 Hui-Juan Zhang
Hui-Juan Zhang Xiao-Mei Hu
Xiao-Mei Hu- Department of Laboratory Medicine, First Affiliated Hospital of Gannan Medical University, Ganzhou, Jiangxi, China
Thalassemia is a heterogeneous group of genetic disorders affecting the hemoglobin genes leading to decrease synthesis of globin chains of hemoglobin and resulting in ineffective erythropoiesis. It usually contains α- and β-thalassemia, most of common mutation types of which can be detected. However, it’s inclined to miss rare thalassemia mutation types. Here, we analyzed the molecular and hematological characteristics of seven cases with rare β-thalassemia -90 (C>T) (HBB: c.-140 C>T) mutation. Five of them carried β-90 (C>T) heterozygous mutation with a β+ thalassemia trait. One case was αSEA/β-90 genotype with decreasing MCV and MCH obviously, and the other was a β+/β0 intermediate thalassemia patient with β-90/βCD17 genotype, presenting with moderate anemia. A pedigree of one case was analyzed subsequently. It was found that the proband’s maternal grandfather and mother were carriers of α3.7/β-90 double heterozygous thalassemia, who presented that MCV and MCH were decreased normally or slightly, and HbA2 was increased. The proband and his aunt were β-90 (C>T) carriers. It’s necessary to point that the MCV and MCH were much higher in carrier of α3.7/β-90 genotype compared with either αSEA/β-90 genotype or β-90 heterozygous mutation. In this study, we explore the genotypes and phenotypes of four diverse β-90、αSEA/β-90、α3.7/β-90、β-90/βCD17 thalassemia mutations, which enriches the gene profile of β-thalassemia mutation in Chinese population.
Introduction
Thalassemia is a heterogeneous grouping of genetic disorders that results from the defect of hemoglobin synthesis (1). The most common type of hemoglobin comprises 2 α-globin and 2 β-globin subunits, which encoded by the α-globin gene on chromosome 16 and the β-globin gene on chromosome 11 respectively (2). It’s known that α-globin gene has 4 alleles to produce 2 α-globin chain and β-globin gene has 2 alleles to produce 2 β-globin chain (3). Once globin gene is defected, it can lead to the diminished or absent production for one or more of the globin chains. Later this imbalance of globin chain impairs the production of normal hemoglobin. At last This impairment causes ineffective erythropoiesis which results in thalassemia disease (4). Thalassemia is widespread in southern China (5). Ganzhou, as a southern city in Jiangxi province, also has a high incident rate of thalassemia. It has been indicated that a higher prevalence of thalassemia with the heterozygote frequency of 9.49% in Ganzhou, whereas the low frequency was found in middle (3.90%) and northern Jiangxi (2.63%) (6). Most of common thalassemia mutation types can be detected, but the rare types of thalassemia mutations are inclined to be missed. Here, we analyzed the molecular and hematological characteristics of seven cases with rare β-thalassemia -90 (C>T) (HBB: c.-140 C>T) mutation. And then a pedigree analysis was further performed.
Subjects and methods
Subjects
Case 1 was admitted due to “lead poisoning”. Routine examination revealed microcytic hypochromic anemia, so hemoglobin electrophoresis and genetic testing were performed. Subsequently, the proband’s grandfather, grandmother, aunt, mother and father were screened for thalassemia and performed for thalassemia genetic diagnosis. Case 2 was admitted due to syncope for 3 times and thrombocytosis for more than 5 months. Thalassemia genetic testing was performed because of microcytic hypochromic anemia, and the patient was also complicated with iron deficiency anemia. Case 3 was hospitalized due to pulmonary infection and combined with old tuberculosis, chronic gastritis and coercive spondylitis. After admission, the patient was found to have microcytic hypochromic anemia and underwent thalassemia genetic detection. Cases 4, 5, 6 and 7 were routinely screened for thalassemia phenotype due to pregnancy supervision or fertility testing, and genetic testing for thalassemia was performed after positive screening. In addition, case 7 underwent splenectomy in her childhood.
Methods
After obtaining the consent of subjects or their guardians, peripheral blood was extracted with EDTA anticoagulant tubes for sample collection. SysmexXN2100 instrument was used for blood cell analysis. The reference range of hemoglobin (Hb) was (120-160) g/L for men and (110-150) g/L for women. The RBC reference range was (4.3-5.8) *1012/L for male and (3.8-5.1) *1012/L for female. The reference range of mean erythrocyte volume (MCV) was (82-100) fL. The mean hemoglobin content (MCH) reference range was (27-34) pg. The hemoglobin electrophoresis was performed by SEBIA capillary electrophoresis. The reference range of HbA was (96.5-97.5) % and the reference range of HbA2 was (2.5-3.5) %.
The whole blood DNA extraction reagent of Tianlong Company was used to extract DNA from peripheral blood on NP968-C extraction instrument of Tianlong Company. The thalassemia genetic detection reagent was produced by Xiamen Zhishan Biology Co. Ltd with melting curve method. The detection range of reagent included three kinds of deletion α thalassemia types (-SEA, -α3.7, -α4.2), three kinds of non-deletion α thalassemia types (αCS/, αQS/, αWS/) and twenty-one β gene mutations (IVS-II-654, -28, -29, -30, -31, -32, CD14-15, CD15-16, CD17, CD30, IVS-I-5, IVS-I-1, CD26, CD27-28, CD41-42, CD43, CD37, CD71-72, IVS-II-5, -73 and -90).
Results
Hematological features, Hb electrophoresis, and genotype analysis of seven cases
Cases 1 to 5 were detected to be -90 (C>T) (HBB: c.-140 C>T) heterozygous mutation with a typical β-thalassemia trait, including decreased MCV, decreased MCH, and increased HbA2. Case 6 has double heterozygotic mutations of the SEA/αα deletion combined with β-thalassemia -90 (C>T) mutation showing decreased MCV and MCH evidently. Case 7 carried compound heterozygous mutation of β-90 and βCD17, with genotype of β-90/βCD17, which showed the characteristics of moderate anemia (Table 1).
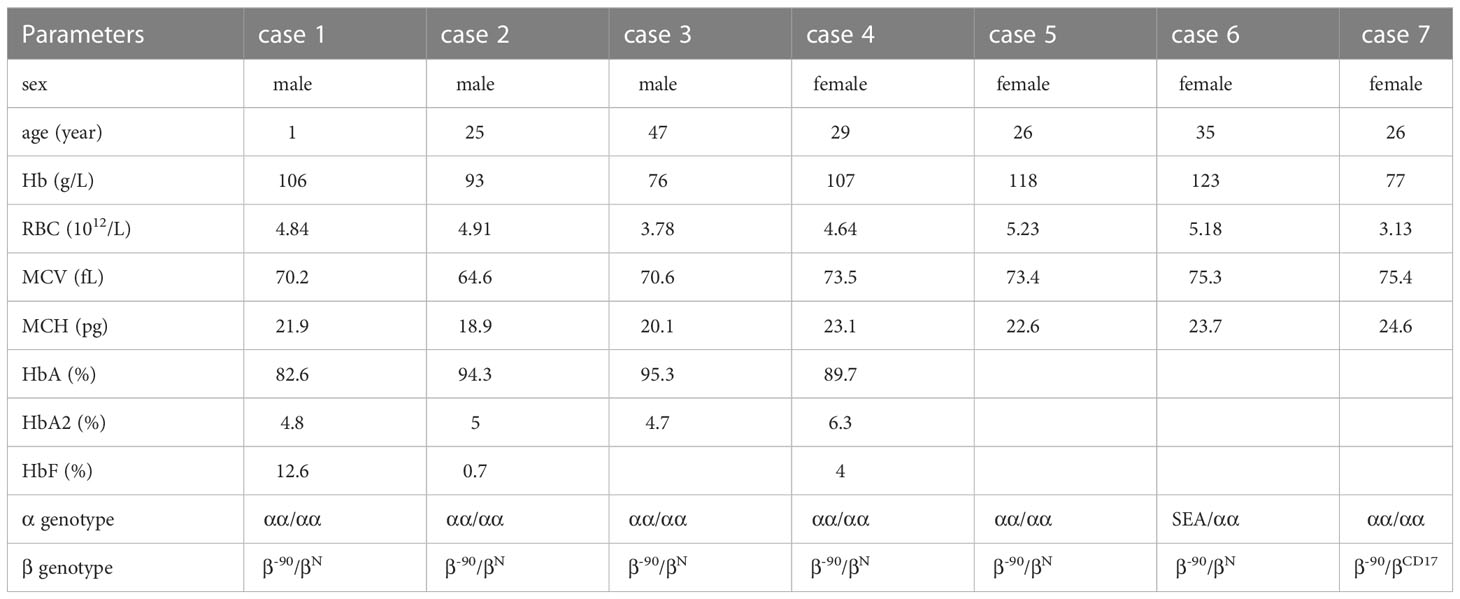
Table 1 The Hematological Profile and Molecular Findings of seven cases.
Pedigree analysis of case 1
The aunt of proband (case 1) carried β-90 heterozygous mutation, which was consistent with the proband. The hematological phenotype showed significant decreases in MCV and MCH and increases in HbA2. However, the maternal grandfather and mother of proband were α3.7/β-90 double heterozygous state of thalassemia, with normal or slightly decreased MCV, mildly reduced MCH, but HbA2 was still elevated (Table 2; Figure 1).
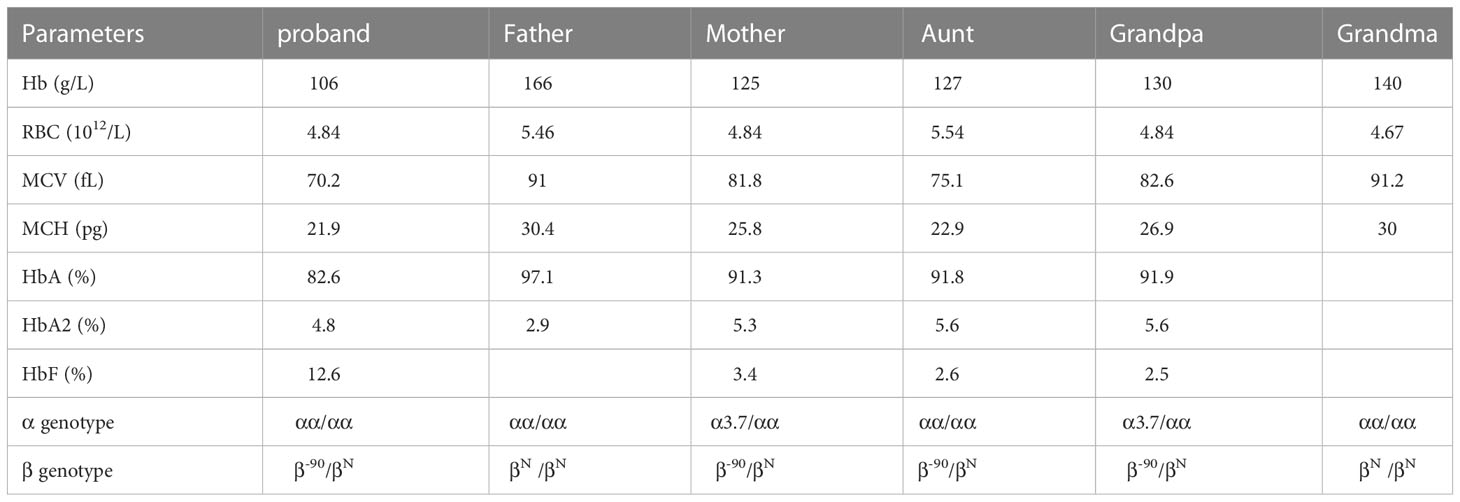
Table 2 The Hematological Profile and Molecular Findings in Pedigree of case 1.

Figure 1 Pedigree analysis of the proband’s family. The arrow indicates the proband (case1, III-1) who carried β-90/βN thalassemia. The corresponding globin genotype of each family member is indicated. “βN” means normal beta globin gene.
Discussion
The mutation of β-thalassemia -90 (C>T) (HBB: c.-140 C>T) is caused by the base transition at -90 position within the upstream promoter of β-globin gene, which located in the CACC box at the binding site of erythroid Kruppel-like factor (EKLF). Due to the mutation of the binding site, the binding affinity between EKLF and the CACCC box decreases, leading to a decline in the transcription level of β-globin gene (7), which is about 60% of the normal transcription level, resulting to the phenotypic characteristics of β+ thalassemia (8).
The β-thalassemia -90 (C>T) mutation was detected in all of seven cases because of microcytic anemia, and delivered in the studied family. The clinical phenotypes of the subjects were consistent with the genetic diagnosis. Simple β-90 carrier was asymptomatic with a β-thalassemia trait, characterized by decreased MCV, decreased MCH, significantly increased HbA2 (>3.5%), indicating a β+ thalassemia phenotype. In this study, the α3.7/β-90 genotype showed normal or slightly decreased MCV, slightly decreased MCH and higher HbA2, while the αSEA/β-90 genotype showed MCV, MCH was decreased significantly. It’s consistent with the published phenotype of αSEA/β-90 patient (9). It indicated that there was no difference of hematological characteristics between β-90/βN and αSEA/β-90 genotypes. However, the MCV and MCH of α3.7/β-90 genotype was much higher than the other two genotypes. It concluded that hematologic phenotype was different once β-90 was combined with different types of α-thalassemia deletion. It needs more evidence to prove the conclusion due to a few cases in α3.7/β-90 and αSEA/β-90 genotypes. Meanwhile, it should be noted that when MCV and MCH are normal or slightly decreased, it may also be double heterozygotes of αβ-thalassemia. The genotype of β-90/βCD17 was β+/β0 intermediate thalassemia, which presented as moderate microcytic hypochromic anemia. The patient had undergone splenectomy at the age of six, which was matched well with the clinical manifestations of moderate β-thalassemia.
β-thalassemia -90 (C>T) mutation was firstly identified in the Portuguese population (10). Later, this rare mutation was found in the India (11), Algeria (12), Pakistan (13) and Thailand (14). With the development of high-throughput second-generation sequencing technology, rare carriers of β-thalassemia -90 (C>T) mutation have been found in many southern regions in China including Guangdong, Guangxi, Hunan, Fujian and so on (15–17). It means that β-thalassemia -90 (C>T) mutation is a relatively common mutation in Chinese rare thalassemia population.
In this research, seven cases and a pedigree associated with β-90 (C>T) rare thalassemia mutation were studied elaborately in China. We found four various genotypes β-90/βN, α3.7/β-90, αSEA/β-90 and β-90/βCD17. This study enriches the gene profile of β-thalassemia mutation in Chinese population, which is of great significance for prevention and control of thalassemia and genetic counseling.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Clinical Ethics Committee of First Affiliated Hospital of Gannan Medical University. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
HZ, XH and DL collected and analyzed the data. HZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the funding from Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control (040201571).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Needs T, Gonzalez-Mosquera LF, Lynch DT. Beta Thalassemia. In: StatPearls. Treasure Island (FL (2023). ineligible companies. Disclosure: Luis Gonzalez-Mosquera declares no relevant financial relationships with ineligible companies. Disclosure: David Lynch declares no relevant financial relationships with ineligible companies.
2. Farid Y, Bowman NS, Lecat P. Biochemistry, Hemoglobin Synthesis. In: StatPearls. Treasure Island (FL (2023). ineligible companies. Disclosure: Nicholas Bowman declares no relevant financial relationships with ineligible companies. Disclosure: Paul Lecat declares no relevant financial relationships with ineligible companies.
3. Bajwa H, Basit H. Thalassemia. In: StatPearls. Treasure Island (FL (2023). ineligible companies. Disclosure: Hajira Basit declares no relevant financial relationships with ineligible companies.
4. Harewood J, Azevedo AM. Alpha Thalassemia. In: StatPearls. Treasure Island (FL (2023). ineligible companies. Disclosure: Alexandre Azevedo declares no relevant financial relationships with ineligible companies.
5. Liang HF, Liang WM, Xie WG, Lin F, Liu LL, Li LJ, et al. The gene spectrum of thalassemia in Yangjiang of western Guangdong Province. Front. Genet. (2023) 14:1126099. doi: 10.3389/fgene.2023.1126099
6. Lin M, Zhong TY, Chen YG, Wang JZ, Wu JR, Lin F, et al. Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. PloS One (2014) 9(7):e101505. doi: 10.1371/journal.pone.0101505
7. Faustino P, Lavinha J, Marini MG, Moi P. beta-Thalassemia mutation at -90C–>T impairs the interaction of the proximal CACCC box with both erythroid and nonerythroid factors. Blood (1996) 88(8):3248–9. doi: 10.1182/blood.V88.8.3248.bloodjournal8883248
8. Li WJ, Lao XW, Jai SQ, Liang FA, Mo QH, Ma JY, et al. A rare transcription mutation (-90 C–>T) in a Chinese family with beta-thalassemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi (2003) 20(6):468–70.
9. Qian H, Huang J, Xu J, Zhao W, Ye X, Liu W. Prenatal diagnosis of a rare beta-thalassemia gene −90 (C>T) (HBB: c.-140 C>T) mutation associated with deletional Hb H disease (–(SEA) /-alpha(4.2) ). Mol. Genet. Genomic Med. (2020) 8(11):e1472. doi: 10.1002/mgg3.1472
10. Faustino P, Osorio-Almeida L, Barbot J, Espirito-Santo D, Goncalves J, Romao L, et al. Novel promoter and splice junction defects add to the genetic, clinical or geographic heterogeneity of beta-thalassaemia in the Portuguese population. Hum. Genet. (1992) 89(5):573–6. doi: 10.1007/BF00219188
11. Achoubi N, Asghar M, Saraswathy KN, Murry B. Prevalence of beta-Thalassemia and hemoglobin E in two migrant populations of Manipur, North East India. Genet. Test Mol. Biomarkers (2012) 16(10):1195–200. doi: 10.1089/gtmb.2011.0373
12. Boudrahem-Addour N, Zidani N, Carion N, Labie D, Belhani M, Beldjord C. Molecular heterogeneity of beta-thalassemia in Algeria: how to face up to a major health problem. Hemoglobin (2009) 33(1):24–36. doi: 10.1080/03630260802626061
13. Moatter T, Kausar T, Aban M, Ghani S, Pal JA. Prenatal screening for beta-thalassemia major reveals new and rare mutations in the Pakistani population. Int. J. Hematol. (2012) 95(4):394–8. doi: 10.1007/s12185-012-1036-7
14. Yamsri S, Singha K, Prajantasen T, Taweenan W, Fucharoen G, Sanchaisuriya K, et al. A large cohort of beta(+)-thalassemia in Thailand: molecular, hematological and diagnostic considerations. Blood Cells Mol. Dis. (2015) 54(2):164–9. doi: 10.1016/j.bcmd.2014.11.008
15. Zhou BY, Wang YX, Xu SS, Gu H, Li MZ. Molecular Spectrum of α- and β-Thalassemia among Young Individuals of Marriageable Age in Guangdong Province, China. Biomed Environ Sci (2021) 34(10):824–9. doi: 10.3967/bes2021.112
16. Lin F, Yang L, Lin M, Zheng X, Lu M, Qiu M, et al. Rare thalassemia mutations among southern Chinese population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi (2017) 34(6):792–6. doi: 10.3760/cma.j.issn.1003-9406.2017.06.002
Keywords: -90 (C>T), rare β -thalassemia, phenotype, genotype, China
Citation: Zhang H-J, Hu X-M and Liu D-D (2023) The research for various genotypes and phenotypes related to rare -90 (C>T) β-thalassemia mutation in Ganzhou city, Southern China. Front. Hematol. 2:1234726. doi: 10.3389/frhem.2023.1234726
Received: 05 June 2023; Accepted: 20 July 2023;
Published: 14 August 2023.
Edited by:
Immacolata Tartaglione, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Zahra Pakbaz, University of California, Irvine, United StatesEfthymia Vlachaki, Aristotle University of Thessaloniki, Greece
Copyright © 2023 Zhang, Hu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui-Juan Zhang, emhqem9lQDE2My5jb20=