 Lorna M. McLeman
Lorna M. McLeman Astrid Glaser
Astrid Glaser Rachel Conyers2,4
Rachel Conyers2,4 Andrew J. Deans
Andrew J. Deans- 1Genome Stability Unit, St Vincent’s Institute, Melbourne, VIC, Australia
- 2Children’s Cancer Centre, Royal Children’s Hospital, Melbourne, VIC, Australia
- 3Department of Medicine, St Vincent’s Health, University of Melbourne, Melbourne, VIC, Australia
- 4Murdoch Children’s Research Institute, Melbourne, VIC, Australia
Fanconi anemia (FA) is the most common inherited bone marrow failure syndrome, characterized by cellular DNA repair deficiency, developmental defects, and a 700-fold increased risk of developing cancer. A bone marrow transplant is the only treatment option for the hematological manifestations of FA, but it can have serious complications. Gene therapy, on the other hand, offers a promising alternative, using cells from the patient that have been corrected ex vivo. However, due to the complexity of cells with a compromised DNA repair pathway, it has been difficult to achieve success in treating FA with gene therapy, despite advancements in the treatment of other blood disorders. This review summarizes all published human trials to date, including a recent study that reported success in treating four pediatric patients with gene therapy, and its interim Phase II study that has successfully treated six further patients. We discuss the key advances, such as improvements in viral vectors, shorter ex vivo transduction protocols, and the use of hypoxia and/or media additives such as N-acetylcysteine or etanercept. We also discuss the potential use of mobilizing agents such as granulocyte-colony stimulating factor (G-CSF) and plerixafor. The data from human trials are systematically reviewed and advances in murine and in vitro studies are discussed.
1 Introduction
Fanconi anemia (FA) is an inherited disorder caused by defective DNA repair. It can be caused by mutations in any of the 22 different “FANC” genes, which normally interact specifically with BRCA1 and BRCA2 breast cancer genes in the repair of stalled DNA replication forks (1). All FA-associated mutations are inherited via an autosomal recessive manner except FANCB, which is x-linked, and FANCR (RAD51), which is autosomal dominant (2, 3). The most common mutations in individuals are in FANCA (64% of FA mutations), FANCC (12%), and FANCG (8%) (4–7). Disruption of the cellular repair pathways causes accumulated replication stalling in hematopoietic stem cells (HSCs), which leads to their progressive depletion. The underlying repair defect also makes the HSCs much more sensitive to DNA interstrand crosslinks—which can arise as byproducts of the metabolism or be caused by chemotherapeutic agents (8, 9). Single-lineage cytopenias, progressing to pancytopenia, occur at a mean age of 8 years (10). Because other stem cell types are also affected during development, FA is additionally characterized by a smaller overall stature, physical abnormalities, reduced fertility, and increased cancer predisposition. Patients can display phenotypic features such as skeletal abnormalities (e.g., a hypoplastic thumb or radius), cutaneous lesions (e.g., café au lait spots), growth abnormalities (e.g., short stature and microcephaly), and eye and genitourinary tract anomalies (11). The cancer risk is high, with over 30% of patients developing myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) (12). Although hematopoietic stem cell transplant (HSCT) outcomes for these conditions have improved in recent years (partly due to the use of fludarabine as a conditioning agent)—up to around 90% 5-year overall survival post-HSCT for patients who have bone marrow failure—there is still a significant reduction to 50% 5-year overall survival for those who develop hematological malignancy (13). The solid tumor risk is also not addressed by transplant, and in fact the risk increases beyond 50% by 20 years post-HSCT (14). There is therefore a significant need to offer alternative curative therapies for these patients.
The only available curative therapy for hematological phenotypes of FA is a bone marrow transplant. Unfortunately, due to the fragile nature of FA patient cells and sensitivity to DNA damage, the risks of chronic graft-versus-host disease and secondary malignancy are high (13). Finding unrelated donor transplants can also be a challenge, and the use of peripheral blood stem cells (which is standard for adult-matched unrelated donors) is also known to increase the risk of graft-versus-host disease and secondary malignancy (14). Overall survival (OS) post-transplant is only 65% at 5 years, reducing to 36% at 20 years (14), compared with, for example, the 5-year OS post-transplant of 95% in pediatric aplastic anemia (15).
Gene therapy has been proposed as a treatment option for FA bone marrow failure and has been studied over the last two decades. In this procedure, a patient’s own (autologous) cells are mobilized and apheresed before being ex vivo transduced with a viral vector containing a wild-type copy of the causative gene. Insertion of the viral gene sequence into the genome of target cells confers normal FA pathway functionality. The cells are then reinfused to the patient, where they have been shown to have a proliferative advantage over non-corrected cells (16–18). Because the modified cells originate from the patient, the use of gene therapy is intended to avoid the complications and potential morbidity of a bone marrow transplant; the procedure removes the laborious search for a human leukocyte antigen (HLA)-haplotype-matched donor, eliminates the requirement for pre-transplant conditioning, maintains existing immunity in the patient, and decreases the risk of graft-versus-host disease. Nonetheless, the difficulties associated with gene therapy for FA have led to disappointment and a process of prolonged in vitro studies aimed at optimizing therapy. Although one successful trial for very young FANCA-associated FA patients with gene therapy has now been reported (19) and also the interim report of the Phase II study (20), significant improvements are still required to make this treatment option a reality for all FA patients. The aim of this systematic review is to summarize the published data from human trials to date, and discuss the advances made in in vitro and murine studies.
2 Methods
Figure 1. Preferred Reporting Items for Systematic Reviews and Meta Analyses (PRISMA) reporting of database searches.
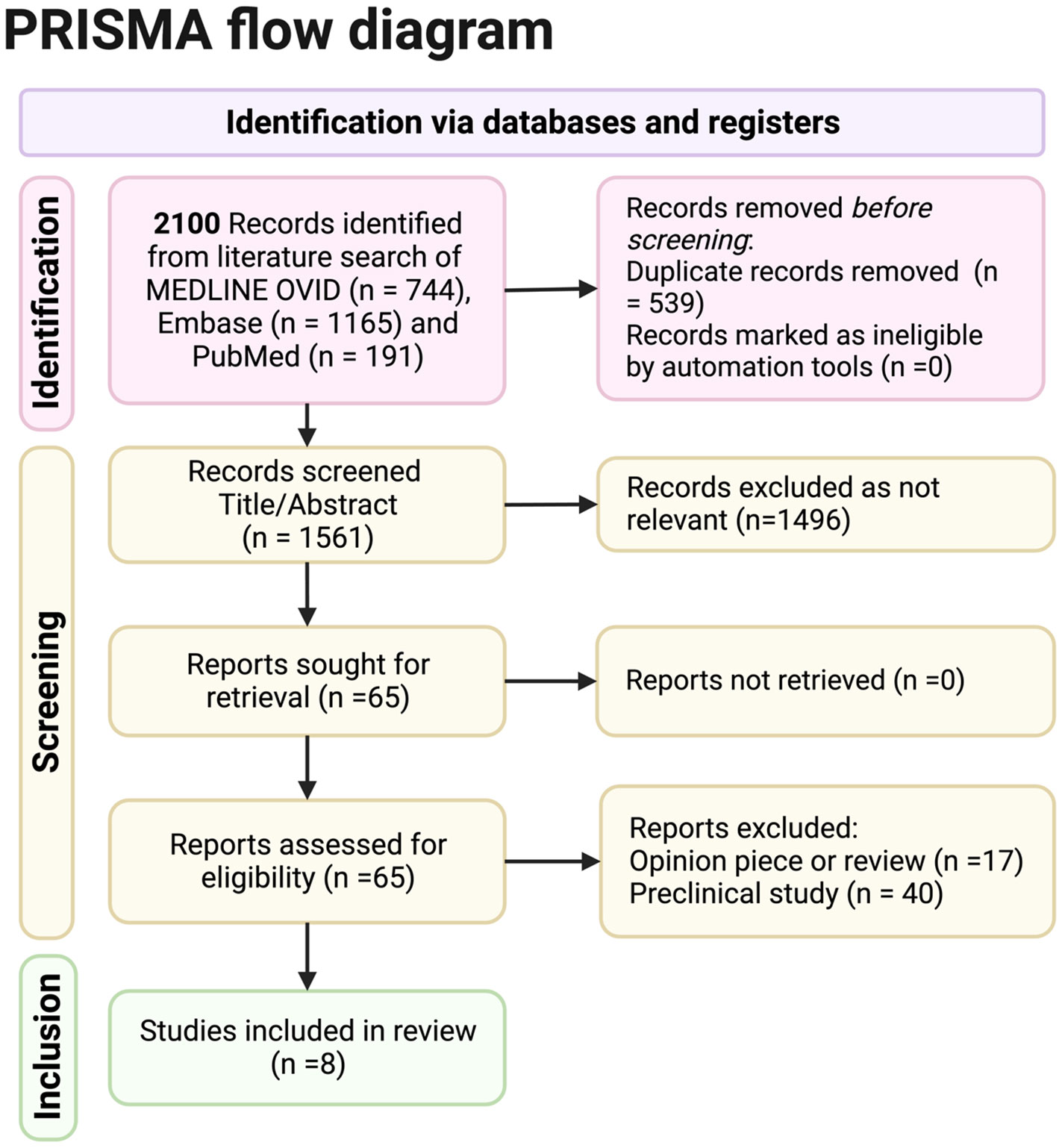
Figure 1 PRISMA reporting of database searches.
The Preferred Reporting Items for Systematic Reviews and Meta Analyses (PRISMA) (2020) guidelines were followed in this systematic review.
2.1 Searches and selection criteria
Searches were conducted in Medline OVID, Embase, and PubMed to identify potential eligible trials and sources of literature on 17 June 2022. Search terms used for Medline and Embase included combinations of “Fanconi anemia” (and term variations) AND “gene editing”/”gene vector”/”hematopoietic stem cell transplantation”/”hematopoietic stem cell”/”genetic therapy”. Terms were limited to English language sources only.
Inclusion criteria were randomized control trials (RCTs), clinical trials, case studies, orcase–control studies, and any participants of any age with FA having been enrolled in any type of gene therapy or editing trial from January 1999 to June 2022.
Exclusion criteria were preclinical, opinion, and review articles, or articles not published in English. Although 40 relevant preclinical studies related to the topic were screened in full text and may be discussed in the discussion part of this article, they were not statistically analyzed.
Data were collected into a structured Microsoft Excel-designed data collection form and where available the following were collected
2.1.1 Study characteristics
Date of data collection, study name, primary author, and publication date
2.1.2 Study methods and participants
Participants’ ages, participants’ sex, country of trial, and FA mutation
2.1.3 Intervention
Whether cell mobilization was used, the vector used, frequency/duration of transductions, and CD34+ cell dose given to patient
2.1.4 Outcomes
Survival, days to engraftment, and genotoxic event.
Missing data will be reported in this study.
2.2 Assessment of risk of bias in the included studies
Two authors planned to work independently to assess for risk of bias using the Cochrane Risk of Bias tool for any RCTs and the Newcastle–Ottawa quality assessment scale for non-randomizedcase–control studies; however, none were present in this review.
2.3 Data synthesis
Homogeneity between studies was assessed to determine whether a meta-analysis could be performed. After discussion with a senior statistician from the University of Melbourne, it was decided that a qualitative analysis was more appropriate.
2.4 Reporting bias assessment
Publication bias was to be assessed with a funnel plot if more than 10 studies were chosen to be included in the review (not in the case of this systematic review). Language bias was a possibility as only English studies were reviewed.
3 Results
3.1 Overall results
The PRISMA flowchart shows the results obtained from the search criteria. From 2,100 initial records selected from a literature search of Medline Ovid, Embase, and PubMed, 1,561 were screened for title and abstract once duplicates were removed. Sixty-five full records were sought for retrieval as relevant, and all were obtained. Out of these records, eight full studies were included. Seventeen opinion pieces were excluded, and 40 preclinical pieces were excluded (although relevant preclinical data and opinion articles by experts will be involved in the discussion).
The four selected trials ran from 1999 to 2022. Overall, 22 patients were enrolled for gene therapy: 15 were treated with lentiviral-based therapy from 2016 onwards (19–21), while the other seven received retroviral-based therapy from 1999 to 2007 (22, 23). Nine had the FANCA mutation and the rest were FANCC.
The youngest patient receiving therapy in any trial was 9 months old and the oldest was 29 years old. These studies are summarised in Table 1.
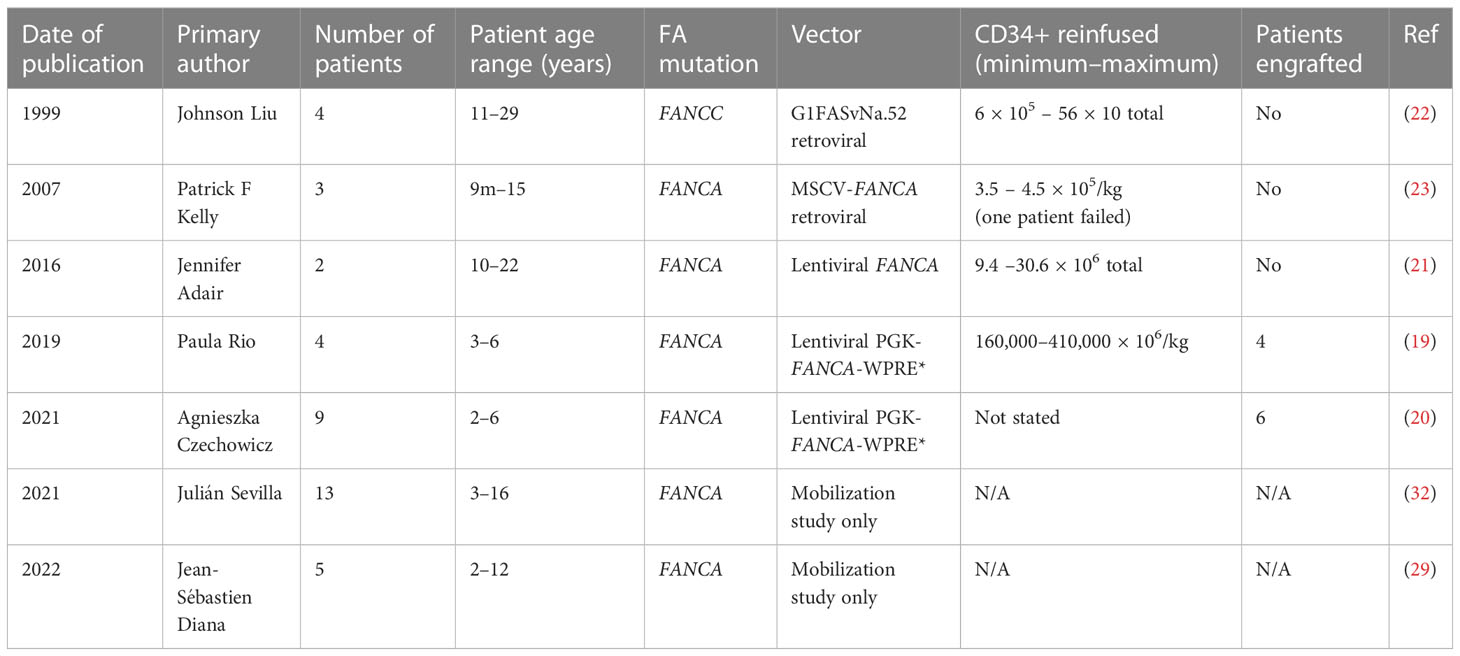
Table 1 Summary of studies included in review.
Successful engraftment of patient cells was demonstrated in only one study (19) and its interim report (20), although resistance to mitomycin C of patient cells was shown in other studies without prolonged hematological responses. The number of reinfused CD34+ cells/kg did not differ significantly between studies, although the number was around 50-fold lower than that being used in other successful gene therapy trials for severe combined immunodeficiency (24). The lower number is likely because FA patients have fewer CD34+ stem cells available for correction (11) and because of their shorter expansion time prior to transplant (25).
Viral vectors differed amongst studies, with lentiviral vectors being favored in the later trials after evidence of safety concerns with retroviral vectors.
Early studies with gammaretroviral vectors showed low levels of success in many trials, and after a patient treated with retroviral gene therapy for x-linked severe combined immunodeficiency developed T-cell acute lymphoblastic leukemia, many groups switched to lentiviral vectors (26, 27). The International Fanconi Anemia Gene Therapy Working Group (28), formed in 2010, also recommended the use of lentiviral vectors over gammaretroviral vectors, as the former could be transduced with shorter protocols, which were less damaging to fragile FA HSCs (24). Subsequent human trials have used a specific lentiviral vector that is self-inactivating and pseudo-typed with vesicular stomatitis virus glycoprotein (VSV-g). This vector is regulated by the human phosphoglycerate kinase (PGK) promoter and encodes the full-length FANCA cDNA.
Two trials have reported on the use of HSC-mobilizing medications, which allow for peripheral blood stem cell collection in FA patients. GCSF at a dose of 6μg/kg or 12μg/kg 12 hourly for days 1–8 and plerixafor at a dose of 240ug/kg 24 hourly for days 4–7 induced mobilization of CD34+ cells; these treatments were safe and effective. Some side effects were observed, such as abdominal pain, vomiting, and fever, but no serious adverse events were reported (29). These methods are beneficial in FA patients because of the reduced number and quality of CD34+ stem cells they produce (11). In both mobilization trials, one-third of the patients—notably the oldest participants—were unable to meet the CD34+ threshold for stem cell collection. This highlights the importance of collecting stem cells early in life, before bone marrow failure develops.
3.2 Detailed analysis of each study
The first FA gene therapy study was published in 1999 (22) by Liu et al. Four FANCC patients—two male and two female, aged 11–29 years—were included in the study. Hematopoietic progenitor cells were collected via bone marrow harvest and transduced ex vivo with the G1FASvNa.52 retroviral vector carrying the FANCC gene. Cells were returned in separate infusions. Patient 1 had 6 × 105 bone marrow CD34+ cells/kg reinfused in four cycles. The FANCC transgene was detectable in peripheral blood (0.01% after infusion one) and bone marrow, for 16 months post infusion. Patient 2 received 56 × 106 peripheral blood CD34+ cells/kg in four infusions and had FANCC vector sequences detected in 1%–3% of peripheral blood mononuclear cells; however, all other samples were negative. Patient 3 was reinfused with a total of 20.4 × 106 peripheral blood CD34+ cells/kg in three infusions and FANCC vector sequences were detectable in <0.01% peripheral blood and bone marrow mononuclear cells, initially dropping to nil after infusion three.
Despite the detection of vectors, the patients only transient improvement in blood counts post each infusion and only short-lived increases in hematopoietic colony numbers in the presence of MMC with subsequent gene therapy cycles. To help determine if the transient improvements in blood counts were related to the gene transfer, the researchers harvested bone marrow from patient 1 to review for FANCC-marked cells at 16 months post-last transfusion: none were found. Again, the researchers reinfused transduced cells 7 months later but were unable to detect evidence of FANCC-marked cells post this infusion.
The only patient to show the sustained presence of FANCC transgene was patient 4. She received 1.13 × 106 bone marrow-derived CD34+ cells/kg in one infusion but was diagnosed with squamous cell carcinoma of the vulva not long after. She underwent pelvic radiation of 45Gy, which induced pancytopenia, resulting in a delay in her continuing radiation treatment. Peripheral blood samples prior to and immediately post radiation did not show any FANCC-marked cells; however, at 220 days post-infusion, as pancytopenia started to recover, the transgene was present and this was confirmed by the RNA PCR assay in peripheral blood mononuclear cells on day 414 (22). This case demonstrated for the first time that the viral expression of a therapeutic transgene results in functional correction and a proliferative advantage over untransduced bone marrow cells in a FA patient.
In 2007, Kelly et al. (23) published their evaluation of the safety and efficacy of gene transfer in seven FANCA patients at Cincinnati Children’s Hospital Medical Center (CCHMC). While all patients had stem cells collected, only three went on to the gene transfer stage (FAAGT 1001–1003) and only two patients (FAAGT 1001 and 1003) had stem cells infused. The cells were collected, pre-stimulated for 36 h, and transduced twice with a murine stem cell virus (MSCV)-FANCA retroviral vector, spending 84 h in culture. The cells were tested via exposure to mitomycin C (MMC) and PCR for MSCV-FANCA vector sequences before reinfusion. Patient FAAGT 1001 received a total of 4.5 × 105 cells nucleated cells/kg and patient FAAGT 1003 received 3.5 × 105 total nucleated cells/kg. FAAGT 1002 had poor cell yield post cryopreservation and poor cell numbers post-transduction so could not be reinfused.
Post-transplant, the reinfusion was well tolerated, but in subsequent blood and bone marrow monitoring patient FAAGT 1001 did not have detectable provirus in blood or bone marrow at any time. In contrast, patient FAAGT 1003 had one to five copies of provirus/105 detected in nucleated peripheral blood cells at 2 and 4 weeks post-infusion but transduced cells were never detected in bone marrow. Both patients had a transient increase in hemoglobin and platelet counts, but no molecular evidence of gene-corrected cells in vivo. No engraftment of transduced gene-corrected cells was shown.
In 2016, an abstract (21) was presented by Jennifer Adair at the First International Fanconi Anemia Working Group Meeting (30) of a Phase I clinical trial (NCT01331018) describing two male patients in Seattle with FANCA mutations who received lentiviral gene therapy. Both patients had low or declining platelets and neutrophils at the time of cell collection and transfusion. No conditioning protocol was included. The first patient was 22 years old and received 9.4 × 106 CD34+ cells from the bone marrow after purification without mobilization. Cells were transduced with a lentiviral vector carrying the FANCA gene in the presence of N-acetylcysteine, resulting in a vector copy number of 0.33 per cell. The second patient was a 10-year-old male who received 30.6 × 106 CD34+ cells from bone marrow. Notably, while the first patient’s cells underwent purification, it resulted in cellular loss, possibly due to low CD34 expression. Subsequently, the second patient underwent only red cell depletion, which led to a higher vector copy number in the transfused product, and higher CFC. This finding indicated an important consideration for FA patients: non-purified cells could more efficiently survive gene transfer, which had also been suggested in previous preclinical studies (31). The transplant was reportedly well tolerated by both patients, although while restoration of MMC resistance was shown in vitro, neither had persistent transduced cells post-infusion. Again, no engraftment of transduced gene-corrected cells was able to be shown in this trial.
The most recently published study investigating gene therapy in FA was published in 2019 by Río et al. (19). This was the first gene therapy trial to show the successful engraftment of gene-corrected cells. In this clinical trial (NCT03157804 FANCOLEN-1), which was part of the larger EUROFANCOLEN collective, four male FANCA patients in Spain underwent mobilized stem cell collection with GCSF 12ug/kg twice daily for 6–7 days and plerixafor 240ug/kg daily for 2–3 days. Cells from patients FA-02002 and FA-02004 were cryopreserved for 20 months and 22 months, respectively, until evidence of bone marrow failure necessitated reinfusion. The remaining two patients received fresh autologous cells that were transduced and reinfused. They collected between 160,000 and 410,000 CD34+ × 106 cells/kg for these patients and calculated an estimated corrected colony-forming cell (CFC) number by calculating the mean vector copy number per cell and analyzing the proportion of MMC-resistant CFCs present. The vector used in this trial was the phosphoglycerate kinase-FANCA-mutated woodchuck hepatitis virus post-transcriptional regulatory element (PGK-FANCA-WPRE*) lentiviral vector (19). Cells were transduced with the virus ex vivo, and then patients were reinfused with between 7,300 cells/kg and 160,000 cells/kg. Patient FA-02002 was infused with 250,000 thawed corrected CD34+ cells/kg equating to 14,000 corrected CFCs/kg. Engraftment of gene-corrected cells was reviewed by analyzing peripheral blood samples every 3 months from 6 months post-infusion. Patient FA-02002 reached 55% of gene-marked cells in peripheral blood leukocytes and 70% in B lymphocytes at 30 months. In bone marrow, they reached 37%–60% of gene-marked cells at 2 years post-infusion and 43.5% of bone marrow CD34+ cells were gene marked at that time.
Patient FA-02004 had 160,000 CD34+ cells/kg equating to 7,300 corrected CFCs/kg infused. They had marked delayed engraftment of gene-marked cells and by 24 months post-infusion the number of corrected cells in the bone marrow was lower at 15%–25%.
The two patients with freshly transduced and infused cells were FA-02006, who received higher cell doses at 410,000 CD34+ cells/kg equating to 160,000 cCFCs/kg, and had 4%–8% of gene-marked cells in the bone marrow at 12 months post-infusion, and patient FA-02005, who had between 10% and 20% of gene-marked cells at 24 months post-infusion. This was slower than engraftment expected from a conditioned bone marrow transplant but was consistently rising in all patients.
Phenotypic correction was assessed ex vivo with survival after MMC exposure and the proportion of chromosomal breaks in peripheral blood T cells after diepoxybutane (DEB) administration. All patient-derived bone marrow samples had progressive increases in MMC resistance over time, and a strong correlation with this and the proportion of corrected bone marrow CD34+ cells, suggests that the level of gene correction was responsible for the phenotypic reversion. Cells collected from patient FA-02002 24 months post-infusion showed up to 70% survival post-MMC exposure, a number similar to healthy donor values. Blood counts also stabilized in three out of four patients (FA-02002, FA-02004, and FA-02006). None of the patients’ bone marrow showed cytogenetic abnormalities via FISH and no clonal cellular populations were found. All patients were alive at the time of follow-up.
In 2021 the interim results of the Phase II study RP-102 clinical trials for nine FA-A patients aged 2–6 years were reported at ASH (20). This followed on from the publication of the successful Phase I/II FANCOLEN-1 trial, with improvements in transduction enhancers, lentiviral vectors, and modifications in cellular processing. The eligibility criteria for the trial included age over or equal to 1 year, with no HLA-matched sibling donor and at least 30 CD34+ cells/μL in the bone marrow. The team used mobilization protocols including GCSF and plerixafor. The viral vector was the previously mentioned lentiviral vector, which also carries the FANCA gene (PGK-FANCA-WPRE). No conditioning was used. The team measured the engraftment of gene-corrected cells with insertion site analysis, including peripheral blood and bone marrow vector copy number (VCN) and MMC resistance in bone marrow colony-forming units as well as improvements in pancytopenia. Engraftment was shown in six patients with peripheral blood VCN at 6 months and two out of three patients who were followed up to 12 months showed increased resistance to MMC in bone marrow colony-forming units. The team reported adverse events: one patient developed further bone marrow failure and required a transplant after an influenza B infection and another patient had a grade 2 infusion reaction to the gene therapy product (20).
3.3 Additional studies assessing mobilization of hematopoietic stem cells in Fanconi anemia patients
In addition to the gene therapy trials, two further studies (29, 32) evaluated the combined use of GCSF and plerixafor to augment stem cell collection in FA patients for the purpose of gene therapy. The first was a multicenter study named FANCOSTEM-1, which enrolled 13 Spanish FANCA patients aged 3 to 16 years. The patients were given GCSF 12 ug/kg twice daily and plerixafor 240 ug/kg up to four doses and apheresis was initiated when CD34+ numbers exceeded 5 CD34+ cells/μL in peripheral blood. Five patients withdrew: two because they did not meet the chromosomal aberration threshold (50%) via diepoxybutane testing for distinguishing FA patients and three who took part in the accompanying gene therapy trial. Mobilization was successful in the remaining cases, and no collections had to be prematurely ceased due to adverse events (32).
The second study (29) was a Phase I/II trial named EUDRACT. Five French FANCA-associated patients were recruited, aged from 2 to 12 years, but one patient was excluded due to cytogenetic abnormalities in their bone marrow. In the remaining four patients, GCSF was used at 6 ug/kg twice daily from days 1 to 8 and plerixafor 240μg/kg each day 2 h prior to apheresis from day 5 onwards. The two youngest patients with evidence of bone marrow failure were able to meet the minimum cellular requirements of 5 × 106/kg CD34+ cells for collection, but the two older patients did not.
The survival number of all studies was 100% at the time of follow-up. Both studies concluded that the combination of GCSF and plerixafor use was safe in FA patients, although increased patient age and certain hematological factors could predict the lower mobilization of CD34+ cells.
4 Discussion
There have been significant advances in gene therapy for FA patients since the first human trial by Johnson Liu et al. in 1999, leading to the successful treatment of 10 pediatric patients in the most recent FANCOLEN-1 trial published in 2019 by Paula Rio et al. (19) and its interim report (20). Further unpublished updates to this ongoing study are dynamic, with 12 total patient enrolments at the time of publication of this review (33). Although analyzing the data in this systematic review with a meta-analysis was not possible given the limited number of studies and lack of similarities between data, there are some key aspects and points to discuss from the qualitative data presented. Firstly, the change in viral vector from gammaretroviral (requiring longer transduction periods and having some safety concerns) to lentiviral made a significant difference to the success of treatment. For example, no patients treated with gammaretroviral vectors showed evidence of gene-corrected cells in vivo, despite showing some initial promising resistance to MMC in vitro. Of the patients treated with lentiviral vectors, two-thirds had a successful outcome. The lentiviral vector was further advanced to include a PGK promoter to express the FANCA cDNA and regulated by a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) (30).
The important potential proliferative advantage of gene-corrected FA cells was highlighted by patient 4 in the first gene therapy trial published in 1999 (22). Although there was no evidence of gene-corrected cells prior to or immediately post her irradiation, she did recover cells with transgene expression upon bone marrow recovery. This proliferative advantage has been thought to be the basis of somatic reversion in some FA patients (34). In a 2017 murine study, where gene-corrected FANCA cells were transplanted into immunodeficient and irradiated mice, there was also a noticeable increase in the proportion of gene-corrected cells that proliferated in vivo, indicating a proliferative advantage post-transplantation (35).
More recently, two studies trialing mobilization protocols for FA patients prior to gene therapy suggest that the procedure should be performed as early in life as possible. This is because older patients, and those with more advanced bone marrow failure, were more difficult to leukapherese to obtain an adequate number of CD34+ HSCs (29, 32). GCSF and plerixafor injections, which work to stimulate the mobilization of HSCs into the bloodstream from the bone marrow, were shown to be excellent and safe. Mobilization is likely to remain a crucial component of the gene therapy protocol of FA patients, given the limited numbers of HSCs compared with non-FA individuals.
Each human trial and numerous in vitro and murine investigation has provided a deeper understanding of the intricate mechanisms underlying gene therapy in FA. As a result, various aspects of the ex vivo cellular handling, transduction, mobilization, and engraftment processes have undergone significant improvements. In this discussion, we will explore the advancements in these crucial steps that have enabled successful treatment outcomes for FA patients.
Most of the in vitro and murine studies focus on improvements to the vector used for transducing FA HSCs. The biggest gains came from switching from gammaretroviral to lentiviral vectors (26, 31, 35–44). In addition to the reduced toxicity of lentivirus in FA cells, lentiviral vectors can also transduce both dividing and quiescent cells, whereas retrovirus is able to transduce dividing cells only. Most HSCs, including those in FA patients, are quiescent (39). Other vector improvements included the addition of mutated WPRE (a 3′ untranslated sequence that increases the stability of expressed transcripts) and a switch to the human PGK promoter, which had been used extensively in non-FA vectors (26). This optimized vector was then the recommendation of the Fanconi Anemia Gene Therapy Working Group and ultimately used in the successful human gene therapy trial discussed in this review. Several trials studying various aspects and improvements to gene therapy in FA are currently active, closed, or recruiting as listed on clinicaltrials.gov (Table 2).
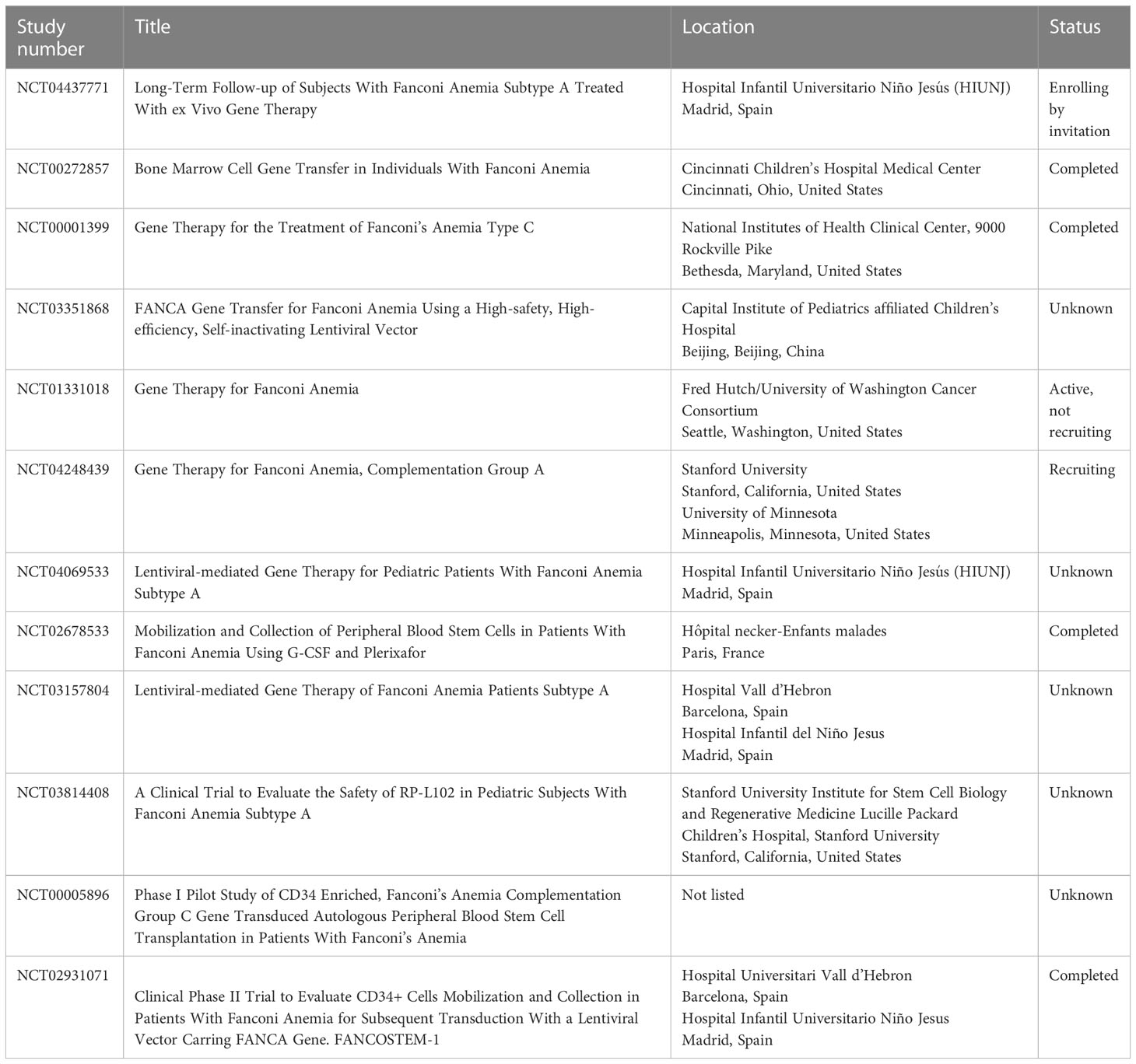
Table 2 Current FA gene therapy studies listed on clinicaltrials.gov.
As FA cells are pro-apoptotic and fragile, several steps in the transduction protocols have been improved to maintain their viability (Table 3). For example, CFCs can be increased by maintaining FA cells ex vivo in conditions of relative hypoxia (5% oxygen compared with 21% oxygen in room air) (31, 41). A similar protective effect has been achieved by maintaining FA cells in the antioxidant N-acetylcysteine (46). Both techniques restrict oxidative DNA damage. It has been shown that the overproduction of the hematopoietic cytokine tumor necrosis factor-alpha (TNF-α) can contribute to bone marrow hypoplasia and stimulate myeloid differentiation in HSCs; therefore, using the TNF-α inhibitor etanercept in cell culture can prevent this and has also been used in ex vivo culture (47). The inclusion of cells found in the bone marrow microenvironment such as mesenchymal stem cells in the transfused product has also been shown to reduce graft failure events in murine models (44). This may suggest not only unhealthy HSCs in FA patients but also a defective microenvironment (24) (Table 3).

Table 3 Improvement factors in in vitro and murine studies.
Future directions in gene therapy for FA are likely to involve the development of gene-editing techniques that differ from traditional viral gene therapy. Major advances in the use of programmable nucleases (such as zinc finger nucleases, TALENs, and the CRISPR/Cas9 system) have been applied in preclinical models to directly target and/or correct causative FA. In 2019, Roman-Rodriguez et al. described using the CRISPR Cas9 non-homologous end-joining approach (which is enhanced in FA cells) to correct function in several different FA subgroups including FANCA, FANCC, FANCD1, and FANCD2 (48). In 2022, Siegner et al. demonstrated direct targeting and conversion of two different mutations (from nonsense to missense) to confer FANCA function using the ABE8e base editor (49). Base editors couple the genome targeting of Cas9 to an enzyme that deaminates DNA nucleotides within a small window close to the CRISPR target site. Although one of the mutations tested showed high levels of unintended off-target mutations within the editing window, both cases demonstrated complete genotypic and phenotypic restoration in the cell lines after 30 days. This highlights the significant possibilities that advances in this technology offer. Gene-editing approaches are likely to be more accurate and have a lower incidence of unwanted insertions and deletions into the genome than viral gene therapy. But important lessons learned from the application of gene therapy, specifically in FA, will also need to be applied in trials using gene editing.
5 Conclusions
Gene therapy for FA has faced certain challenges that have slowed its progress in comparison to some other monogenic conditions. These challenges include difficulties in harvesting and mobilizing sufficient CD34+ cells for transduction, maintaining fragile FA cells ex vivo with minimal cellular loss, and ensuring successful engraftment in a complex bone marrow microenvironment. Overcoming these complexities has proven to be a significant hurdle in the advancement of gene therapy for FA. Improvements have included changes in viral vectors, shortening and enhancement of ex vivo transduction procedures, and the potential for use of mobilizing agents such as GCSF and plerixafor from the human trials.
In good news for patients, there are now several trials recruiting new participants, and positive results observed in those undergoing long-term follow-up. Early indications suggest that the treatment is well tolerated and can delay or prevent hematopoietic decline for several years and likely longer.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
LM performed the analysis of articles for review and wrote the first draft of the manuscript. AD, LM, RC, and AG contributed to manuscript revision. All authors read and contributed and approved the final submitted version.
Funding
LM is a recipient of a grant-in-aid (MRV0027) from Maddie Riewoldt’s Vision. AG is the inaugural Captain Courageous Fellow from Maddie Riewoldt’s Vision (MRV0034). AD is supported by grants from the National Health and Medical Research Council (Australia, GNT1181110), the US Department of Defense Bone Marrow Failure Research Program (USA, BM210032), the Medical Research Future Fund Stem Cell Therapies Initiative (Australia, MRF2024395), and the Victorian Government OIS program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tan W, Deans AJ. A defined role for multiple Fanconi anemia gene products in DNA-damage-associated ubiquitination. Exp Hematol (2017) 50:27–32. doi: 10.1016/j.exphem.2017.03.001
2. Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet (2004) 36(11):1219–24. doi: 10.1038/ng1458
3. Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun (2015) 6:8829. doi: 10.1038/ncomms9829
4. Joshi G, Arthur NBJ, Geetha TS, Datari PVR, Modak K, Roy D, et al. Comprehensive laboratory diagnosis of Fanconi anaemia: comparison of cellular and molecular analysis. J Med Genet (2023) 60:801–9. doi: 10.1136/jmg-2022-108714
5. Galvez E, Vallespin E, Arias-Salgado EG, Sanchez-Valdepenas C, Gimenez Y, Navarro S, et al. Next-generation sequencing in bone marrow failure syndromes and isolated cytopenias: experience of the spanish network on bone marrow failure syndromes. Hemasphere (2021) 5(4):e539. doi: 10.1097/HS9.0000000000000539
6. Maung KZY, Leo PJ, Bassal M, Casolari DA, Gray JX, Bray SC, et al. Rare variants in Fanconi anemia genes are enriched in acute myeloid leukemia. Blood Cancer J (2018) 8(6):50. doi: 10.1038/s41408-018-0090-7
7. Nepal M, Che R, Zhang J, Ma C, Fei P. Fanconi anemia signaling and cancer. Trends Cancer (2017) 3(12):840–56. doi: 10.1016/j.trecan.2017.10.005
8. Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, et al. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol Cell (2015) 60(1):177–88. doi: 10.1016/j.molcel.2015.08.020
9. Sharp MF, Bythell-Douglas R, Deans AJ, Crismani W. The Fanconi anemia ubiquitin E3 ligase complex as an anti-cancer target. Mol Cell (2021) 81(11):2278–89. doi: 10.1016/j.molcel.2021.04.023
10. Brosh RM, Bellani M, Liu Y, Seidman MM. Fanconi Anemia: A DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Res Rev (2017) 33:67–75. doi: 10.1016/j.arr.2016.05.005
11. Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood (2014) 123(1):26–34. doi: 10.1182/blood-2013-09-427740
12. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood (2003) 101(3):822–6. doi: 10.1182/blood-2002-05-1498
13. Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in Fanconi anemia: current evidence, challenges and recommendations. Expert Rev Hematol (2017) 10(1):81–97. doi: 10.1080/17474086.2016.1268048
14. Peffault de Latour R, Porcher R, Dalle JH, Aljurf M, Korthof ET, Svahn J, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood (2013) 122(26):4279–86. doi: 10.1182/blood-2013-01-479733
15. Samarasinghe S, Steward C, Hiwarkar P, Saif MA, Hough R, Webb D, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol (2012) 157(3):339–46. doi: 10.1111/j.1365-2141.2012.09066.x
16. Soulier J, Leblanc T, Larghero J, Dastot H, Shimamura A, Guardiola P, et al. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood (2005) 105(3):1329–36. doi: 10.1182/blood-2004-05-1852
17. Gregory JJ Jr., Wagner JE, Verlander PC, Levran O, Batish SD, Eide CR, et al. Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci USA (2001) 98(5):2532–7. doi: 10.1073/pnas.051609898
18. Lo Ten Foe JR, Kwee ML, Rooimans MA, Oostra AB, Veerman AJ, van Weel M, et al. Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur J Hum Genet (1997) 5(3):137–48. doi: 10.1159/000484749
19. Río P, Navarro S, Wang W, Sánchez-Domínguez R, Pujol RM, Segovia JC, et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med (2019) 25(9):1396–401. doi: 10.1038/s41591-019-0550-z
20. Czechowicz A, Sevilla J, Agarwal R, Booth C, Zubicaray J, Río P, et al. Gene therapy for fanconi anemia [Group A]: interim results of RP-L102 clinical trials. Blood (2021) 138 supplement 1:3968–2969. doi: 10.1182/blood-2021-147071
21. Adair JE, Becker PS, Chandrasekaran D, Choi G, Woolfrey AE, Burroughs L, et al. Gene therapy for fanconi anemia in seattle: clinical experience and next steps. Blood (2016) 128(22):3510. doi: 10.1182/blood.V128.22.3510.3510
22. Liu JM, Kim S, Read EJ, Futaki M, Dokal I, Carter CS, et al. Engraftment of hematopoietic progenitor cells transduced with the Fanconi anemia group C gene (FANCC). Hum Gene Ther (1999) 10(14):2337–46. doi: 10.1089/10430349950016988
23. Kelly PF, Radtke S, von Kalle C, Balcik B, Bohn K, Mueller R, et al. Stem cell collection and gene transfer in Fanconi anemia. Mol Ther (2007) 15(1):211–9. doi: 10.1038/sj.mt.6300033
24. Adair JE, Sevilla J, De Heredia CD, Becker PS, Kiem HP, Bueren J. Lessons learned from two decades of clinical trial experience in gene therapy for fanconi anemia. Curr Gene Ther (2016) 16(5):338–48.
25. Diez B, Genovese P, Roman-Rodriguez FJ, Alvarez L, Schiroli G, Ugalde L, et al. Therapeutic gene editing in CD34+ hematopoietic progenitors from Fanconi anemia patients. EMBO Mol Med (2017) 9:1574–88. doi: 10.15252/emmm.201707540
26. Gonzalez-Murillo A, Lozano ML, Alvarez L, Jacome A, Almarza E, Navarro S, et al. Development of lentiviral vectors with optimized transcriptional activity for the gene therapy of patients with Fanconi anemia. Hum Gene Ther (2010) 21(5):623–30. doi: 10.1089/hum.2009.141
27. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest (2008) 118(9):3132–42. doi: 10.1172/JCI35700
28. International Fanconi Anemia Gene Therapy Working Group. Available at: https://fanconihope.org/research-information/current-research-projects/international-fa-gene-therapy-working-group/.
29. Diana JS, Manceau S, Leblanc T, Magnani A, Magrin E, Bendavid M, et al. A new step in understanding stem cell mobilization in patients with Fanconi anemia: A bridge to gene therapy. Transfusion (2022) 62(1):165–72. doi: 10.1111/trf.16721
30. Tolar J, Adair JE, Antoniou M, Bartholomae CC, Becker PS, Blazar BR, et al. Stem cell gene therapy for fanconi anemia: report from the 1st international Fanconi anemia gene therapy working group meeting. Mol Ther (2011) 19(7):1193–8. doi: 10.1038/mt.2011.78
31. Jacome A, Navarro S, Rio P, Yanez RM, Gonzalez-Murillo A, Lozano ML, et al. Lentiviral-mediated genetic correction of hematopoietic and mesenchymal progenitor cells from Fanconi anemia patients. Mol Ther (2009) 17(6):1083–92. doi: 10.1038/mt.2009.26
32. Sevilla J, Navarro S, Rio P, Sanchez-Dominguez R, Zubicaray J, Galvez E, et al. Improved collection of hematopoietic stem cells and progenitors from Fanconi anemia patients for gene therapy purposes. Mol Ther Methods Clin Dev (2021) 22:66–75. doi: 10.1016/j.omtm.2021.06.001
33. Czechowicz1 A, Sevilla J, Booth C, Navarro S, Agarwal1 R, Zubicaray J, et al. Lentiviral-mediated gene therapy for fanconi anemia [Group A]: results from global RP-L102 clinical trials. ASGCT (2023) 140(Supplement 1):10646–7. doi: 10.1182/blood-2022-168342
34. Ramirez MJ, Pujol R, Trujillo-Quintero JP, Minguillon J, Bogliolo M, Rio P, et al. Natural gene therapy by reverse mosaicism leads to improved hematology in Fanconi anemia patients. Am J Hematol (2021) 96(8):989–99. doi: 10.1002/ajh.26234
35. Rio P, Navarro S, Guenechea G, Sanchez-Dominguez R, Lamana ML, Yanez R, et al. Engraftment and in vivo proliferation advantage of gene-corrected mobilized CD34+ cells from Fanconi anemia patients. Blood (2017) 130(13):1535–42. doi: 10.1182/blood-2017-03-774174
36. Habi O, Girard J, Bourdages V, Delisle MC, Carreau M. Correction of fanconi anemia group C hematopoietic stem cells following intrafemoral gene transfer. Anemia (2010) 2010:947816. doi: 10.1155/2010/947816
37. Yamada K, Olsen JC, Patel M, Rao KW, Walsh CE. Functional correction of fanconi anemia group C hematopoietic cells by the use of a novel lentiviral vector. Mol Ther (2001) 3(4):485–90. doi: 10.1006/mthe.2001.0287
38. Galimi F, Noll M, Kanazawa Y, Lax T, Chen C, Grompe M, et al. Gene therapy of Fanconi anemia: preclinical efficacy using lentiviral vectors. Blood (2002) 100(8):2732–6. doi: 10.1182/blood-2002-04-1245
39. Yamada K, Ramezani A, Hawley RG, Ebell W, Arwert F, Arnold LW, et al. Phenotype correction of Fanconi anemia group A hematopoietic stem cells using lentiviral vector. Mol Ther (2003) 8(4):600–10. doi: 10.1016/S1525-0016(03)00223-5
40. Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, Barrero MJ, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature (2009) 460(7251):53–9. doi: 10.1038/nature08129
41. Becker PS, Taylor JA, Trobridge GD, Zhao X, Beard BC, Chien S, et al. Preclinical correction of human Fanconi anemia complementation group A bone marrow cells using a safety-modified lentiviral vector. Gene Ther (2010) 17(10):1244–52. doi: 10.1038/gt.2010.62
42. Osborn MJ, Gabriel R, Webber BR, DeFeo AP, McElroy AN, Jarjour J, et al. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum. Gene Ther (2015) 26(2):114–26. doi: 10.1089/hum.2014.111
43. Chakkaramakkil Verghese S, Goloviznina NA, Kurre P. Phenotypic correction of Fanconi anemia cells in the murine bone marrow after carrier cell mediated delivery of lentiviral vector. Stem Cell Res Ther (2016) 7(1):170. doi: 10.1186/s13287-016-0431-z
44. Fernandez-Garcia M, Luisa Lamana M, Hernando-Rodriguez M, Sanchez-Dominguez R, Bueren J, Yanez R. Improved hematopoietic gene therapy in a mouse model of fanconi anemia mediated by mesenchymal stromal cells. Hum Gene Ther (2018) 29(3):327–36. doi: 10.1089/hum.2017.076
45. Rio P, Meza NW, Gonzalez-Murillo A, Navarro S, Alvarez L, Surralles J, et al. In vivo proliferation advantage of genetically corrected hematopoietic stem cells in a mouse model of Fanconi anemia FA-D1. Blood (2008) 112(13):4853–61. doi: 10.1182/blood-2008-05-156356
46. Cohen-Haguenauer O, Peault B, Bauche C, Daniel MT, Casal I, Levy V, et al. In vivo repopulation ability of genetically corrected bone marrow cells from Fanconi anemia patients. Proc Natl Acad Sci U S A (2006) 103(7):2340–5. doi: 10.1073/pnas.0510613103
47. Li J, Sejas DP, Zhang X, Qiu Y, Nattamai KJ, Rani R, et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest (2007) 117(11):3283–95. doi: 10.1172/JCI31772
48. Roman-Rodriguez FJ, Ugalde L, Alvarez L, Diez B, Ramirez MJ, Risueno C, et al. NHEJ-mediated repair of CRISPR-cas9-induced DNA breaks efficiently corrects mutations in HSPCs from patients with fanconi anemia. Cell Stem Cell (2019) 25(5):607–21 e7. doi: 10.1016/j.stem.2019.08.016
Keywords: Fanconi anemia, gene editing, gene vector, hematopoietic stem cell transplantation, hematopoietic stem cell, genetic therapy
Citation: McLeman LM, Glaser A, Conyers R and Deans AJ (2023) A systematic review investigating advances in gene therapy for Fanconi anemia over the last three decades. Front. Hematol. 2:1216596. doi: 10.3389/frhem.2023.1216596
Received: 04 May 2023; Accepted: 10 July 2023;
Published: 03 August 2023.
Edited by:
Andrew J Innes, Imperial College London, United KingdomReviewed by:
Stefan Meyer, The University of Manchester, United KingdomMehmet Erman Karasu, ETH Zürich, Switzerland
Copyright © 2023 McLeman, Glaser, Conyers and Deans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorna M. McLeman, bG1jbGVtYW5Ac3ZpLmVkdS5hdQ==; Andrew J. Deans, YWRlYW5zQHN2aS5lZHUuYXU=