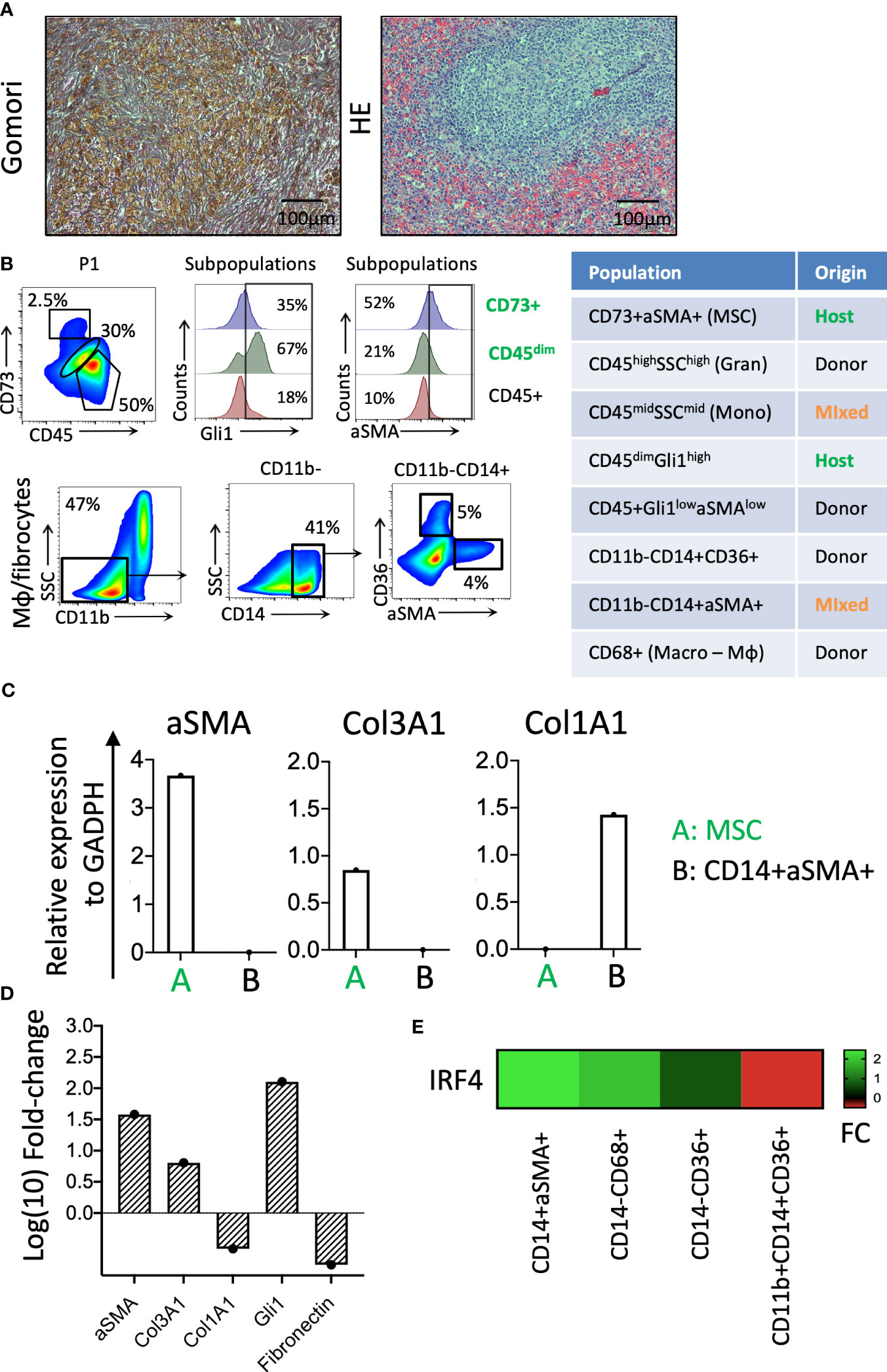
Figure 4 Identification of cellular components of splenic fibrosis. (A) Gömöri silver stain reveals dense reticulin fibers and focal collagen deposition in the splenic tissue. Hematoxylin and eosin staining indicate the myeloid cell proliferation loci in the splenic tissue. (B) The phenotypic characterization and chimerism of mesenchymal and hematopoietic-origin fibrocytes. (C) The relative expression of the fibrosis-related genes in the patient-origin MSC and donor-derived fibrocyte populations of the spleen. (D) The relative fibrotic gene expression levels of fibrotic spleen patient MSCs compared with a control BM donor TFGβ/FGF-fibrinogen-induced MSC culture. (E) The IRF4 expression levels in fibrocytes (CD14+αSMA+), Mφ (CD14−CD68+), erythroid cells (CD14−CD36+), and monocytes (CD11b+CD14+CD36+) of the splenic tissue (FC, fold change).
Results
The immunophenotyping analysis of patient PB at the sAML stage (before undergoing HSCT) showed increased numbers of circulating Lin-CD133+CD34+CD90+ neoplastic HSCs that are related to disease initiation and transformation (33% of PBMCs), as previously published (Figure 1B) (4, 7). The PB blasts determined to be CD45dim cells were identified as CD33+ and/or CD11b+ cells (32% of PBMCs), and the monocytic circulating cells were identified as CD45highSSClow(CD36+CD61+CD31+) cells (5% of PBMCs) (Figure 1B).
The genotyping of the aforementioned sorted fractions, as depicted with respective allelic frequencies in Figure 1C, indicates that sAML myelopoiesis is dominated by CALR-K385fs*-mutated clones originating from the HSCs (Lin-CD133+CD34+CD90+) fraction. Within this circulating HSC population, additional CBL-C381F mutated clones were detected, whereas no such mutation was detected in the other circulating cells of myeloid or lymphoid lineages.
After HSCT, the hematopoiesis in the enlarged spleen (as described in Figures 2A–D) was primarily of donor origin (Figure 2F). The donor-derived populations include HSPCs, progenitors of the erythroid and megakaryocytic lineages, and innate immune cells involved in the initial stage of immune reconstitution, such as granulocytes and early lymphoid progenitors. Interestingly, a donor-derived fibrocyte monocytic population expressing CD45/αSMA was also observed (Figures 2B, F).
The splenic HSCPs (Lin-CD38−CD133+CD34+CD90+/−, 0.4%) are exclusively of donor origin (Figures 2A, F). Their transcriptomic signature differs from that of mob blood and BM HSCs, as shown in the PCA analysis of Figure 3A. The donor-derived splenic HSCs exhibited an upregulation in the cell cycle- (CDKN1A) and homing-related (ITGB2 and CXCR4) genes in comparison with their BM or mob blood counterparts. In contrast, they exhibited a significant downregulation in the HSC self-renewal-related TERT gene and the Thy1 stem cell marker gene in comparison with the BM HSCs (Figure 3B). The signaling pathway analysis of the upregulated gene clusters of splenic HSCs in comparison with their control BM counterparts pointed to the presence of trans-endothelial migration-related genes (six genes) and the occurrence of commitment to antigen-presenting cell (16 genes)/T17 helper cell differentiation (10 genes) (Figure 3C).
Principal component analysis (PCA) of the transcriptome profiles of the dominant graft-derived lymphoid population (Lin-CD34−CD38+) revealed that donor hematopoiesis in the patient spleen likely involves cells that exhibit similarities to PB-naive B cells. This finding became apparent through the comparison of the transcriptomic data to the PB control donor cell fractions available from the Blueprint Consortium (Figure 3D).
The residual patient hematopoiesis in the spleen (determined primarily by low donor chimerism and the detection of germline patient SNPs in the TET2 gene) is driven by common myeloid progenitors (CMPs) and predominantly includes monocytic cells (Figure 2F) found expressing high levels of the Gli1 gene (CD45dimGli1high; Figure 4B). The detected host-dominant granulocyte–monocyte progenitor (GMP) population and host-derived monocytic cells (Figures 2F, 4B) do not have any mutations of the sAML stage before HSCT.
The fibrosis of the spleen was determined by increased thick reticulin fiber deposition in the red pulp filled with diffused Gamna–Gandy bodies (as visualized via Gömöri staining; Figure 4A). The chimerism analysis of the mesenchymal origin CD73+/αSMA+ myofibroblasts and CD45+/CD14+/αSMA+ monocytic origin myofibroblasts indicated they were of host and donor origin, respectively (Figure 4B). The qPCR for fibrosis-related genes showed that these cells are responsible for the production of different types of collagen, with the mesenchymal host-derived ones producing primarily Col3A1 and the monocytic fibrocytes from the donor Col1A1 (Figure 4C). The mesenchymal CD73+ cells of the patient’s spleen expressed significantly higher levels of fibrosis-related genes (i.e., αSMA, Col3A1, and Gli1) than the control BM MSCs expanded with fibrinogenic factors in vitro (Figure 4D). The donor-derived fibrocytes (CD14+αSMA+) expressed very high levels of the IRF4 gene, a regulator of M2 macrophage differentiation responsible for resolving inflammation and new extracellular matrix production (8) (Figure 4E).
Discussion
In the case of the patient included in our study, hypersplenism appears to be the result of extensive EMH and fibrosis. The EMH primarily consisted of graft cells and the residual myelopoiesis from the patient. Splenic fibrosis is the component of persistent resident myofibroblast activation due to local inflammation and insufficient donor cell fibrocyte conversion toward the physiological wound healing remodeling phase.
The residual disease in the spleen of our patient consisted of myeloid progenitors and Gli1-expressing monocytes, indicating that the Hedgehog pathway plays an important role in the splenic survival of residual clones in MPN, such as in Gli1+ dormant leukemic cells in AML (9). A lack of previously detected genetic lesions of BM cells in the spleen of MPN patients has been reported before (1). In our HSCT case, this might be a result of malignant clone clearance in extramedullary sites or implies that splenic niches provide a residence for different disease-related clones than the BM. Given that stromal survival signals maintain myeloid malignancies by activating the Hedgehog pathway, it can be inferred that the fibrotic spleen creates a microenvironment that promotes distinct clonal dynamics compared with those seen in the BM.
In addition, we identified activated macrophages of host origin within the splenic macrophage population, which express CD31 and play a crucial role in regulating iron metabolism and erythrocyte destruction. Previous studies have reported that these phagocytic cells reside within the splenic tissue before birth and are distinct from the circulating monocytes (10). This finding may account for the detection of germline patient SNPs in splenic host macrophages and of mutations specifically related to the disease not being detected.
The spleen typically functions as a reservoir for the monocytes that are dispatched to the sites of inflammation (11), thus providing a plausible explanation for the presence of donor-derived monocytes that reside in this organ in circulation. The fate of monocytes is determined by a complex interplay of environmental signals that modulate transcriptional regulation, enabling these cells to adapt to the specific secretome of different tissues (12). Donor-derived monocytes are expected to mount an alloreactive response toward the site of inflammation, generating phagocytes that aid in the clearance of the spleen. Remarkably, a subset of the donor-derived monocytic population that engrafts in the spleen transforms into fibrocytes (CD14+αSMA+Col1A1+). Such αSMA-expressing monocytes–macrophages have been reported to preserve primitive hematopoietic cells in the BM (13). The existence of these cells in the patient’s spleen also suggests that they played a role in promoting the engraftment of donor-derived HSCs and in supporting the peripheral survival of host-residual clones, in turn sustaining a self-perpetuating cycle of tissue inflammation and exacerbating organ enlargement.
The migration of donor HSCs to the spleen appears to be governed by the CXCR4/CXCL12 axis, similar to HSC homing in the BM (14), as evidenced by the elevated levels of expression of CXCR4 in splenic HSCs relative to those in the BM and mob blood (Figure 3B). The distinctive tissue-related transcriptomic signatures of donor HSCs found in the spleen suggested that they have a limited self-renewal capacity and a propensity for innate immunity/regulatory T-cell development, which contrasts with earlier research on splenic HSCs/LSCs (15). Given that Th17 cells primarily regulate the production of neutrophils and phagocytes as the early drivers of the graft vs. leukemia effect, it was expected that the priming of donor progenitors toward this effect would also occur in the context of HSCT in the spleen. Their relatively immature development during the initial year of immune reconstitution, however, more likely provoked an inflammatory reaction from innate immune cells, thus promoting myofibroblast proliferation rather than facilitating the alloreactive response and clearance of fibrosis.
During later HSCT stages, it has been hypothesized that adapted immune cells, such as the naive B cells identified here (Lin-CD34−CD38+), will emerge from the graft. It has also been hypothesized that these cells would attempt to restructure the white pulp by expanding in the plasmablast residence, the spleen (16).
Surprisingly, in addition to the BM, the spleen seems to serve as a site for the megakaryopoiesis and erythropoiesis of donor cells. The observation of donor megakaryocytes in a patient with a complete histologic response in the BM achieved after transplant is a curious finding, given that megakaryopoiesis in the spleen is typically restricted to patients with MPN and is eliminated during the process of graft clearance in a successful HSCT (3). It seems that the splenic microenvironment played a crucial role in determining the developmental trajectories of donor progenitor cells in the presented HSCT case, leading them to exhibit EMH phenotypes that resemble the early stages of MPN progression and fibrosis development in the BM.
Megakaryopoiesis in the spleen has been reported in cases of sepsis due to MEP progenitors egressing from the BM (17). Splenic megakaryocytes are tissue-imprinted and produce platelets exhibiting an immune regulatory function through their antigen presentation and neutrophil activation (17). These findings shed light on the immunomodulatory role of donor platelets produced in the spleen in our HSCT case. More specifically, active donor megakaryopoiesis and the presence of a mixed genotype of platelets in the splenic tissue could enhance red pulp macrophage activation of both host and donor origin. It has been reported that the resident splenic macrophages help to clear IgG-opsonized platelets in cases of immune thrombocytopenic purpura (ITP) (18). In ITP patients, the loss of immunological tolerance to autoantigens due to an imbalance of the Th1-to-Th2 cell ratio and dysfunctional B-cell autoantibody production against platelets in the spleen are the primary components of ITP pathology (19). Splenectomy can be an effective last-resort treatment option for these patients (18).
In the context of immune reconstitution following HSCT, it appears that we are witnessing a comparable phenomenon to ITP, which is caused by malfunctioning adaptive immune cells within the spleen. Their presence could constantly impair donor megakaryopoiesis in both the spleen and BM, ultimately resulting in graft exhaustion.
Compromised megakaryopoiesis and malfunctioning platelets within the spleen perpetuate a vicious cycle of fibrosis development, involving not only mesenchymal cells of host origin but also a distinct population of fibrocytes derived from the donor. The latter cells appear to have acquired the characteristics of M2 macrophages and could be responsible for malignant clone survival in the spleen either due to their anti-inflammatory and immunosuppressive functions as tumor-associated macrophages (TAMs) (20, 21) or as wound-healing M2 macrophages (22).
Our analysis was constrained to an in-depth profiling of the isolated splenic cells obtained from the fresh tissue of a single patient. Unfortunately, the precise nature of cellular interactions could not be fully elucidated within the scope of this study. To gain deeper insights into the intricacies of cellular interactions, additional spatial transcriptomic methodologies are warranted. This could be achieved by investigating the cellular topology in preserved spleen samples from more extensive cohorts of splenectomized MPN patients, which in turn would enable a broader perspective on this subject.
In this report, we present a compelling case of splenomegaly following a successful HSCT. Through our investigation, we have discovered that there are distinct donor-origin populations that directly and indirectly promote spleen enlargement, in addition to two types of fibrocytes that contribute to splenic fibrosis by producing different collagen types. Notably, we found that one of these fibrocyte types originated from the graft, highlighting the crucial role of the disease microenvironment in shaping malignant clones.
Our patient’s splenectomy proved to be a vital intervention in breaking the cycle of graft–leukemia interactions and in preventing donor cell depletion in their extramedullary organs. This finding sheds new light on the complex mechanisms of disease eradication by HSCT, underscoring the importance of continued research in this field.
The benefits of employing pre-transplant conditioning spleen treatments, such as splenectomy or irradiation, have been the subject of prolonged discussion. The recent findings from two extensive patient cohort studies provide insights into the safety of splenectomy as a preparatory measure before transplantation in myelofibrosis patients (23, 24). Furthermore, advantageous outcomes have been reported, including improved non-relapsed mortality and overall survival, associated with pretransplant splenectomy for patients with progressive disease coupled with massive splenomegaly (> 15 cm) (24). It is noteworthy, however, that these investigations do not include patients with sAML arising from MPN within their subsets of progressive disease, as was the case in our patient.
For myelofibrosis patients carrying a substantial tumor burden due to massive splenomegaly, pre-transplant splenectomy could potentially emerge as a viable and safe intervention, offering a means of reducing the risk of graft failure during HSCT.
In a limited retrospective study involving patients with massive splenomegaly receiving JAK2 inhibitors, the discussion highlighted the encouraging outcomes associated with the inclusion of spleen irradiation in the conditioning regimen (25).
These reports underscore the urgent need to further investigate the efficacy of spleen-directed interventions (such as splenectomy, irradiation, and pharmacological organ reduction) within the context of conditioning regimens for MPN HSCT, specifically targeting advanced-stage-disease patients, including those with spleen size exceeding 15 cm and sAML transformation.
Patient perspective
Following the splenectomy, the patient experienced not only an objective improvement in their blood count but also a notable subjective improvement in their general condition, which was characterized by transfusion independence.
Data availability statement
All the sequencing data used in the manuscript has been submitted to the European Nucleotide Archive under the Primary accession number PRJEB71465 and will be private until 28.02.2025. To access the data before this date, please contact the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Ärtzekammer Hamburg, Hamburg, Germany. The studies were conducted in accordance with local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
EB: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft. OB: Formal analysis, Investigation, Methodology, Writing – original draft. AY: Formal analysis, Investigation, Methodology, Data curation, Project administration, Writing – original draft. AS: Data curation, Formal analysis, Methodology, Software, Validation, Writing – original draft. SK: Data curation, Formal analysis, Methodology, Validation, Writing – original draft. VF: Data curation, Formal analysis, Software, Writing – original draft. IT: Data curation, Formal analysis, Software, Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Deutsche José Carreras Leukämie Stiftung (DJCLS 19R/201), and the Else Kröner-Fresenius-Stiftung (2016_A105) to IT.
Acknowledgments
The authors would like to thank the personnel of the Core Facility in University Medical Center Hamburg Eppendorf for their assistance in FACS sorting experiments. The patient agreed to the donation of biological material (spleen and blood) and signed informed written consent under an approved protocol from the Ethics Committee of the Ärztekammer Hamburg.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Guy A, Bidet A, Ling A, Caumont C, Boureau L, Viallard J-F, et al. Novel findings of splenic extramedullary hematopoiesis during primary myelofibrosis, post-essential thrombocythemia, and post-polycythemia vera myelofibrosis. Virchows Arch (2021) 479(4):755–64. doi: 10.1007/s00428-021-03110-9
2. Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int (2015) 87:292–307. doi: 10.1038/ki.2014.287
3. Pizzi M, Gergis U, Chaviano F, Orazi A. The effects of hematopoietic stem cell transplant on splenic extramedullary hematopoiesis in patients with myeloproliferative neoplasm-associated myelofibrosis. Hematol. Oncol. Stem Cell Ther (2016) 9(3):96–104. doi: 10.1016/j.hemonc.2016.07.002
4. Triviai I, Zeschke S, Rentel J, Spanakis M, Scherer T, Gabdoulline R, et al. ASXL1/EZH2 mutations promote clonal expansion of neoplastic HSC and impair erythropoiesis in PMF. Leukemia (2019) 33(1):99–109. doi: 10.1038/s41375-018-0159-0
5. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
6. Ulgen E, Ozisik O, Sezerman OU. PathfindR: An R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front Genet (2019) 10:858. doi: 10.3389/fgene.2019.00858
7. Triviai I, Stübig T, Neibuhr B, Hussein K, Tsiftsolgou A, Fehse B, et al. CD133 marks a stem cell population that drives human primary myelofibrosis. Haematologica (2015) 100(6):768–79. doi: 10.3324/haematol.2014.118463
8. Lehtonen A, Veckman V, Nikula T, Lahesmaa R, Kinnunen L, Matikainen S. Differential expression of IFn Regulatory Factor 4 Gene in human monocyte-derived dendritic cells and macrophages. J Immunol (2005) 175:6570–9. doi: 10.4049/jimmunol.175.10.6570
9. Lainez-González D, Serrano-López J, Alonso-Domínguez JM. Understanding the Hedgehog Signaling Pathway in Acute Myeloid Leukemia Stem Cells: a necessary step toward a cure. Biol (Basel) (2021) 10(4):255. doi: 10.3390/biology10040255
10. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals the origins and dynamics of monocytes and tissue macrophages in homeostasis. Immunity (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
11. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science (2009) 325(5940):612–6. doi: 10.1126/science.1175202
12. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol (2015) 15(12):731–44. doi: 10.1038/nri3920
13. Ludin A, Itkin T, Gur-Cohen S, Mildner A, Shezen A, Golan K, et al. Monocytes-macrophages that express a-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol (2012) 13(11):1072–82. doi: 10.1038/ni.2408
14. Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood (2005) 106(6):1901–10. doi: 10.1182/blood-2005-04-1417
15. Bührer ED, Amrein MA, Forster S, Isringhausen S, Schürch CM, Bhate SS, et al. Splenic red pulp macrophages provide a niche for CML stem cells and induce therapy resistance. Leukemia (2022) 36:2634–46. doi: 10.1038/s41375-022-01682-2
16. Mebius R, Kraal G. Structure and function of the spleen. Nat Rev Immunol (2005) 5:606–16. doi: 10.1038/nri1669
17. Valet C, Magnen M, Qiu L, Cleary SJ, Wang KM, Ranucci S, et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. JCI (2022) 132(7):e153920. doi: 10.1172/JCI153920
18. Nagelkerke SQ, Bruggeman CW, den Haan JMM, Mul EPJ, van den Berg TK, van Bruggen R, et al. Red pulp macrophages in the human spleen are a distinct population with a unique expression of Fc-g receptors. Blood Adv (2018) 2:941–53. doi: 10.1182/bloodadvances.2017015008
19. Marini I, Backhoul T. Pathophysiology of Autoimmune Thrombocytopenia: current insight with a focus on Thrombopoiesis. Hämostaseologie (2019) 39:227–37. doi: 10.1055/s-0039-1678732
20. Cencini E, Fabri A, Sicuranza A, Gozzetti A, Bocchia M. The role of tumor-associated macrophages in hematologic Malignancies. Cancers (Basel) (2021) 13(14):3597. doi: 10.3390/cancers13143597
21. Al-Matary YS, Botezatu L, Opalka B, Hönes JM, Lams RF, Thivakaran A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia-supporting state in a Growth factor independence 1 dependent manner. Haematologica (2016) 101:1216–27. doi: 10.3324/haematol.2016.143180
22. Kotwal G, Chien S. Macrophage differentiation in normal and accelerating wound healing. Results Probl Cell Differ (2017) 62:353–64. doi: 10.1007/978-3-319-54090-0_14
23. Bossard J-B, Beuscart J-B, Robin M, Mohty M, Barraco F, Chevallier P, et al. Splenectomy before allogeneic hematopoietic cell transplantation for myelofibrosis: a French nationwide study. Am J Hematol (2020) 96(1):80–8. doi: 10.1002/ajh.26034
24. Polverelli N, Mauff K, Kröger N, Robin M, Beelen D, Beauvais D, et al. Impact of spleen size and splenectomy on outcomes of allogeneic hematopoietic cell transplantation for myelofibrosis: A retrospective analysis by the chronic Malignancies working party on behalf of European Society for blood and marrow transplantation. Am J Hematol (2020) 96(1):69–79. doi: 10.1002/ajh.26020
Keywords: hypersplenism, myeloproliferative neoplasia (MPN), secondary AML, fibrosis, hematopoietic (stem) cell transplantation (HCST)
Citation: Besiridou E, Bhatt O, Yurukova A, Sahu A, Kinkley S, Fischer V and Triviai I (2024) Persistent hypersplenism following successful hematopoietic stem cell transplantation: unraveling the cellular components of extramedullary residual disease and donor hematopoiesis—a case report. Front. Hematol. 2:1328503. doi: 10.3389/frhem.2023.1328503
Received: 26 October 2023; Accepted: 11 December 2023;
Published: 09 January 2024.
Edited by:
Pontus Lundberg, Roche, SwitzerlandReviewed by:
Ram Babu Undi, University of Oklahoma Health Sciences Center, United StatesZhongbo Hu, St. Jude Children’s Research Hospital, United States
Copyright © 2024 Besiridou, Bhatt, Yurukova, Sahu, Kinkley, Fischer and Triviai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioanna Triviai, dHJpdmlhaUBtb2xnZW4ubXBnLmRl