 Mayumi Hirayama1
Mayumi Hirayama1 Hirotaka Matsui
Hirotaka Matsui- 1Laboratory of Transcriptional Regulation in Leukemogenesis, International Research Center for Medical Sciences, Kumamoto University, Kumamoto, Japan
- 2Department of Molecular Laboratory Medicine, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan
- 3Department of Laboratory Medicine, National Cancer Center Hospital, Tokyo, Japan
- 4Department of Medical Oncology and Translational Research, Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan
An R-loop is a nucleic acid structure consisting of a DNA : RNA hybrid and single-stranded DNA. It is formed physiologically in normal cells and is involved in transcription, replication, and gene rearrangement; in particular, it has multiple roles including in mitochondrial DNA replication and class switch recombination of immunoglobulin genes in B cells. However, accumulating evidence indicates aberrant R-loop formation in various malignancies, including hematopoietic neoplasms. The accumulation of such inappropriate R-loops can cause conflicts between transcription and DNA replication. This exacerbates genomic instability through the generation of DNA replication stress, that, in turn, leads to cellular phenotypic changes and disease progression. When RNAs are synthesized during transcription they hybridize with template DNA in cis, giving rise to R-loops. In addition, it was recently revealed that noncoding RNAs also form R-loops when bound to genomic DNA in trans. Together with such observations, new roles for the R-loop in disease development have been proposed. The relationship between inflammation and the R-loop has also attracted much attention. In this review, we will focus on the mechanisms of R-loop formation in various hematopoietic neoplasms and introduce the important findings from recent studies. Therapeutic concepts for targeting R-loop accumulation in hematopoietic neoplasms will also be discussed.
1 Introduction
An R-loop is a nucleic acid structure that consists of a DNA : RNA hybrid and single-stranded DNA (ssDNA). It is formed when RNA hybridizes to a strand of genomic DNA (in cis or trans configuration) and the remaining DNA strand is displaced as a single strand (1). R-loops are found in as much as 5% of the entire genome and are thought to serve the opposing functions of maintaining genomic integrity and inducing DNA damage, i.e., both physiological and pathological R-loops exist (Figure 1). Physiologically, an R-loop is formed during mitochondrial DNA (mtDNA) replication and the immunoglobulin class switch recombination of immunoglobulin genes in B lymphocytes, and in other processes (1, 2). R-loops are also involved in the repair of DNA damage, thus contributing to the maintenance of genome integrity. For instance, it has been shown that R-loops are formed at sites where DNA double-strand breaks (DSBs) occur and that RAD52 recognizes and resolves R-loops, thereby initiating homologous recombination repair (3).
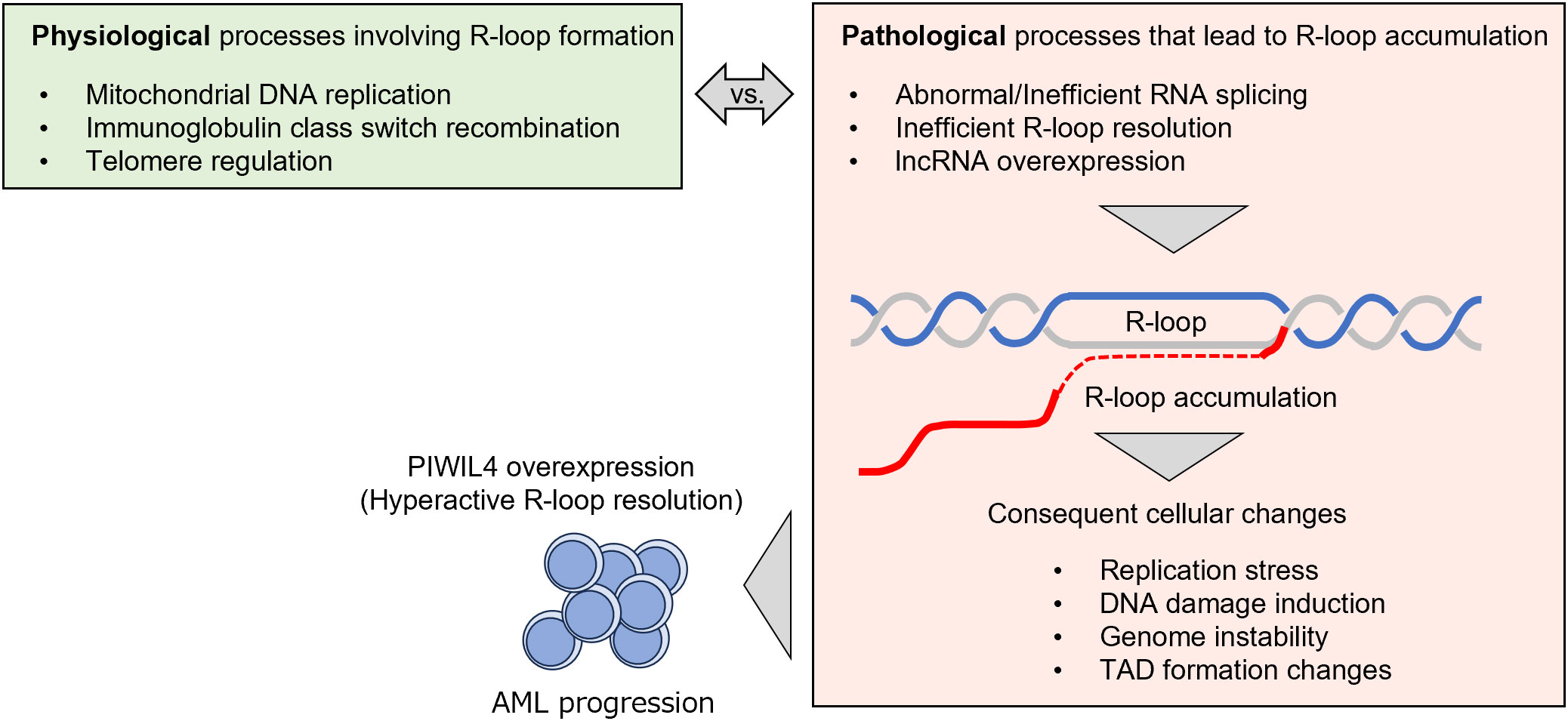
Figure 1 Graphical summary depicting the role of R-loop formation in hematopoietic neoplasms presented in this manuscript. An R-loop is a structure composed of a DNA : RNA hybrid and single strand DNA. R-loop is formed physiologically in various phases (green rectangle), while excessive accumulation of R-loop is observed in malignancies, including hematopoietic neoplasms. This leads to DNA replication stress and DNA damage, leading to genomic instability (orange rectangle). Some hematopoietic neoplasms are considered to acquire additional mechanisms to resolve R-loops, thereby overcoming the cellular damage associated with R-loop accumulation and gaining proliferative potential.
However, R-loops that aberrantly accumulate in the genome can be harmful to cells. The number of R-loops increases if transcriptional activity is inappropriately high or, conversely, when polymerases are prone to pausing, especially in malignancies (4). Once R-loops accumulate in the genome, they provoke collisions with replication forks formed during the DNA replication process; they are particularly prone to cause excess DNA damage in regions where transcription and replication proceed in opposite directions. For this reason, cells are equipped with various safeguards to resolve R-loops. RNase H1 and H2 are nucleases that directly digest DNA : RNA hybrids. RNase H2 acts in a cell-cycle–dependent manner whereas RNase H1 acts independently of the cell cycle (5). The endonucleases, XPG and XPF, are involved in transcription-coupled nucleotide excision repair (TC-NER) and are thought to cleave nucleotides that make up R-loops (6, 7). THO/TREX are protein complexes required for exporting synthesized mRNA out of the nucleus; they are considered to indirectly suppress R-loop accumulation since R-loops increase when they do not function properly (8). Recent studies have also revealed a role of APOBEC3B in editing single-stranded DNA formed in the R-loop structure (9, 10). The findings from these investigations suggest that APOBEC3B-mediated DNA editing is implicated in the induction of genetic mutations, particularly in tumor cells. For information on the many other factors that regulate R-loops, see the review by Petermann et al. (11).
Recent significant advancements in R-loop research can be largely attributed to the development of technical infrastructure that facilitates more precise and comprehensive detection of R-loops. Immunofluorescence staining and dot blotting using the S9.6 antibody, which recognizes DNA : RNA hybrids, have been primary methods for R-loop detection. Additionally, a technique for the comprehensive analysis of R-loops in nuclei through immunoprecipitation using the antibody has been developed (12). However, it is noteworthy that the S9.6 antibody’s specificity has been a point of consideration, as it may also recognize ribosomal RNA in the cytoplasm and nucleolus, in addition to R-loops (13, 14). To address this concern, an RNase H1 mutant with binding affinity to R-loops but lacking enzymatic activity has recently been employed for visualizing the sites of R-loop formation and for comprehensive R-loop sequencing (15–17). Furthermore, a method for the comprehensive detection of single-strand DNA, a component of R-loops, has also been developed (18). With the evolution of these technologies, the pathological significance of R-loop accumulation is gradually becoming clearer.
This review will first briefly introduce the normal cellular processes that require R-loop formation, using mtDNA replication, immunoglobulin class switch recombination, and telomere regulation as examples. Next, the roles of R-loops aberrantly formed in hematopoietic neoplasms and their pathological significance will be discussed.
2 Physiological functions of R-loops
2.1 R-loops in mitochondrial DNA replication
Mitochondrial DNA is composed of double circular strands, termed heavy (H)- and light (L)-strands, each of which has an independent replication start site (19). The replication of mtDNA begins when POLRMT, a mitochondrial RNA polymerase, initiates transcription from the L-strand promoter (Figure 2). Transcription terminates within a conserved sequence block (CSB) after approximately 120 bases, and the transcribed RNA hybridizes with template DNA to form an R-loop structure. RNase H1, a DNA : RNA hybrid–specific endonuclease, then digests the DNA : RNA hybrid to create a 3’ end of RNA that serves as a primer for DNA synthesis. Such a primer is used by DNA polymerase γ to initiate the synthesis of a nascent H-strand beginning at the H-strand replication initiation site (shown as OH in Figure 2) on the parental L-strand (20). Replication of the nascent H-strand proceeds as topoisomerase and Twinkle, a mtDNA helicase, unwind the double strand and release the parent H-strand as a single strand. When the L-strand replication initiation site (shown as OL in Figure 2) on the parental H-strand is exposed as a single strand, a primer RNA is also synthesized from this site, where the synthesis of the nascent L-strand is initiated. Although the details of such processes remain debatable (19–25), the formation of an R-loop is considered essential for mtDNA replication, since cells lacking RNase H1 are unable to replicate mtDNA (20). Additionally, in light of research indicating that mutations in RNase H1 lead to mitochondrial disorders (26), it has been proposed that dysregulation of R-loops in mitochondria is linked to the onset of diseases, particularly demonstrated in certain central nervous system diseases (27). However, at present, the association between hematopoietic neoplasms and dysregulation of mitochondrial R-loops remains unclear.
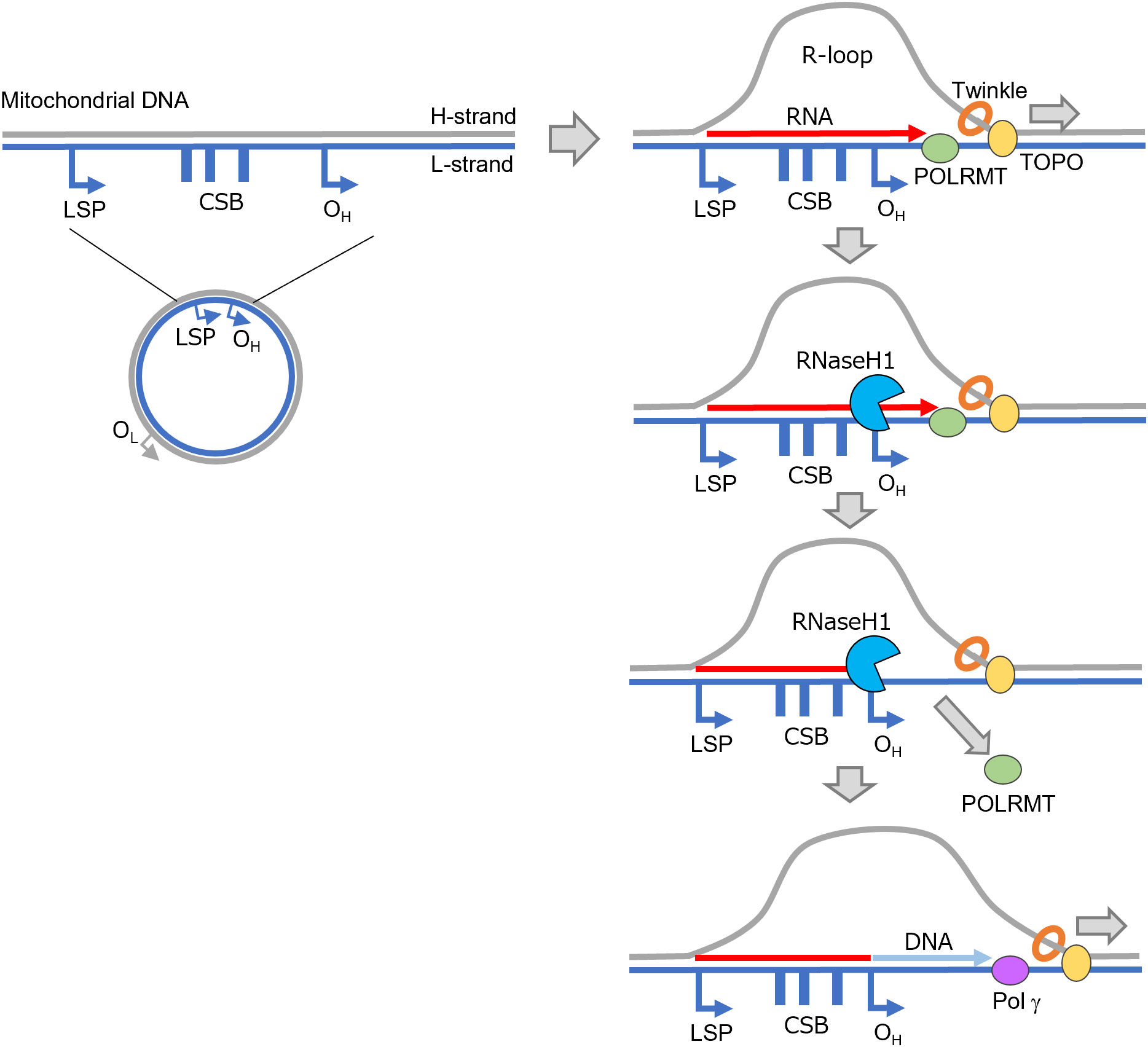
Figure 2 An R-loop is formed during mtDNA replication. Transcription by POLRMT is initiated at the LSP and an R-loop is formed. RNase H1 subsequently digests the RNA, and replication by POL γ is initiated from OH. CSB, conserved sequence block; LSP, light strand promoter; OH, H-strand replication initiation site (OriH); OL, L-strand replication initiation site (OriL); POL γ, DNA polymerase γ; POLRMT, mitochondrial RNA polymerase; TOPO, topoisomerase.
2.2 R-loops in immunoglobulin class switch recombination
B lymphocytes expressing membrane-bound IgM and IgD on the cell surface become activated and proliferate upon binding to their specific antigens. Mature B lymphocytes further differentiate into antibody-secreting cells upon being activated by specific antigens or other stimuli. This process results in the increased production of secretory IgG, IgA or IgE immunoglobulins while keeping the specificity of IgM for the antigen. This is called class switch recombination. The heavy chain of an antibody consists of variable, diversity, and joining segments involved in antigen specificity, and a constant (C) region that determines the class of the antibody. Upon class switch recombination, the Cμ region in the IgM gene (the constant region for IgM) is replaced with a CH region encoding other classes of antibodies; recombination of the CH region involves the noncoding I exon and switch (S) regions located upstream of each C region (28, 29). The binding of transcription factors to the I region initiates the synthesis of RNA, termed a germline transcript (GLT), which is complementary to the target S region that becomes recombined with the Sμ region (Figure 3). A GLT partially forms a DNA : RNA hybrid and displaces ssDNA in the S region. Activation-induced cytidine deaminase (AID) then acts on the ssDNA, replacing dC in the DNA with dU; the dU is then recognized and removed by uracil-N-glycosylase, forming a nick at that position, ultimately resulting in DSBs. This occurs simultaneously at two locations in the Sμ and S regions to be recombined, with the intervening region being removed as an excision circle. Finally, the cleaved DNA surfaces are joined together by nonhomologous end joining to complete recombination.
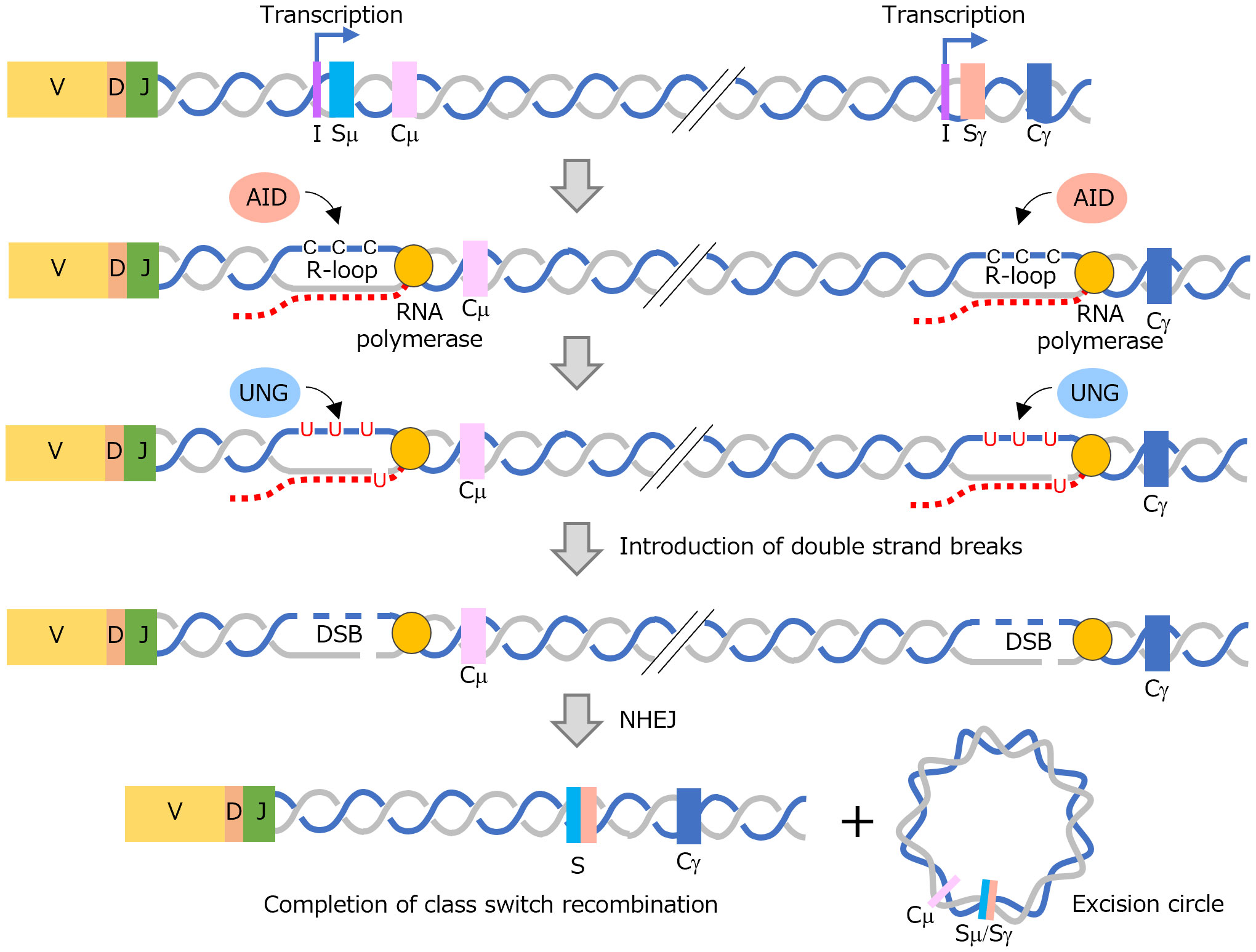
Figure 3 Formation of R-loops is associated with immunoglobulin class switch recombination. An R-loop is formed in the S region at two locations, and a DSB is induced by the action of AID and UNG. Finally, cleaved DNA binds and class switch reconstitution takes place. AID, activation-induced cytidine deaminase; DSB, DNA double-strand break; NHEJ, nonhomologous end joining; UNG, uracil-N-glycosylase.
It has been reported that RNase H2 and Senataxin play a role in class switch recombination in B cells (30). Furthermore, APOBEC3B, an enzyme possessing cytidine deaminase activity similar to AID, has been documented to participate in DNA editing within R-loops, extending beyond the regions associated with class switch recombination (9, 10). This observation suggests that APOBEC3B could potentially serve as a therapeutic target. When these abnormalities do not function properly, genomic instability may be heightened in the immunoglobulin heavy-chain locus or other genomic regions. As our knowledge in this field advances, it is possible that a connection between the abnormal regulation of R-loops and the development of malignancies and immunological diseases is revealed.
2.3 R-loops in telomere regulation
Ribonucleic acid has also been shown to bind to DNA as a trans-acting factor, forming R-loops in the telomere region of chromosomes. At the end of a telomere region, the shelterin protein complex binds to the telomere and forms a T-loop – a loop structure specific to this region (31). In comparison, telomeric repeat-containing RNA (TERRA), a long noncoding RNA (lncRNA), is transcribed from a sub-telomeric to telomeric region in some chromosomes. TERRA binds complementarily to telomere DNA as a trans-acting factor to form an R-loop that is involved in the maintenance of telomeric chromatin structure (32–35) (Figure 4). In the centromere region, RNAs derived from α-satellite repeats have been shown to bind to centromeres as cis-acting factors and form R-loops (36, 37). Ribonucleic acids in the centromere region are considered to be required for centromere proteins (CENP)-A and CENP-C to interact with the centromere and, perhaps indirectly, contribute to the proper binding of kinetochores to microtubules and chromosome segregation (38, 39). Recent studies have shed light on the participation of factors associated with the repair of DNA damage, such as BRCA1 and RAD51, along with RNA regulatory factors like THO complex, in the mechanism of telomere regulation by TERRA (40–42). This intricate interplay has recently been paid significant attention.
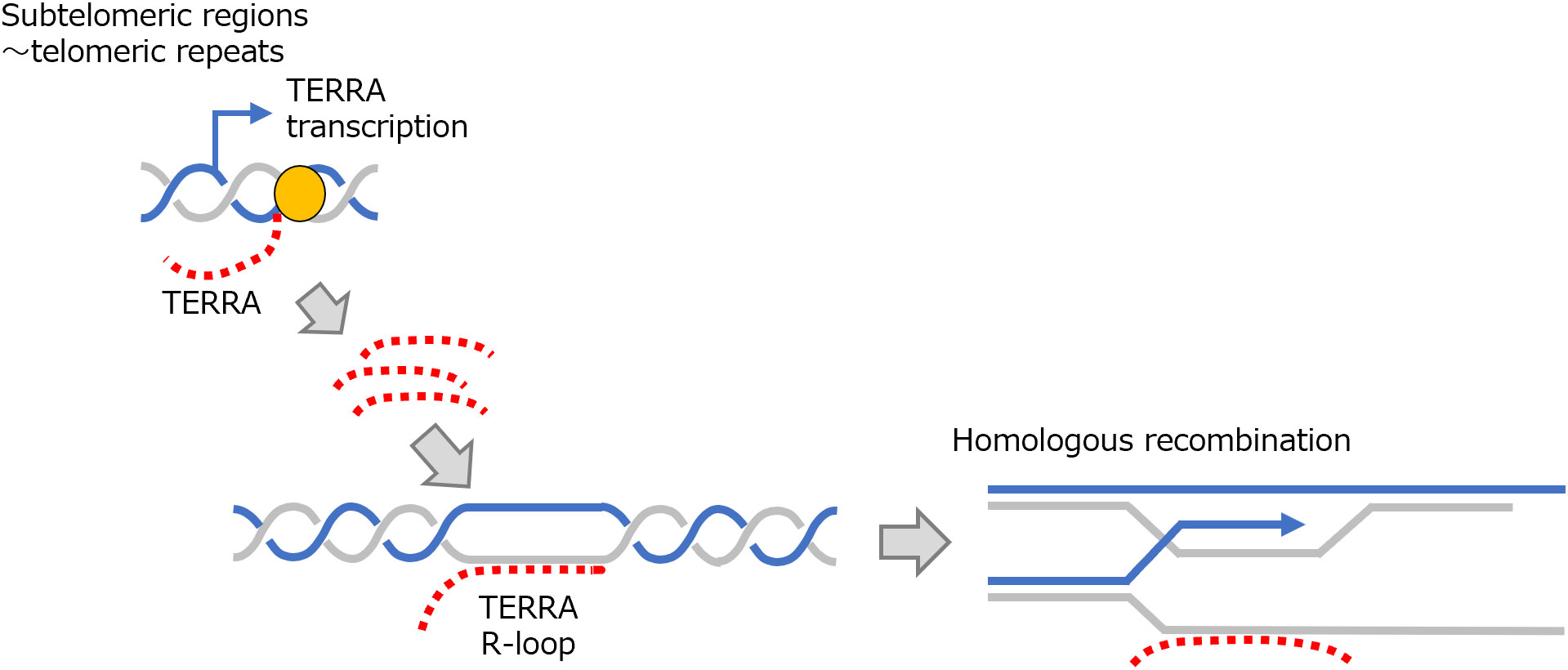
Figure 4 R-loop formation and telomere regulation by TERRA. TERRA is transcribed from subtelomeric to telomeric regions and in trans forms an R-loop in telomeric regions. This process is involved in HR in telomeric regions. HR, homologous recombination; TERRA, telomeric repeat-containing RNA.
3 Aberrant R-loop formation in hematopoietic neoplasms
Although R-loops have been shown to form during several specific physiological processes, R-loop accumulation is also often observed in malignancies and neurodegenerative diseases and has been implicated in the pathogenesis of such diseases (11, 43). In recent years, the accumulation of R-loops has been successively reported to be involved in the pathogenesis of various hematopoietic neoplasms, as described below (44).
3.1 Excessive R-loop formation due to mutations in myeloid neoplasm–related RNA splicing factors
3.1.1 R-loop accumulation caused by mutations in U2 small nuclear ribonucleoprotein–related RNA splicing factors
Somatic mutations in genes encoding RNA splicing factors are detected at a high frequency in more than 50% of myelodysplastic syndromes (MDS) (45, 46). In hematopoietic neoplasms, mutations in more than 30 genes that are thought to be involved in RNA splicing have been observed. Of these, representative gene mutations associated with MDS are found in SF3B1, SRSF2, and U2AF1.
Intronic sequences in transcribed protein-coding RNA generally contain a branch point (BP) several dozen bases upstream from the 3’ splice site (SS). Splicing factor 3b subunit 1 (SF3B1) recognizes and binds to the BP together with U2 small nuclear RNA (snRNA) (47). The BP serves as a binding site for 5’ SS that is cleaved during the first step of the splicing reaction to form an intron lariat. SF3B1 also indirectly interacts with U2 snRNA auxiliary factor 2 (U2AF2), which binds to the 3’ SS via SURP and G-patch domain containing 1 (SUGP1) (48, 49). In MDS, p.K700E is the most frequent mutation in SF3B1; the binding of SF3B1 to SUGP1 is impaired by this mutation, leading to disruption of the interaction between SF3B1 and U2AF2. As a result, BPs that should be recognized are ignored, and, instead, cryptic 3’ SS located upstream of the canonical 3’ SS are used (50, 51). This indicates that mutations in SF3B1 have a significant impact on the selection of 3’ SS in RNA splicing. SF3B1 mutations are found not only in myeloid neoplasms represented by MDS, but also in chronic lymphocytic leukemia (52, 53) and solid cancers, such as uveal melanoma (54, 55), with frequencies ranging from 5–17% and 14–29%, respectively.
Serine and arginine rich splicing factor 2 (SRSF2) is an RNA splicing factor that belongs to the serine and arginine rich protein family and recognizes the exonic splicing enhancer (ESE) (56). ESE is a splicing regulatory cis-element that positively regulates RNA splicing (57, 58). As far as currently known, SRSF2 recognizes and binds GGNG and CCNG motifs present in ESE with equal affinity. Mutations in SRSF2 are found in about 15% of patients with MDS. A mutational hot spot is found at p.P95 in the hinge region, between an RNA recognition motif and arginine/serine-rich domain, and typical mutations are p.P95R and p.P95H. Mutant SRSF2 has a higher affinity for the CCNG compared to GGNG motif, resulting in altered preferential usage of cassette exons (59–61).
U2 small nuclear RNA auxiliary factor 1 (U2AF1), together with U2AF2, constitute the U2AF complex, which recognizes the 3’ SS of U2-type introns and recruits U2 small nuclear ribonucleoprotein (snRNP) (62–64). Mutations of U2AF1, p.S34F/Y and p.Q157P/Q are often detected in MDS; p.S34 mutants have been shown to promote splicing of exons possessing a U at position -3 in the 3’ SS, while tending to include a C at the position. In comparison, p.Q157 mutants preferentially splice exons possessing an A at a +1 position of the 3’ SS, while keeping those with a G (65, 66). Figure 5A illustrates the RNA splicing factors involved in 3’ SS recognition.
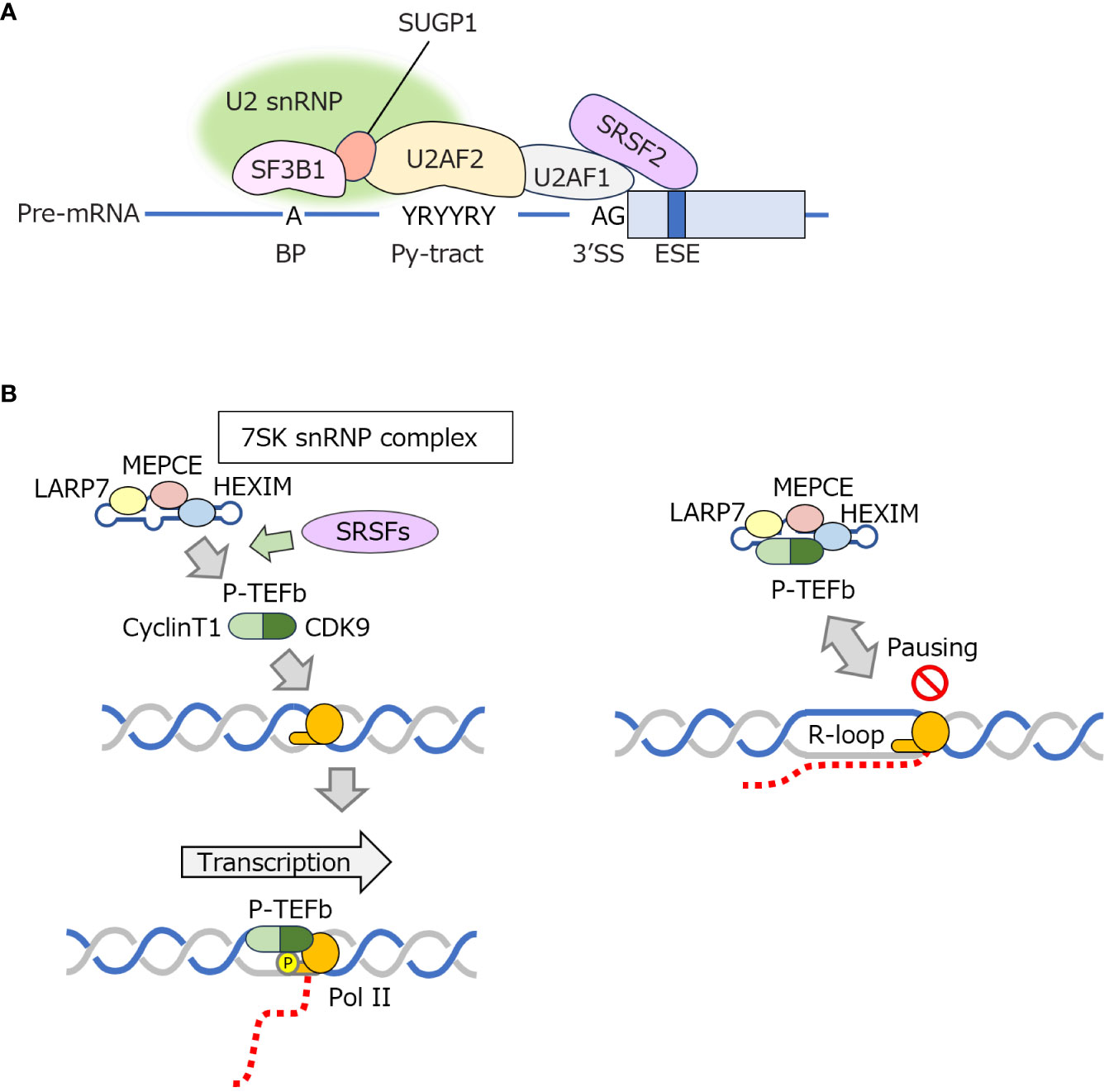
Figure 5 Roles of SRSF2 in regulating 3′ SS recognition and P-TEFb–mediated transcriptional elongation (A). SRSF2, SF3B1, U2AF1, and other factors cooperatively recognize the 3’ SS of pre-mRNA. (B) The P-TEFb complex phosphorylates the CTD of Pol II to promote transcription elongation. SRSF proteins dissociate P-TEFb from 7SK snRNPs to promote transcription elongation. An R-loop is more likely to form when transcriptional elongation is paused. BP, branch point; CDK9, cyclin-dependent kinase 9; CTD, c-terminal domain of RNA polymerase II; ESE, exonic splicing enhancer; HEXIM, hexamethylene-bis-acetamide-inducible protein in vascular smooth muscle cells; LARP7, La RibonucleoProtein domain family member 7; MEPCE, methylphosphate capping enzyme; Pol II, RNA polymerase II; P-TEFb, positive transcription elongation factor b; Py-tract, polypyrimidine tract; SF3B1, splicing factor 3b subunit 1; snRNP, small nucleolar ribonucleoprotein; SRSFs, serine and arginine rich splicing factor proteins including SRSF1 and SRSF2; 3’ SS, 3’ splice site; SUGP1, SURP and G-patch domain containing 1; U2AF1, U2 small nuclear RNA auxiliary factor 1.
Despite different patterns of splicing changes, depending on the mutated splicing factor involved, such mutations commonly cause MDS. However, the mechanisms by which they uniformly cause myeloid neoplasms have not been elucidated until recently. In this context, it was reported in 2018 that MDS with mutations in SRSF2 and U2AF1 commonly exhibits R-loop accumulation (67). Another group subsequently reported a similar phenomenon in SF3B1-mutated MDS (68). R-loops do not necessarily increase at the position where splicing changes occur; rather, it is likely that the pausing of RNA polymerase II (Pol II) correlates with R-loop accumulation (4). In the case of SRSF2, the ability of the protein to facilitate dissociation of positive transcription elongation factor b (P-TEFb), a protein complex consisting of cyclin-dependent kinase 9 and cyclin T, from 7SK snRNP has been implicated in R-loop formation. P-TEFb phosphorylates the C-terminal domain of Pol II, which is required for efficient transcriptional elongation by Pol II (Figure 5), but cannot phosphorylate Pol II when bound to 7SK snRNP. In P-TEFb activation, SRSF1 and SRSF2 dissociate P-TEFb from the 7SK complex (69). Pol II pauses once transcription is initiated. Pol II is then phosphorylated by P-TEFb to be released from its pause so that it can proceed with transcriptional elongation. Mutations in SRSF2 impair this process, making it difficult for transcriptional elongation to proceed properly, leading to a conflict between transcription and replication that facilitates R-loop formation (67). However, since R-loop accumulation is also observed in myeloid neoplasms with mutations in other RNA splicing factors, it is possible that R-loop formation is suppressed by mechanisms other than through P-TEFb regulation; however, the detailed mechanisms remain unclear. Cells expressing SF3B1 or U2AF1 mutants show reduced nonsense-mediated decay (NMD) activity as well as impaired DNA replication and increased DNA damage. All such phenotypes are alleviated by the enforced expression of RNase H1 (70), suggesting an interplay exists between NMD regulation and R-loop formation. Mutations in SRSF2 have also been implicated in NMD (71).
Regarding treatment intervention, studies have suggested the potential effectiveness of ATR inhibitors in MDS characterized by the accumulation of R-loops due to mutations in RNA splicing factors (72, 73). This effectiveness is likely associated with the activation of the pathways involved in DNA damage response, triggered by increased DNA damage linked to R-loop accumulation. Additionally, the collision of the R-loop with the replication machinery may activate the ATR pathway (74). However, to date, the efficacy of ATR inhibitors in clinical settings has not yet been validated. Further studies are clearly needed to elucidate the pathophysiological roles of R-loop accumulation in myeloid neoplasms with mutations in RNA splicing factors and to identify therapeutic target molecules and pathways relevant to this phenomenon.
3.1.2 R-loop accumulation in myeloid neoplasms with RNA helicase mutations
In recent years, myeloid neoplasms caused by DDX41 mutations have attracted considerable attention. DDX41 was recently found to be one of the genes responsible for hereditary myeloid neoplasms (75–77), including MDS and acute myeloid leukemia (AML). The diseases are characterized by late onset, less proliferating phenotype, and the acquisition of a somatic DDX41 variant in individuals with a pathogenic germline variant before overt disease manifestation. Furthermore, a recent large-scale mutational analysis revealed that CUX1 mutations coexist almost exclusively in myeloid neoplasms with DDX41 mutations, demonstrating the specificity of hematopoietic neoplasms due to this mutation (78).
Recent reviews outline our current understanding of the roles of DDX41 mutations in myeloid neoplasms (79–82), which is also briefly elucidated in the subsequent texts. DDX41 probably has multiple biological functions; for more than a decade, it has been suggested that DDX41 is a nucleic acid sensor in the cytoplasm (83, 84). In comparison, DDX41 has recently been shown to serve as an RNA splicing factor, particularly in hematopoietic stem cells (HSCs) and myeloid progenitor cells (75, 85). However, whereas RNA splicing factors frequently mutated in MDS, such as SRSF2 and SF3B1, are known to be involved in 3’ SS recognition, DDX41 has been shown to interact with 5’SS (85). It has also been shown that DDX41 is incorporated into the spliceosome at a late stage of the c-complex, when 5’ and 3’ SS recognition has been completed and the spliceosome is activated. In addition, as noted above, disease phenotype and coexisting mutations in DDX41-mutated myeloid neoplasms differ from those of SRSF2 and SF3B1 mutations. Therefore, at this stage, DDX41 mutations should be discussed separately from such mutations.
Recent reports have implicated DDX41 mutations in R-loop accumulation (85–87). A study using zebrafish as a model showed that R-loops increased and hematopoietic stem and progenitor cell (HSPC) production was impaired in cells with a homozygous deletion of Ddx41 (87). In addition, activation of the cGAS-STING pathway was observed in HSPCs lacking Ddx41, possibly due to increased R-loop formation. However, how DDX41 suppresses R-loop accumulation and how increased R-loops trigger inflammatory responses have not been thoroughly tested.
Another recent study suggested that DDX41 directly resolves R-loops (Figure 6A) (86). Proteins in the vicinity of the R-loop were comprehensively explored and it was found that multiple RNA helicases were present in close proximity to R-loops. Of these RNA helicases, only DDX41 and Aquarius were found to increase DNA damage when expression was suppressed. The conclusion was that DDX41 specifically regulates R-loops in the promoter region and functions as an R-loop–resolving enzyme that suppresses DNA replication stress. Indeed, a study examining the RNA helicase activity of DDX41 in vitro also showed that DDX41 exhibits DNA : RNA hybrid-unwinding activity (83). However, it is still unclear how DDX41 specifically recognizes the R-loops in promoter regions. In addition, considering that DDX41-repressed cells show apparent DNA damage signals after mitosis (85), the accumulation of R-loops during the S-phase might not directly lead to increased DNA damages; This implies that DDX41 may not function solely as an RNA helicase directly resolving R-loops, but may instead indirectly contribute to R-loop resolution through other mechanisms. The report further suggests that DDX41 coordinates RNA splicing and transcriptional elongation, thus preventing R-loop formation (Figure 6B). Therefore, the mechanism of how loss of DDX41 increases R-loop formation still needs to be carefully investigated (Figure 6C).
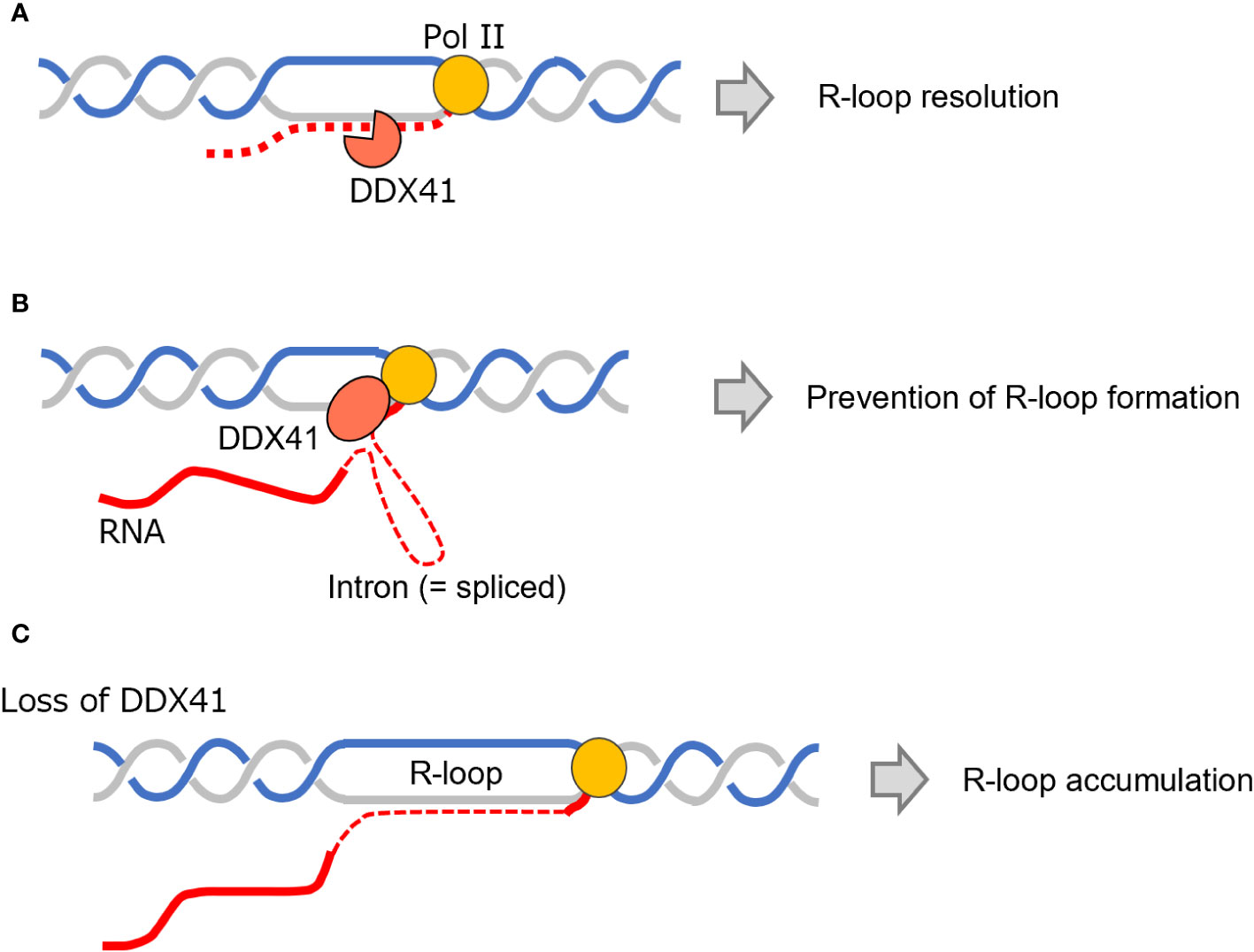
Figure 6 Biological roles of DDX41 and the relationship of DDX41 to R-loops. (A, B) DDX41 functions as an enzyme that can directly resolve an R-loop (A) or coordinates RNA splicing and transcription elongation, thus preventing R-loop accumulation (B). (C) R-loops accumulate upon DDX41 deficiency. DDX41, DEAD-box RNA helicase 41; Pol II, RNA polymerase II.
For other RNA helicases, mutations in DHX15, encoding DEAH-box type RNA helicase 15, are detected at a frequency of about 6% in AML with t(8:21) (88). The mutations are concentrated in p.R222G, which is located between motifs Ia and Ib within the RecA-like domain of DHX15. This suggests that it affects RNA binding, although how this mutation alters DHX15 function has not been studied to date. DHX15 is thought to play a role in the removal of intron lariat, the final step of RNA splicing (89). Recently, however, it was found that DHX15 interacts with the G-patch domain of SUGP1, causing cells with DHX15 mutations to exhibit RNA splicing changes partially similar to SF3B1 mutations (90). Another group also described how DHX15 is involved in BP recognition (91). These studies suggest that DHX15 mutations may be involved in an increase in R-loops through a mechanism similar to that observed in MDS, as described above (67, 68).
In addition, lower levels of DHX9 expression have been shown to be associated with the progression of MDS to high-risk MDS and AML (92). The study further described how the suppression of DHX9 expression induced DNA damage via R-loop formation. Another group also reported the involvement of DHX9 in R-loop regulation (93). Mutations and premature termination due to aberrant RNA splicing in DHX34 are also found in myeloid neoplasms (94, 95), but the causative link between the mutations and R-loop formation is so far unknown.
3.2 AML cell growth by PIWIL4 overexpression through inhibition of R-loop accumulation
It is known that as much as 45% of the human genome is occupied by repetitive sequences derived from transposing elements, called transposons (96, 97). Transposons modify the genome, leading to genetic diversity, and are therefore involved in the evolution of organisms. The DNA transposons cleave themselves from genomic DNA, while retrotransposons synthesize DNA from transcribed RNA, respectively, and invade other genomic regions. Since inappropriate overactivation of transposons can disrupt genes or confer new unwanted functions onto translated proteins, organisms possess systems to prevent the aberrant activation of transposons. These include P-element-induced wimpy testis (PIWI)–interacting RNA (piRNA), which interacts complementarily with transposon RNA. In humans, piRNAs form a complex with PIWI-like (PIWIL) proteins, translated from eight PIWIL genes, that cleave transposon RNA and inhibit the transcription of transposons (98, 99). Although piRNAs are expressed specifically in germ cells, PIWIL proteins are known to be upregulated in various malignancies (99, 100). PIWIL4 was recently found to be overexpressed in AML, possibly in a MYC-dependent manner (101).
Intriguingly, PIWIL4, a PIWIL protein, appears to have a function distinct from piRNA regulation. This observation stems from the fact that only 6% of PIWIL4 protein overexpressed in AML binds to piRNAs, whereas 48% interacts with protein-encoding RNAs, especially those involved in cancer-related pathways (101). In addition, the inhibition of PIWIL4 expression reduces the stem cell activity of AML cells and, along with increased DNA replication stress, results in increased DNA damage, leading to activation of ataxia telangiectasia and the Rad3-related (ATR) pathway. Since PIWIL4 belongs to the RNase H superfamily, and the binding sites of PIWIL4 contain guanine-rich sequences, the relationship between PIWIL4 and R-loops was also examined. It was found that PIWIL4 has R-loop digestion activity and that this action is required for the maintenance of the proliferative activity of AML cells (Figure 7) (101).
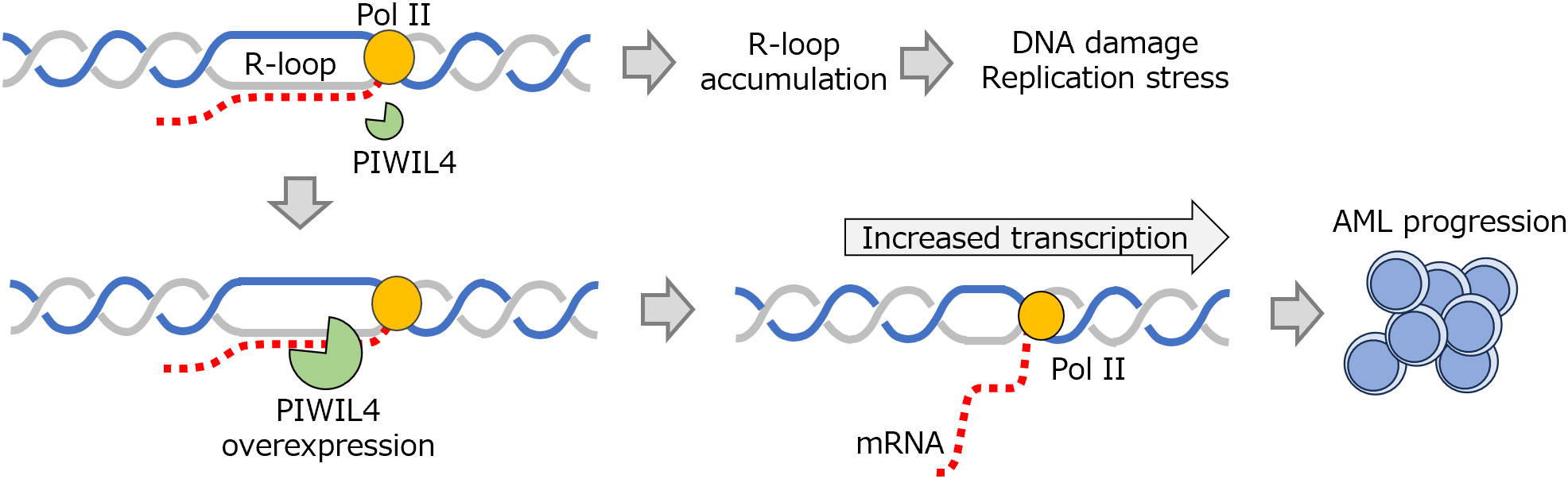
Figure 7 AML progression via R-loop regulation by PIWIL4. PIWIL4 has enzymatic activity that digests R-loops, and its overexpression in AML promotes transcription by eliminating R-loops, thus contributing to disease progression. AML, acute myeloid leukemia; PIWIL4, P-element–induced wimpy testis-like protein 4; Pol II, RNA polymerase II.
Acute myeloid leukemia cells have higher transcriptional activity due to oncogene activation, and, as a result, are prone to accumulating R-loops that potentially increase replication stress and genomic instability. Such cells are required to attenuate these disturbances in proliferation. Therefore, highly proliferative AML cells are likely to be equipped with, and be dependent on, higher R-loop resolving activity.
From a therapeutic perspective, the high dependence of AML cells on PIWIL4, unlike normal cells, suggests expectations of a combination of ATR inhibition and PIWIL4 suppression for the induction of specific damage to AML cells. Many unclear points are yet to be addressed: This includes the mechanism by which PIWIL4 is selectively induced in AML, how RNA is targeted by PIWIL4, and which RNases, including RNase H1, H2 and PIWIL4, are selectively used in AML cells to resolve R-loops. However, the concept that tumor cells acquire a system that maintains survival by suppressing excessive R-loops deserves further study.
3.3 R-loop regulation by lncRNA: involvement in AML pathophysiology
In recent years, lncRNAs, functional noncoding RNAs longer than 200 bases, have been shown to be involved in the pathophysiology of various diseases. HOTTIP is a 3764-base lncRNA located at the 5’ end of the HOXA cluster; its name derives from “HOXA transcript at distal tip” (102, 103). Eleven HOXA genes, clustering at the mammalian HOXA locus, are expressed in a concentration gradient manner and act as master regulators that determine the anterior–posterior axis and segmentation of tissues during development. HOTTIP is transcribed under the same regulation of expression as HOXA genes and coordinates the activation of 5’ HOXA genes by recruiting WD-repeat containing protein 5/mixed lineage leukemia protein-1 (Figure 8A) (102). The HOTTIP/HOXA9 pathway is transcriptionally upregulated in many malignancies (103).
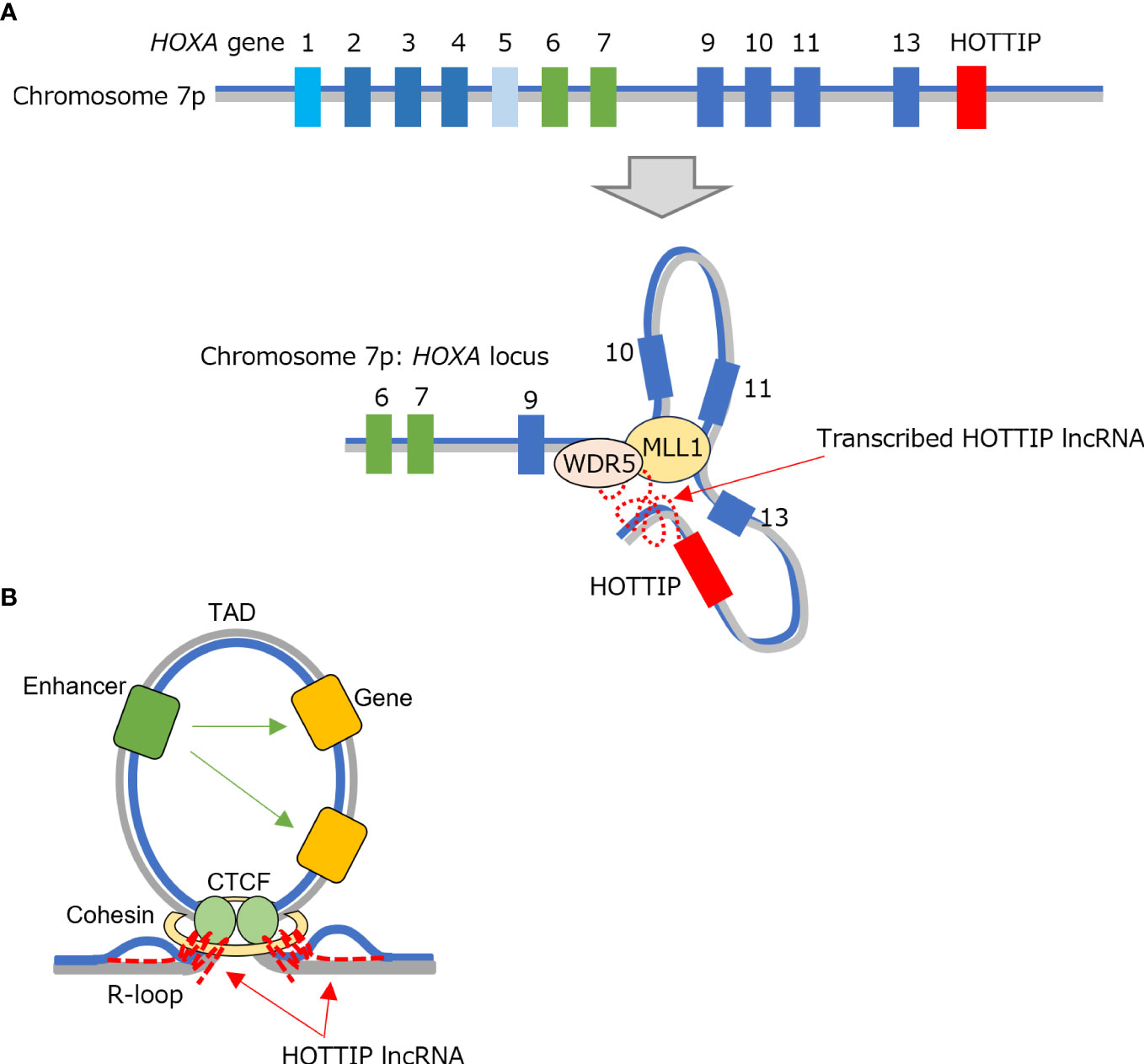
Figure 8 TAD formation involving HOTTIP. (A) HOTTIP is located at the end of the HOXA region and undergoes expression regulation in common with HOXA genes. HOTTIP recruits WDR5/MLL1 complexes to the 5’ HOXA locus to maintain the locus in an active state. (B) The transcribed HOTTIP also co-localizes with CTCF/cohesin factor and participates in TAD formation while adopting an R-loop structure. CTCF, CCCTC-binding factor; HOTTIP, HOXA transcript at the distal tip; MLL1, mixed lineage leukemia protein-1; TAD, topologically associating domain; WDR5, WD-repeat containing protein 5.
In hematopoietic neoplasms, HOTTIP is specifically upregulated in AML with a NPM1 mutation or MLL rearrangement. It plays a role in the self-renewal of AML cells through HOTTIP-dependent topologically associating domain (TAD) formation (104). After screening for factors that interact with HOTTIP, the same research group found it interacted with a CCCTC-binding factor (CTCF)-binding domain to form an R-loop structure (Figure 8B). This showed that HOTTIP binds both CTCF/cohesin factors and proteins that regulate R-loops (Figure 8B), although its binding was observed at only some CTCF boundaries (105). The study group further showed that R-loop formation by HOTTIP in the CTCF within Wnt/β-catenin genes regulates their expression by controlling TAD formation in the region.
Another lncRNA, lnc530, indirectly suppresses R-loop accumulation in mouse embryonic stem cells by forming a complex with R-loop regulators such as DDX5, a DEAD-box type RNA helicase, and TDP-43, an RNA-binding protein related to amyotrophic lateral sclerosis (106). This suggests that R-loop regulation by lncRNA is not limited to HOTTIP. However, HOTTIP appears to be unique in that it directly constitutes an R-loop as a TAD component. Further detailed studies are needed on the recognition mechanism of the R-loop formation site (i.e., TAD boundaries) by HOTTIP and its interaction with CTCF-related factors. More recently, there has been a growing recognition of the involvement of transposon elements and enhancer RNAs, a class of lncRNA transcribed from enhancer regions, in chromatin regulation based on such TAD structures. This concept has garnered increased attention (107, 108).
3.4 Involvement of DNA methylation–associated factors in R-loop regulation
The ten–eleven translocation (TET) enzymes are involved in DNA demethylation. Three members exist: TET1, TET2, and TET3, which convert a methylated cytosine, termed 5-methylcytosine, to 5-hydroxymethylcytosine (5hmC), thus competing with DNA methylation. Such enzymes serve as one of the key mechanisms regulating gene transcription, and, in fact, regulation of DNA methylation by TET proteins has been shown to be involved in cell differentiation and lineage determination (109). In maintaining DNA integrity, abundant 5hmC at sites where DNA DSBs occur (110) suggests that TET proteins contribute to the stabilization of genomic DNA by regulating DNA methylation. However, TET proteins have roles in keeping cell identity by inducing ssDNA breaks at cell type–specific enhancers (111).
Loss-of-function mutations in TET2 are observed with high frequency in myeloid neoplasms (112), especially in MDS, and show a frequency from 20 to 35% (113, 114); TET2 mutations are also frequent in certain lymphoid neoplasms such as nodal T-follicular helper cell lymphoma (nTFHL) angioimmunoblastic-type (nTFHL-AI), nTFHL not otherwise specified, and diffuse large B cell lymphoma (DLBCL) (115–117). Somatic mutations in genes encoding DNA methylation regulators (such as DNMT3A and TET2), which are acquired in HSCs in an age-dependent manner, confer a selective advantage on HSCs and cause clonal proliferation (118). Myelodysplastic syndromes and nTFHL-AI are considered to arise from such clonal hematopoietic cells and develop when these cells acquire secondary or tertiary mutations that favor further clonal expansion.
Recently, DLBCL-like lymphoma has been shown to develop in mice lacking both Tet2 and Tet3, along with strongly induced DNA damage in lymphoma cells (119). In B cells lacking TET proteins, increased R-loops and G-quadruplex structures were observed around the gene promoter and 5’ untranslated region (Figure 9), suggesting that a transcriptional pausing occurs in such regions. In addition, 5hmC was found to be decreased especially around such regions.
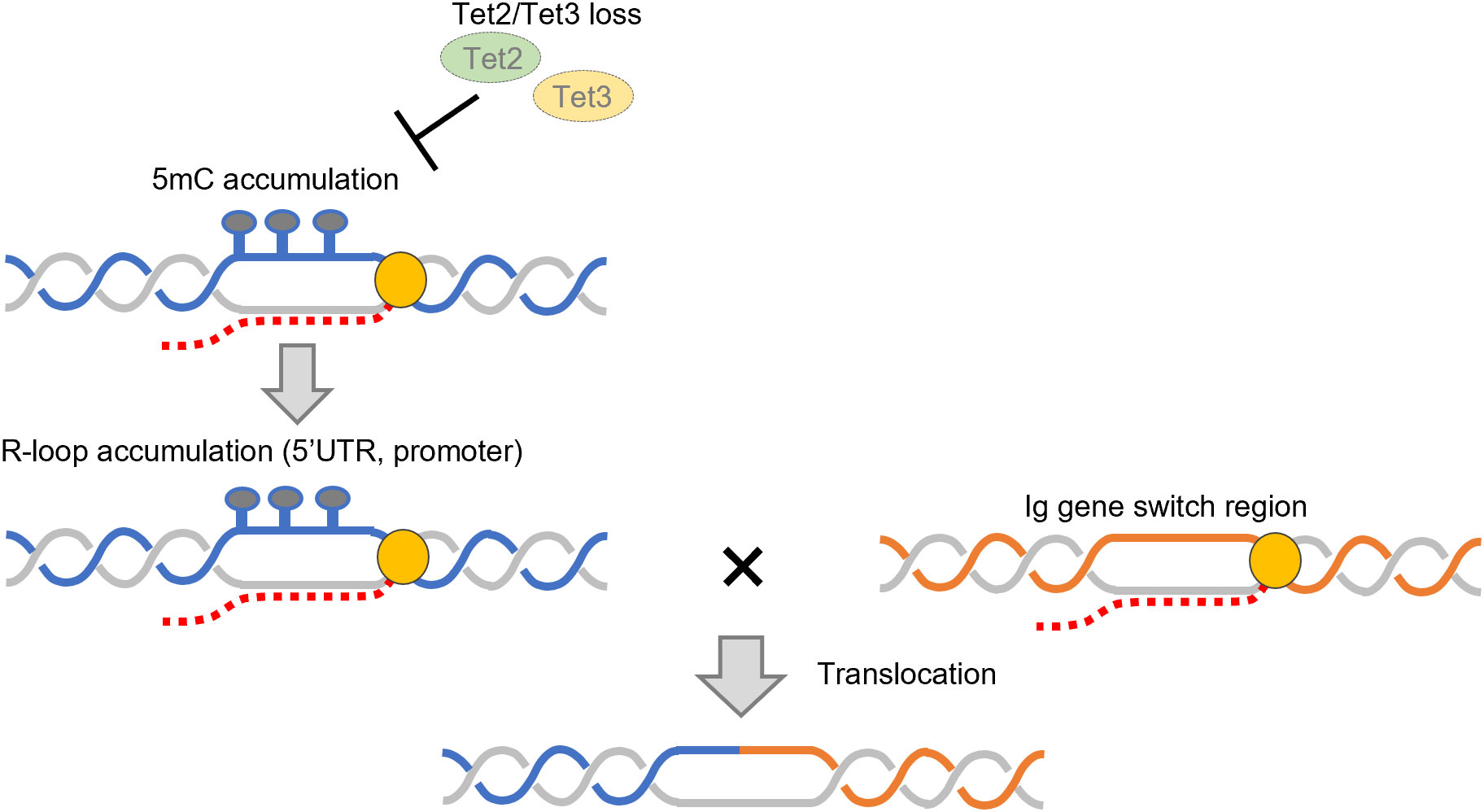
Figure 9 Accumulation of R-loops and increased translocation upon deletion of TET2/3. Deletion of TET2/3 causes an increase in R-loops in the 5’ UTR and promoter regions. Translocations between these and immunoglobulin gene regions increase upon loss of TET2/3. 5mC: 5-methylcytosine, TET, ten–eleven translocation enzyme; 5’ UTR, 5’ untranslated region.
B cells form R-loops in immunoglobulin gene loci and undergo class switch recombination (see subsection 2.2). Intriguingly, Tet2/3-deficient B cells are prone to translocation between immunoglobulin gene loci and R-loop/G-quadruplex–forming regions in the genome (119) (Figure 9). However, further deletion of Dnmt1 in this condition suppressed aberrant R-loop formation. These findings suggest that regulation of the DNA methylation state by TET proteins may prevent excessive DNA damage caused by R-loop cleavage and lymphomagenesis. Another study showed that methyl-CpG–binding protein 2, which binds to methylated DNA, inhibits R-loop accumulation (120). Yet another study showed that the presence of an R-loop inhibits the binding of DNA methyltransferase to DNA (121); regulation of R-loops by TET proteins may also be mediated through this system. However, the detailed mechanism by which DNA methylation inhibits R-loop formation remains largely unknown and requires further investigation.
3.5 R-loop accumulation via activation of the NF-κB signaling pathway by human T-cell leukemia virus type 1
Acute T-cell lymphoma/leukemia (ATL) is a lymphoproliferative disease caused by infection of CD4-positive T lymphocytes with human T-cell leukemia virus type 1 (HTLV-1) retrovirus. A latent period of 30–50 years exists from infection to disease onset, and 5–10% of carriers develop the disease during their lifetime. Once ATL arises, the disease is intractable, with a 4-year survival rate of approximately 17% for the acute type (122). Since many ATL cells overexpress C-C chemokine receptor-4 (123), mogamulizumab, a monoclonal antibody drug targeting this receptor, was approved for the treatment of ATL in 2012 (124). Also, lenalidomide was approved in 2017 (125). HTLV-1 strongly activates the nuclear factor kappa light-chain–enhancer of activated B cells (NF-κB) pathway (126). In contrast, lenalidomide binds to cereblon and induces ubiquitination and subsequent proteasomal degradation of Ikaros and Aiolos, thereby inhibiting activation of the NF-κB pathway (127–129). Further application of such molecular-targeted therapies is expected to improve the outcomes of this disease.
Initial activation of NF-κB by HTLV-1 is considered to be mediated by Tax protein encoded by a Tax gene in the pX region of the HTLV-1 genome (130). However, given that cells infected with HTLV-1 and expressing Tax do not proliferate due to senescence (131) and that Tax expression is often transient, Tax-independent constitutive activation of NF-κB is thought to occur in ATL cells.
Recently, it was found that NF-κB activation in Tax-expressing cells leads to R-loop accumulation, that, in turn, induces DNA DSBs and senescence (131). During transcription, RNA polymerases activate the TC-NER pathway when transcription is paused at DNA damage sites, including R-loops (132, 133). He et al. showed that R-loops that accumulated via activation of NF-κB by Tax are processed into an ssDNA gap and further to a DSB by TC-NER factors, including XPF, XPG, and CSB, leading to the induction of senescence (Figure 10) (131). They further found that inactivation of the TC-NER pathway occurs in ATL cell lines and patient-derived ATL cells. This means that cells with accumulated genomic damage, including R-loops, are inhibited from proliferating by the TC-NER pathway, but, once the TC-NER pathway is suppressed, cells begin to proliferate despite the accumulated R-loops. However, the mechanism by which activation of the NF-κB pathway leads to increased R-loops is still elusive. Helicobacter pylori infection has been shown to activate NF-κB in gastric mucosal epithelial cells via the alpha kinase 1/TRAF interacting forkhead-associated protein A signaling pathway, which in turn increases R-loops (134). However, even in this case, the relationship between R-loop formation and NF-κB activation is unclear.
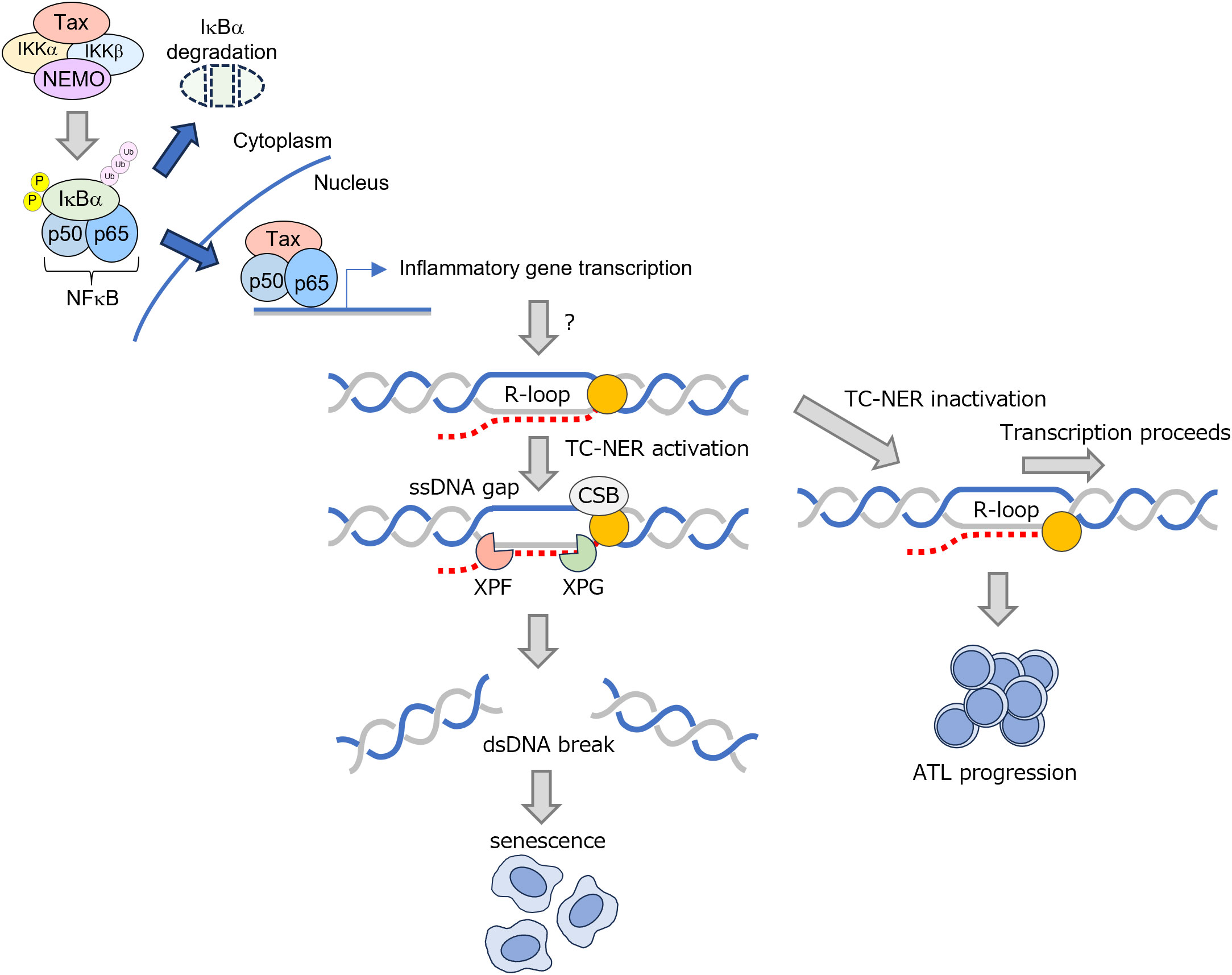
Figure 10 Increased R-loop formation mediated by Tax-induced inflammation and changes during ATL transition. Tax protein encoded by HTLV-I increases R-loops, which are eliminated by TC-NER. Upon progression to ATL, the TC-NER pathway is inactivated, allowing transcriptional elongation to proceed in the presence of R-loops. ATL, acute T-cell lymphoma/leukemia; CSB, conserved sequence block; dsDNA, double-stranded DNA; HTLV-1, human T-cell leukemia virus type I; IKK, Ikappa B kinase; NEMO, NF-kB essential modulator; ssDNA, single-stranded DNA; TC-NER, transcription-coupled nucleotide excision repair; XPF and XPG, TC-NER endonucleases.
4 Conclusion
This review summarizes recent papers on the formation and regulation of R-loops and the involvement of R-loop accumulation in hematopoietic neoplasms. Although the reasons for R-loop accumulation vary among cell types and diseases, DNA replication stress and subsequent DNA damage are undoubtedly involved in the pathophysiology of the hematopoietic neoplasms described in this paper. A recently published database, R-loopBase, comprehensively covers factors involved in R-loop regulation (135). While the specific associations of the factors in the database with hematopoietic neoplasms are not clear at this time, it remains a possibility that some factors listed in this database could be implicated in the pathogenesis of hematopoietic neoplasms. Also, new relationships with the disease phenotype of R-loop may emerge as research progresses.
It has also been shown that dysregulated activation of the innate immune signaling pathways occurs within MDS cells, which is associated with disease phenotypes of MDS (136, 137). However, the detailed mechanism by which this phenomenon occurs remains elusive. In this context, it has recently been reported that R-loops, which were originally thought to be processed only in the nucleus, are transported from the nucleus to the cytoplasm, leading to activation of the innate immune response (138, 139). This is of interest when considering the relationship between R-loop accumulation and disease phenotypes. McLemore et al. found that in MDS, R-loops formed through transcription by RNA polymerase III can be exported from the nucleus to cytoplasm. This, in turn, triggers an innate immune response mediated by a cGAS–STING–NOD-, LRR- and pyrin domain-containing protein 3 inflammasome pathway (138). They further suggest that this innate immune response causes degradation of GATA-binding factor 1 by caspase-1, resulting in a differentiation skewing of cells toward a granulocyte lineage. Crossley et al. also showed that a portion of each R-loops is cleaved by XPG and XPF endonucleases and is transported to the cytoplasm (139); the R-loops existing the cytoplasm are directly detected by cGAS and toll-like receptor 3, thus triggering an innate immune response. In addition, recent findings demonstrate that transcripts transcribed from transposon regions can activate an intracellular nucleic acid sensor, melanoma differentiation-associated protein 5, thus inducing innate immune responses (140). Coupled with the observed dysregulation of transposons in hematopoietic neoplasms, this mechanism may contribute to the excessive induction of innate immune responses observed in MDS and other hematopoietic neoplasms.
Moreover, excess R-loops in mtDNA have been implicated in the pathogenesis of diseases. Breast cancer type 2 susceptibility protein (BRCA2) is involved in the regulation of R-loop formation in the nucleus (141, 142). The suppression of BRCA2 expression has been shown to cause not only nuclear damage but also mitochondrial oxidative stress and replication abnormalities that lead to increased mitochondrial R-loops (143). Currently, mutations in the gene encoding RNase H1 have been implicated in mitochondrial encephalomyopathy (27). While the exact connection between the development of hematopoietic neoplasms and dysregulation of mitochondrial R-loop regulation is not yet fully understood, the observation of mitochondrial fragmentation in MDS (144), suggests a potential link. It is possible that accumulation of mitochondrial R-loops in hematopoietic neoplasms may also contribute to the formation of disease phenotypes.
In malignancies with increased R-loops, defects in DNA replication and consequent increased DNA damage are observed; the ATR signaling pathway, in particular, seems to be activated in response to these defects. Thus, further accumulation of R-loops, perhaps with agents directed against DNA damage response pathways, may be an attractive therapeutic target to control disease (74, 145). Additionally, although not yet confirmed in hematopoietic neoplasms, it has been demonstrated that PARP1 is implicated in the establishment of protein interaction networks involving R-loops, and inhibiting PARP1 results in an increase in R-loop levels (146). This implies that inhibiting PARP1 in diseases where R-loops accumulate may induce synthetic lethality, suggesting that targeting PARP1 in diseases characterized by R-loop accumulation could be a potential therapeutic approach in the future.
Author contributions
MH: Writing – original draft, Writing – review & editing. SS: Writing – review & editing. HM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Japanese Society for the Promotion of Science Grants-in-Aid for Scientific Research No. 22K17187 (MH) and No. 21K08419 (HM).
Acknowledgments
The authors would like to express our sincere appreciation to all those who supported our review, especially Ms. Michie Yoshikawa for her clerical work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. García-Muse T, Aguilera A. R loops: from physiological to pathological roles. Cell (2019) 179:604–18. doi: 10.1016/j.cell.2019.08.055
2. Niehrs C, Luke B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol (2020) 21:167–78. doi: 10.1038/s41580-019-0206-3
3. Yasuhara T, Kato R, Hagiwara Y, Shiotani B, Yamauchi M, Nakada S, et al. Human rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell (2018) 175:558–70.e11. doi: 10.1016/j.cell.2018.08.056
4. Xu C, Li C, Chen J, Xiong Y, Qiao Z, Fan P, et al. R-loop-dependent promoter-proximal termination ensures genome stability. Nature (2023) 621:610–9. doi: 10.1038/s41586-023-06515-5
5. Lockhart A, Pires VB, Bento F, Kellner V, Luke-Glaser S, Yakoub G, et al. RNase H1 and H2 are differentially regulated to process RNA-DNA hybrids. Cell Rep (2019) 29:2890–900.e5. doi: 10.1016/j.celrep.2019.10.108
6. Cristini A, Ricci G, Britton S, Salimbeni S, Huang SN, Marinello J, et al. Dual processing of R-loops and topoisomerase I induces transcription-dependent DNA double-strand breaks. Cell Rep (2019) 28:3167–81.e6. doi: 10.1016/j.celrep.2019.08.041
7. Goulielmaki E, Tsekrekou M, Batsiotos N, Ascensão-Ferreira M, Ledaki E, Stratigi K, et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat Commun (2021) 12:3153. doi: 10.1038/s41467-021-23505-1
8. Gómez-González B, García-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marín A, et al. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J (2011) 30:3106–19. doi: 10.1038/emboj.2011.206
9. McCann JL, Cristini A, Law EK, Lee SY, Tellier M, Carpenter MA, et al. APOBEC3B regulates R-loops and promotes transcription-associated mutagenesis in cancer. Nat Genet (2023) 55:1721–34. doi: 10.1038/s41588-023-01504-w
10. Zong C, Zhang Z, Gao L, He J, Wang Y, Li Q, et al. APOBEC3B coordinates R-loop to promote replication stress and sensitize cancer cells to ATR/Chk1 inhibitors. Cell Death Dis (2023) 14:348. doi: 10.1038/s41419-023-05867-0
11. Petermann E, Lan L, Zou L. Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat Rev Mol Cell Biol (2022) 23:521–40. doi: 10.1038/s41580-022-00474-x
12. Alecki C, Francis NJ. Identification of R-loop-forming sequences in drosophila melanogaster embryos and tissue culture cells using DRIP-seq. Bio Protoc (2021) 11:e4011. doi: 10.21769/BioProtoc.4011
13. König F, Schubert T, Längst G. The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PloS One (2017) 12:e0178875. doi: 10.1371/journal.pone.0178875
14. Smolka JA, Sanz LA, Hartono SR, Chédin F. Recognition of RNA by the S9.6 antibody creates pervasive artifacts when imaging RNA:DNA hybrids. J Cell Biol (2021) 220:e202004079. doi: 10.1083/jcb.202004079
15. Chen JY, Zhang X, Fu XD, Chen L. R-ChIP for genome-wide mapping of R-loops by using catalytically inactive RNASEH1. Nat Protoc (2019) 14:1661–85. doi: 10.1038/s41596-019-0154-6
16. Zhang X, Hao Y, Fu XD. Mapping R-loops using catalytically inactive RNaseH1 (R-chIP). Methods Mol Biol (2022) 2528:359–72. doi: 10.1007/978-1-0716-2477-7_24
17. Cerritelli SM, Sakhuja K, Crouch RJ. RNase H1, the gold standard for R-loop detection. Methods Mol Biol (2022) 2528:91–114. doi: 10.1007/978-1-0716-2477-7_7
18. Xu D, Huang Y, Luo L, Tang L, Lu M, Cao H, et al. Genome-wide profiling of endogenous single-stranded DNA using the SSiNGLe-P1 method. Int J Mol Sci (2023) 24:12062. doi: 10.3390/ijms241512062
20. Misic J, Milenkovic D, Al-Behadili A, Xie X, Jiang M, Jiang S, et al. Mammalian RNase H1 directs RNA primer formation for mtDNA replication initiation and is also necessary for mtDNA replication completion. Nucleic Acids Res (2022) 50:8749–66. doi: 10.1093/nar/gkac661
21. Fernández-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol (2003) 88:41–56. doi: 10.1113/eph8802514
22. Holmes JB, Akman G, Wood SR, Sakhuja K, Cerritelli SM, Moss C, et al. Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc Natl Acad Sci USA (2015) 112:9334–9. doi: 10.1073/pnas.1503653112
23. Posse V, Al-Behadili A, Uhler JP, Clausen AR, Reyes A, Zeviani M, et al. RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PloS Genet (2019) 15:e1007781. doi: 10.1371/journal.pgen.1007781
24. Pearce S, Nezich CL, Spinazzola A. Mitochondrial diseases: translation matters. Mol Cell Neurosci (2013) 55:1–12. doi: 10.1016/j.mcn.2012.08.013
25. Young MJ. Off-target effects of drugs that disrupt human mitochondrial DNA maintenance. Front Mol Biosci (2017) 4:74. doi: 10.3389/fmolb.2017.00074
26. Akman G, Desai R, Bailey LJ, Yasukawa T, Dalla Rosa I, Durigon R, et al. Pathological ribonuclease H1 causes R-loop depletion and aberrant DNA segregation in mitochondria. Proc Natl Acad Sci USA (2016) 113:E4276–85. doi: 10.1073/pnas.1600537113
27. Reyes A, Melchionda L, Nasca A, Carrara F, Lamantea E, Zanolini A, et al. RNASEH1 mutations impair mtDNA replication and cause adult-onset mitochondrial encephalomyopathy. Am J Hum Genet (2015) 97:186–93. doi: 10.1016/j.ajhg.2015.05.013
28. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu. Rev Immunol (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
29. Stavnezer J, Schrader CE. IgH chain class switch recombination: mechanism and regulation. J Immunol (2014) 193:5370–8. doi: 10.4049/jimmunol.1401849
30. Zhao H, Hartono SR, de Vera KMF, Yu Z, Satchi K, Zhao T, et al. Senataxin and RNase H2 act redundantly to suppress genome instability during class switch recombination. Elife (2022) 11:e78917. doi: 10.7554/eLife.78917
31. Engin AB, Engin A. The connection between cell fate and telomere. Adv Exp Med Biol (2021) 1275:71–100. doi: 10.1007/978-3-030-49844-3_3
32. Fernandes RV, Feretzaki M, Lingner J. The makings of TERRA R-loops at chromosome ends. Cell Cycle (2021) 20:1745–59. doi: 10.1080/15384101.2021.1962638
33. Pérez-Martínez L, Wagner T, Luke B. Telomere interacting proteins and TERRA regulation. Front Genet (2022) 13:872636. doi: 10.3389/fgene.2022.872636
34. Gong Y, Liu Y. R-loops at chromosome ends: from formation, regulation, and cellular consequence. Cancers (Basel) (2023) 15:2178. doi: 10.3390/cancers15072178
35. Gambelli A, Ferrando A, Boncristiani C, Schoeftner S. Regulation and function of R-loops at repetitive elements. Biochimie (2023) 214:141–55. doi: 10.1016/j.biochi.2023.08.013
36. Giunta S, Hervé S, White RR, Wilhelm T, Dumont M, Scelfo A, et al. CENP-A chromatin prevents replication stress at centromeres to avoid structural aneuploidy. Proc Natl Acad Sci USA (2021) 118:e2015634118. doi: 10.1073/pnas.2015634118
37. Racca C, Britton S, Hédouin S, Francastel C, Calsou P, Larminat F. BRCA1 prevents R-loop-associated centromeric instability. Cell Death Dis (2021) 12:896. doi: 10.1038/s41419-021-04189-3
38. Zhang C, Wang D, Hao Y, Wu S, Luo J, Xue Y, et al. LncRNA CCTT-mediated RNA-DNA and RNA-protein interactions facilitate the recruitment of CENP-C to centromeric DNA during kinetochore assembly. Mol Cell (2022) 82:4018–32.e9. doi: 10.1016/j.molcel.2022.09.022
39. Perea-Resa C, Blower MD. Centromere biology: transcription goes on stage. Mol Cell Biol (2018) 38:e00263–18. doi: 10.1128/mcb.00263-18
40. Vohhodina J, Goehring LJ, Liu B, Kong Q, Botchkarev VV Jr., Huynh M, et al. BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage. Nat Commun (2021) 12:3542. doi: 10.1038/s41467-021-23716-6
41. Feretzaki M, Pospisilova M, Valador Fernandes R, Lunardi T, Krejci L, Lingner J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature (2020) 587:303–8. doi: 10.1038/s41586-020-2815-6
42. Fernandes RV, Lingner J. The THO complex counteracts TERRA R-loop-mediated telomere fragility in telomerase+ cells and telomeric recombination in ALT+ cells. Nucleic Acids Res (2023) 51:6702–22. doi: 10.1093/nar/gkad448
43. Richard P, Manley JL. R loops and links to human disease. J Mol Biol (2017) 429:3168–80. doi: 10.1016/j.jmb.2016.08.031
44. Lee SY, Miller KM, Kim JJ. Clinical and mechanistic implications of R-loops in human leukemias. Int J Mol Sci (2023) 24:5966. doi: 10.3390/ijms24065966
45. Saez B, Walter MJ, Graubert TA. Splicing factor gene mutations in hematologic Malignancies. Blood (2017) 129:1260–9. doi: 10.1182/blood-2016-10-692400
46. Pellagatti A, Boultwood J. Splicing factor mutant myelodysplastic syndromes: Recent advances. Adv Biol. Regul (2020) 75:100655. doi: 10.1016/j.jbior.2019.100655
47. Will CL, Schneider C, MacMillan AM, Katopodis NF, Neubauer G, Wilm M, et al. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J (2001) 20:4536–46. doi: 10.1093/emboj/20.16.4536
48. Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, et al. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol Cell (2019) 76:82–95.e7. doi: 10.1016/j.molcel.2019.07.017
49. Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, Rabadan R. Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci USA (2020) 117:10305–12. doi: 10.1073/pnas.1922622117
50. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun (2016) 7:10615. doi: 10.1038/ncomms10615
51. Tang Q, Rodriguez-Santiago S, Wang J, Pu J, Yuste A, Gupta V, et al. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev (2016) 30:2710–23. doi: 10.1101/gad.291872.116
52. Wan Y, Wu CJ. SF3B1 mutations in chronic lymphocytic leukemia. Blood (2013) 121:4627–34. doi: 10.1182/blood-2013-02-427641
53. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365:2497–506. doi: 10.1056/NEJMoa1109016
54. Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discovery (2013) 3:1122–9. doi: 10.1158/2159-8290.Cd-13-0330
55. Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet (2013) 45:133–5. doi: 10.1038/ng.2523
56. Liu HX, Chew SL, Cartegni L, Zhang MQ, Krainer AR. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol (2000) 20:1063–71. doi: 10.1128/mcb.20.3.1063-1071.2000
57. Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science (2002) 297:1007–13. doi: 10.1126/science.1073774
58. Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Res (2006) 34:3494–510. doi: 10.1093/nar/gkl498
59. Masaki S, Ikeda S, Hata A, Shiozawa Y, Kon A, Ogawa S, et al. Myelodysplastic syndrome-associated SRSF2 mutations cause splicing changes by altering binding motif sequences. Front Genet (2019) 10:338. doi: 10.3389/fgene.2019.00338
60. Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci USA (2015) 112:E4726–34. doi: 10.1073/pnas.1514105112
61. Liang Y, Tebaldi T, Rejeski K, Joshi P, Stefani G, Taylor A, et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia (2018) 32:2659–71. doi: 10.1038/s41375-018-0152-7
62. Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3' splice site AG by the splicing factor U2AF35. Nature (1999) 402:832–5. doi: 10.1038/45590
63. Tronchère H, Wang J, Fu XD. A protein related to splicing factor U2AF35 that interacts with U2AF65 and SR proteins in splicing of pre-mRNA. Nature (1997) 388:397–400. doi: 10.1038/41137
64. Zhao Y, Cai W, Hua Y, Yang X, Zhou J. The biological and clinical consequences of RNA splicing factor U2AF1 mutation in myeloid Malignancies. Cancers (Basel) (2022) 14:4406. doi: 10.3390/cancers14184406
65. Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, et al. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing. In Vivo Cancer Cell (2015) 27:631–43. doi: 10.1016/j.ccell.2015.04.008
66. Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia (2015) 29:909–17. doi: 10.1038/leu.2014.303
67. Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, et al. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol Cell (2018) 69:412–25.e6. doi: 10.1016/j.molcel.2017.12.029
68. Singh S, Ahmed D, Dolatshad H, Tatwavedi D, Schulze U, Sanchi A, et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia (2020) 34:2525–30. doi: 10.1038/s41375-020-0753-9
69. Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, et al. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell (2013) 153:855–68. doi: 10.1016/j.cell.2013.04.028
70. Cheruiyot A, Li S, Nonavinkere S Srivatsan, Ahmed T, Chen Y, Lemacon DS, et al. Nonsense-mediated RNA decay is a unique vulnerability of cancer cells harboring SF3B1 or U2AF1 mutations. Cancer Res (2021) 81:4499–513. doi: 10.1158/0008-5472.Can-20-4016
71. Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O, Krainer AR. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev (2020) 34:413–27. doi: 10.1101/gad.332270.119
72. Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, et al. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res (2018) 78:5363–74. doi: 10.1158/0008-5472.Can-17-3970
73. Flach J, Jann JC, Knaflic A, Riabov V, Streuer A, Altrock E, et al. Replication stress signaling is a therapeutic target in myelodysplastic syndromes with splicing factor mutations. Haematologica (2021) 106:2906–17. doi: 10.3324/haematol.2020.254193
74. Rinaldi C, Pizzul P, Longhese MP, Bonetti D. Sensing R-loop-associated DNA damage to safeguard genome stability. Front Cell Dev Biol (2020) 8:618157. doi: 10.3389/fcell.2020.618157
75. Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell (2015) 27:658–70. doi: 10.1016/j.ccell.2015.03.017
76. Li P, White T, Xie W, Cui W, Peker D, Zeng G, et al. AML with germline DDX41 variants is a clinicopathologically distinct entity with an indolent clinical course and favorable outcome. Leukemia (2022) 36:664–74. doi: 10.1038/s41375-021-01404-0
77. Quesada AE, Routbort MJ, DiNardo CD, Bueso-Ramos CE, Kanagal-Shamanna R, Khoury JD, et al. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am. J Hematol (2019) 94:757–66. doi: 10.1002/ajh.25486
78. Makishima H, Saiki R, Nannya Y, Korotev S, Gurnari C, Takeda J, et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood (2023) 141:534–49. doi: 10.1182/blood.2022018221
79. Weinreb JT, Bowman TV. Clinical and mechanistic insights into the roles of DDX41 in haematological Malignancies. FEBS Lett (2022) 596:2736–45. doi: 10.1002/1873-3468.14487
80. Kim K, Ong F, Sasaki K. Current understanding of DDX41 mutations in myeloid neoplasms. Cancers (Basel) (2023) 15:344. doi: 10.3390/cancers15020344
81. Badar T, Chlon T. Germline and somatic defects in DDX41 and its impact on myeloid neoplasms. Curr Hematol Malig Rep (2022) 17:113–20. doi: 10.1007/s11899-022-00667-3
82. Shinriki S, Matsui H. Unique role of DDX41, a DEAD-box type RNA helicase, in hematopoiesis and leukemogenesis. Front Oncol (2022) 12:992340. doi: 10.3389/fonc.2022.992340
83. Singh RS, Vidhyasagar V, Yang S, Arna AB, Yadav M, Aggarwal A, et al. DDX41 is required for cGAS-STING activation against DNA virus infection. Cell Rep (2022) 39:110856. doi: 10.1016/j.celrep.2022.110856
84. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol (2011) 12:959–65. doi: 10.1038/ni.2091
85. Shinriki S, Hirayama M, Nagamachi A, Yokoyama A, Kawamura T, Kanai A, et al. DDX41 coordinates RNA splicing and transcriptional elongation to prevent DNA replication stress in hematopoietic cells. Leukemia (2022) 36:2605–20. doi: 10.1038/s41375-022-01708-9
86. Mosler T, Conte F, Longo GMC, Mikicic I, Kreim N, Möckel MM, et al. R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat Commun (2021) 12:7314. doi: 10.1038/s41467-021-27530-y
87. Weinreb JT, Ghazale N, Pradhan K, Gupta V, Potts KS, Tricomi B, et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev Cell (2021) 56:627–40.e5. doi: 10.1016/j.devcel.2021.02.006
88. Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet (2016) 48:1551–6. doi: 10.1038/ng.3709
89. Wen X, Tannukit S, Paine ML. TFIP11 interacts with mDEAH9, an RNA helicase involved in spliceosome disassembly. Int J Mol Sci (2008) 9:2105–13. doi: 10.3390/ijms9112105
90. Zhang J, Huang J, Xu K, Xing P, Huang Y, Liu Z, et al. DHX15 is involved in SUGP1-mediated RNA missplicing by mutant SF3B1 in cancer. Proc Natl Acad Sci USA (2022) 119:e2216712119. doi: 10.1073/pnas.2216712119
91. Maul-Newby HM, Amorello AN, Sharma T, Kim JH, Modena MS, Prichard BE, et al. A model for DHX15 mediated disassembly of A-complex spliceosomes. Rna (2022) 28:583–95. doi: 10.1261/rna.078977.121
92. Huang N, Song Y, Shi W, Guo J, Zhang Z, He Q, et al. DHX9-mediated pathway contributes to the Malignant phenotype of myelodysplastic syndromes. iScience (2023) 26:106962. doi: 10.1016/j.isci.2023.106962
93. Cristini A, Groh M, Kristiansen MS, Gromak N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep (2018) 23:1891–905. doi: 10.1016/j.celrep.2018.04.025
94. Rio-Machin A, Vulliamy T, Hug N, Walne A, Tawana K, Cardoso S, et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun (2020) 11:1044. doi: 10.1038/s41467-020-14829-5
95. Rivera OD, Mallory MJ, Quesnel-Vallières M, Chatrikhi R, Schultz DC, Carroll M, et al. Alternative splicing redefines landscape of commonly mutated genes in acute myeloid leukemia. Proc Natl Acad Sci USA (2021) 118:e2014967118. doi: 10.1073/pnas.2014967118
96. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet (2009) 10:691–703. doi: 10.1038/nrg2640
97. Kazazian HH Jr., Moran JV. Mobile DNA in health and disease. N Engl J Med (2017) 377:361–70. doi: 10.1056/NEJMra1510092
98. Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol (2011) 12:246–58. doi: 10.1038/nrm3089
99. Wang X, Ramat A, Simonelig M, Liu MF. Emerging roles and functional mechanisms of PIWI-interacting RNAs. Nat Rev Mol Cell Biol (2023) 24:123–41. doi: 10.1038/s41580-022-00528-0
100. Yao J, Xie M, Ma X, Song J, Wang Y, Xue X. PIWI-interacting RNAs in cancer: Biogenesis, function, and clinical significance. Front Oncol (2022) 12:965684. doi: 10.3389/fonc.2022.965684
101. Bamezai S, Pulikkottil AJ, Yadav T, Naidu VM, Mueller J, Mark J, et al. doi: 10.1182/blood.2022018651
102. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature (2011) 472:120–4. doi: 10.1038/nature09819
103. Ghafouri-Fard S, Dashti S, Taheri M. The HOTTIP (HOXA transcript at the distal tip) lncRNA: Review of oncogenic roles in human. BioMed Pharmacother (2020) 127:110158. doi: 10.1016/j.biopha.2020.110158
104. Luo H, Zhu G, Xu J, Lai Q, Yan B, Guo Y, et al. HOTTIP lncRNA promotes hematopoietic stem cell self-renewal leading to AML-like disease in mice. Cancer Cell (2019) 36:645–59.e8. doi: 10.1016/j.ccell.2019.10.011
105. Luo H, Zhu G, Eshelman MA, Fung TK, Lai Q, Wang F, et al. HOTTIP-dependent R-loop formation regulates CTCF boundary activity and TAD integrity in leukemia. Mol Cell (2022) 82:833–51.e11. doi: 10.1016/j.molcel.2022.01.014
106. Gong D, Wang L, Zhou H, Gao J, Zhang W, Zheng P. Long noncoding RNA Lnc530 localizes on R-loops and regulates R-loop formation and genomic stability in mouse embryonic stem cells. Stem Cell Rep (2023) 18:952–68. doi: 10.1016/j.stemcr.2023.02.003
107. Choudhary MNK, Quaid K, Xing X, Schmidt H, Wang T. Widespread contribution of transposable elements to the rewiring of mammalian 3D genomes. Nat Commun (2023) 14:634. doi: 10.1038/s41467-023-36364-9
108. Bai X, Li F, Zhang Z. A hypothetical model of trans-acting R-loops-mediated promoter-enhancer interactions by Alu elements. J Genet. Genomics (2021) 48:1007–19. doi: 10.1016/j.jgg.2021.07.005
109. Lio CJ, Yue X, Lopez-Moyado IF, Tahiliani M, Aravind L, Rao A. TET methylcytosine oxidases: new insights from a decade of research. J Biosci (2020) 45:21. doi: 10.1007/s12038-019-9973-4
110. Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, et al. 5-hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep (2016) 14:1283–92. doi: 10.1016/j.celrep.2016.01.035
111. Wang D, Wu W, Callen E, Pavani R, Zolnerowich N, Kodali S, et al. Active DNA demethylation promotes cell fate specification and the DNA damage response. Science (2022) 378:983–9. doi: 10.1126/science.add9838
112. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med (2009) 360:2289–301. doi: 10.1056/NEJMoa0810069
113. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia (2014) 28:241–7. doi: 10.1038/leu.2013.336
114. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet (2009) 41:838–42. doi: 10.1038/ng.391
115. Lio CJ, Yuita H, Rao A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid Malignancies. Blood (2019) 134:1487–97. doi: 10.1182/blood.2019791475
116. Lemonnier F, Couronné L, Parrens M, Jaïs JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood (2012) 120:1466–9. doi: 10.1182/blood-2012-02-408542
117. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
118. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
119. Shukla V, Samaniego-Castruita D, Dong Z, González-Avalos E, Yan Q, Sarma K, et al. TET deficiency perturbs mature B cell homeostasis and promotes oncogenesis associated with accumulation of G-quadruplex and R-loop structures. Nat Immunol (2022) 23:99–108. doi: 10.1038/s41590-021-01087-w
120. Marchena-Cruz E, Camino LP, Bhandari J, Silva S, Marqueta-Gracia JJ, Amdeen SA, et al. DDX47, MeCP2, and other functionally heterogeneous factors protect cells from harmful R loops. Cell Rep (2023) 42:112148. doi: 10.1016/j.celrep.2023.112148
121. Grunseich C, Wang IX, Watts JA, Burdick JT, Guber RD, Zhu Z, et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Mol Cell (2018) 69:426–37.e7. doi: 10.1016/j.molcel.2017.12.030
122. Imaizumi Y, Iwanaga M, Nosaka K, Ishitsuka K, Ishizawa K, Ito S, et al. Prognosis of patients with adult T-cell leukemia/lymphoma in Japan: A nationwide hospital-based study. Cancer Sci (2020) 111:4567–80. doi: 10.1111/cas.14658
123. Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, Izawa D, et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood (2002) 99:1505–11. doi: 10.1182/blood.v99.5.1505
124. Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol (2014) 32:1157–63. doi: 10.1200/jco.2013.52.0924
125. Ishida T, Fujiwara H, Nosaka K, Taira N, Abe Y, Imaizumi Y, et al. Multicenter phase II study of lenalidomide in relapsed or recurrent adult T-cell leukemia/lymphoma: ATLL-002. J Clin Oncol (2016) 34:4086–93. doi: 10.1200/jco.2016.67.7732
126. Leung K, Nabel GJ. HTLV-1 transactivator induces interleukin-2 receptor expression through an NF-kappa B-like factor. Nature (1988) 333:776–8. doi: 10.1038/333776a0
127. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science (2010) 327:1345–50. doi: 10.1126/science.1177319
128. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science (2014) 343:305–9. doi: 10.1126/science.1244917
129. Fink EC, Ebert BL. The novel mechanism of lenalidomide activity. Blood (2015) 126:2366–9. doi: 10.1182/blood-2015-07-567958
130. Giam CZ, Jeang KT. HTLV-1 Tax and adult T-cell leukemia. Front Biosci (2007) 12:1496–507. doi: 10.2741/2163
131. He Y, Pasupala N, Zhi H, Dorjbal B, Hussain I, Shih HM, et al. NF-κB-induced R-loop accumulation and DNA damage select for nucleotide excision repair deficiencies in adult T cell leukemia. Proc Natl Acad Sci USA (2021) 118:e2005568118. doi: 10.1073/pnas.2005568118
132. Mullenders L. DNA damage mediated transcription arrest: Step back to go forward. DNA Repair (Amst) (2015) 36:28–35. doi: 10.1016/j.dnarep.2015.09.005
133. Duan M, Speer RM, Ulibarri J, Liu KJ, Mao P. Transcription-coupled nucleotide excision repair: New insights revealed by genomic approaches. DNA Repair (Amst) (2021) 103:103126. doi: 10.1016/j.dnarep.2021.103126
134. Bauer M, Nascakova Z, Mihai AI, Cheng PF, Levesque MP, Lampart S, et al. The ALPK1/TIFA/NF-κB axis links a bacterial carcinogen to R-loop-induced replication stress. Nat Commun (2020) 11:5117. doi: 10.1038/s41467-020-18857-z
135. Lin R, Zhong X, Zhou Y, Geng H, Hu Q, Huang Z, et al. R-loopBase: a knowledgebase for genome-wide R-loop formation and regulation. Nucleic Acids Res (2022) 50:D303–d15. doi: 10.1093/nar/gkab1103
136. Gañán-Gómez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia (2015) 29:1458–69. doi: 10.1038/leu.2015.69
137. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med (2021) 218:e20201544. doi: 10.1084/jem.20201544
138. McLemore AF, Hou HA, Meyer BS, Lam NB, Ward GA, Aldrich AL, et al. Somatic gene mutations expose cytoplasmic DNA to co-opt the cGAS/STING/NLRP3 axis in myelodysplastic syndromes. JCI Insight (2022) 7:e159430. doi: 10.1172/jci.insight.159430
139. Crossley MP, Song C, Bocek MJ, Choi JH, Kousorous J, Sathirachinda A, et al. R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature (2023) 613:187–94. doi: 10.1038/s41586-022-05545-9
140. Clapes T, Polyzou A, Prater P, Sagar, Morales-Hernández A, Ferrarini MG, et al. Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration. Nat Cell Biol (2021) 23:704–17. doi: 10.1038/s41556-021-00707-9
141. Shivji MKK, Renaudin X, Williams ÇH, Venkitaraman AR. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep (2018) 22:1031–9. doi: 10.1016/j.celrep.2017.12.086
142. Wang Y, Ma B, Liu X, Gao G, Che Z, Fan M, et al. ZFP281-BRCA2 prevents R-loop accumulation during DNA replication. Nat Commun (2022) 13:3493. doi: 10.1038/s41467-022-31211-9
143. Renaudin X, Lee M, Shehata M, Surmann EM, Venkitaraman AR. BRCA2 deficiency reveals that oxidative stress impairs RNaseH1 function to cripple mitochondrial DNA maintenance. Cell Rep (2021) 36:109478. doi: 10.1016/j.celrep.2021.109478
144. Aoyagi Y, Hayashi Y, Harada Y, Choi K, Matsunuma N, Sadato D, et al. Mitochondrial fragmentation triggers ineffective hematopoiesis in myelodysplastic syndromes. Cancer Discovery (2022) 12:250–69. doi: 10.1158/2159-8290.Cd-21-0032
145. Kim JJ, Lee SY, Gong F, Battenhouse AM, Boutz DR, Bashyal A, et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev (2019) 33:1751–74. doi: 10.1101/gad.331231.119
Keywords: DNA methylation, long noncoding RNA, lymphoid neoplasms, myeloid neoplasms, Piwil4, R-loop, RNA splicing
Citation: Hirayama M, Shinriki S and Matsui H (2023) R-loops in normal and malignant hematopoiesis. Front. Hematol. 2:1297657. doi: 10.3389/frhem.2023.1297657
Received: 20 September 2023; Accepted: 05 December 2023;
Published: 19 December 2023.
Edited by:
Emanuele Azzoni, University of Milano Bicocca, ItalyReviewed by:
Anastasia Conti, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), ItalyDiu T. T. Nguyen, Queen Mary University of London, United Kingdom
Copyright © 2023 Hirayama, Shinriki and Matsui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hirotaka Matsui, aG1hdHN1aUBuY2MuZ28uanA=