 Rui Bergantim1
Rui Bergantim1 Sérgio Chacim2Alexandra Monteiro3Ana M. Macedo4,5
Sérgio Chacim2Alexandra Monteiro3Ana M. Macedo4,5 Gonçalo M. C. Rodrigues6
Gonçalo M. C. Rodrigues6 Maria Gomes da Silva7*
Maria Gomes da Silva7*- 1Hematology Department, Centro Hospitalar Universitário de São João, Porto, Portugal
- 2Hematology Department, Instituto Português de Oncologia do Porto, Porto, Portugal
- 3Hematology Department, Centro Hospitalar Universitário de Lisboa Central – Hospital Santo António dos Capuchos, Lisbon, Portugal
- 4Evidenze, Lisbon, Portugal
- 5Faculty of Medicine and Biomedical Sciences, University of Algarve, Faro, Portugal
- 6Medical Department – Hematology, Janssen-Cilag, S.A., Lisbon, Portugal
- 7Hematology Department, Instituto Português de Oncologia Lisboa Francisco Gentil, Lisbon, Portugal
Clinical features of Waldenström Macroglobulinemia (WM) are variable, often leading to heterogeneous decisions regarding patients’ diagnosis, risk stratification, and treatment. This study assessed the agreement rates on WM diagnosis, risk stratification, and active treatment strategies to capture how this heterogeneity may influence national practices among hematologists. A two-round Delphi-like Panel with 22 national hematologists experienced in WM was conducted online, where 33 statements were classified using a 4-point Likert scale. For each statement, the consensus level was set at 70% for “fully agree/disagree”; the majority level was defined as >70% in agreement or disagreement. After two rounds, no statements were categorized as consensus, and 15 out of 33 failed to obtain a qualified majority. Globally, the experts could not reach a qualified majority in approximately half of the sentences from each category (diagnosis, risk assessment, and therapeutic decision), indicating that contradictory opinions are transversal to all the topics involving WM. A lack of consensus in diagnosing and managing WM among Portuguese hematologists became evident. These results illustrate heterogeneity in clinical practices, and future research initiatives should be considered to improve and reinforce accepted guidelines for diagnosing, assessing, and treating WM patients.
1 Introduction
Waldenström’s macroglobulinemia (WM) is a B-cell lymphoproliferative disease characterized by the presence of a monoclonal immunoglobulin (Ig) M gammopathy and bone marrow (BM) infiltration by lymphoplasmacytic lymphoma (LPL) (1, 2). This rare disease accounts for 1 to 2% of all lymphomas, with a genetic predisposition of up to 20%, and may be associated with autoimmune diseases, such as Sjögren syndrome and autoimmune hemolytic anemia. Its prevalence is higher among Caucasian males, with a median age at diagnosis ranging from 63 to 73 years (3). Although a substantial proportion of patients are initially asymptomatic, around 40 to 70% develop symptoms within 3 to 10 years of diagnosis. Initial clinical manifestations include anemia, fatigue, malaise, nonspecific B-symptoms of fever and weight loss, hyperviscosity, and neuropathy (1, 3, 4).
The diagnosis is based on the histopathological confirmation of BM infiltration by LPL and the identification of circulating monoclonal IgM protein (3). Additionally, testing for specific gene mutations through allele-specific polymerase chain reaction (PCR) or next-generation sequencing techniques can be helpful for the differential diagnosis of other morphologically similar diseases (5). The myeloid differentiation primary response MYD88L265P gene mutation is found in the tumor cells of about 90% of patients with WM (3). However, this mutation is not per se diagnostic of WM, as patients may have wild-type MYD88, and MYD88 mutations can be present in other lymphoid malignancies (3). Furthermore, around 30% to 40% of patients have mutations in the activating C-X-C chemokine receptor type 4 (CXCR4) gene, and more than forty CXCR4 mutations have already been identified (6). MYD88 and CXCR4 mutational status has been associated with different clinical presentations, including disease burden, extramedullary disease, serum IgM levels, symptomatic status at diagnosis, and overall survival (4).
WM is incurable, although treatable, and due to its prolonged course, multiple treatment regimens may be used over time to control the disease and its symptoms (7). Active surveillance is a reasonable approach for asymptomatic patients, and therapy is reserved only for symptomatic disease (5). Several factors are considered when selecting an adequate and timely-appropriate treatment to relieve WM patients’ symptoms, including the clinical and genetic features, patient and physician preferences and comorbidities, and the regimen’s efficacy and toxicity profile (5). Most common treatment indications include anemia (hemoglobin ≤10.0 g/dL), thrombocytopenia (platelet count <100x109/L), constitutional symptoms, peripheral neuropathy and symptomatic hyperviscosity, extramedullary disease, symptomatic cryoglobulinemia, symptomatic cold agglutinins, and/or amyloidosis (5).
The 5-year survival estimates for symptomatic patients have increased to 96%, 90%, and 81% for low-, intermediate-, and high-risk WM, respectively (8, 9). Risk categorization was proposed by the International Prognostic Scoring System for Waldenström Macroglobulinemia (ISSWM) and was based on the presence of 5 covariates, namely age >65 years, hemoglobin ≤11.5 g/dL, platelet count ≤100 × 109/L, β2-microglobulin >3 mg/L and monoclonal IgM concentration >7.0 g/dL (8, 10). More recently, a revised version of this system has been proposed, and five risk categories (instead of the previous three) can now be assessed by including serum albumin (<3.5 gr/dL) and LDH (≥250 IU/L) as covariates, together with age (≤ 65 vs. 66 – 75 vs. ≥ 76 years) and b2-microglobulin (≥ 4 mg/L) (11).
Preferred treatment options at first-line and relapse include several combinations based on anti-CD20 monoclonal antibodies, particularly rituximab (12, 13), alkylating agents (bendamustine and cyclophosphamide (14, 15)), and proteasome inhibitors, such as bortezomib (16). They include DRC (dexamethasone, rituximab, cyclophosphamide), BR (bendamustine, rituximab), or even BDR (bortezomib, dexamethasone, rituximab). Another treatment strategy relies on oral Bruton’s tyrosine kinase inhibitors (BTKis, ibrutinib, and zanubrutinib (17), both approved by FDA and EMA but none reimbursed in Portugal up until early 2023) in monotherapy or even the combination of ibrutinib with rituximab for the treatment of frontline and relapse/refractory patients (3, 7, 18).
The increased understanding of the underlying biology can lead to new genomically-driven therapeutic approaches in WM. Although recurrent mutations in MYD88 and CXCR4 genes shape the landscape of WM, other mutations have been identified, representing potential targets for developing more focused therapeutic approaches (19). Hence, the individual genetic profile of patients has proven to be important in determining the most effective strategies for WM. In fact, patients lacking MYD88 mutations show morphologically similar disease to MYD88-mutated patients but may present a more aggressive profile, decreased overall survival, higher risk of disease transformation, and a worse response to ibrutinib (20). Patients with CXCR4 mutations display a significantly lower rate of adenopathy, and those with nonsense mutations have increased BM infiltration and serum IgM levels (21, 22).
Currently, WM patients in Portugal suffer the consequences of the absence of an implemented referral model, specialized centers specifically dedicated to studying and treating the disease, national therapeutic guidelines, and published national epidemiological data. To the best of our knowledge, the unique available information arises from a regional study that estimated a 2.57% incidence of LPL among mature B-cell non-Hodgkin lymphomas (Silva MG, personal communication), which aligns with other Western countries’ estimates (23–25). Overall, this clinical scenario leads to several distinctive opinions regarding the most suitable approach to WM diagnosis, risk stratification, and decision on active therapeutic strategies that might compromise the overall outcomes of WM patients. To better characterize the approach to WM in Portugal, a two-round Delphi-like Panel (DlP) with a group of Portuguese hematologists experienced in WM was conducted online. The aim was to assess this heterogeneity and discuss real-life practices that may contribute to the standardization of the strategies and, eventually, form the basis for generating practical recommendations to improve the care of Portuguese WM patients.
2 Methods
This study assessed national hematologists’ agreement level regarding diagnosis, risk assessment, and treatment of WM. A group of four experts experienced in WM management was assembled in a focus group to define statements deemed critical for WM. The list of 33 proposed statements was divided into three main categories: I. WM Diagnosis (overall, immunohistochemistry or immunophenotyping by flow cytometry and genetics); II. WM Risk Assessment; and III. WM Therapeutic Decision (first-line and relapse/refractory disease). Following the focus group, a two-round DlP was conducted (Figure 1).

Figure 1 Delphi-like panel methodology.
2.1 Round 1
A group of 40 national hematologists regularly engaged in the management of WM treatment, affiliated with academic and non-academic centers, was invited to anonymously respond to the questionnaire, specifically to categorize the previously defined 33 statements using a 4-point Likert scale: “fully disagree”, “disagree”, “agree”, and “fully agree”. The consensus agreement level was set at 70% of responses “fully disagree” or “fully agree”. Combined levels of at least 70% in terms of agreement (i.e., “agree” and “fully agree”) or disagreement (i.e., “disagree” and “fully disagree”) were categorized as a qualified majority. The statements that did not reach the consensus or the qualified majority agreement level were then selected for a second round.
2.2 Round 2
The group of national hematologists invited in the first round was invited again by e-mail to answer an anonymized online questionnaire and categorize the remaining statements using the same 4-point Likert scale. The consensus agreement level was set again at 70% of responses “fully disagree” or “fully agree”, and combined levels of at least 70% in terms of agreement or disagreement were categorized as a qualified majority. In this study, no statistical analysis was conducted to assess the data. The responses (n = 22) were analyzed by frequency distribution through the four response options (Table 1).
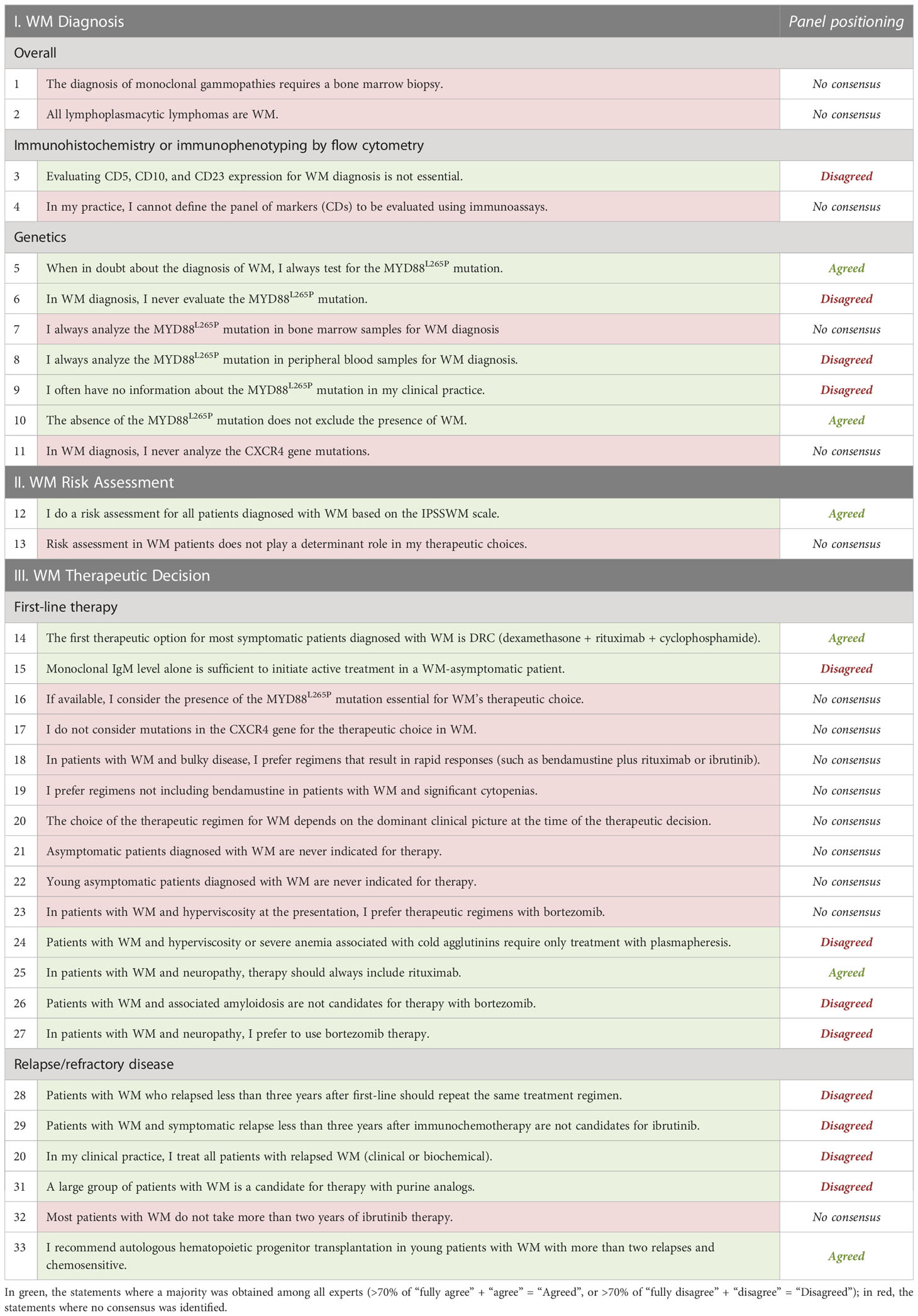
Table 1 Statements defined for the Delphi-like Panel.
3 Results
In this DlP, no statements were categorized as consensus, 18 gathered a qualified majority (Figure 2), and the remaining 15 failed to reach it (Figure 3).
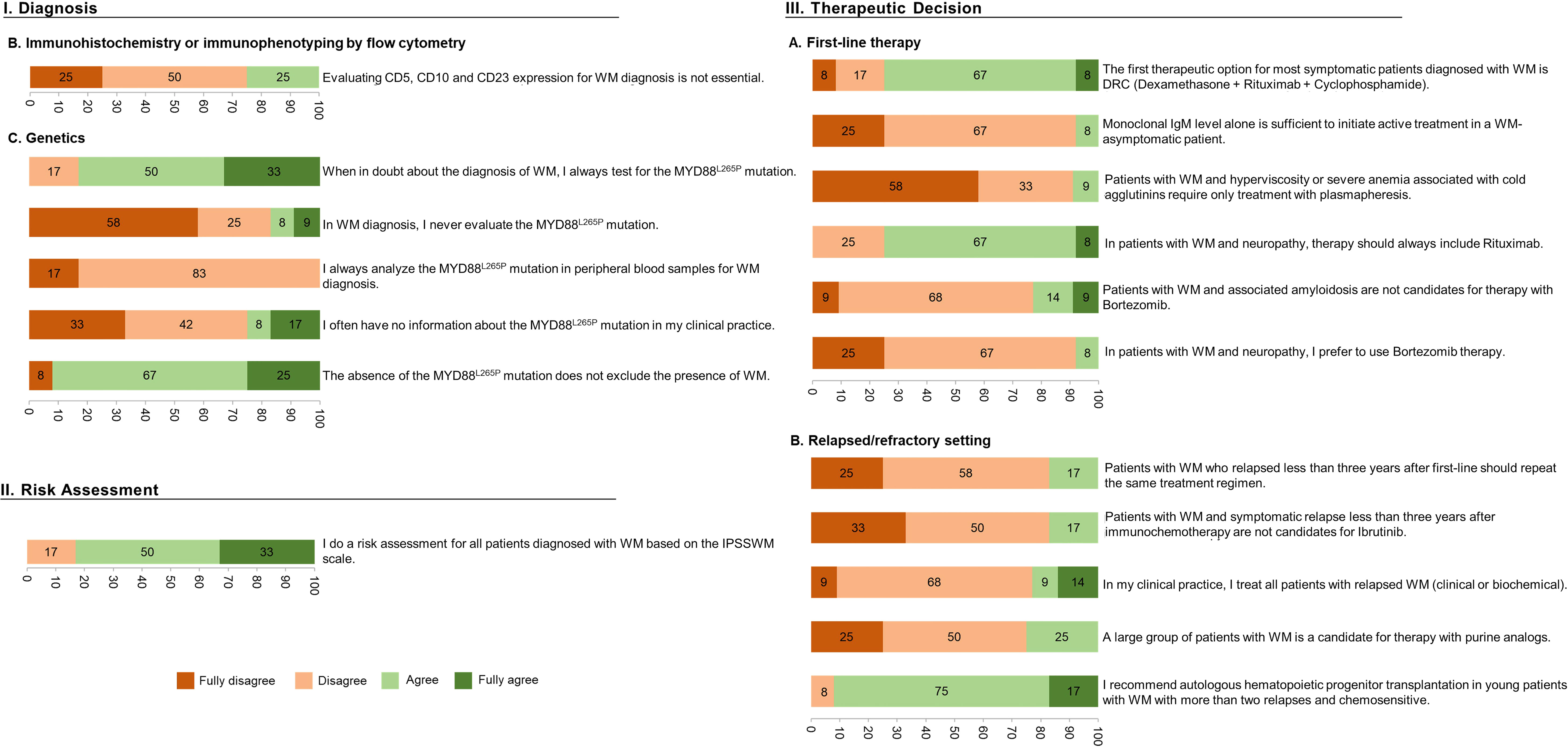
Figure 2 Characterization of the statements (per %) achieving a qualified majority after the 2-round Delphi-like Panel.
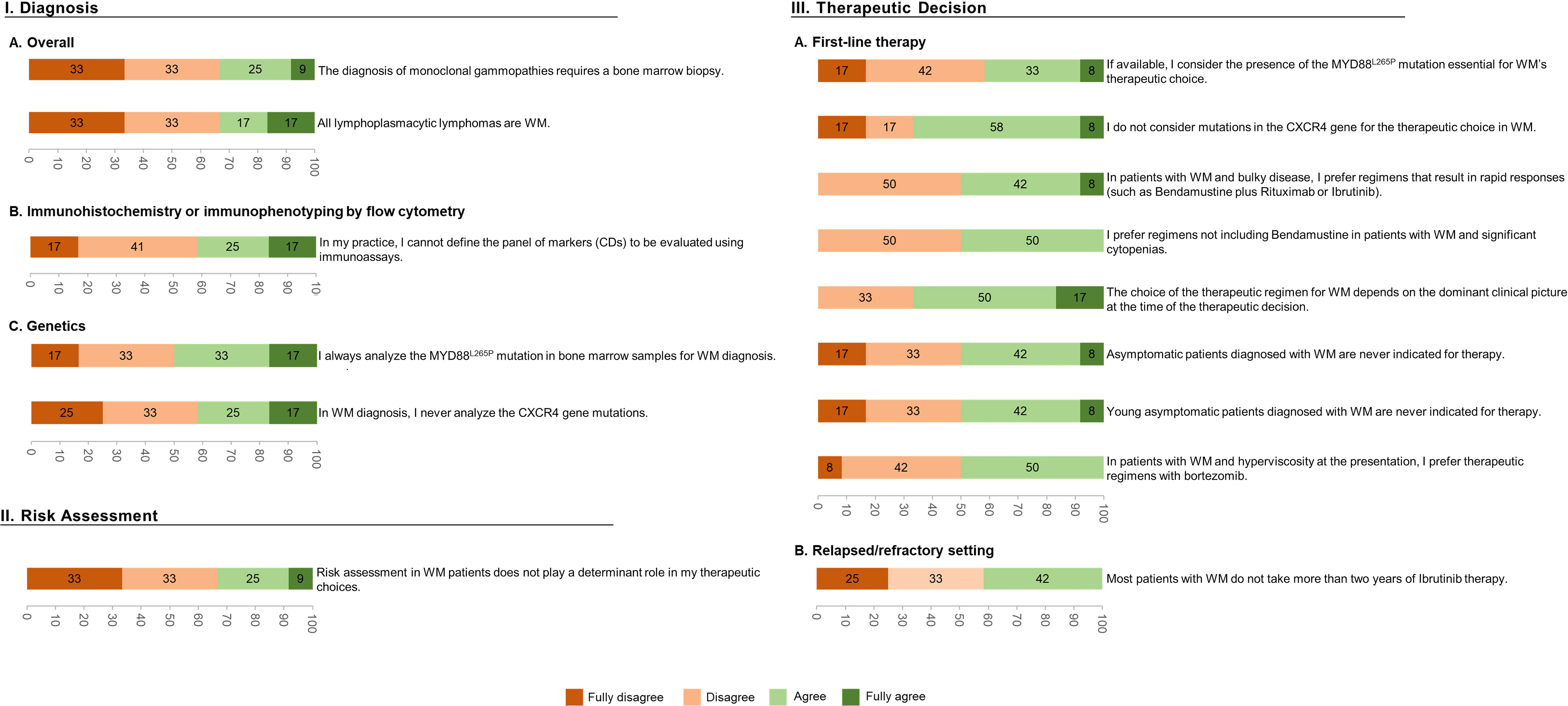
Figure 3 Characterization of the statements (per %) that failed to reach a qualified majority after the 2-round Delphi-like Panel.
Among the 11 statements related to WM Diagnosis, the experts could not agree on 5, including “The diagnosis of monoclonal gammopathies requires a bone marrow biopsy” and “All lymphoplasmacytic lymphomas are WM”. Concerning the statements on practices for assessing WM-specific mutations, the specialists were more concordant. A qualified majority was obtained, for example, when agreeing with the analysis of the MYD88L265P mutation to be essential (83%) and when agreeing that the absence of MYD88L265P mutation does not exclude the presence of WM (92%). A qualified majority was also achieved when disagreeing with the statement: “I never evaluate the MYD88L265P mutation” (83%), when disagreeing with the analysis of the MYD88L265P mutation from peripheral blood samples (100%), and when disagreeing with not having information on the MYD88L265P mutation in the clinical practice (75%). Still, a qualified majority could not be achieved in two related statements, specifically, “I always analyze the MYD88L265P mutation in bone marrow samples for WM diagnosis” and “In WM diagnosis, I never analyze the CXCR4 gene mutations”.
Regarding the “WM Risk Assessment” category, most of the WM experts (83%) agreed about performing a risk assessment using the IPSSWM scale (Figure 2, Risk Assessment). Nonetheless, most experts could not concur with the sentence, “Risk assessment in WM patients does not play a determinant role in my therapeutic choices”.
Considering the last category of this DlP, related explicitly to WM Therapeutic decisions, the experts could not reach a qualified majority in 9/20 statements, mainly associated with first-line therapy. Among these are genetic landscape-related topics: “If available, I consider the presence of the MYD88L265P mutation to be essential for the therapeutic choice of WM”, and “I do not consider mutations in the CXCR4 gene for the therapeutic choice in WM”. The same happened with clinically focused questions: “The choice of the therapeutic regimen for WM depends on the dominant clinical picture at the time of the therapeutic decision” and “Young asymptomatic patients diagnosed with WM never have an indication for therapy”. In the remaining 11 statements where a majority was reached, almost all the experts agreed on DRC being the first therapeutic option for most symptomatic patients in first-line therapy and on including rituximab in case of neuropathy. On the contrary, a qualified majority of hematologists expectedly disagreed with the fact that: 1) patients with WM and hyperviscosity or severe anemia associated with cold agglutinins only require treatment with plasmapheresis, 2) patients with associated amyloidosis not being candidates for therapy with bortezomib, and 3) in case of associated neuropathy, a proteasome inhibitor being preferentially selected. Unexpectedly the panel also disagreed with the statement that “IgM level alone is sufficient to initiate treatment in asymptomatic patients”, despite ESMO’s guidelines endorsing that very high monoclonal IgM levels alone are sufficient to initiate active treatment in a WM-asymptomatic patient. The statements where no qualified majority was obtained but are supported by current ESMO guidelines are “In patients with WM and bulky disease, I prefer regimens that result in rapid responses (such as bendamustine plus rituximab or ibrutinib)”, “In patients with WM and significant cytopenia, I prefer regimens that do not include bendamustine”, and “In patients with WM and hyperviscosity at presentation, I prefer therapeutic regimens with bortezomib”.
Regarding the relapse/refractory setting, a qualified majority was obtained in 5/6 sentences. Most expert panel members recommended autologous stem cell transplantation (ASCT) in young patients with WM with more than two relapses and chemosensitivity, while expectedly disagreeing with 1) if a relapse episode occurs less than three years after the first-line of treatment, the therapeutic scheme should be repeated, 2) if a symptomatic relapse occurs less than three years after CIT, the patient is a non-candidate to ibrutinib, 3) all patients in relapse are to be treated, and 4) a large group of patients being candidates for therapy with purine analogs. This is in agreement with the 2018 ESMO guidelines (3).
4 Discussion
This study assessed the agreement rates on clinical practices related to WM diagnosis, risk stratification, and active treatment among national hematologists. A two-round DlP with 22 Portuguese hemato-oncologists was conducted online to address this goal, where 33 statements were classified using a 4-point Likert scale.
No statement reached consensus, set at 70% of “fully agree/disagree”. Additionally, combined levels of at least 70% agreement (i.e., “agree” and “fully agree”) or disagreement (i.e., “disagree” and “fully disagree”) were categorized as a qualified majority. Accordingly, 45% (15 out of 33) of the statements in this DlP could not obtain a qualified majority. Overall, the results indicate heterogeneity among national clinical practices, emphasizing the need to explore this subject and develop practical guidelines to address these inconsistencies. Thus, we will focus on statements with no qualified majority and heterogeneous classifications since the remaining are aligned with current guidelines.
Of 11 statements concerning WM diagnosis, 50% did not obtain consensus or a qualified majority. The experts could not agree on whether a trephine BM biopsy should be performed to diagnose monoclonal gammopathies. This could result from believing that lymphoplasmacytic cells’ characteristic BM infiltration may be detected less aggressively by performing BM aspirates. Nonetheless, BM trephines may be used for mutational screening, particularly in samples with a high percentage of infiltrating tumor cells (26), being especially useful when the analysis has not been performed on fresh BM aspirate samples (27). BM aspiration and biopsy with immunohistochemistry are both required for WM diagnosis (3). The experts also failed to reach a qualified majority on all LPL being WM. ESMO clearly states that, although in most LPL, the abnormal immunoglobulin is IgM – so defining WM – an abnormal production of IgG can occur in 1 out of 20 LPL cases and, in some cases, no monoclonal gammopathy is present; in those cases, WM cannot be diagnosed (28).
In the genetic studies chapter, the panel agreed that detecting MYD88L265P mutation helps to differentiate WM from morphologically similar lymphomas or IgM myeloma and that its presence alone is not diagnostic of WM (29). Accordingly, while the MYD88 mutation can be identified in 90% of patients with WM, it has been demonstrated that the absence of MYD88L265P gene mutation does not exclude WM, and a few WM patients can have non-L265P MYD88 mutations (30). Dogliotti I. et al. recently published a European consensus statement from the Consortium of WM, which recommends that patients with a high probability of WM diagnosis but failed detection of MYD88L265P in the BM should undergo reliable and sensitive methods, such as by Sanger or NGS, to complete the MYD88 gene sequencing, especially if BTK inhibitors are to be used (27). The relevance of the analysis of CXCR4 genetic mutations for WM diagnosis was an issue that failed unanimity, perhaps due to existing technical challenges for routinely sequencing this gene. Guidance on these WM markers and other diagnostic recommendations have also been elegantly described by the same authors, where detailed laboratory requirements are suggested (27). Specifically, it is disclosed that novel methods with improved sensitivity (Cast-PCR, high-resolution melting analysis, among others) could be used as alternatives for detecting the MYD88L265P and CXCR4 mutations.
While the experts agreed that risk assessment of WM patients based on the IPSSWM scale is important, they were not unanimous in considering that it should play a role in the therapeutic choices. Nonetheless, and according to previous recommendations, as the risk assessment reflects the tumor burden, it can help to decide the most suitable treatment (29).
In the topic “First-line therapy”, in the scope of WM Therapeutic Decision, nearly 60% of the statements failed to obtain a qualified majority, a result that could be expected given the number of available options in this setting (31). Though there was no consensus when debating the choice of therapeutic regimens in dominant clinical scenarios, most experts recommended these to be considered to decide on treatment. Moreover, this is in agreement with the ESMO guidelines, mainly if the tumor burden is a part of the clinical scenario. Thus, the dominant clinical picture is essential and should be considered when choosing the most appropriate therapy.
The statement that WM asymptomatic patients should never be treated but rather be followed without clinical intervention also divided the experts’ opinions. Still, according to the ESMO “laboratory indications for initiation of therapy”, there might be factors that can contribute to the appearance of symptoms, including anemia, thrombocytopenia, and IgM concentrations that might result in fatigue, bruising, and hyperviscosity symptoms, respectively. Overall, while the clinical intervention of asymptomatic patients is not fully established, there is a growing inclination to perform BM assessment as it may provide prognostic information on the risk of progression (32).
Notwithstanding, a qualified majority of experts from the panel disagreed with IgM levels alone being sufficient to initiate treatment in asymptomatic patients, as IgM level per se may not correlate with the WM clinical manifestations. While ESMO guidelines also consider that the isolated level of monoclonal IgM alone is not a valid indicator for initiating treatment, the scenario might be altered when IgM levels are ≥ 60 g/L, as it correlates with symptomatic hyperviscosity. Overall, the treatment must be chosen considering the specific aims of the therapy, the urgency of rapidly controlling the disease, and the risk of treatment-related neuropathy, immunosuppression, and secondary malignancies (33).
The experts could not unify their opinions regarding MYD88L265P and CXCR4 mutations being essential for therapeutic choices in WM. Although this only impacts the choice of BTK inhibitors, CXCR4 might stand out as a valuable biomarker for future therapies. Indeed, the assessment of WM genomic markers has been significantly evolving, including CXCR4 analysis. Although previous recommendations stated that CXCR4 analysis should be optional and not used for clinical decisions (29), more recent ones suggest that MYD88 and CXCR4 mutation status may be helpful for future treatment selection (34, 35). Furthermore, these authors suggest using treatment algorithms that rely on CXCR4 and MYD88 testing when approaching symptomatic untreated and previously treated patients (34, 35).
Regimens that lead to rapid responses in patients with bulky disease, such as bendamustine plus rituximab or ibrutinib, are supported by ESMO, despite the lack of unanimity herein, probably due to the access restrictions to ibrutinib in Portugal. There was no global agreement for patients with significant cytopenias on adopting regimens that do not include bendamustine, contrasting with ESMO recommendations. BR remains the frontline standard of care for patients with WM and high tumor burden (such as massive organomegaly/lymphadenopathy and hyperviscosity), even with the development of second-generation BTK inhibitors. Nonetheless, in WM patients with cytopenias, DRC, bortezomib/rituximab (± dexamethasone) or ibrutinib/rituximab may be preferable because of lower myelotoxicity (29). Due to its favorable safety profile, DRC remains the first therapeutic option for most symptomatic WM patients. Though no agreement was obtained concerning the use of bortezomib in WM patients with hyperviscosity, ESMO guidelines endorse the adoption of this agent with low-dose dexamethasone and rituximab (BDR) or even alone. In the relapsed/refractory setting, a qualified majority was obtained in 5/6 sentences. This higher agreement rate, compared to other areas of the panel, reflects a greater consensus regarding the treatment choice according to the time of relapse and the role of ASCT in young patients with more than two relapses and chemosensitivity, in agreement with ESMO guidelines. The exception was the sentence related to the fact that most patients do not take ibrutinib for more than two years, where no agreement was achieved. This contrasts with results from clinical trials and real-world studies, where more than 50% of patients remain on treatment after two years, even considering toxicity-related discontinuations (18, 30, 36, 37).
Lastly, it is essential to acknowledge a limitation of this study. Specifically, it would be interesting to disclose the potential causes for the lack of consensus herein and eventually relate it to the hematologists’ scientific background, practice setting, or geographical location. Still, the anonymity subjacent to the methodology used impairs this assumption. Nonetheless, and according to our perspective, the lack of consensus may stem from a lack of information/education in a population of hematologists who are not exclusively dedicated to WM.
5 Conclusions
The results obtained in this DlP revealed an evident lack of consensus in the diagnosis, risk assessment, and management of WM among Portuguese hematologists. Though some of the statements obtaining a qualified majority are in line with the recommendations from a multidisciplinary panel of experts (38), it was clear that management decisions for WM patients hinge on many variables. Even so, it was possible to identify some confluence points regarding diagnosis and management. Although WM remains incurable, with a relentless propensity for relapse, it has witnessed several practice-altering advances in recent years. Due to the availability of a new array of therapies, and the patients’ intrinsic variability, the management approaches have become increasingly complex, underlining the need to implement updated guidelines that can actively impact future treatment decisions.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
This study did not require written informed consent for participation under national legislation and institutional requirements.
Author contributions
All authors contributed significantly to the design, organization, and preparation of this work, interpreted the data, and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The project received financial support from Janssen for the logistics of expert meetings and editorial support.
Acknowledgments
The authors would like to thank Ana L. Torres and Maria João Santos, from Evidenze Portugal, for providing medical writing and editorial support to this manuscript and all the hematologists who participated in this study, providing their answers to the questionnaire.
Conflict of interest
RB: Consulting or Advisory Role: Janssen-Cilag, Amgen, BMS, Takeda; Speakers' Bureau: Janssen Cilag, Amgen, BMS, Takeda; Research Funding: Amgen, BMS. SC: Speakers’ Bureau: Janssen Cilag, Celgene and BMS; Research Funding: BMS. MGS: Relationship type: company or organization (last three years); employment: none. Consultancy: Janssen, Roche, Gilead Sciences, MSD, Celgene, Takeda, Novartis, ADC Therapeutics. Research Funding: Gilead Genesis, AstraZeneca; Honoraria: No. Speakers Bureau: Janssen; Stock in publicly-traded company: No; Stock in private company: No; Patents & Royalties: No. SC, AM, AMM, and GMCR: none to declare.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kapoor P, Ansell SM, Braggio E. Waldenstrom macroglobulinemia: genomic aberrations and treatment. Cancer Treat Res. (2016) 169:321–61. doi: 10.1007/978-3-319-40320-5_16
2. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
3. Kastritis E, Leblond V, Dimopoulos MA, Kimby E, Staber P, Kersten MJ, et al. Waldenstrom’s macroglobulinaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. (2018) 29(Suppl 4):iv41–50. doi: 10.1093/annonc/mdy146
4. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in waldenstrom macroglobulinemia. Blood (2014) 123(18):2791–6. doi: 10.1182/blood-2014-01-550905
5. Castillo JJ, Treon SP. Toward personalized treatment in waldenstrom macroglobulinemia. Hematol. Am. Soc. Hematol. Educ. Program (2017) 2017(1):365–70. doi: 10.1182/asheducation-2017.1.365
6. Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in waldenstrom’s macroglobulinema: chances and challenges. Leukemia (2021) 35(2):333–45. doi: 10.1038/s41375-020-01102-3
7. Kastritis E, Dimopoulos MA. Proteasome inhibitors in waldenstrom macroglobulinemia. Hematol. Oncol. Clin. North Am. (2018) 32(5):829–40. doi: 10.1016/j.hoc.2018.05.011
8. Morel P, Duhamel A, Gobbi P, Dimopoulos MA, Dhodapkar MV, McCoy J, et al. International prognostic scoring system for waldenstrom macroglobulinemia. Blood (2009) 113(18):4163–70. doi: 10.1182/blood-2008-08-174961
9. Buske C, Sadullah S, Kastritis E, Tedeschi A, García-Sanz R, Bolkun L, et al. Treatment and outcome patterns in European patients with waldenström’s macroglobulinaemia: a large, observational, retrospective chart review. Lancet Haematol (2018) 5(7):e299–309. doi: 10.1016/S2352-3026(18)30087-5
10. Dimopoulos MA, Kastritis E, Delimpassi S, Zomas A, Kyrtsonis MC, Zervas K. The international prognostic scoring system for waldenstrom’s macroglobulinemia is applicable in patients treated with rituximab-based regimens. Haematologica (2008) 93(9):1420–2. doi: 10.3324/haematol.12846
11. Kastritis E, Morel P, Duhamel A, Gavriatopoulou M, Kyrtsonis MC, Durot E, et al. A revised international prognostic score system for waldenström’s macroglobulinemia. Leukemia (2019) 33(11):2654–61. doi: 10.1038/s41375-019-0431-y
12. Dimopoulos MA, Hamilos G, Efstathiou E, Siapkaras I, Matsouka C, Gika D, et al. Treatment of waldenstrom’s macroglobulinemia with the combination of fludarabine and cyclophosphamide. Leuk Lymphoma (2003) 44(6):993–6. doi: 10.1080/1042819031000077025
13. Zheng YH, Xu L, Cao C, Feng J, Tang HL, Shu MM, et al. Rituximab-based combination therapy in patients with waldenstrom macroglobulinemia: a systematic review and meta-analysis. Onco Targets Ther. (2019) 12:2751–66. doi: 10.2147/OTT.S191179
14. Yang K, Tan J, Wu T. Alkylating agents for waldenstrom’s macroglobulinaemia. Cochrane Database Syst. Rev. (2009) 1:Cd006719. doi: 10.1002/14651858.CD006719.pub3
15. Buske C. Alkylating agents in the treatment of waldenström macroglobulinemia. Hematol. Oncol. Clin. North Am. (2018) 32(5):821–7. doi: 10.1016/j.hoc.2018.05.009
16. Oza A, Rajkumar SV. Waldenstrom macroglobulinemia: prognosis and management. Blood Cancer J. (2015) 5(3):e394–4. doi: 10.1038/bcj.2015.28
17. Tam CS, Opat S, D'Sa S, Jurczak W, Lee HP, Cull G, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic waldenström macroglobulinemia: the ASPEN study. Blood (2020) 136(18):2038–50. doi: 10.1182/blood.2020006844
18. Buske C, Tedeschi A, Trotman J, García-Sanz R, MacDonald D, Leblond V, et al. Ibrutinib plus rituximab versus placebo plus rituximab for waldenström’s macroglobulinemia: final analysis from the randomized phase III iNNOVATE study. J. Clin. Oncol. (2022) 40(1):52–62. doi: 10.1200/JCO.21.00838
19. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P) -directed survival signalling in waldenström macroglobulinaemia cells. Br. J. Haematol (2015) 168(5):701–7. doi: 10.1111/bjh.13200
20. Hunter ZR, Xu L, Tsakmaklis N, Demos MG, Kofides A, Jimenez C, et al. Insights into the genomic landscape of MYD88 wild-type waldenström macroglobulinemia. Blood Adv. (2018) 2(21):2937–46. doi: 10.1182/bloodadvances.2018022962
21. Schmidt J, Federmann B, Schindler N, Steinhilber J, Bonzheim I, Fend F, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br. J. Haematol (2015) 169(6):795–803. doi: 10.1111/bjh.13361
22. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated waldenstrom’s macroglobulinemia. N Engl. J. Med. (2015) 372(15):1430–40. doi: 10.1056/NEJMoa1501548
23. McMaster ML. The epidemiology of waldenstrom macroglobulinemia. Semin. Hematol. (2023) 60:65–72. doi: 10.1053/j.seminhematol.2023.03.008
24. Thandra KC, Barsouk A, Saginala K, Padala SA, Barsouk A, Rawla P, et al. Epidemiology of non-hodgkin’s lymphoma. Med. Sci. (Basel) (2021) 9(1). doi: 10.3390/medsci9010005
25. Groves FD, Travis LB, Devesa SS, Ries LA, Fraumeni JF Jr. Waldenström’s macroglobulinemia. Cancer (1998) 82(6):1078–81. doi: 10.1002/(SICI)1097-0142(19980315)82:6<1078::AID-CNCR10>3.0.CO;2-3
26. Willenbacher E, Willenbacher W, Wolf DG, Zelger B, Peschel I, Manzl C, et al. Digital PCR in bone marrow trephine biopsies is highly sensitive for MYD88(L265P) detection in lymphomas with plasmacytic/plasmacytoid differentiation. Br. J. Haematol (2019) 186(1):189–91. doi: 10.1111/bjh.15792
27. Dogliotti I, Jiménez C, Varettoni M, Talaulikar D, Bagratuni T, Ferrante M, et al. Diagnostics in waldenstrom’s macroglobulinemia: a consensus statement of the European consortium for waldenstrom’s macroglobulinemia. Leukemia (2022) 37:388–95. doi: 10.1038/s41375-022-01762-3
28. King RL, Gonsalves WI, Ansell SM, Greipp PT, Frederick LA, Viswanatha DS, et al. Lymphoplasmacytic lymphoma with a non-IgM paraprotein shows clinical and pathologic heterogeneity and may harbor MYD88 L265P mutations. Am. J. Clin. Pathol. (2016) 145(6):843–51. doi: 10.1093/ajcp/aqw072
29. Dimopoulos MA, Kastritis E. How I treat waldenstrom macroglobulinemia. Blood (2019) 134(23):2022–35. doi: 10.1182/blood.2019000725
30. Ogunbiyi O, Arulogun SO, Patel A, Rismani A, Kyriakou C, D'Sa S, et al. Real world experience on the use of ibrutinib monotherapy in waldenström macroglobulinemia. Blood (2021) 138(Supplement 1):4507–7. doi: 10.1182/blood-2021-153714
31. Cao XX, Yi SH, Jiang ZX, He JS, Yang W, Du J, et al. Treatment and outcome patterns of patients with waldenstrom’s macroglobulinemia: a large, multicenter retrospective review in China. Leuk Lymphoma (2021) 62(11):2657–64. doi: 10.1080/10428194.2021.1938030
32. Pratt G, El-Sharkawi D, Kothari J, D'Sa S, Auer R, McCarthy H, et al. Diagnosis and management of waldenstrom macroglobulinaemia-a British society for haematology guideline. Br. J. Haematol (2022) 197(2):171–87. doi: 10.1111/bjh.18036
33. Mazzucchelli M, Frustaci AM, Deodato M, Cairoli R, Tedeschi A. Waldenstrom’s macroglobulinemia: an update. Mediterr J. Hematol. Infect. Dis. (2018) 10(1):e2018004. doi: 10.4084/MJHID.2018.004
34. Treon SP, Xu L, Liu X, Hunter ZR, Yang G, Castillo JJ, et al. Genomic landscape of waldenstrom macroglobulinemia. Hematol. Oncol. Clin. North Am. (2018) 32(5):745–52. doi: 10.1016/j.hoc.2018.05.003
35. Despina F, Meletios Athanasios D, Efstathios K. Emerging drugs for the treatment of waldenstrom macroglobulinemia. Expert Opin. Emerg. Drugs (2020) 25(4):433–44. doi: 10.1080/14728214.2020.1822816
36. Trotman J, Buske C, Tedeschi A, Matous JV, MacDonald D, Tam CS, et al. Single-agent ibrutinib for rituximab-refractory waldenstrom macroglobulinemia: final analysis of the substudy of the phase III Innovate(TM) trial. Clin. Cancer Res. (2021) 27(21):5793–800. doi: 10.1158/1078-0432.CCR-21-1497
37. Castillo JJ, Gustine JN, Meid K, Flynn CA, Demos MG, Guerrera ML, et al. Response and survival outcomes to ibrutinib monotherapy for patients with waldenstrom macroglobulinemia on and off clinical trials. Hemasphere (2020) 4(3):e363. doi: 10.1097/HS9.0000000000000363
Keywords: hematological malignancy, non-Hodgkin lymphoma, Waldenström Macroglobulinemia, Delphi panel, diagnosis, risk stratification, treatment
Citation: Bergantim R, Chacim S, Monteiro A, Macedo AM, Rodrigues GMC and da Silva MG (2023) Waldenström Macroglobulinemia diagnosis, risk assessment and treatment in Portugal – results from a Delphi-like Panel. Front. Hematol. 2:1203369. doi: 10.3389/frhem.2023.1203369
Received: 10 April 2023; Accepted: 08 June 2023;
Published: 26 June 2023.
Edited by:
Guido Gini, Azienda Ospedaliero Universitaria Ospedali Riuniti, ItalyCopyright © 2023 Bergantim, Chacim, Monteiro, Macedo, Rodrigues and da Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Gomes da Silva, bWdzaWx2YUBpcG9saXNib2EubWluLXNhdWRlLnB0