 Suparno Chakrabarti
Suparno Chakrabarti Sarita Rani Jaiswal
Sarita Rani Jaiswal- 1Cellular Therapy and Immunology, Manashi Chakrabarti Foundation, New Delhi, India
- 2Department of Blood and Marrow Transplantation, Dharamshila Narayana Super-Speciality Hospital, New Delhi, India
Despite advances in transplantation techniques and immunosuppressive therapies, graft-versus-host disease (GVHD) remains a significant cause of morbidity and mortality, necessitating the use of innovative strategies for its prevention. T-cell activation plays a crucial role in the pathogenesis of GVHD, and T-cell costimulation blockade (COSBL) has emerged as a promising approach to prevent this devastating condition. This review aims to explore the concept of COSBL and its potential as a paradigm-shifting strategy in the prevention of GVHD, in the context of the existing modalities for the prevention of GVHD and the preclinical and clinical studies on COSBL. The unique property of abatacept (CTLA4Ig) is not just limited to dampening T-cell activation. The salutary effect of abatacept on natural killer (NK) cells and Tregs alike provides a unique opportunity to dissociate T-cell-mediated GVHD from NK cell-mediated graft-versus-leukemia. Further research is warranted to explore other modalities of COSBL, determine the optimal dosing and combinations for COSBL, and identify predictive biomarkers for patient stratification, ultimately paving the way for improved outcomes in hematopoietic cell transplantation recipients.
1 Introduction
Despite several advances in the field of medicine, allogeneic hematopoietic cell transplantation (HCT) remains the only curative option for a vast majority of hematological disorders, both benign and malignant. Since its successful clinical implementation in 1969, courtesy of a series of diligent canine experiments carried out by Thomas and colleagues, certain paradigms established at the outset remain the cornerstone of the success in the field of HCT (1–7). One such paradigm is the inevitable immunological confrontation between immune cells of the donor graft and those of the host, with the ultimate goal being the establishment of immune tolerance. Although the process of conditioning directed at myeloablation and immunosuppression could result in successful engraftment of donor hematopoietic stem cells (HSCs) in the majority of recipients who are fully matched at major histocompatibility complex (MHC) class 1 and 2 genes of the donor, the donor lymphocytes accompanying the graft would invariably mount an alloreactive response directed at the mismatched minor histocompatibility antigens of the host, manifesting as graft-versus-host disease (GVHD). Pharmacological interventions directed at the prevention of activation and the proliferation of alloreactive donor T lymphocytes were deemed to be essential, as evident from canine studies (7). Thus, the pharmacological prevention of GVHD was established as an inseparable component of any allogeneic HCT protocol employing unmanipulated donor graft.
2 Preventing GVHD—an unfulfilled promise
2.1 Pathogenesis—the more we know…
There are three absolute requirements in the occurrence of acute GVHD, as described by Billingham in 1966 (8). First is the presence of immunologically competent cells in the graft, the second is the disparity in the major or minor histocompatibility antigens between the donor and the host, and the third is the inability of the host to eliminate the alloreactive immunocompetent cells of the graft. GVHD is typically viewed as the result of a three-step process (9, 10), wherein conditioning regimen-induced tissue damage and disruption of gut microbes results in the release of damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). This results in an inflammatory cascade wherein the antigen-presenting cells (APCs) in the host are matured and activated, enabling the presentation of host antigens via activated host APCs to the alloreactive T cells in the graft. Antigen recognition is the prerequisite, but not sufficient, for T-cell activation. Additional costimulatory signals are essential to the initiation of T-cell activation. Once activated, the alloreactive T cells home to the target organs, such as the skin, gut, and liver, where the abundance of APCs and host antigens result in a cascading process of T-cell activation, proliferation, and tissue damage, which can be either cytokine mediated or directly cytotoxic by way of cytotoxic T cells (CTL) (10). Once the alloreactive process is let loose in its full vigor, restoration of normal T-cell homeostasis, as witnessed in the case of pathogen-mediated T-cell activation, rarely happens. The abatement of this process almost always requires a pharmacological intervention and might proceed uncontrolled despite every available intervention being implemented, resulting in a fatal outcome. Such is the power of uncontrolled alloreactivity. On the other hand, donor T cells are the key effectors of the antitumor effect directed against leukemia or the underlying malignancy following HCT. This is termed as the graft-versus-leukemia/tumor (GVL/GVT) response. The GVL effect is often inseparable from a generalized GVH effect. The past 50 years have seen untiring efforts from transplant immunologists to separate the GVL effect from GVHD, but with little success. Although this might be the paradigm in the approach to HCT in malignant diseases, one can afford to focus less on the preservation of a GVL effect in non-malignant diseases (NMDs). However, aggressive attempts at the elimination of GVHD in NMD patients are often complicated by delayed immune reconstitution and serious opportunistic infection. This puts in context the primacy of an effective GVHD prophylaxis protocol in allogeneic HCT, which does not compromise the GVL effect or adversely affect immune reconstitution.
3 Pharmacological prevention—the choice is not easy
Corticosteroids have been the agent of choice in subduing inflammation and lymphocyte activation and were considered for GVHD prophylaxis in the early days. However, the introduction of calcineurin inhibitors (CNIs), such as cyclosporine (CSA), a cyclic peptide derived from a soil fungus in 1969, combined with a short course of intermittent low-dose methotrexate (MTX), a folate antagonist used as an anticancer agent, was shown to be superior to either drug alone and became the gold standard of GVHD prophylaxis (11, 12). The incidence of grade 2–4 acute GVHD ranged from 30% to 50%, depending on the donor source, and the incidence of chronic GVHD ranged from 50% to 70%.
Several modifications to this approach have been attempted over the last five decades. Tacrolimus (FK506), a fungus-derived macrolide antibiotic found to prevent T-cell activation via the NFAT pathway, which is similar to CSA, was introduced in the 1980s, with its potency being 100 times that of CSA. At the same time, another fungus-derived product, mycophenolate mofetil (MMF), was found to be a potent inhibitor of T and B cells via the inhibition of purine metabolism pathways. Tacrolimus has been employed in combination with either MMF or MTX in related and unrelated HCT, with no significant reduction in the incidence of either acute or chronic GVHD as compared with CSA/MTX (13). Another derivative from a fungal species, sirolimus, was found to be an immunosuppressant (ISA) by coincidence. Sirolimus bears structural similarities to FK506 and often binds to similar intracellular proteins. Yet, sirolimus stands out by dint of its effect through distinct pathways. Sirolimus dampens signaling through the mammalian target of rapamycin (mTOR) pathway in effector T cells (Teffs) and restricts the cell cycle pathways; however, the availability of alternate pathways, via phosphatidylinositol mannoside 2 (PIM2) and STAT5 for cell cycle and metabolism, allow the proliferation of Tregs (14). However, none of these drugs in combination was significantly superior to CSA/MTX, and the combinations of these agents are often guided by the host comorbidities vis-à-vis the toxicity profile. For example, sirolimus, when used with myeloablative doses of busulfan, increased the risk of hepatic sinusoidal obstruction syndrome (SOS), and there was an increase in the risk of thrombotic microangiopathy when tacrolimus and sirolimus were combined (15, 16).
4 T-cell depletion—one way or the other
Antibodies against T cells, either in the form of polyclonal horse- or rabbit-derived antibodies, known as antithymocyte globulin (ATG) (17, 18), or monoclonal antibodies, such as alemtuzumab (anti-CD52) (19) or OKT3 (anti-CD3) (20), have been used both for host immunosuppression and/or lymphodepletion of the infused graft in vivo. Despite numerous clinical studies showing a greater reduction in both acute and chronic GVHD, the dosing of such agents remains empirical and outcomes remain unpredictable. The concerns that are associated with the use of ATG or alemtuzumab are the risks of serious viral reactivations and, in cases of malignant diseases, relapse (21, 22).
A more predictable and measurable approach to the elimination of donor-derived T lymphocytes has been physical manipulation of the graft with depletion of T lymphocytes or its subsets and/or selection of CD34+ HSCs (23). With these processes being much refined over the last two decades, the immunomagnetic depletion of TCRαβ+ or CD45RA+ T cells from the graft remain the most common approach to graft manipulation (24). Such approaches have drastically reduced the incidence of acute and chronic GVHD, but often at the cost of morbidity and mortality arising out of viral reactivations (22). Moreover, T-cell depletion of the graft might be a preferred option in NMDs, but relapse remains a major concern in those with advanced malignancies (25).
5 Post-transplantation cyclophosphamide—simple yet elegant
The existing approaches to serotherapy or immunomagnetic depletion of T cells are largely non-selective if the aim of the process is to restrict the alloreactive T-cell population alone. In a series of preclinical studies, groups from Japan and Johns Hopkins University in the USA have shown that high-dose cyclophosphamide (Cy), if administered 2–3 days after graft infusion, can result in long-term tolerance in mismatched mice models (26, 27). Cy and its active metabolite are rapidly metabolized by aldehyde dehydrogenase (ALDH) and cells that lack ALDH are far more susceptible to Cy-induced death. On the other hand, cells endowed with high amounts of ALDH are inherently protected from Cy-induced damage. In general, quiescent cells are Cy-resistant as they possess sufficient amounts of ALDH, and rapidly proliferating cells remain susceptible due to the scarcity of the enzyme (28). Following infusion of the graft in the absence of pharmacological immunosuppression as in standard GVHD prophylaxis, alloreactive T cells, both from the donor graft and the host, unaffected by the conditioning regimen, would be activated and proliferate in such an unhinged immune-enabling environment. This proliferation peaks at 72 h and if Cy is administered around this time, they might spare the non-alloreactive T cells and HSCs and selectively target proliferating alloreactive T cells of both donor and host origin.
Based on this principle, the Johns Hopkins University group initiated a clinical trial with non-myeloablative conditioning followed by two doses of post-transplantation Cy (PTCy) on days +3 and +4, followed by FK506 and MMF, in patients receiving a HLA-haploidentical family donor (HFD) graft. The results were astounding in terms of both engraftment and GVHD, with over 90% of patients having sustained engraftment and less than 20% of patients experiencing severe GVHD, acute or chronic (29, 30). Subsequently, PTCy-based approaches have become the standard of care in HFD-HCT, and are currently expanding their sway in matched and mismatched unrelated donor (UD) HCT (31).
6 Has PTCy ended five decades of strife?
With a unique mechanism of action, along with ease of access and delivery, PTCy seems to have broken the glass ceiling in alternative-donor HCT, particularly those with HLA-mismatched donors, both related and unrelated. However, delayed engraftment, infectious complications, high-dose Cy-associated bladder and cardiac toxicities, and the increased risk of disease relapse remain some of the major concerns associated with the PTCy approach. Although PTCy has definitely made HFD-HCT a global possibility, severe GVHD or other forms of alloreactivity remain a concern, particularly in children with NMDs who have been heavily transfused (32). Apart from GVHD, aberrant alloreactivity manifests as hemophagocytic syndrome (HPS) or macrophage activation syndrome (MAS) in the immediate post-transplant period in children with NMDs receiving HFD-HCT. Our group was the first to report a high incidence of post-transplant HPS (PTHPS) following HFD-HCT in children, in addition to early-onset severe acute GVHD (33). We had postulated that GVHD and HPS were two ends of the spectrum of T-cell alloreactivity. When T-cell cytotoxicity is dominant, the manifestation is that of classic GVHD, but abortive cytotoxicity in the face of APC-driven alloreactivity manifests primarily as PTHPS. Both conditions, if manifesting early and progressing unabated, result in high rates of mortality.
It was hypothesized that children possibly metabolized Cy differently than adults and PTCy along with CNI/MMF might not suffice as an optimum GVHD prophylaxis regimen in children with NMDs undergoing HFD-HCT (32, 34, 35).
7 Unraveling the T-cell costimulation pathway, and a serendipitous discovery
An understanding of T-cell biology effectively began in the 1980s, with recognition of the T-cell receptor (TCR) complex as the putative protein critical to ascribing specificity in antigen recognition. However, contemporaneous studies also highlighted the insufficiency of antigen recognition as the sole trigger for T-cell activation. It was realized that if antigen recognition is the first step in the process of T-cell activation, a second step is necessary in imparting fruition to the process (36). This was recognized as the “costimulation pathway”, without whose engagement T cells are not activated, despite recognition and ligation with the cognate antigen taking place. With the advent of monoclonal technology in the 1980s, a search for costimulatory molecules brought the CD28 receptor into the limelight, the extracellular domain of which was homologous to the immunoglobulin (Ig)V region. CD28 was found to be constitutively expressed in both CD4+ and CD8+ T cells (37). The ligands of CD28 were soon identified on APCs and named B7/BB-1 or B7-1 (later named CD80) (38, 39). Interestingly, antibodies directed at CD80 failed to prevent T-cell activation, leading to speculation regarding the existence of other ligands for CD28. This prompted further studies and the identification of another ligand B7-2 (CD86) (40). Similar to CD28, CD86 is constitutively expressed on APCs and is responsible for initiation of the costimulation process. CD80, on the other hand, is induced on activation of APCs and is involved more in the sustenance and modulation of the T-cell activation process.
Antigen recognition and costimulation could result in a process of uncontrolled activation, which is not what happens in physiological conditions. Hence, a pathway for putting the brakes on the process of T-cell activation and proliferation had to exist. Moreover, soluble CD28 fusion proteins developed at the time were found to be of low affinity, making it unfeasible as a therapeutic proposition, as immense amounts of antibody would be required for any meaningful biological activity (41).
A molecule homologous to CD28 was discovered on screening the murine cytolytic T-cell library. The gene for this molecule, named cytotoxic T-lymphocyte-associated antigen protein 4 (CTLA4), was mapped to the same chromosomal region as CD28 and found to be expressed on activated T cells. The close resemblance to CD28 in every aspect prompted Linsley and colleagues from the BMS Research Institute to create a fusion protein consisting of an extracellular domain of CTLA4 and an IgG1 heavy chain (42). They found that this fusion protein, named CTLA4Ig, had a much higher affinity and binding avidity to B7 ligands than CD28 and was a potent inhibitor of immune responses involving T and B lymphocytes. The function of CTLA4 was yet to be discerned, but the potential of CTLA4Ig to block the CD28-B7 pathway was recognized.
8 CTLA4 and the coinhibitory pathways—when saying “NO” is not enough
The interaction between CD28 and B7 ligands results in the upregulation of the PI3K pathway, the antiapoptotic pathway, and increased IL2 production. CTLA4 was found to bind to the same ligands with 20 times greater avidity than CD28, resulting in abrogation of the activation pathway, thus tempering the process of T-cell activation and proliferation (43, 44). However, in contrast to CD28, CTLA4 is not constitutively expressed on conventional T cells (Tconv) and is upregulated only on the ligation of CD28 to B7 ligands. This is probably due its higher binding affinity to B7 ligands and the resultant competitive displacement of CD28, as CTLA4 is internalized rapidly following cell surface expression (45).
In addition to the competitive inhibition of CD28-B7 pathway, CTLA4 was found to act in a unique cell-extrinsic and cell-intrinsic manner (46). CD28 was found to mediate the trogocytosis of CD80 and CD86 along with MHC class 2 peptides to the T-cell surface and provide stimulation to fellow T cells in a cis manner. This process results in the autostimulation of T cells independent of APCs (47). CTLA4 can inherently interrupt this process by the trans-endocytosis of B7 molecules on APCs, in addition to molecules trogocytosed via the CD28–B7 interaction (47, 48). These pathways, which have been uncovered recently, focus on the critical role of CTLA4 in the process of T-cell regulation.
Although we have focused primarily on the CD28/CTLA4-B7 pathways, there are several other costimulatory and coinhibitory pathways. We shall discuss the relevance and therapeutic implication of each pathway in the clinical context in subsequent sections.
9 Costimulation blockade and transplantation tolerance— the other story
As we have discussed earlier, T-cell activation is a two-step process. Antigen-primed T cells, in the absence of costimulation, become anergic to the specific antigen. Hence, COSBL excited transplant immunologists in pursuit of lasting transplant tolerance. The serendipitous discovery of CTLA4Ig allowed overcoming the disappointment of CD28-directed antibodies. Early studies in xenogeneic models showed long-term allograft survival with CTLA4Ig treatment (49, 50). This initial hype was tempered by later studies showing COSBL-resistant graft rejection, particularly in non-human primates (51). This called for the engagement of other costimulatory pathways in the act of transplant tolerance.
Simultaneously, and subsequently, several other costimulation pathways were discovered. The most studied of them is the CD40-CD40L (CD154) pathway, where CD40 is constitutively expressed on APCs, including B cells and the ligand, and CD154 is expressed predominantly on CD4+ T cells, resulting in the reverse costimulation of the APCs (52, 53). This pathway is the prime driver of T-cell-dependent B-cell maturation, and, hence, deemed to be an attractive target in solid organ transplantation (SOT). Although blocking the CD40-CD154 pathway alone prevented acute rejection in organ transplantation models, it did not result in durable tolerance (54). This was, however, achieved by the co-administration of CTLA4Ig with or without rapamycin (55, 56). The encouraging preclinical findings prompted introduction of anti-CD154 antibodies in a phase 1 clinical trial. Unfortunately, serious thromboembolic complications were noted, resulting in the abandonment of the trial (57). It was realized that CD154 is also expressed on activated platelets with CD40 being constitutively expressed on the endothelia, which resulted in collateral damage (58).
Most of the costimulatory molecules discovered belong to either the Ig superfamily or the tumor necrosis factor (TNF) receptor (TNFR) superfamily. Inducible costimulatory molecule (ICOS), OX-40 (CD134), and 41BB (CD137), all belonging to the TNFR group, are inducible receptors on activated T cells and/or memory T cells (59). These pathways synergize with the CD28-B7 pathway in upregulating T-cell activation and/or B-cell maturation along with class switch recombination. 41BB activates CD8 T cells in a CD28-B7-independent manner, and all these costimulation pathways seem to drive CTLA4Ig-resistant T-cell activation (60).
10 CTLA4Ig and the dilemma of anergy, tolerance, and regulatory suppression
Although the terms anergy and tolerance are often used interchangeably, they are distinct functional states. The classic example of anergy is the absence of costimulation following binding to the cognate antigen, as in classic COSBL with monoclonal antibodies (61). This is believed to be the primary modus operandi of CTLA4Ig. Anergy is a dysfunctional state akin to functional paralysis of T-cell function, which does not require persistence of the antigen and can be reversed via IL2 and restoration of the PI3/AKT/NFAT pathway (62). Adaptive tolerance, on the other hand, is a more permanent state of antigen-specific unresponsiveness, which is not reversible with IL2 and does require persistence of the antigen. Thus, COSBL combined with CTLA4Ig leads to a state of anergy that might prevent and delay graft rejection in SOT, but it is not the same as tolerance (63). Even though a state of anergy is induced by COSBL, conversion to a state of tolerance would require invocation of the regulatory pathways. Despite identification of several regulatory pathways, the one involving regulatory T cells (Tregs) remains the most widely understood.
Tregs are best described as thymic or natural (tTregs) and peripheral or induced (pTregs). The tTregs arise out of thymic selection, where CD4+T cells with intermediate affinity to self-antigens go on to constitutively express a high-affinity IL2 receptor (CD25) and forkhead box protein 3 (FoxP3), which are defining characteristics (64, 65). On the other hand, CD4+CD25+ T cells can convert to CD4+CD25+FoxP3+ T cells, which are referred to as pTregs, on antigen and IL2 stimulation in an anti-inflammatory cytokine milieu of IL10, and transforming growth factor beta (TGFβ) (66).
When CTLA4Ig blocks the CD28-B pathway in Tconv, CTLA4 expression is not induced and the state of anergy does not convert to a state of tolerance due to a lack of CTLA4 binding with B7 ligands. Unlike in Tconv, where CTLA4 is induced following T-cell activation via the CD28-B7 pathway, it is constitutively expressed on Tregs, and this determines and often initiates the regulatory cascade (67). However, PD1 is also upregulated in Tregs and the PD1-PDL1 pathway takes primacy when CTLA4 ligation is attenuated due to occupation of B7 ligands by CTLA4Ig (68). Tregs mirror Tconvs in terms of receptor expression and antigen-activated effector phenotypes, but the functions vary widely (69). In fact, the functional implications of many of these pathways remain poorly understood.
It is a widely held view that the survival of Tregs depends on the availability of IL2, and in an environment where availability of IL2 is limited, Tregs mop up all available IL2, starving the Teffs (70). It is indeed ironic that the apoptosis of Tregs in an IL2-depleted environment is driven by FoxP3, the defining transcription factor (71). Thus, the thymic microenvironment only supports CD4+FoxP3+ cells which have a strong expression of CD25. The situation is further complicated by the plasticity of Tregs in the periphery, where the balance between IL6 and TGFβ decides the functional fate, that is whether CD4+CD25+ T cells will take a Th17 or Treg pathway (72).
The CD28/CTLA4-B7 and PD1-PDL1 axes both play critical and differential roles in functions of Tconv and Tregs (73). CD28 ligation is critical to the activation of the majority of CD4+ and most CD8+ T cells. However, this pathway is not an absolute requirement for Treg function, except during the initial stage of thymic selection. CTLA4 on Tregs plays a critical role both in the competitive inhibition of CD28-B7 pathway for Tconvs and the depletion of B7 molecules and MHC class II-derived peptides from the APC surface via-endocytosis (47, 74). PD1-PDL1 is a coinhibitory pathway for Teff cells and PD1 is upregulated on Teffs following chronic antigen exposure. In contrast, in Tregs, the upregulation of PD1 is critical both to its survival and suppressive activity, particularly when it is exposed to CTLA4Ig, that is, when natural CTLA4 and B7 ligation is inhibited (68). PD1 expression on Tregs was associated with reduced apoptosis and greater suppression in presence of low-dose IL2 (75). In fact, higher levels of expression of PD1 on Tregs was associated with an improved clinical response in patients with chronic GVHD treated with low-dose IL2. In addition, PD1 expression on Tregs was found to be critical in allograft tolerance in CTLA4Ig-mediated COSBL blockade (68). Preclinical models have demonstrated a synergistic effect of CTLA4, PD1, and ICOS on Treg-mediated suppression of Teffs (68, 76).
Finally, it has been demonstrated in recent times that CD80 and PDL1 molecules on APC surfaces can form heterodimers in a cis interaction. This makes both molecules unavailable to the respective inhibitory receptors CTLA4 and PD1. Interestingly, CD80 can engage with CD28 in its heterodimerized form and simultaneously protects itself from trans-endocytosis by CTLA4 (77). This rescues activated Teff from inhibitory pathways. However, CTLA4Ig can effectively bind to CD80 and prevent its dimerization with PDL1, thus preventing CD28-CD80 ligation and facilitating PD1–PDL1 interaction at the same time.
CTLA4Ig has led to divergent outcomes in experimental mice models. For example, in NOD mice, exposure to CTLA4Ig has resulted in the earlier onset of diabetes and concomitant reduction in Tregs (78, 79). The same has been reported in preclinical SOT models (80). This might be attributable to the blockade of CD28-mediated costimulation, which has been shown to induce the chromatin modifying enzyme, enhancer of zeste homolog 2 (Ezh2). Ezh2 has been shown to play a key role in the stability of Tregs post activation in knockout mice models (81). The CD28 pathway has also been shown to be critical for peripheral homeostasis of tTregs (82). Thus, it is possible that by blocking the CD28 pathway, CTLA4Ig might inadvertently compromise the stability of tTregs and result in adverse outcomes in certain autoimmune diseases (AIDs) and SOTs.
This anti-Treg effect of CTLA4Ig is probably limited to tTregs, as a substantial body of evidence demonstrates the induction of pTregs by CTLA4Ig (83). This differential effect probably arises due to the fact that, in contrast to tTregs, pTregs are not entirely dependent on CD28 ligation (84). CTLA4Ig has been shown to convert CD4+CD25+ T cells to FoxP3+CD4+CD25+ Tregs in murine lymph nodes (83). TCR ligation is a prerequisite for this process and it is APC dependent, mediated primarily via CD86 and not CD80. This process is also not dependent on high doses of IL2. Although, this study did not find this process to be dependent on TGFβ, another study demonstrated that the suppressive effect of CTLA4Ig on Teff is dependent on the presence of both Tregs and TGFβ (85). Furthermore, CTLA4-Ig monotherapy in a fully mismatched heart transplant model (BALB/c onto C57BL/6), administered on days 0, 4, 14, and 28 and every 4 weeks thereafter was shown to be dependent on Tregs to maintain its suppressive effect at a dose of 10 mg/kg (86).
Thus, anergy which is achieved by CTLA4Ig exposure, can translate to durable tolerance only in the presence of continued Treg-mediated suppression in an antigen-specific manner.
11 CTLA4Ig—from bench to bedside
Durable tolerance in the absence of continued immunosuppression has been the holy grail of SOT. The concept of COSBL was indeed appealing in terms of the prevention of T-cell activation and graft rejection. Although long-term graft survival was achievable in smaller animals with CTLA4Ig, this was not quite achieved in non-human primates (51). Combined therapy with CTLA4Ig and anti-CD154 antibodies seemed to provide superior results than either alone (56). However, the protocol could not be escalated to the clinic due to the abandonment of phase 1 trials involving anti-CD154 antibodies, as discussed above. The problem was partly addressed by modifying CTLA4Ig to LEA29Y, which had greater binding to CD86. Subsequently, in 2011, LEA29Y was approved in renal transplantation as belatacept (Nulojix®) (87, 88). The parent molecule, CTLA4Ig, on the other hand, was extensively explored in a whole host of preclinical AID models. Although it showed excellent results in some, it also resulted in paradoxical worsening in a few (89–93). In due course, it was realized that CTLA4Ig would be effective in conditions where the pathophysiology is driven by the CD28-B7 pathway. It might be counterproductive in situations where the disease progression is dependent on insufficiency of a coinhibitory pathway, where CTLA4Ig would further downregulate CTLA4 expression. Finally, through extensive phase 3 clinical trials demonstrating long-term benefit, the parent CTLA4Ig molecule [abatacept (Orencia®)] was approved for patients with rheumatoid arthritis (94–96).
The inherent caveat in CTLA4Ig-mediated competitive inhibition of CD28-B7 ligation could abort T-cell activation upfront, but was found to decrease the population of Tregs in SOT models. This was ascribed to the failure of upregulation of CTLA4 in Teffs, as this is an event consequent to CD28–B7 interaction. In addition, CTLA4 is constitutively expressed on Tregs, but without binding to B7 molecules occupied by CTLA4Ig, they fail to execute its regulatory function (97, 98). In an attempt to address these problems, antibodies against both CD80 and CD86 were developed, but this did not translate to the clinic. TGN1412, a partial-agonist antibody, was developed against CD28, aimed at the expansion of Tregs. This was found to be safe in preclinical models. However, in the first eight healthy controls in a phase 1 trial, rapid polyclonal expansion of T cells was noted with disastrous consequences, bringing the development of CD28-directed treatment to a halt (99). Thus, CTLA4Ig remained the only safe and effective agent for COSBL, either in transplantation or in autoimmunity.
12 CTLA4Ig in HCT—the preclinical promise
Several preclinical models showed the efficacy of CTLA4Ig alone or in combination with anti-CD154 and anti-T cell antibodies in inducing engraftment across major MHC mismatch without thymic irradiation (56). This has been achieved in several models without employing myelotoxic agents. Durable donor chimerism was observed and the mechanism of tolerance was interrogated with various gene knockout models. Observations made through a series of experiments, which were subsequently summarized by Wekerle and Sykes (80), were as follows:
1) In SOT, COSBL can prevent early rejections and sustain a graft. However, long-term tolerance was a rarity if the thymus was intact.
2) COSBL-resistant graft rejection was observed across all models of SOT with CTLA4Ig and/or anti-CD154.
3) Bilateral tolerance can be established across major MHC-mismatched HCT with CTLA4Ig and anti-CD154, if CD8+ T cells were depleted. This was not achievable with CD4+ T-cell depletion, indicating the dependence on CD4+ T cells for anergy-associated tolerance.
4) Thymic irradiation was not essential to establish long-term tolerance, if CD8 depletion of the graft was carried out. The peripheral clonal deletion of alloreactive T cells was deemed more important.
5) Escape from the state of anergy was possible with either IL2 exposure or the alternative activation pathway of the costimulatory pathway and forced entry into the cell cycle pathway.
6) Establishment of anergy was abrogated if viral infection was experienced during the induction of tolerance (100).
13 CTLA4Ig in HCT—the proof of principle
The group from the Dana–Farber Cancer Institute carried out the first clinical trial employing COSBL with CTLA4Ig. In a series of in vitro experiments, the researchers demonstrated that in mixed-leukocyte reactions (MLRs), T-cell proliferation was inhibited by over 90% if both CD80 and CD86 were blocked by either individual antibodies or CTLA4Ig (101). Although the same was achievable with CSA or the blockade of adhesion pathways, antigen-specific anergy could only be established by COSBL. This was demonstrated when T-cell proliferation remained muted on rechallenge to donor APCs but not to third-party antigens for CTLA4Ig-treated cells, and not for those exposed to CSA or adhesion blockade. They also identified that the minimum time needed to achieve an anergic state with CTLA4Ig in MLR conditions was 36 h. Donor precursor helper T lymphocytes (pHTLs) were detected at 10–4 in HLA-matched donors and one log higher in HLA-mismatched donors. Although CSA reduced T-cell proliferation in MLR, it did not affect the pHTLs. However, pHTLs were reduced below the critical threshold for the alloreactivity of 10–5 by COSBL in both matched and mismatched settings. A key observation was the absolute necessity for allorecognition prior to COSBL, which was impaired in the presence of CSA or adhesion blockade. Another critical observation was the reversal of the state of anergy with continued exposure to suprathreshold amounts of IL2. Thus, allorecognition, blockade of both CD80 and CD86, and lack of signaling through common gamma (Ɣ) chains via IL2, were deemed to be the prerequisites for COSBL-based HCT (101). Based on these observations, the group undertook a study on haploidentical HCT with alloanergized marrow following myeloablative conditioning in 12 young patients with acute leukemia, refractory (n = 7) or multiply relapsed (n = 3) (102). The marrow was incubated for 36 h with CTLA4Ig with irradiated host mononuclear cells and subsequently infused unmodified along with a standard GVHD prophylaxis of CSA/MTX. The median CD3+, CD4+, and CD8+ T cell counts were 16, 8, and 6 million/kg, respectively, with pHTL frequency having a median value of 0.0005. Although the absolute number of T cells and its subsets increased after incubation, the frequency of pHTL reduced by three logs.
Only one patient had graft failure. One patient had grade 3 GVHD, with another two patients experiencing grade 2 gut GVHD, which was responsive to steroids. Five early-non-relapse mortality (NRM)) cases were observed in this heavily pretreated cohort. The most notable fact was a low incidence of viral infections, along with the rapid recovery of CD4+ T cells and immunoglobulin levels. The proliferative response of engrafted T cells to recipient and third-party antigens was monitored in a few patients after the discontinuation of CSA. The alloresponse to the host was 6–12 times lower than the third-party response. In addition, antiviral T-cell response was also detectable. This was the first demonstration of successful establishment of antigen-specific anergy via COSBL and preceded the landmark publication on megadose CD34-selected haploidentical HCT by an Italian research group in 1998 (103). The subsequent lack of enthusiasm in pursuing this approach might seem baffling in retrospect, but the muted enthusiasm in pursuing the parent molecule of CTLA4Ig among SOT immunologists might have acted as a dampener. The same group reported on another 12 patients transplanted on this protocol one decade later (104). The use of myeloablative conditioning in poor-risk older patients accounted for a treatment-related mortality (TRM) in excess of 50%, even though the incidence of GVHD remained low, particularly in younger patients, who had a much lower mortality as well. This experience goes on to highlight the complexity of a HCT protocol, where success in one aspect of the procedure, that is, the prevention of GVHD, can be overshadowed by the adverse impact of other aspects, which were patient selection and regimen-related toxicity (RRT) in this case. Nonetheless, the proof of principle was there in the clinic that COSBL can effectively induce anergy and prevent GVHD in HLA-MM HCT.
14 CTLA4Ig (abatacept)—from rejection in SOT to approval in HCT
Abatacept received approval for treatment of rheumatoid arthritis in 2005. Soon thereafter, based on the preceding preclinical studies, Kean and colleagues adopted a schedule of abatacept at 10 mg/kg on days 0, + 5, +15, and +30, as GVHD prophylaxis along with conventional CSA/MTX. In 2013, the results of the first trial on the use of abatacept in vivo in unrelated donor (URD)-HCT in 10 patients with acute leukemia following myeloablative conditioning were published (105). Four of these were 8/8 HLA-matched and the rest were 7/8 HLA-matched. All were engrafted, with two patients developing grade 2–4 acute GVHD by 100 days and another patient after 100 days. All patients were steroid responsive, and five developed cGVHD. Two died of relapsed leukemia and another died of multiple causes, partly contributed by cGVHD of the liver. Most importantly, consistent peak and trough levels of abatacept were achieved in this four-dose schedule, with no drug-related toxicity. Only half of the patients developed cytomegalovirus (CMV) reactivation, and definite instances of Epstein–Barr virus (EBV) lymphoproliferative disease (LPD) were observed.
Buoyed by the results of the phase 1 study, establishing the general feasibility and safety of abatacept, a phase 2 study (ABA2) was carried out using the same schedule of GVHD prophylaxis in both 8/8 and 7/8 HLA-matched URD-HCT (106). A total of 142 patients were randomized in the 8/8 HLA-matched cohort to receive or not receive abatacept along with CNI/MTX. The abatacept group had a 20% reduction in grade 2–4 acute GVHD at 100 days (43.1% vs. 62.1%; p = 0.006) and 180 days (44.8% vs. 63.7%: p = 0.006). Severe aGVHD-free survival (SGFS) at 180 days was 93.2% in the abatacept group (vs. 82%; p = 0.05), with an overall survival of 74.3% at 2 years (vs. 64%; p = 0.15). Randomization in the 7/8 HLA-matched cohort in this study was aborted due to reluctance on the part of the participating centers to subject patients with HLA-MM donors to conventional CNI/MTX prophylaxis, as the expected rates of grade 3–4 GVHD and TRM were in excess of 35%–45%. Hence, for the 7/8 cohort, a single-arm open-label design was adopted, with a comparator arm consisting of 127 age-matched patients from the CIBMTR registry, who had received 7/8 URD-HCT without ATG.
The outcomes in the 7/8 abatacept cohort were quite exceptional in that no grade 3–4 aGVHD were witnessed in those who received the study drug (n = 39), and 2.3% in the intention-to-treat (ITT) analysis, compared with 30.2% in the comparator arm (p < 0.001). This held true for all grades of aGVHD across all the study time points, translating to a better SGFS, TRM, and OS in the 7/8 abatacept group. Based on these encouraging results, abatacept became the first drug to be approved for GVHD prophylaxis in URD-HCT in December 2021. Furthermore, a post hoc analysis of these trial data showed a reduced incidence of aGVHD and a comparable survival in the 7/8 HLA-mismatched abatacept cohort when compared with an 8/8-matched cohort who received only CNI/MTX (107). In addition, an analysis of 50 patients not enrolled in the ABA2 trial, but treated on the same protocol, yielded very similar results (108) (Table 1).
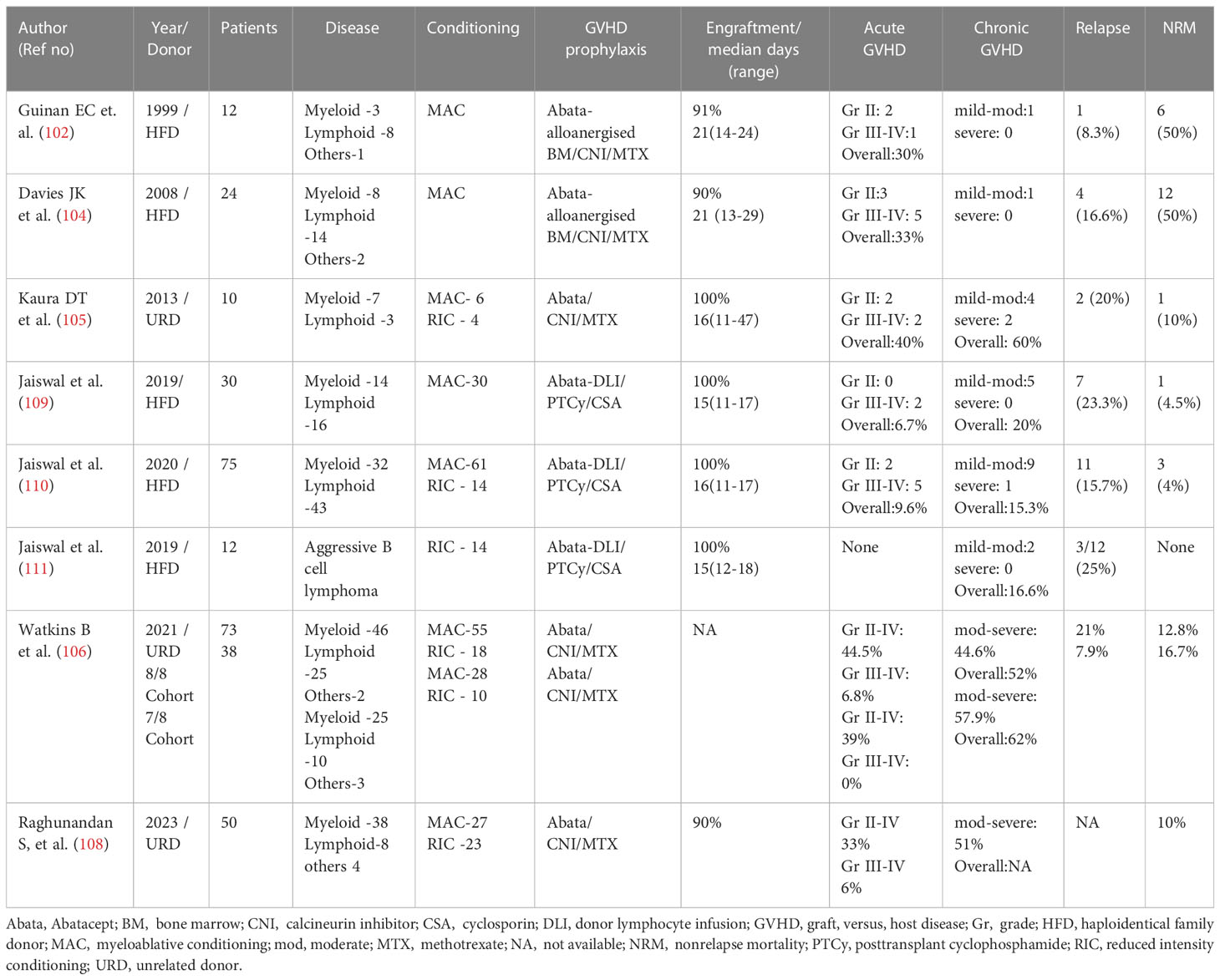
Table 1 Abatacept for GVHD prophylaxis in Malignant diseases.
15 Abatacept in HFD-HCT—finding the perfect partner for durable tolerance
HFD-HCT was attempted in the 1980s using standard GVHD prophylaxis, with disastrous consequences (112). However, the Perugia group established the feasibility of engrafting positively selected CD34+ HSC at a megadose from HFD in patients following intense myeloablation and immunoablation, with the near-complete elimination of GVHD occurring in mid-1990 (103). Even though the proof of principle was established, very high TRM was reported due to opportunistic infections resulting from the complete absence of T cells in the graft and the extended recovery of the adaptive immune system from the engrafted donor cells. Although the first trial of a CTLA4Ig-induced alloanergized graft took place around the same time, with a pattern of immune recovery superior to that reported in the CD34 megadose trial, this approach was not met with much acceptance (102). The downside of non-selective removal of T cells, both naive and memory, was shown to be detrimental to the overall outcome. Particularly, when only a small fraction of T cells is actually alloreactive, the blanket removal of all T cells seems to be overkill. Incubation with CTLA4Ig in the presence of host APCs has shown that donor-specific anergy can be achieved without interrupting the third-party response (101).
As highlighted earlier, although PTCy had vastly improved the outcome of HFD-HCT, concerns remained associated with its use in children with non-malignant diseases and those at high risk of relapse (25, 32). Introducing the same schedule of abatacept with CNI/MTX, as was used in the ABA2 trial, was deemed inadequate by our group for HFD-HCT employing peripheral blood stem cell (PBSC) graft. Instead, we conceptualized the combination of COSBL with PTCy along with sirolimus in HFD-HCT for NMD patients. The rationale for this combination was as follows:
15.1 Abatacept was to be administered to the patient in vivo prior to infusion of the graft
This should be able to interrupt the primary costimulatory CD28-B7 pathway and induce anergy in the large majority of alloreactive T cells. However, COSBL-resistant T cells could possibly escape this intervention; these could be CD28-negative memory T cells (113, 114) or Th17-Teff cells (115), which are less dependent on the CD28-B7 pathway. Such cells would proliferate in response to the host’s alloantigens and vice versa in the next 72 h, when the administration of PTCy would essentially take care of COSBL-resistant alloreactive pathways. Given the large quantum of T cells administered with an unmanipulated peripheral blood stem cell (PBSC) graft, the synergistic efficacy of abatacept and PTCy might be the ideal platform to selectively target the alloreactive T cells in an HLA-MM HCT.
15.2 Establishment of anergy, as discussed above, is not synonymous with tolerance
Anergy-induced hyperresponsiveness to donor antigen can be abrogated by inflammatory milieu and ɣ-chain signaling (101), which is typically present in the immediate post-conditioning phase. Hence, the early and repeated administration of abatacept for COSBL was deemed important in maintaining a continued state of anergy. Equally important is the need to avoid any early viral reactivation or acquired viral infections, which could also abrogate the achievement and maintenance of anergy (100).
15.3 Ex vivo alloanergization with CTLA4Ig or the administration of abatacept in vivo with CNI/MTX did not reduce the incidence of cGVHD
This clearly indicates that the early anergy achieved with COSBL does not translate to long-term anergy or tolerance when used in combination with CNI. The engagement of TCR and the MHC allopeptide, that is, allorecognition, is impeded by CNI. However, allorecognition is a prerequisite to successful anergy following COSBL. Therefore, CNI in combination with CTLA4Ig was associated with a lack of durable tolerance in preclinical studies, explaining the lack of impact of COSBL with CNI on cGVHD (116, 117). On the other hand, preclinical models have amply demonstrated the synergy between mTOR inhibition with sirolimus and COSBL in the achievement of durable tolerance without thymic irradiation (118, 119). Pilat and Wekerle demonstrated in a mismatched mice model that the addition of sirolimus to CTLA4Ig can override COSBL-resistant rejection. More importantly, the addition of sirolimus rendered the CD40 pathway redundant, allowing mixed chimerism and the acceptance of fully mismatched cardiac allograft. This was primarily driven by the sirolimus-induced upregulation of the Treg pathway (120). Sirolimus can induce FoxP3 expression on naive CD4+ T cells in the periphery through both dependent- and independent-TGFβ-SMAD3 pathways (14). In addition, some evidence exists regarding protection against herpes viruses when this combination was employed (121). We employed this principle in our protocol, combining sirolimus with abatacept and PTCy. In addition, abatacept was planned for prolonged admission, that is, in a 4-weekly schedule for 6 months, with sirolimus tapering after 9 months.
15.4 Finally, it is necessary to acknowledge that tolerance following HCT is driven by regulatory pathways
One of the reasons for not being able to induce tolerance in a COSBL-CNI-based protocol is the downregulation of Tregs (122, 123). On the other hand, sirolimus has a salutary effect on Tregs, both in terms of number and function (124–126). Although the conventional wisdom in SOT is that CTLA4Ig has a negative impact on tTregs, this is less than linear in the setting of HCT, which is primarily related to the amount of Tregs infused in the graft and its lineage plasticity. In the presence of TGFβ, abatacept has been shown to promote T-cell suppression, which could be translated in the setting of HCT as discussed below. Abatacept with sirolimus has the potential to convert peripheral CD4+CD25− T cells to CD4+CD25+FoxP3+ Tregs, further augmenting the regulatory network (127). In addition, PTCy seems to critically spare the Tregs (128), accounting for further synergy in the combination of abatacept, PTCy, and sirolimus. Hence, in contrast to AIDs and SOT, finding the right partner for abatacept might actually promote and induce long-term tolerance via a Treg-driven regulatory pathway.
The above considerations translated to the clinical protocol combining abatacept, PTCy, and sirolimus (AbaCyS) for HFD-HCT in NMD patients with a PBSC graft (Figure 1A).
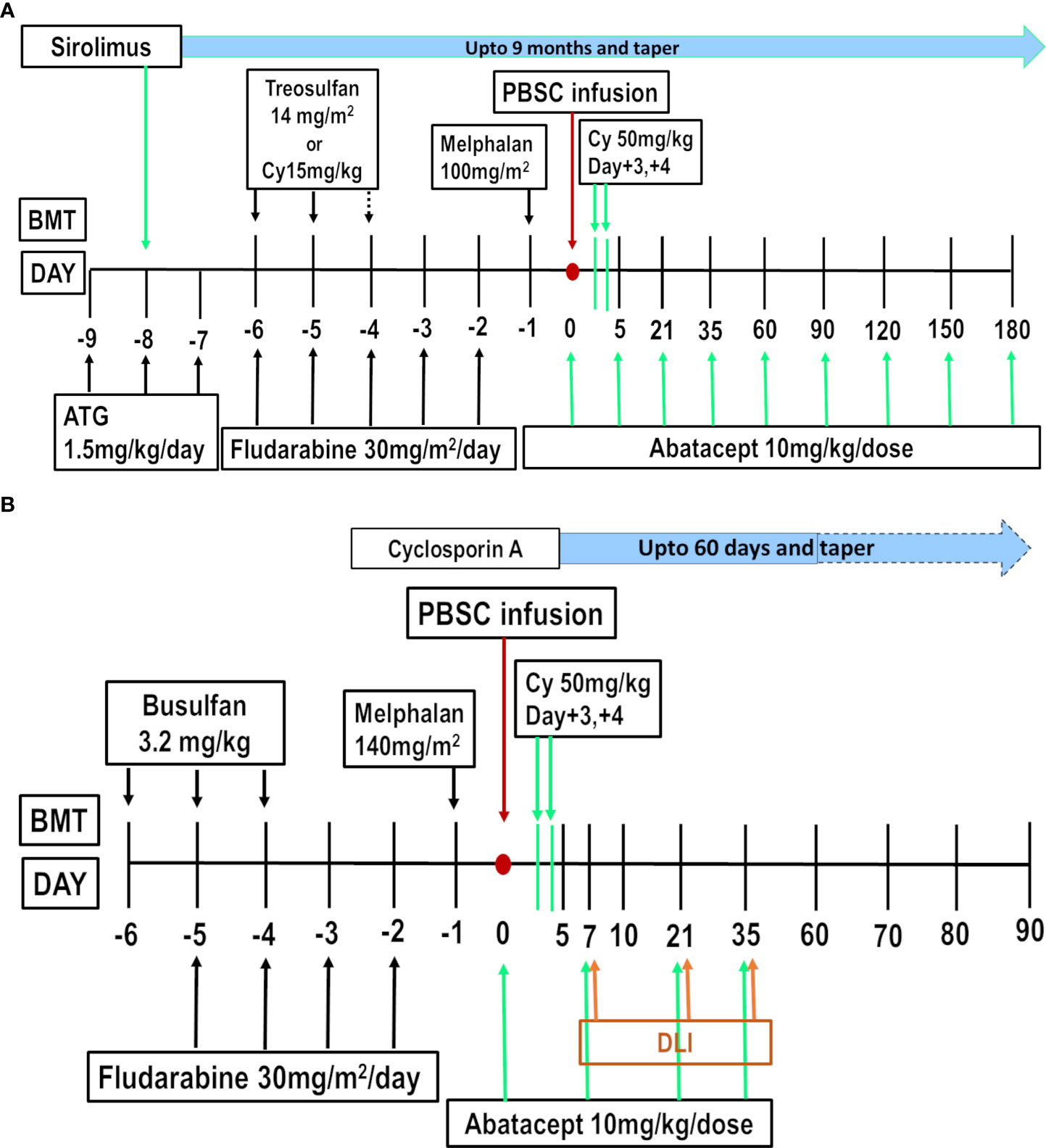
Figure 1 (A) Abatacept, PTCy, Sirolimus (AbaCyS) protocol for Non-malignant Disease. (B) Abatacept-DLI, PTCy, Cyclosporine (AbaDCyC) protocol for Malignant Disease.
16 Abatacept, PTCy, and sirolimus (AbaCyS)—translation into the clinic
The first study on extended abatacept, PTCy, and sirolimus was reported by our group in 2016 (129). In 10 patients with severe aplastic anemia (SAA), this protocol was associated with 90% OS and 10% incidence of GF, aGVHD, and cGVHD. Similar results were obtained in a pilot study on 10 patients with thalassemia and sickle cell anemia, with an extremely low incidence of complications (i.e., < 10%) (130). In addition, the significant attenuation of PTHPS was observed. In an extended study on 40 patients, with a median follow-up of 4.4 years, NMD patients treated on a AbaCyS protocol showed a GVHD and event-free survival (GEFS) of 82%, with aGVHD in 2.6%, and cGVHD in 14.5% (131). The incidence of both CMV and adenovirus (AdV) reactivation was low, with a NRM of 5%. The quality of life was excellent for all patients at 1 year and beyond, with late infections witnessed almost exclusively in patients with cGVHD. In a multicenter retrospective analysis of children and young adults with SAA, a comparison of 15 patients receiving AbaCyS with 64 patients receiving standard PTCy found lower incidences of TRM, aGVHD, and cGVHD, and improved GEFS and OS, in the AbaCyS cohort as compared with the standard PTCy group (132).
Following these studies, a similar extended schedule of abatacept was employed in URD-HCT for sickle cell disease, resulting in the reduction of cases of severe GVHD, both acute and chronic, with improved survival (133). However, the protocol employed abatacept in combination with CNI, and the overall incidences of aGVHD and cGVHD were 28.6% and 57%, respectively, probably highlighting the failure of the combination of CNI and abatacept to induce tolerance, as witnessed with the AbaCyS protocol. The same protocol in 10 children with bone marrow failure has been reported on recently, with very similar results (134) (Table 2).
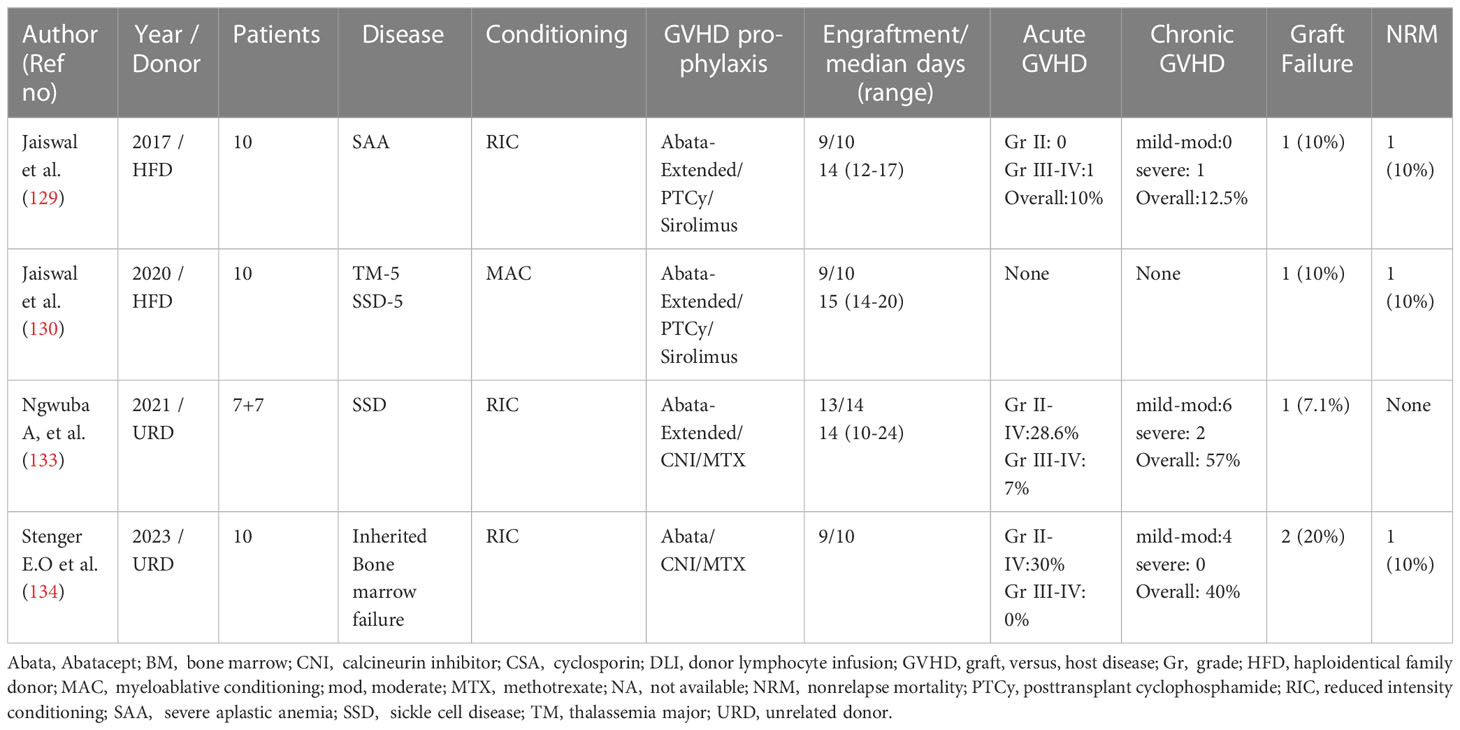
Table 2 Abatacept for GVHD prophylaxis in Non-Malignant diseases.
17 Early and sustained reconstitution of Tregs—the key to GVHD-free survival
Immune reconstitution was studied quite extensively in the patients on the AbaCyS protocol, and the findings can be summarized as follows:
17.1 T-cell recovery was not affected with the AbaCyS protocol.
CD4+ T cell recovery to over 200 cells/μL was achieved by 3 months post HCT, with predominantly memory phenotype, suggesting peripheral expansion of infused donor cells, followed by the subsequent recovery of naive T cells, both CD4+ and CD8+ T cells (130, 131). The more rapid recovery of both CD4+ and CD8+ T cells was seen in patients with an ex vivo CTLA4Ig-alloanergized graft. In the ABA2 study on URD-HCT, CD4+ T cell recovery was delayed in the initial months but was comparable to the standard CNI/MTX group at 3 months and beyond (106).
17.2 Contrary to the ABA study, the recovery of Tregs was much more rapid with the AbaCyS protocol
The percentage of CD4 + 25 + 127dimFoxP3+ T cells was significantly higher in the AbaCyS group at days +30, +60, and +90, when compared with the non-abatacept cohort in the first 10 patients (129). This trend was sustained in the larger cohort of 40 patients (131). Furthermore, earlier and better Treg recovery correlated with a lower incidence of chronic GVHD.
17.3 Davies and Guinan reported on the effect of CTLA4Ig-alloanergized HFD-HCT on recovery of Tregs in five long-term survivors
Similar to the AbaCyS study, the authors showed a rapid and sustained rise in Tregs (135). In addition, they documented in the MLR assays that alloanergization in vitro increased the frequency of Tregs, which suppress Teff proliferation. In addition, it was also demonstrated that the suppression of the alloresponse of donor-derived Tregs against the host was antigen specific. Both of these studies, therefore, establish the primacy of abatacept in augmenting donor Tregs in vitro and in vivo.
Thus, early and sustained donor-specific anergy was possible with the AbaCyS protocol in NMD patients undergoing HFD-HCT. The long-term survival without cGVHD was possibly influenced by the early expansion of infused Tregs from the donor followed by sustenance of high levels of Tregs, inducing donor-specific tolerance, which was not witnessed with the abatacept- and CNI-based protocols.
18 Can intravenous immunoglobulin enhance the suppressive effect of abatacept?
TGFβ and Tregs have been shown to be critical in maintaining the suppressive effects of CTLA4Ig in murine models. In addition, TGFβ is critical in diverting naive T cells down the Treg pathway following antigen exposure (127, 136–138). In fact, in the absence of TGFβ and abundance of IL6, even CD4+CD25+ T cells can differentiate to Th17 cells (136). Thus, we hypothesized that the efficacy of CTLA4Ig in vivo could be optimized in the presence of TGFβ. Unfortunately, TGFβ is not commercially available for clinical use, but has been shown to be present in substantial amounts in intravenous immunoglobulin (IVIG) preparations, and increased levels of different isoforms of TGFβ were documented immediately following administration of IVIG in children with AIDs (139). In addition, IVIG has been shown to upregulate Tregs either directly or via the induction of tolerogenic DCs (140, 141).
Based on these considerations, we have modified the AbaCyS protocol to administer IVIG at 500 mg/kg 6 hours before abatacept. The results of this protocol are still pending, but no short-term adverse effects have been reported so far.
19 Abatacept and relapse in malignant diseases—a twist in the tale
In contrast to other interventions, which result in the non-selective depletion of T cells, abatacept has not been associated with increase in the relapse risk. In the initial cohort of patients undergoing HFD-HCT with CTLA4Ig-alloanergized graft (102), three out of four patients in second or third complete remission (CR) remained disease free. In those transplanted with persistent disease, only one experienced disease progression and two remained in long-term remission. The rest of the five died of infections or RRT. Interestingly, autopsies conducted on four of them did not show evidence of residual leukemia. This trend was maintained in the extended cohort of 24 patients reported a decade later (104).
In the ABA study of 2013, only 2 out of 10 patients with acute leukemia had relapsed (105). In the extended ABA2 trial, only 7.9% patients in the ABA-ITT arm of the 7/8 cohort relapsed compared with 21.4% in the comparator group (106). In the 8/8 group, the relapse rate was 21.5% in the abatacept group, compared with 23.6% in the placebo group. The effect of abatacept on relapse could be corroborated by preclinical studies showing the abrogation of GVL effect in murine models by blocking the CD40-40L pathway, but CTLA4Ig-mediated COSBL left the GVL effect intact (142).
Thus, these clinical studies uniformly concluded in an understated manner that the relapse risk was not increased with the use of abatacept. However, our group had a different interpretation of these findings.
20 CTLA4Ig (abatacept) and natural killer cells—an affair lost in serendipity
While the researchers working on COSBL in SOT had focused largely on COSBL-resistant T-cell subsets, those indulging in preclinical HCT models made a different observation. CTLA4Ig was employed as a part of non-myeloablative protocols in HLA-MM models. The group from Seattle observed that CTLA4Ig with 200 Gy radiation was able to overcome the T-cell barrier in haploidentical canine pairs, but NK cells remained unaffected, resulting in eventual rejection (143). In murine models, when COSBL was combined with non-myeloablative doses of busulfan, donor engraftment failed to occur without large doses of BM cells. However, depletion of NK cells in the host with the anti-NK1.1 antibody resulted in a striking increase in the stable engraftment of donor cells. Further assays showed no involvement of the CD28 and CD40 pathways in NK cell cytotoxicity and the blocking of LFA1 was associated with reversal of NK-mediated rejection (144). These studies clearly indicate that NK cells were resistant to CTLA4Ig-mediated suppression.
However, the lack of an increase in malignancies observed in patients receiving CTLA4Ig for AIDs, prompted a group from China to explore the underlying mechanism, if any (145). They showed that CTLA4Ig reduced tumor metastasis and prolonged survival in B16 mice melanoma models. Interestingly, this effect was abrogated by the NK-cell depletion of the host mice. CTLA4Ig enhanced the antitumor effect of NK cells in vitro with increased expression of CD86, which ligates with CTLA4Ig. In addition, the antitumor cytotoxicity of NK cells was blocked by blocking CD86. The effect of CTLA4Ig on NK cells, which thus far was perceived to be only a sparing effect, could actually be a potentiator of NK-cell cytotoxicity.
21 Abatacept-dissociating T-cell-mediated alloreactivity from NK cell cytotoxicity
NK cells are probably the most potent antitumor cells, apart from antigen-directed CTLs. NK cells are endowed with a plethora of activating receptors, which can bind to a diverse array of ligands, many of which are upregulated with stress, viral infections, or the malignant transformation of cells. Due to such an extensive armory of activating receptors and the ability to mount direct cytotoxic and cytolytic attack against target cells, NK cells are kept under inhibitory control via the interaction of inhibitory KIRs and corresponding self-MHC class 1 ligands.
GVL was conceived as a T cell-mediated effect, until the Perugia group demonstrated a potent antileukemia effect mediated via NK cells following CD34-selected HFD-HCT. This was primarily observed in patients with myeloid leukemia receiving grafts from donors who were mismatched at NK cell KIR ligands in the GVH direction. This is called NK-ligand mismatch or NK-alloreactivity, which is demonstrable only in the presence of HLA class 1-mismatched HCT. NK cells are the first lymphocyte subset to recover after an allogeneic HCT. In contrast to T cells, NK cells do not drive an organ-directed GVH reaction. In fact, the cytotoxicity is directed toward the hematopoietic compartment. Thus, NK cells are uniquely placed to mount a GVL response without invoking generalized alloreactivity. Following HCT with a T cell-replete graft, NK cells surge briefly at around 3–4 weeks post HCT. However, following a T-cell-depleted (TCD) HCT, NK cells remain the dominant lymphocyte population until T-cell recovery. In a TCD HCT, post-transplant immunosuppression is generally not necessary. Under these circumstances, in the absence of T cells, NK cells gain a unique opportunity to proliferate and exert cytotoxicity against leukemia cells, a phenomenon that is not witnessed in T-replete HCT.
However, NK cells traverse through stages of maturation and can only deliver the optimum GVL effect if they have achieved a fully mature state with CD56dimCD16bright phenotypes and if they are released from inhibitory control by losing the expression of NKG2A receptors with the expression of requisite activating and inhibitory molecules in the right cytokine milieu. Despite NK ligand mismatch, donor-derived NK cells seemed to lack these capabilities in the first few months after HCT, as observed following CD34-selected HFD-HCT. Thus, the mere presence of NK cells is not sufficient for a strong GVL effect. In addition, immature CD56bright NK cells could promote T-cell-mediated alloreactivity in the context of a T-replete HLA-MM HCT. We thus hypothesized that the serendipitous sparing and potentiation of NK cells by abatacept while abrogating T-cell-mediated alloreactivity might provide a unique opportunity to explore the NK cell-mediated GVL effect without invoking the wrath of GVHD.
Abatacept was shown to anergize alloreactive T cells in both preclinical and clinical studies, without impeding response against pathogens or third-party antigens and inducing long-term tolerance in combination with PTCy and sirolimus in NMD patients. However, the tolerance-inducing protocol, AbaCyS, was not deemed to be ideal in HCT for advanced malignancies, where a strong GVL effect would be necessary. It was contemplated that it might be best to optimize the GVL effect via the unique NK cell-sparing/-promoting property of abatacept and abrogate T-cell-mediated alloreactivity through COSBL. While designing the abatacept-based protocol for malignant diseases, we deliberated on the following:
1. Sirolimus adversely affects NK cell proliferation and cytotoxicity (146, 147). CNI, on the other hand, does not restrict the NK cell pathway. Hence, if NK-mediated GVL was to be optimized, sirolimus needs to be replaced with CNI. This would be carried out with full cognizance of the fact that the incidence of cGVHD might increase as compared with the AbaCyS protocol. In ex vivo experiments, Comoli and colleagues demonstrated that the combination of CTLA4Ig and low-dose CSA is able to induce anergy without compromising antiviral and antileukemia-specific CTLp. However, the NK cell-mediated antileukemia effect was not looked into in this study. Thus, based on these considerations, CSA was planned at a lower dose targeting a trough level of 50–150 ng/mL, with tapering planned at day +60.
2. The choice of the third agent, if abatacept and CNI were to be combined, would be either MTX or MMF, if one considers the ABA2 study. However, the success of the AbaCyS protocol prompted us to consider abatacept and PTCy as the backbone of the protocol, along with cyclosporine as the CNI of choice. Both MMF (146) (not MTX) and PTCy would affect NK cell proliferation (148), but the effect of PTCy would be limited to the early post-HCT period alone. PTCy eliminates the majority of proliferating NK cells immediately after its infusion, resulting in immature NK cells repopulating the NK cell pool in the early post-HCT period, compromising the NK cell-mediated GVL effect (148). MMF significantly attenuates both NK cell proliferation and cytotoxicity in a continuous and protracted manner, seriously compromising early NK cell recovery. It might be apt to state that the perseverance of the transplant community with CNI/MTX could be attributed to the salutary impact on disease relapse. Both the agents at lower doses spare NK cells in an understated manner, which might contribute to the early GVL effects.
3. Thus, to override the adverse impact of PTCy and optimize NK cell-mediated GVL effect early after HCT, we developed the concept of “CTLA4Ig (abatacept)-primed donor lymphocyte infusion (DLI)”, where donor lymphocytes capped by CD3+ T-cell dose are administered 6 h following the administration of abatacept. The dose capping and scheduling of DLI were developed based on earlier studies on unmanipulated DLIs aliquoted and cryopreserved from a granulocyte colony-stimulating factor (G-CSF)-mobilized PBSC collection (mDLI) and CD56+DLI following HFD-HCT (149, 150). Twenty-one patients with relapsed/refractory myeloid leukemia were administered mDLI at days +21, +35, and +60, with significant improvement in DFS of 61.9% at 2 years, compared with one of 25% in those on an identical protocol without DLI. This was not associated with an increase in aGVHD. However, cGVHD was observed in 41% of patients in the mDLI group. In a pilot study on 10 patients with relapsed/refractory myeloid leukemia, CD56+DLI isolated from a lymphapheresis product was administered on day +7 (150). This resulted in excellent NK cell reconstitution. Despite the presence of CD3+CD56− T cells up to 1 × 106/kg, no acute GVHD grade 2–4 was observed. Based on the observations from these studies, abatacept-DLI was planned for days +7, +21, and +35. The first dose on day +7 was aimed at replenishing NK cells in the window of a PTCy-induced IL15 surge (151) and aimed to achieve the maximum NK-GVL effect at the peak of lymphopenia and minimum tumor burden.
The above considerations resulted in a protocol (AbaDCyC) consisting of abatacept on day 0; abatacept with DLI on days +7, +21, and +35; and PTCy on days +3 and +4, along with low-dose, short-course CSA, from day +5 to day +60 (Figure 1B).
22 AbaDCyC (CTLA4Ig-primed DLI)—realizing the promise of dissociating GVHD and GVL?
In the first 30 patients with advanced leukemia treated on this protocol, the incidences of aGVHD, cGVHD, and NRM were 6.7%, 21%, and 4.5%, respectively, with a relapse risk (RR) of 23.3% and an OS of 79% at 18 months (109). Almost all patients received the day +7 dose of DLI without any acute toxicity. The pattern of early mature NK cell recovery mimicked that observed with CD56+DLI. In addition, early surges of CD56dim16+ NK cells with lower levels of NKG2A expression were associated with a lower relapse rate. Interestingly, CD86 was upregulated on mature NK cells in this cohort, suggesting a direct effect of CTLA4Ig on NK cells.
A further 75 patients receiving AbaDCyC were compared with 50 patients receiving mDLI (110). Acute and chronic cases of GVHD in the AbaDCyC group were 9.6% and 15.3%, respectively, compared with 18.8% and 36.5%, respectively, in the mDLI group. Both NRM (4% vs. 14.4%) and RR (15.7% vs. 31.1%) were lower in the AbaDCyC CTLA4Ig-DLI group. Distinct patterns of immune recovery were observed in the two groups. Although an early and sustained recovery of T-cell subsets correlated with a reduced RR in the mDLI group, this correlated with mature NK cell recovery in the AbaDCyC group. It is worth noting that the hallmark of NK cell recovery after both PTCy and CD34-selected HFD-HCT has been a distinctly immature phenotype with a high level of expression of the inhibitory receptor, NKG2A (148, 152). NK cell recovery after abatacept-DLI, on the other hand, was marked by the early recovery of CD56dimKIR+NKG2Alow phenotypes. Early T-cell recovery in the AbaDCyC group was characterized by the dominance of memory phenotypes. The same trend was observed in 12 patients with relapsed/refractory lymphoma treated on this protocol (111). No instances of aGVHD were reported, with only two patients developing mild-to-moderate GVHD. The DFS at 2 years was 75%.
Another notable observation was a superior recovery of Tregs in the AbaDCyC cohort and this did not adversely affect the RR. In addition, there was the rapid and sustained recovery of NKG2C+NKG2A-adaptive NK cells (ANK), which expands in response to CMV infection in a normal physiological state (153). These ANK cells have been shown to be associated with a reduced RR of leukemia by Miller and colleagues (154). Similar observations were made in the AbaDCyC cohort, albeit with a more prompt and greater number of these cells than previously observed.
Another observation from these studies with AbaDCyC in HFD-HCT relates to the lower incidence of cGVHD than that reported in patients receiving Abata-CNI/MTX following URD-HCT (106, 110). The use of mDLI following PTCy was associated with a higher incidence of cGVHD than the conventional PTCy protocol (149). However, this was not observed in the AbaDCyC cohort, where cGVHD was strongly associated with lower numbers of ANK cells and vice versa (155). The association between NKG2C+ANK cells and reduced GVHD has been reported earlier (156). However, the mechanistic explanation for this phenomenon remains unclear. One explanation could be the possible coexistence of Tregs and ANK cells in this protocol. In contrast to conventional NK cells, ANK cells are not downregulated by Tregs via IL37 (157). Thus, both Tregs and ANK cells can rapidly reconstitute, with Tregs offering protection from T cell-mediated alloreactivity and ANK cells mediating a sustained GVL effect. Further studies are ongoing to understand the mechanistic pathway behind the bonhomie of ANK cells and Tregs in the context of HFD-HCT receiving AbaDCyC (CTRI : REF/2021/08/046552), and these might herald a paradigm shift in our approach to dissociation of GVHD and GVL (Figure 2).
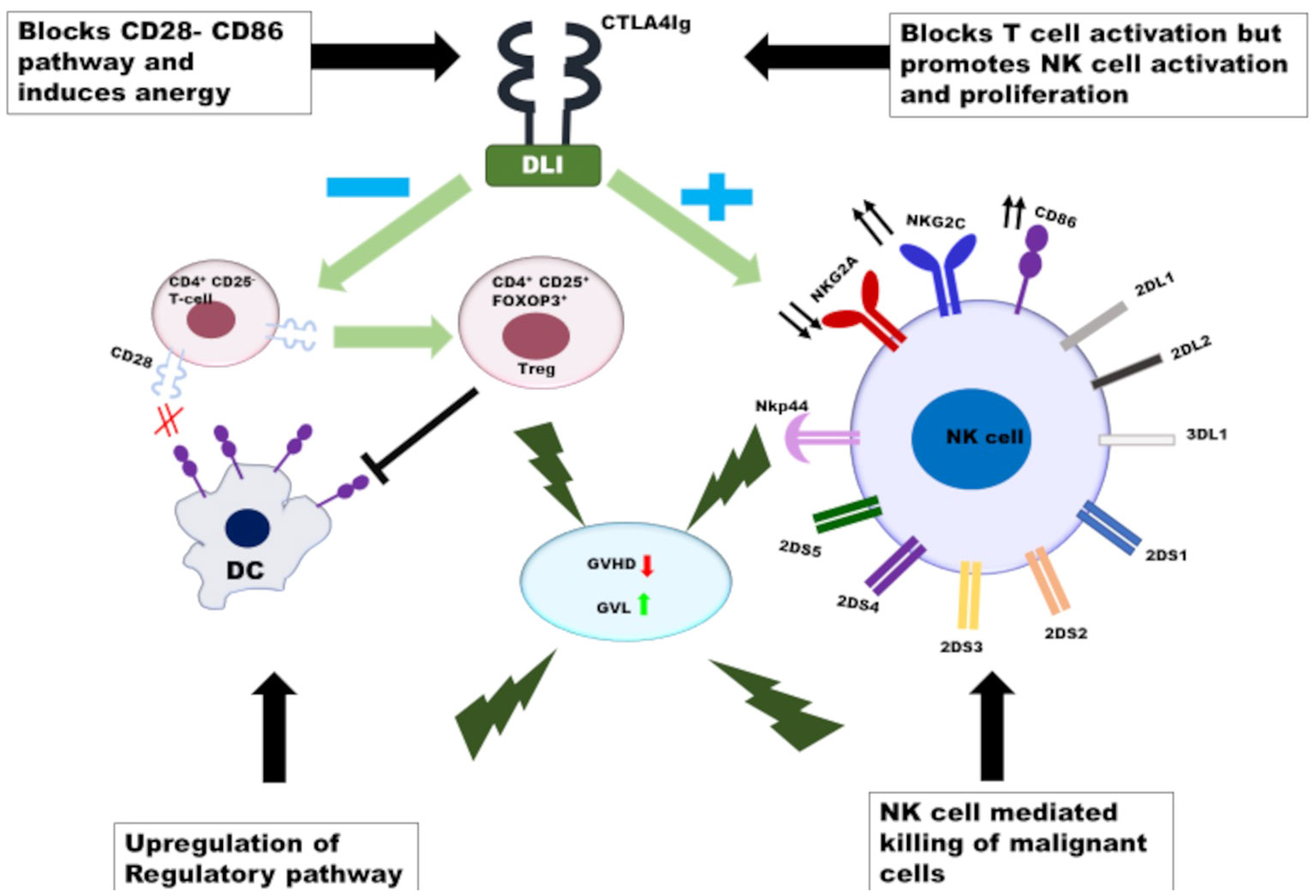
Figure 2 The proposed mechanistic pathway of abatacept-mediated dissociation of GVHD and GVL: CTLA4Ig anergises the alloreactive T cells in the donor graft as well as the donor lymphocytes infusion in-vivo and induces conversion of CD4+CD25-T cells to Tregs. At the same time CTLA4Ig promotes the activation and proliferation of NK cells infused with DLI following administration of PTCy, resulting in selective killing of tumor cells without induction of GVHD.  - decreases,
- decreases,  - increases,
- increases,  - no interaction,
- no interaction,  - addition and
- addition and  - no addition.
- no addition.
23 Abatacept has arrived—but questions remain
Although the incidence of aGVHD was reduced in the URD-HCT and HFD-HCT groups, we observed certain disconcerting patterns in cases of severe aGVHD, as and when it happened and however rare it might be. The onset of aGVHD was early (by day +30), severe, and restricted to the gut. This has been alluded to by the group using CTLA4Ig-alloanergized grafts and was not discussed in the ABA2 study. The reason for selective trafficking of COSBL-resistant alloreactive T cells to the gut remains unclear at this time. One of the observations has been severe infective enterocolitis during the early post-HCT period, in those who went on to develop severe gut GVHD. This might instigate both the mobilization of alloreactive donor T cells to the gut and the abrogation of COSBL-induced anergy. Another hypothesis is that COSBL-resistant T cells are most likely of Th17 lineage, which has an inherent propensity for trafficking to the gut. However, if this pattern is consistently observed, exploration of the mechanistic pathways and early biomarkers would be warranted.
In this context, we analyzed the influence of immunogenetic factors on the outcome of 136 patients undergoing HFD-HCT on the AbaDCyC protocol (158). The heterozygous deletion of KLRC2 (the gene encoding for NKG2C) in the donor was associated with a higher incidence of aGVHD and NRM.
Another interesting observation is that derived from the pharmacokinetics of abatacept in the ABA2 study (159). A trough level of less than 39 μg/mL after the first dose of abatacept correlated with a higher incidence of aGVHD grade 2–4. Those with higher trough levels of abatacept had a significantly lower incidence of aGVHD without any adverse impact on relapse or viral reactivations.
Based on the above observations, the following questions need to be addressed:
● Should abatacept dosing be guided by trough levels after the first dose, as suggested by the exposure response observation from the ABA2 study? Alternatively, should a higher dose be recommended for those deemed to be at a higher risk of aGVHD, as higher trough levels were not associated with serious side effects?
● Should belatacept be explored in HCT? If so, then how and when? We feel the time is ripe for randomized studies comparing belatacept and abatacept in both NMDs and malignant diseases.
● Should COSBL for other pathways, such as CD40-CD154 or OX40-OX40L, be explored in combination with abatacept to address COSBL-resistant GVHD in NMD patients, instead of PTCy?
● Should we refine donor selection criteria for abatacept-based HFD-HCT, as suggested in a recent study (158)?
24 Conclusion
Five decades of allogeneic HCT have been based on certain basic principles established in the canine experiments carried out by Thomas and colleagues. However, GVHD remains a constant nemesis thwarting the progress of allogeneic HCT. Although CNI/MTX and variations thereof have been the cornerstone of GVHD prevention, it has never been enough, particularly for alternative-donor HCT. Attempts at breaking the glass ceiling have ranged from T-cell-directed serotherapy and graft manipulation, to the simple yet elegant concept of PTCy. COSBL with abatacept has finally found its way to the bedside after decades of preclinical prevarications, setting a new paradigm for the prevention of GVHD. The safety profile of abatacept in the context of HCT, coupled with its unique effect on NK cells and Tregs, open up new avenues for expanding the scope of allogeneic HCT, particularly from HLA-MM donors. Despite COSBL with abatacept providing a new direction for the simultaneous prevention of GVHD and maintenance of a GVL effect, the final destination remains distant. The challenge lies in our understanding and flexibility in innovating strategies to optimize COSBL and choosing the right partners for abatacept based on the disease and donor. What is most important, however, is the willingness to adapt to shifting paradigms.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors SC and SJ declare that they were an editorial board member of Frontiers at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Storb R, Epstein RB, Ragde H, Bryant J, Thomas ED. Marrow engraftment by allogeneic leukocytes in lethally irradiated dogs. Blood (1967) 30:805–11. doi: 10.1182/blood.V30.6.805.805
2. Storb R, Epstein RB, Rudolph RH, Thomas ED. Allogeneic canine bone marrow transplantation following cyclophosphamide. Transplantation (1969) 7:378–86. doi: 10.1097/00007890-196905000-00007
3. Storb R, Epstein RB, Thomas ED. Marrow repopulating ability of peripheral blood cells compared to thoracic duct cells. Blood (1968) 32:662–7. doi: 10.1182/blood.V32.4.662.662
4. Storb R, Kolb HJ, Graham TC, Leblond R, Kolb H, Lerner KG, et al. Marrow grafts between histoincompatible canine family members. Rev Eur Etud Clin Biol (1972) 17:680–5.
5. Storb R, Rudolph RH, Kolb HJ, Graham TC, Mickelson E, Erickson V, et al. Marrow grafts between DL-A-matched canine littermates. Transplantation (1973) 15:92–100. doi: 10.1097/00007890-197301000-00014
6. Storb R, Thomas ED. Human marrow transplantation. Transplantation (1979) 28:1–3. doi: 10.1097/00007890-197907000-00001
7. Storb R, Thomas ED. Graft-versus-host disease in dog and man: the Seattle experience. Immunol Rev (1985) 88:215–38. doi: 10.1111/j.1600-065X.1985.tb01160.x
10. Antin JH, Ferrara JL. Cytokine dysregulation and acute graft-versus-host disease. Blood (1992) 80:2964–8. doi: 10.1182/blood.V80.12.2964.2964
11. Deeg HJ, Storb R, Appelbaum FR, Kennedy MS, Graham TC, Thomas ED. Combined immunosuppression with cyclosporine and methotrexate in dogs given bone marrow grafts from DLA-haploidentical littermates. Transplantation (1984) 37:62–5. doi: 10.1097/00007890-198401000-00017
12. Storb R, Deeg HJ, Whitehead J, Appelbaum F, Beatty P, Bensinger W, et al. Methotrexate and cyclosporine compared with cyclosporine alone for prophylaxis of acute graft versus host disease after marrow transplantation for leukemia. N Engl J Med (1986) 314:729–35.
13. Inamoto Y, Flowers ME, Appelbaum FR, Carpenter PA, Deeg HJ, Furlong T, et al. A retrospective comparison of tacrolimus versus cyclosporine with methotrexate for immunosuppression after allogeneic hematopoietic cell transplantation with mobilized blood cells. Biol Blood Marrow Transplant (2011) 17:1088–92. doi: 10.1016/j.bbmt.2011.01.017
14. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol (2009) 9:324–37.
15. Furlong T, Kiem HP, Appelbaum FR, Carpenter PA, Deeg HJ, Doney K, et al. Sirolimus in combination with cyclosporine or tacrolimus plus methotrexate for prevention of graft-versus-host disease following hematopoietic cell transplantation from unrelated donors. Biol Blood Marrow Transplant (2008) 14:531–7. doi: 10.1016/j.bbmt.2008.02.009
16. Cutler C, Antin JH. Sirolimus for GVHD prophylaxis in allogeneic stem cell transplantation. Bone Marrow Transplant (2004) 34:471–6. doi: 10.1038/sj.bmt.1704604
17. Kroger N, Zabelina T, Kruger W, Renges H, Stute N, Durken M, et al. Anti-thymocyte-globulin as part of the preparative regimen prevents graft failure and severe graft versus host disease (GvHD) in allogeneic stem cell transplantation from unrelated donors. Ann Hematol (2001) 80:209–15. doi: 10.1007/s002770000269
18. Spyridonidis A, Labopin M, Brissot E, Moiseev I, Cornelissen J, Choi G, et al. Should anti-thymocyte globulin be added in post-transplant cyclophosphamide based matched unrelated donor peripheral blood stem cell transplantation for acute myeloid leukemia? A study on behalf of the Acute Leukemia Working Party of the EBMT. Bone Marrow Transplant (2022) 57:1774–80. doi: 10.1038/s41409-022-01816-1
19. Chakrabarti S, Hale G, Waldmann H. Alemtuzumab (Campath-1H) in allogeneic stem cell transplantation: where do we go from here? Transplant Proc (2004) 36:1225–7. doi: 10.1016/j.transproceed.2004.05.067
20. Filipovich AH, Krawczak CL, Kersey JH, McGlave P, Ramsay NK, Goldman A, et al. Graft-versus-host disease prophylaxis with anti-T-cell monoclonal antibody OKT3, prednisone and methotrexate in allogeneic bone-marrow transplantation. Br J Haematol (1985) 60:143–52. doi: 10.1111/j.1365-2141.1985.tb07395.x
21. Chakrabarti S, Mackinnon S, Chopra R, Kottaridis PD, Peggs K, O'Gorman P, et al. High incidence of cytomegalovirus infection after nonmyeloablative stem cell transplantation: potential role of Campath-1H in delaying immune reconstitution. Blood (2002) 99:4357–63. doi: 10.1182/blood.V99.12.4357
22. Zaia J, Baden L, Boeckh MJ, Chakrabarti S, Einsele H, Ljungman P, et al. Viral disease prevention after hematopoietic cell transplantation. Bone Marrow Transplant (2009) 44:471–82. doi: 10.1038/bmt.2009.258
23. Aversa F. T-cell depletion: from positive selection to negative depletion in adult patients. Bone Marrow Transplant (2015) 50 Suppl 2:S11–3. doi: 10.1038/bmt.2015.88
24. Handgretinger R, Arendt AM, Maier CP, Lang P. Ex vivo and in vivo T-cell depletion in allogeneic transplantation: towards less or non-cytotoxic conditioning regimens. Expert Rev Clin Immunol (2022) 18:1285–96. doi: 10.1080/1744666X.2022.2134857
25. Jaiswal SR, Chakrabarti S. Haploidentical transplantation in children with acute leukemia: the unresolved issues. Adv Hematol (2016) 2016:3467672. doi: 10.1155/2016/3467672
26. Mayumi H. A review of cyclophosphamide-induced transplantation tolerance in mice and its relationship with the HLA-haploidentical bone marrow transplantation/post-transplantation cyclophosphamide platform. Front Immunol (2021) 12:744430. doi: 10.3389/fimmu.2021.744430
27. Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood (2001) 98:3456–64. doi: 10.1182/blood.V98.12.3456
28. Luznik L, Fuchs EJ. High-dose, post-transplantation cyclophosphamide to promote graft-host tolerance after allogeneic hematopoietic stem cell transplantation. Immunol Res (2010) 47:65–77.
29. O'Donnell PV, Luznik L, Jones RJ, Vogelsang GB, Leffell MS, Phelps M, et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol Blood Marrow Transplant (2002) 8:377–86. doi: 10.1053/bbmt.2002.v8.pm12171484
30. Luznik L, O'Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic Malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant (2008) 14:641–50. doi: 10.1016/j.bbmt.2008.03.005
31. Rappazzo KC, Zahurak M, Bettinotti M, Ali SA, Ambinder AJ, Bolanos-Meade J, et al. Nonmyeloablative, HLA-mismatched unrelated peripheral blood transplantation with high-dose post-transplantation cyclophosphamide. Transplant Cell Ther (2021) 27:909 e1–6. doi: 10.1016/j.jtct.2021.08.013
32. Jaiswal SR, Chakrabarti A, Chatterjee S, Ray K, Chakrabarti S. Haploidentical transplantation in children with unmanipulated peripheral blood stem cell graft: The need to look beyond post-transplantation cyclophosphamide in younger children. Pediatr Transplant (2016) 20:675–82. doi: 10.1111/petr.12724
33. Jaiswal SR, Chakrabarti A, Chatterjee S, Bhargava S, Ray K, Chakrabarti S. Hemophagocytic syndrome following haploidentical peripheral blood stem cell transplantation with post-transplant cyclophosphamide. Int J Hematol (2016) 103:234–42.
34. Yule SM, Boddy AV, Cole M, Price L, Wyllie R, Tasso MJ, et al. Cyclophosphamide metabolism in children. Cancer Res (1995) 55:803–9.
35. Yule SM, Boddy AV, Cole M, Price L, Wyllie R, Tasso MJ, et al. Cyclophosphamide pharmacokinetics in children. Br J Clin Pharmacol (1996) 41:13–9.
36. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med (1987) 165:302–19.
37. June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today (1990) 11:211–6.
38. Freeman GJ, Freedman AS, Segil JM, Lee G, Whitman JF, Nadler LM. B7, a new member of the Ig superfamily with unique expression on activated and neoplastic B cells. J Immunol (1989) 143:2714–22. doi: 10.4049/jimmunol.143.8.2714
39. Gimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, et al. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci USA (1991) 88:6575–9.
40. Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr., Lombard LA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science (1993) 262:909–11. doi: 10.1126/science.7694363
41. Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, et al. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med (1994) 180:2049–58.
42. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174:561–9.
43. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-X
44. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature (1987) 328:267–70. doi: 10.1038/328267a0
45. Iida T, Ohno H, Nakaseko C, Sakuma M, Takeda-Ezaki M, Arase H, et al. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J Immunol (2000) 165:5062–8. doi: 10.4049/jimmunol.165.9.5062
46. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol (2011) 11:852–63.
47. Xu X, Dennett P, Zhang J, Sherrard A, Zhao Y, Masubuchi T, et al. CTLA4 depletes T cell endogenous and trogocytosed B7 ligands via cis-endocytosis. J Exp Med (2023) 220. doi: 10.1084/jem.20221391
48. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
49. Weber CJ, Hagler MK, Chryssochoos JT, Larsen CP, Pearson TC, Jensen P, et al. CTLA4-Ig prolongs survival of microencapsulated rabbit islet xenografts in spontaneously diabetic Nod mice. Transplant Proc. (1996) 28:821–3.
50. Graser E, Risch K, Linsley PS, Hancock WW, Muller A, Brock J, et al. Synergism of CTLA4-IG and anti-CD4 monoclonal antibody treatment in a rat kidney transplant model. Transplant Proc (1997) 29:1307–9. doi: 10.1016/S0041-1345(96)00624-0
51. Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, et al. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol (1997) 159:5187–91. doi: 10.4049/jimmunol.159.11.5187
52. Noelle RJ, Mackey M, Foy T, Buhlmann J, Burns C. CD40 and its ligand in autoimmunity. Ann N Y Acad Sci (1997) 815:384–91.
54. Meng L, Guo Z, Kim O, He G, Hart J, Szot GL, et al. Blockade of the CD40 pathway fails to prevent CD8 T cell-mediated intestinal allograft rejection. Transplant Proc (2001) 33:418–20.
55. Subbotin V, Sun H, Chen C, Aitouche A, Valdivia L, Fung JJ, et al. Combined blockade of CD28/B7 and CD40/CD40L costimulatory pathways prevents the onset of chronic rejection. Transplant Proc (1998) 30:941–2.
56. Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA (1997) 94:8789–94.
57. Kirk AD, Burkly LC, Batty DS, Baumgartner RE, Berning JD, Buchanan K, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med (1999) 5:686–93.
58. Langer F, Ingersoll SB, Amirkhosravi A, Meyer T, Siddiqui FA, Ahmad S, et al. The role of CD40 in CD40L- and antibody-mediated platelet activation. Thromb Haemost (2005) 93:1137–46. doi: 10.1160/TH04-12-0774
59. Pilat N, Sayegh MH, Wekerle T. Costimulatory pathways in transplantation. Semin Immunol (2011) 23:293–303.
60. Wang J, Guo Z, Dong Y, Kim O, Hart J, Adams A, et al. Role of 4-1BB in allograft rejection mediated by CD8+ T cells. Am J Transplant (2003) 3:543–51.
61. Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci USA (1993) 90:6586–90.
62. Schwartz RH. T cell anergy. Annu Rev Immunol (2003) 21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110
63. Sykes M. Mechanisms of transplantation tolerance in animals and humans. Transplantation (2009) 87:S67–9. doi: 10.1097/TP.0b013e3181a2a6b8
64. Blazar BR, Taylor PA. Regulatory T cells. Biol Blood Marrow Transplant (2005) 11:46–9. doi: 10.1016/j.bbmt.2004.11.005
65. Fuchs A, Gliwinski M, Grageda N, Spiering R, Abbas AK, Appel S, et al. Minimum Information about T Regulatory Cells: A Step toward Reproducibility and Standardization. Front Immunol (2017) 8:1844.
66. Chen ZM, O'Shaughnessy MJ, Gramaglia I, Panoskaltsis-Mortari A, Murphy WJ, Narula S, et al. IL-10 and TGF-beta induce alloreactive CD4+CD25- T cells to acquire regulatory cell function. Blood (2003) 101:5076–83. doi: 10.1182/blood-2002-09-2798
67. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
68. Borges TJ, Murakami N, Lape IT, Gassen RB, Liu K, Cai S, et al. Overexpression of PD-1 on T cells promotes tolerance in cardiac transplantation via ICOS-dependent mechanisms. JCI Insight (2021) 6. doi: 10.1172/jci.insight.142909
69. Hall BM, Tran GT, Verma ND, Plain KM, Robinson CM, Nomura M, et al. Do natural T regulatory cells become activated to antigen specific T regulatory cells in transplantation and in autoimmunity? Front Immunol (2013) 4:208.
70. Malek TR. The main function of IL-2 is to promote the development of T regulatory cells. J Leukoc Biol (2003) 74:961–5.
71. Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, et al. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity (2013) 38:1116–28. doi: 10.1016/j.immuni.2013.02.022
72. Zhao L, Qiu DK, Ma X. Th17 cells: the emerging reciprocal partner of regulatory T cells in the liver. J Dig Dis (2010) 11:126–33. doi: 10.1111/j.1751-2980.2010.00428.x
73. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53.
74. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat Immunol (2019) 20:218–31.
75. Asano T, Meguri Y, Yoshioka T, Kishi Y, Iwamoto M, Nakamura M, et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood (2017) 129:2186–97. doi: 10.1182/blood-2016-09-741629
76. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci (2018) 15:666–73.
77. Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity (2019) 51:1059–1073 e9. doi: 10.1016/j.immuni.2019.11.003
78. Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest (2004) 114:979–87. doi: 10.1172/JCI200420483
79. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12:431–40. doi: 10.1016/S1074-7613(00)80195-8
80. Wekerle T, Kurtz J, Bigenzahn S, Takeuchi Y, Sykes M. Mechanisms of transplant tolerance induction using costimulatory blockade. Curr Opin Immunol (2002) 14:592–600.
81. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42:227–38. doi: 10.1016/j.immuni.2015.01.007
82. Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol (2003) 171:3348–52. doi: 10.4049/jimmunol.171.7.3348
83. Razmara M, Hilliard B, Ziarani AK, Chen YH, Tykocinski ML. CTLA-4 x Ig converts naive CD4+CD25- T cells into CD4+CD25+ regulatory T cells. Int Immunol (2008) 20:471–83.
84. Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol (2005) 6:152–62.
85. Deppong CM, Bricker TL, Rannals BD, Van Rooijen N, Hsieh CS, Green JM. CTLA4Ig inhibits effector T cells through regulatory T cells and TGF-beta. J Immunol (2013) 191:3082–9.
86. Schwarz C, Unger L, Mahr B, Aumayr K, Regele H, Farkas AM, et al. The immunosuppressive effect of CTLA4 immunoglobulin is dependent on regulatory T cells at low but not high doses. Am J Transplant (2016) 16:3404–15.
87. Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant (2005) 5:443–53.
88. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med (2016) 374:333–43.
89. Adams AB, Ford ML, Larsen CP. Costimulation blockade in autoimmunity and transplantation: the CD28 pathway. J Immunol (2016) 197:2045–50. doi: 10.4049/jimmunol.1601135
90. Viglietta V, Bourcier K, Buckle GJ, Healy B, Weiner HL, Hafler DA, et al. CTLA4Ig treatment in patients with multiple sclerosis: an open-label, phase 1 clinical trial. Neurology (2008) 71:917–24. doi: 10.1212/01.wnl.0000325915.00112.61
91. Khoury SJ, Rochon J, Ding L, Byron M, Ryker K, Tosta P, et al. ACCLAIM: A randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult Scler (2017) 23:686–95. doi: 10.1177/1352458516662727
92. Rachid O, Osman A, Abdi R, Haik Y. CTLA4-Ig (abatacept): a promising investigational drug for use in type 1 diabetes. Expert Opin Investig Drugs (2020) 29:221–36.
93. Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest (1999) 103:1243–52. doi: 10.1172/JCI5857
94. Kremer JM, Genant HK, Moreland LW, Russell AS, Emery P, Abud-Mendoza C, et al. Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: a randomized trial. Ann Intern Med (2006) 144:865–76.
95. Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med (2005) 353:1114–23.
96. Kremer JM, Russell AS, Emery P, Abud-Mendoza C, Szechinski J, Westhovens R, et al. Long-term safety, efficacy and inhibition of radiographic progression with abatacept treatment in patients with rheumatoid arthritis and an inadequate response to methotrexate: 3-year results from the AIM trial. Ann Rheum Dis (2011) 70:1826–30. doi: 10.1136/ard.2010.139345
97. Riella LV, Liu T, Yang J, Chock S, Shimizu T, Mfarrej B, et al. Deleterious effect of CTLA4-Ig on a Treg-dependent transplant model. Am J Transplant (2012) 12:846–55.
98. Charbonnier LM, Vokaer B, Lemaitre PH, Field KA, Leo O, Le Moine A. CTLA4-Ig restores rejection of MHC class-II mismatched allografts by disabling IL-2-expanded regulatory T cells. Am J Transplant (2012) 12:2313–21.
99. Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, et al. "Cytokine storm" in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol (2007) 179:3325–31.
100. Williams MA, Onami TM, Adams AB, Durham MM, Pearson TC, Ahmed R, et al. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade-based regimen. J Immunol (2002) 169:5387–91. doi: 10.4049/jimmunol.169.10.5387
101. Gribben JG, Guinan EC, Boussiotis VA, Ke XY, Linsley L, Sieff C, et al. Complete blockade of B7 family-mediated costimulation is necessary to induce human alloantigen-specific anergy: a method to ameliorate graft-versus-host disease and extend the donor pool. Blood (1996) 87:4887–93. doi: 10.1182/blood.V87.11.4887.bloodjournal87114887
102. Guinan EC, Boussiotis VA, Neuberg D, Brennan LL, HIrano N, Nadler LM, et al. Transplantation of anergic histoincompatible bone marrow allografts. N Engl J Med (1999) 340:1704–14.
103. Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med (1998) 339:1186–93.
104. Davies JK, Gribben JG, Brennan LL, Yuk D, Nadler LM, Guinan EC. Outcome of alloanergized haploidentical bone marrow transplantation after ex vivo costimulatory blockade: results of 2 phase 1 studies. Blood (2008) 112:2232–41. doi: 10.1182/blood-2008-03-143636
105. Koura DT, Horan JT, Langston AA, Qayed M, Mehta A, Khoury HJ, et al. In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol Blood Marrow Transplant (2013) 19:1638–49. doi: 10.1016/j.bbmt.2013.09.003
106. Watkins B, Qayed M, McCracken C, Bratrude B, Betz K, Suessmuth Y, et al. Phase II trial of costimulation blockade with abatacept for prevention of acute GVHD. J Clin Oncol (2021) 39:JCO2001086. doi: 10.1200/JCO.20.01086
107. Qayed M, Watkins B, Gillespie S, Bratrude B, Betz K, Choi SW, et al. Abatacept for GVHD prophylaxis can reduce racial disparities by abrogating the impact of mismatching in unrelated donor stem cell transplantation. Blood Adv (2022) 6:746–9. doi: 10.1182/bloodadvances.2021005208
108. Raghunandan S, Gorfinkel L, Graiser M, Bratrude B, Suessmuth Y, Gillespie S, et al. Abatacept for the prevention of GVHD in patients receiving mismatched unrelated transplants: A real-world analysis. Blood Adv (2023). doi: 10.1182/bloodadvances.2023010225
109. Jaiswal SR, Bhakuni P, Joy A, Kaushal S, Chakrabarti A, Chakrabarti S. CTLA4Ig primed donor lymphocyte infusion: A novel approach to immunotherapy after haploidentical transplantation for advanced leukemia. Biol Blood Marrow Transplant (2019) 25:673–82. doi: 10.1016/j.bbmt.2018.12.836
110. Jaiswal SR, Bhakuni P, Bhagawati G, Aiyer HM, Soni M, Sharma N, et al. CTLA4Ig-primed donor lymphocyte infusions following haploidentical transplantation improve outcome with a distinct pattern of early immune reconstitution as compared to conventional donor lymphocyte infusions in advanced hematological Malignancies. Bone Marrow Transplant (2021) 56:185–94. doi: 10.1038/s41409-020-01002-1
111. Jaiswal SR, Aiyer HM, Rawat G, Gera A, Chakrabarti S. CTLA4Ig-based reduced intensity conditioning and donor lymphocyte infusions for haploidentical transplantation in refractory aggressive B-cell lymphoma relapsing after an autograft: Early results from a pilot study. Exp Hematol (2019) 77:26–35 e1. doi: 10.1016/j.exphem.2019.08.002
112. Powles RL, Morgenstern GR, Kay HE, McElwain TJ, Clink HM, Dady PJ, et al. Mismatched family donors for bone-marrow transplantation as treatment for acute leukaemia. Lancet (1983) 1:612–5. doi: 10.1016/S0140-6736(83)91793-2
113. Mathews DV, Wakwe WC, Kim SC, Lowe MC, Breeden C, Roberts ME, et al. Belatacept-resistant rejection is associated with CD28(+) memory CD8 T cells. Am J Transplant (2017) 17:2285–99.
114. Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK. CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J Immunol (2008) 181:3906–14. doi: 10.4049/jimmunol.181.6.3906
115. Krummey SM, Cheeseman JA, Conger JA, Jang PS, Mehta AK, Kirk AD, et al. High CTLA-4 expression on Th17 cells results in increased sensitivity to CTLA-4 coinhibition and resistance to belatacept. Am J Transplant (2014) 14:607–14.
116. Perico N, Amuchastegui S, Bontempelli M, Remuzzi G. CTLA4Ig alone or in combination with low-dose cyclosporine fails to reverse acute rejection of renal allograft in the rat. Transplantation (1996) 61:1320–2. doi: 10.1097/00007890-199605150-00006
117. Jordan SC, Matsumara Y, Zuo XJ, Marchevsky A, Linsley P, Matloff J. Donor-specific transfusions enhance the immunosuppressive effects of single-dose cyclosporine A and CTLA4-Ig but do not result in long-term graft acceptance in a histoincompatible model of rat lung allograft rejection. Transpl Immunol (1996) 4:33–7. doi: 10.1016/S0966-3274(96)80030-X
118. Pilat N, Klaus C, Schwarz C, Hock K, Oberhuber R, Schwaiger E, et al. Rapamycin and CTLA4Ig synergize to induce stable mixed chimerism without the need for CD40 blockade. Am J Transplant (2015) 15:1568–79.
119. Wu T, Sozen H, Luo B, Heuss N, Kalscheuer H, Lan P, et al. Rapamycin and T cell costimulatory blockade as post-transplant treatment promote fully MHC-mismatched allogeneic bone marrow engraftment under irradiation-free conditioning therapy. Bone Marrow Transplant (2002) 29:949–56. doi: 10.1038/sj.bmt.1703574
120. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
121. Pinelli DF, Wakeman BS, Wagener ME, Speck SH, Ford ML. Rapamycin ameliorates the CTLA4-Ig-mediated defect in CD8(+) T cell immunity during gammaherpesvirus infection. Am J Transplant (2015) 15:2576–87.
122. Coenen JJ, Koenen HJ, van Rijssen E, Kasran A, Boon L, Hilbrands LB, et al. Rapamycin, not cyclosporine, permits thymic generation and peripheral preservation of CD4+ CD25+ FoxP3+ T cells. Bone Marrow Transplant (2007) 39:537–45. doi: 10.1038/sj.bmt.1705628
123. Miroux C, Morales O, Carpentier A, Dharancy S, Conti F, Boleslowski E, et al. Inhibitory effects of cyclosporine on human regulatory T cells in vitro. Transplant Proc (2009) 41:3371–4.
124. Singh K, Kozyr N, Stempora L, Kirk AD, Larsen CP, Blazar BR, et al. Regulatory T cells exhibit decreased proliferation but enhanced suppression after pulsing with sirolimus. Am J Transplant (2012) 12:1441–57.
125. Chen YB, Efebera YA, Johnston L, Ball ED, Avigan D, Lekakis LJ, et al. Increased foxp3(+)Helios(+) regulatory T cells and decreased acute graft-versus-host disease after allogeneic bone marrow transplantation in patients receiving sirolimus and RGI-2001, an activator of invariant natural killer T cells. Biol Blood Marrow Transplant (2017) 23:625–34. doi: 10.1016/j.bbmt.2017.01.069
126. Shin HJ, Baker J, Leveson-Gower DB, Smith AT, Sega EI, Negrin RS. Rapamycin and IL-2 reduce lethal acute graft-versus-host disease associated with increased expansion of donor type CD4+CD25+Foxp3+ regulatory T cells. Blood (2011) 118:2342–50. doi: 10.1182/blood-2010-10-313684
127. Moreira-Teixeira L, Resende M, Devergne O, Herbeuval JP, Hermine O, Schneider E, et al. Rapamycin combined with TGF-beta converts human invariant NKT cells into suppressive Foxp3+ regulatory cells. J Immunol (2012) 188:624–31.
128. Kanakry CG, Ganguly S, Zahurak M, Bolanos-Meade J, Thoburn C, Perkins B, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med (2013) 5:211ra157. doi: 10.1126/scitranslmed.3006960
129. Jaiswal SR, Bhakuni P, Zaman S, Bansal S, Bharadwaj P, Bhargava S, et al. T cell costimulation blockade promotes transplantation tolerance in combination with sirolimus and post-transplantation cyclophosphamide for haploidentical transplantation in children with severe aplastic anemia. Transpl Immunol (2017) 43-44:54–9. doi: 10.1016/j.trim.2017.07.004
130. Jaiswal SR, Bhakuni P, Aiyer HM, Soni M, Bansal S, Chakrabarti S. CTLA4Ig in an extended schedule along with sirolimus improves outcome with a distinct pattern of immune reconstitution following post-transplantation cyclophosphamide-based haploidentical transplantation for hemoglobinopathies. Biol Blood Marrow Transplant (2020) 26:1469–76. doi: 10.1016/j.bbmt.2020.05.005
131. Jaiswal SR, Saifullah A, Arunachalam J, Lakhchaura R, Agarwal M, Baligar P, et al. 73 - long-term outcome of abatacept-ptcy-sirolimus based haploidentical HCT in younger patients with nonMalignant diseases. Transplant Cell Ther (2023) 29:S61–2. doi: 10.1016/S2666-6367(23)00142-2
132. Kharya G, Jaiswal SR, Bhat S, Raj R, Yadav SP, Dua V, et al. Impact of conditioning regimen and graft-versus-host disease prophylaxis on the outcome of haploidentical peripheral blood stem cell transplantation for high-risk severe aplastic anemia in children and young adults: A report from the pediatric severe aplastic anemia consortium of India. Transplant Cell Ther (2023) 29:199 e1–199 e10. doi: 10.1016/j.jtct.2022.12.010
133. Ngwube A, Shah N, Godder K, Jacobsohn D, Hulbert ML, Shenoy S. Abatacept is effective as GVHD prophylaxis in unrelated donor stem cell transplantation for children with severe sickle cell disease. Blood Adv (2020) 4:3894–9.
134. Stenger EO, Watkins B, Rogowski K, Chiang KY, Haight A, Leung K, et al. Abatacept GVHD prophylaxis in unrelated hematopoietic cell transplantation for pediatric bone marrow failure. Blood Adv (2023) 7:2196–205. doi: 10.1182/bloodadvances.2022008545
135. Davies JK, Nadler LM, Guinan EC. Expansion of allospecific regulatory T cells after anergized, mismatched bone marrow transplantation. Sci Transl Med (2009) 1:1ra3. doi: 10.1126/scitranslmed.3000153
136. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178:6725–9. doi: 10.4049/jimmunol.178.11.6725
137. Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol (2008) 29:429–35. doi: 10.1016/j.it.2008.06.005
138. Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM, et al. TGF-beta-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol (2014) 44:1728–36.
139. Rissmann A, Pieper S, Adams I, Brune T, Wiemann D, Reinhold D. Increased blood plasma concentrations of TGF-beta1 and TGF-beta2 after treatment with intravenous immunoglobulins in childhood autoimmune diseases. Pediatr Allergy Immunol (2009) 20:261–5.
140. Kessel A, Ammuri H, Peri R, Pavlotzky ER, Blank M, Shoenfeld Y, et al. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol (2007) 179:5571–5. doi: 10.4049/jimmunol.179.8.5571
141. Kaufman GN, Massoud AH, Dembele M, Yona M, Piccirillo CA, Mazer BD. Induction of regulatory T cells by intravenous immunoglobulin: A bridge between adaptive and innate immunity. Front Immunol (2015) 6:469. doi: 10.3389/fimmu.2015.00469
142. Ohata J, Sakurai J, Saito K, Tani K, Asano S, Azuma M. Differential graft-versus-leukaemia effect by CD28 and CD40 co-stimulatory blockade after graft-versus-host disease prophylaxis. Clin Exp Immunol (2002) 129:61–8.
143. Chen Y, Fukuda T, Thakar MS, Kornblit BT, Storer BE, Santos EB, et al. Immunomodulatory effects induced by cytotoxic T lymphocyte antigen 4 immunoglobulin with donor peripheral blood mononuclear cell infusion in canine major histocompatibility complex-haplo-identical non-myeloablative hematopoietic cell transplantation. Cytotherapy (2011) 13:1269–80. doi: 10.3109/14653249.2011.586997
144. Kean LS, Hamby K, Koehn B, Lee E, Coley S, Stempora L, et al. NK cells mediate costimulation blockade-resistant rejection of allogeneic stem cells during nonmyeloablative transplantation. Am J Transplant (2006) 6:292–304.
145. Peng Y, Luo G, Zhou J, Wang X, Hu J, Cui Y, et al. CD86 is an activation receptor for NK cell cytotoxicity against tumor cells. PloS One (2013) 8:e83913. doi: 10.1371/journal.pone.0083913
146. Eissens DN, van der Meer A, Van CB, Preijers FW, Joosten I, Rapamycin and MPA. but not CsA, impair human NK cell cytotoxicity due to differential effects on NK cell phenotype. Am J Transplant (2010) 10:1981–90.
147. Wai LE, Fujiki M, Takeda S, Martinez OM, Krams SM. Rapamycin, but not cyclosporine or FK506, alters natural killer cell function. Transplantation (2008) 85:145–9. doi: 10.1097/01.tp.0000296817.28053.7b
148. Russo A, Oliveira G, Berglund S, Greco R, Gambacorta V, Cieri N, et al. NK cell recovery after haploidentical HSCT with posttransplant cyclophosphamide: dynamics and clinical implications. Blood (2018) 131:247–62. doi: 10.1182/blood-2017-05-780668
149. Jaiswal SR, Zaman S, Chakrabarti A, Sen S, Mukherjee S, Bhargava S, et al. Improved outcome of refractory/relapsed acute myeloid leukemia after post-transplantation cyclophosphamide-based haploidentical transplantation with myeloablative conditioning and early prophylactic granulocyte colony-stimulating factor-mobilized donor lymphocyte infusions. Biol Blood Marrow Transplant (2016) 22:1867–73. doi: 10.1016/j.bbmt.2016.07.016
150. Jaiswal SR, Zaman S, Nedunchezhian M, Chakrabarti A, Bhakuni P, Ahmed M, et al. CD56-enriched donor cell infusion after post-transplantation cyclophosphamide for haploidentical transplantation of advanced myeloid Malignancies is associated with prompt reconstitution of mature natural killer cells and regulatory T cells with reduced incidence of acute graft versus host disease: A pilot study. Cytotherapy (2017) 19:531–42. doi: 10.1016/j.jcyt.2016.12.006
151. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
152. Nguyen S, Dhedin N, Vernant JP, Kuentz M, Al JA, Rouas-Freiss N, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood (2005) 105:4135–42. doi: 10.1182/blood-2004-10-4113
153. Jaiswal SR, Chakraborty S, Lakhchaura R, Shashi P, Mehta A, Soni M, et al. Early and sustained expansion of adaptive natural killer cells following haploidentical transplantation and CTLA4Ig-primed donor lymphocyte infusions dissociate graft-versus-leukemia and graft-versus-host effects. Transplant Cell Ther (2021) 27:144–51. doi: 10.1016/j.jtct.2020.10.005
154. Cichocki F, Taras E, Chiuppesi F, Wagner JE, Blazar BR, Brunstein C, et al. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight (2019) 4. doi: 10.1172/jci.insight.125553
155. Jaiswal SR, Bhakuni P, Bhagwati G, Aiyar HM, Chakrabarti A, Chakrabarti S. Alterations in NKG2A and NKG2C subsets of natural killer cells following epstein-barr virus reactivation in CTLA4Ig-based haploidentical transplantation is associated with increased chronic graft-versus-host disease. Transplantation (2020) 104:e23–30. doi: 10.1097/TP.0000000000002941
156. Kordelas L, Steckel NK, Horn PA, Beelen DW, Rebmann V. The activating NKG2C receptor is significantly reduced in NK cells after allogeneic stem cell transplantation in patients with severe graft-versus-host disease. Int J Mol Sci (2016) 17. doi: 10.3390/ijms17111797
157. Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol Res (2018) 6:766–75.
158. Jaiswal SR, Saifullah A, Arunachalam J, Lakhchaura R, Agarwal M, Baligar P, et al. 556 - immunogenetic determinants of relapse and non-relapse mortality following haploidentical HCT with ptcy and T cell costimulation blockade with abatacept. Transplant Cell Ther (2023) 29:S415–7. doi: 10.1016/S2666-6367(23)00624-3
Keywords: costimulation blockade, abatacept, graft-versus-host disease (GVHD), regulatory T cells (Tregs), hematopoietic cell transplantation, natural killer (NK) cells, haploidentical
Citation: Chakrabarti S and Jaiswal SR (2023) Abatacept and T-cell costimulation blockade—shifting the paradigm in the prevention of graft-versus-host disease. Front. Hematol. 2:1243247. doi: 10.3389/frhem.2023.1243247
Received: 20 June 2023; Accepted: 17 July 2023;
Published: 03 August 2023.
Edited by:
Hong Zheng, The Pennsylvania State University, United StatesReviewed by:
Govindarajan Thangavelu, University of Minnesota Twin Cities, United StatesAkshay Sharma, St. Jude Children’s Research Hospital, United States
Copyright © 2023 Chakrabarti and Jaiswal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suparno Chakrabarti, c3VwY2hha0BnbWFpbC5jb20=