 Ritsuko Shimizu
Ritsuko Shimizu Masayuki Yamamoto
Masayuki Yamamoto- 1Department of Molecular Hematology, Tohoku University Graduate School of Medicine, Sendai, Japan
- 2Tohoku Medical Megabank Organization, Tohoku University, Sendai, Japan
GATA1 is an essential master regulator of erythropoiesis and megakaryopoiesis. Accumulating lines of evidence have shown that dynamic changes in GATA1 gene expression levels during erythropoiesis are crucial for proper erythroid differentiation. Since GATA1 is an X-chromosome gene, GATA1 knockout leads to embryonic lethal dyserythropoiesis in male mice, while heterozygous female mice can survive. In the past decade, it has become clear that germline GATA1 gene mutations leading to structural changes in the GATA1 protein are involved in congenital dyserythropoiesis in males. In contrast, decreased GATA1 expression levels, which cause embryonic lethal dyserythropoiesis in male mice, increase the risk of erythroleukemia development in female mice, while female GATA1-knockout mice do not show substantial phenotypic alterations in erythroid or megakaryocyte lineages. In this review, we summarize the recent progress in elucidating the roles of GATA1 in normal and pathogenetic erythropoiesis and discuss the possible mechanisms of pathogenesis of dyserythropoiesis and erythroleukemia.
Introduction
GATA1 is an essential master regulator of erythropoiesis and several other hematopoietic pathways, including megakaryopoiesis (1–4). GATA1 belongs to the hematopoietic GATA transcription factor family, along with GATA2 and GATA3 (5). The regulation of Gata1 gene expression has been robustly investigated using mice. The expression of Gata1 begins in common myeloid progenitors and gradually increases during erythroid differentiation (6). In contrast, GATA2 plays indispensable roles in generating and maintaining hematopoietic stem and progenitor cells (HSPCs) (7). While GATA2 is abundantly expressed in HSPCs, the Gata1 gene is not actively expressed, as the region upstream of the Gata1 promoter is highly methylated, which prevents Gata1 gene expression in HSPCs (8). When this DNA hypermethylation is removed in HSPCs, GATA2 drives Gata1 expression by binding to the open chromatin of the promoter (8). Subsequently, Gata2 expression is gradually reduced, and Gata1 expression is elevated toward erythroid development by self-activation of GATA1, resulting in the dynamic change in GATA factor expression from GATA2 to GATA1 referred to as “GATA factor switching” (9, 10).
The negative regulation of GATA1 in HSPCs is functionally important for hematopoiesis. In fact, genetic ablation of the region upstream of the Gata1 promoter, referred to as the Gata1 methylation-determining region (G1MDR), results in abundant expression of GATA1 in HSPCs in mice (11). This ectopic expression of GATA1 suppresses Gata2 gene expression in HSPCs, resulting in imbalanced GATA2/GATA1 expression in HSPCs and disturbance of hematopoietic homeostasis (11).
After reaching a maximum level in the proerythroblast stage, the expression level of Gata1 gradually decreases during erythroid maturation (12). This downregulation of Gata1 expression is also an important feature of erythroid homeostasis. In fact, ectopic expression of GATA1 in late erythroid lineages again disturbs normal erythropoiesis (13). It has been shown that multiple enhancers scattered around the Gata1 gene participate cooperatively and independently in Gata1 gene regulation during distinct steps of erythropoiesis (6, 14). However, the molecular basis of Gata1 gene downregulation during mature erythroid differentiation remains unclear. Considering many lines of evidence in this regard, we propose that the dynamics of Gata1 expression are crucial in ensuring proper erythroid differentiation (6, 15). Changes in the expression level of GATA1 either act as regulatory cues for normal erythropoiesis or block differentiation at a variety of stages during erythroid differentiation.
Structure of GATA1
GATA1 is highly conserved between humans and mice, and the production of the 413-amino acid GATA1 protein is directed by the GATA1 gene, which is comprised of five coding exons in both species (5). One (human) or two (mouse) main noncoding first exons along with a few minor alternative first exons precede these coding exons (16–20).
The GATA1 protein has four functional domains, all of which are required for a fully functional GATA1. Two transactivation domains, located in the N- and C-terminal regions, contribute independently and cooperatively to the full transcriptional activity of GATA1 (21). GATA1 has two zinc-finger domains aligned in the middle of the protein. The C-terminal zinc-finger (C-finger) domain is required to recognize a target site conforming to the consensus GATA motif, while the N-terminal zinc finger (N-finger) domain is utilized in bivalent binding to two GATA motifs aligned in a palindromic configuration in collaboration with the C-finger domain (22). In addition, two C-finger domains in homodimerized GATA1 molecules bivalently bind to two GATA motifs aligned in the same orientation (22).
The C-finger and N-finger domains are also important for interacting with a variety of coactivators and corepressors by which GATA1 function is properly tuned (23–26). In particular, interaction with FOG1 (Friend of GATA1) through the N-finger domain plays important roles in GATA1 activity (23). While a conventional expression plasmid assay revealed that the N-finger domain does not contribute to the transactivation activity of GATA in cultured cells, more sophisticated analyses utilizing genetically engineered mice revealed that GATA1 lacking the N-finger domain cannot support embryonic erythropoiesis in vivo (27), demonstrating that the N-finger domain is required for proper erythropoiesis. The domain structures of GATA1 and the constituent amino acid motifs within the functional domains appear to play specific roles in the integrated function of GATA1. Mutations within these motifs seem to provoke specific defects in GATA1 function in mammals.
In addition to the canonical transcripts in humans, which include one noncoding and five coding exons, a noncanonical transcript variant skipping exon 2 has been identified (28). The variant transcript yields a short form of the GATA1 protein (named GATA1s) by utilizing the second ATG codon on exon 3 (Figure 1) (28). Since GATA1s lacks 83 amino acids present in the N-terminus of full-length GATA1 (GATA1fl), the function of the N-terminus transactivation domain is strongly disrupted, whereas the functions of the other three functional domains, i.e., the C-terminus transactivation domain and two zinc finger domains, are preserved. Therefore, GATA1s is predicted to play different roles as a transcription factor than GATA1fl. How the expression of this transcriptional isoform is regulated and how GATA1s contributes to hematopoiesis remain to be clarified.
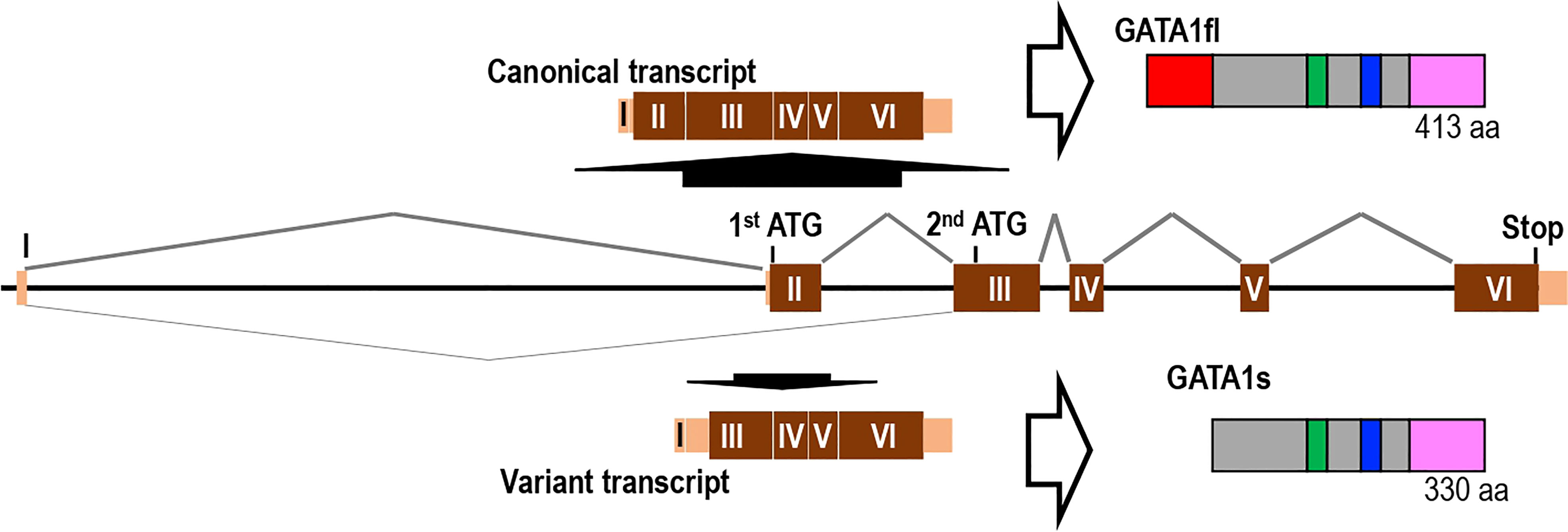
Figure 1 Gene structure and splicing isoforms of the GATA1 gene. The human GATA1 gene that produces full-length GATA1 (GATA1fl) consists of six exons (one noncoding and five coding exons). Short form GATA1 (GATA1s) lacking the N-terminal transactivation domain is produced by alternative splicing that skips exon 2.
GATA1 mutations with quantitative protein deficits and diseases
Mice lacking GATA1 expression die in early to mid-gestation due to defects in primitive erythropoiesis (29). Of note, transgenic expression of GATA1fl under proper transcriptional regulation rescues the embryonic lethal phenotype of GATA1-knockout mice (30), indicating that the abnormal and pathologic features leading to embryonic lethality in GATA1-deficient mice arise from an insufficient quantity of GATA1fl protein.
Multiple Gata1 gene knockout and knockdown mouse lines have been established by means of gene deletion and disruption of the cis-regulation of the Gata1 gene, respectively, and mouse lines with various levels of Gata1 expression have been generated (29, 31, 32). The severity of the phenotypes observed in the affected animals corresponds well to the expression level of GATA1. Mice bearing a 4- to 5-fold decrease in Gata1 expression (Gata1-low) either die in utero or are born alive with accompanying phenotypes in erythroid and megakaryocytic lineages, although live mice show a variety of phenotypes depending on the background of the mouse strain (32–34). A reduction in Gata1 expression to 5% (Gata1.05) of the endogenous level or Gata1-null mutation is embryonic lethal, as this level of Gata1 cannot sustain embryonic erythropoiesis (29, 31). It should be noted that mice with low-level Gata1 expression show distinct pathological characteristics from those with complete Gata1 deletion. Mice conditionally deleted for the Gata1 gene, i.e., no GATA1 expression, develop erythroid aplasia, while mice bearing conditionally deleted noncoding exon 1 of Gata1, in which a small amount of GATA1 is expressed from the accessory noncoding first exon, show the accumulation of erythroid progenitors (35, 36). A low level of GATA1 function is sufficient to promote the development of progenitor cells to the proerythroblast stage, but more abundant GATA1 appears to be required for further differentiation.
Since GATA1 is an X-chromosome gene, a male with inherited GATA1 variants/mutations exhibits the symptoms conferred by the variant. In contrast, a female carrying the altered gene on one of her two X-chromosomes is usually asymptomatic or shows only a mild phenotype in accordance with the level of X-chromosome inactivation bias (37, 38). In females, two types of erythroid and megakaryocyte progenitors exist in hematopoietic organs: normally developing progenitors with an activated GATA1 gene in the intact X-chromosome and progenitors with abnormal GATA1 function.
We previously found that heterozygous female mice carrying the Gata1.05 allele (Gata1.05/X) were prone to developing leukemia at 3-6 months after birth, in accord with the pathogenesis of human pure erythroleukemia cases (39, 40). However, Gata1-null heterozygous mice (Gata1-null/X) never develop leukemia (39), although both Gata1.05/Y and Gata1-null/Y are embryonic lethal. Since GATA1 is important for the promotion of differentiation, control of proliferation, and inhibition of apoptosis in erythroid cells, immature erythroid progenitors in Gata1-null situation are excluded by apoptosis (41). Therefore, erythroid progenitors with activated Gata1-null alleles are aplastic and have less potential to transform into leukemic cells (39, 41, 42). In contrast, immature erythroid progenitors carrying Gata1.05 mutation (in which Gata1 expression is reduced to 5% of endogenous levels) are alive as they are escaping from apoptosis due to the 5% expression of GATA1 to endogenous level, but those progenitors are losing the potential to differentiate into mature erythrocytes and are continuing the proliferation. Abnormally accumulated erythroid progenitors are prone to acquire additional genetic events and finally transform into leukemic cells. Thus, these results demonstrate that quantitative differences in GATA1 levels, i.e., 0%, 5% or 100%, determine the fate of erythroid progenitors.
A hemizygous boy carrying a germline deletion of 4,108 bp comprising the GATA1 3’ enhancer and exons 1-4 of the neighboring HDAC6 gene reportedly showed a persistently low platelet count with abnormal α-granule content (43). In this case, GATA1 expression in megakaryocytic lineage cells was decreased to approximately 50% of that in control individuals, while his mother showed thrombocyte markers within the normal range. The boy also suffered from mild dyserythropoiesis (43). To the best of our knowledge, this is the first human case with germline mutations causing a quantitative defect in GATA1 expression.
GATA1 mutations with qualitative protein defects and diseases
Apart from those diseases associated with a decrease in GATA1 protein levels or quantitative defects of GATA1, there are many inherited GATA1-related diseases caused by the production of truncated or mutant GATA1 proteins. The mutations resulting in qualitative defects in GATA function can be divided into two categories depending on the structure of the variant proteins. One is mutations that produce a single amino acid replacement in the GATA1 protein due to mutations in one or several nucleotides. The other is mutations that result in a substantially changed GATA1 protein due to frameshift, nonsense or splicing mutations. The mutations that produce single amino acid replacement will be discussed in this section. The sole single-amino-acid-deletion mutant that has been reported (A59del) is also included in this category.
As summarized in Figure 2, more than 136 variants/mutations impacting a single amino acid in GATA1 exist, according to the data in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and jMorp (Japanese multiomics reference panel) and further original searches of the literature (references cited in the Supplement Table). jMorp is a multiomics database (https://jmorp.megabank.tohoku.ac.jp/) generated by the Tohoku Medical Megabank Project (TMM), which includes a reference panel of genetic polymorphisms collected from 38,000 participants (44). The TMM comprises approximately 157,000 participants in Miyagi and Iwate prefectures in Japan who have been recruited for cohort studies and examined in health check studies in combination with genomic and omics research (45).
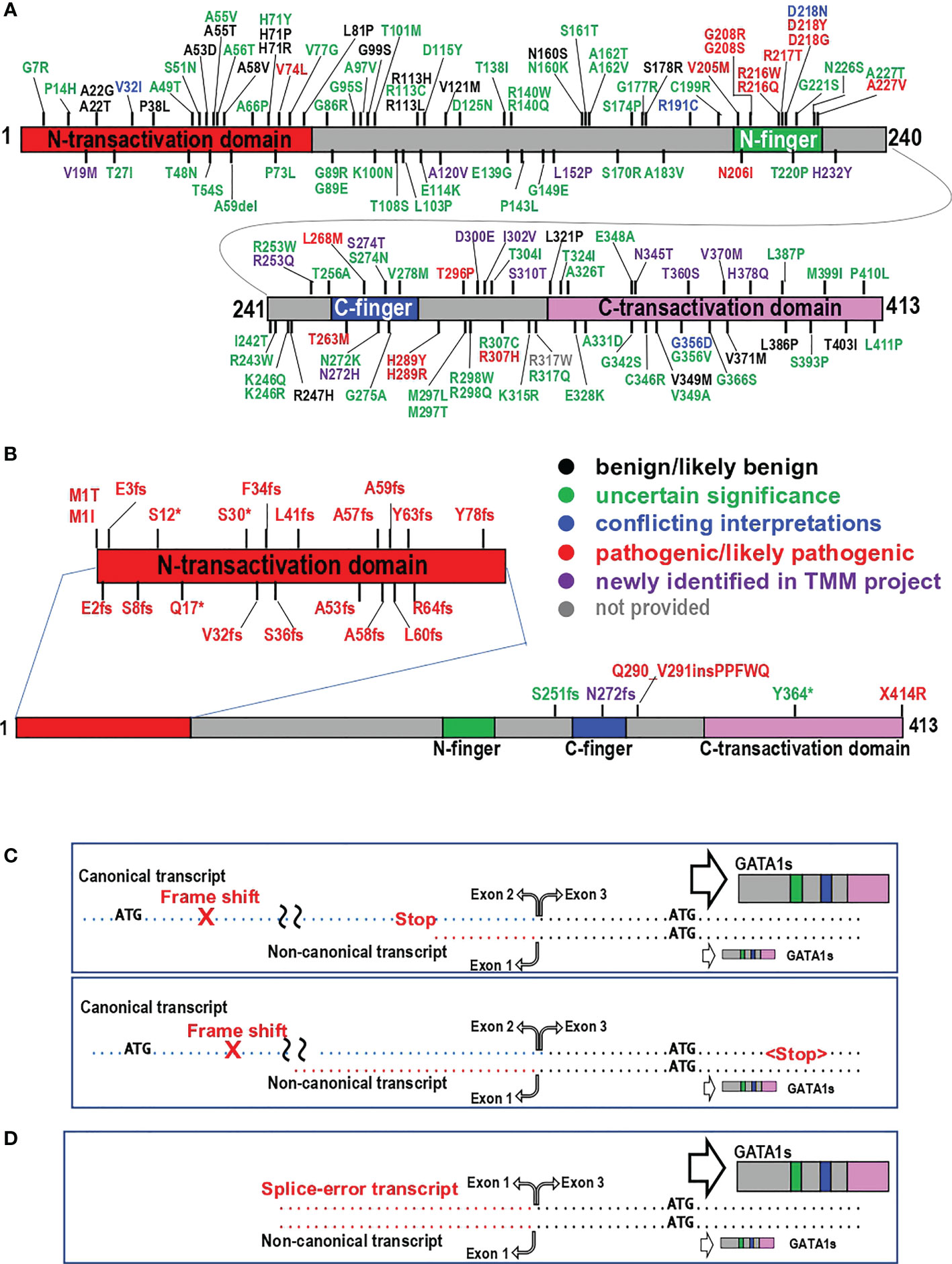
Figure 2 Mutations in the GATA1 protein. (A, B) Positions of missense and deletion mutations (A) and structural mutations (B) in the GATA1 protein are shown. Data obtained from ClinVar and jMorp on February 14, 2023, were used in addition to the data described in the literature. Variants in black, green, blue and red letters are categorized as “benign/likely benign”, “uncertain significance”, “conflicting interpretations”, and “pathogenic/likely pathogenic”, respectively. Newly identified variants in the TMM project are indicated by purple letters. Note that information about R317W is not provided in ClinVar. (C, D) Two major types of GATA1 gene variants causing exclusive production of GATA1s. GATA1 gene variants that cause exclusive production of GATA1s are divided into two groups in accordance with the level of GATA1s produced. (C) Frameshift mutations. If the in-frame stop codon marks the termination of translation upstream of the alternative initiation codon (upper panel), posttermination ribosomes can reinitiate translation at codon 84 of the canonical transcript and produce GATA1s utilizing the alternative initiation codon in exon 3. In contrast, neither GATA1fl nor GATA1s is produced from the canonical transcript if the premature stop codon does not appear upstream of the alternative initiation codon (lower panel). (D) Transcript originating from abnormal splicing that skips exon 2 produces GATA1s together with an original noncanonical transcript.
As shown in Figure 2A, pathological substitutions of amino acid residues are most prevalent in the N-finger region of GATA1. The first germline mutation in the GATA1 gene is the V205M mutation (37). The N-finger has two important functions: one is the association with the cofactors FOG1 and LMO2 (23, 46), while the other is DNA binding to the palindromic GATA motif (22, 47, 48). Elaborate structure−function analyses of the N-finger have clarified the amino acid residues responsible for both functions (46, 49). Mutations associated with binding to FOG1 or to DNA lead to bleeding disorders due to thrombocytopenia and/or abnormal platelets and mild to moderate dyserythropoiesis. The reported phenotypes caused by mutations in the N-finger vary slightly by case in the literature (37, 50–56). Cell-based assay have clarified that the differences in disease phenotype and severity caused by mutations in the N-finger are largely correlated with how the GATA1 mutation affects the association of GATA1 with cofactors FOG1 and LOM2 (57). We surmise that ethnic and/or environmental influences in addition to the characteristics of the substituted amino acid residues produce complexly diversified phenotypes in human.
Of note, several substitutions in the C-finger and downstream region have also been identified (Figure 2A) and have intriguing pathological impacts (38, 56, 58–62). Affected males carrying either L268M, H289Y or T296P mutations suffer from bleeding disorders caused by platelet dysfunction, while platelet numbers indicate borderline or mild-to-moderate thrombocytopenia. Red blood cell parameters are only marginally influenced (56, 58, 59). The clinical characteristics of the H289R mutation are not significant in either males or females. However, coinheritance of this mutation with a heterozygous PKLR gene mutation causes overt congenital dyserythropoietic anemia (CDA) (60). PKLR encodes pyruvate kinase and is the most common causative gene for congenital hemolytic anemia. Heterozygous PKLR mutation is considered to modify clinical symptoms in other red blood cell diseases (63). In contrast, R307H and R307C mutations cause severe anemia in males (61, 62). Heterozygous females carrying the T263M mutation appear to suffer from anemia and thrombocytopenia, but no hemizygous males were identified in this family (38). These diverse observations indicate that the phenotypes of the substitution mutations in the C-finger and related regions vary by case, reflecting the importance of the contributions of the substituted amino acid residues. Further structure−function analyses are required to clarify the pathologic basis of these mutations.
GATA1 mutations with large structure changes and diseases
Large structural changes in the GATA1 protein are the second type of structural mutation that has been identified. We identified mutations/variants categorized in this group through searches in ClinVar and jMorp and a literature search. These structural changes are generated mostly by frameshift, nonsense, and splicing mutations. These mutations, excluding splicing mutations in exon 2, are summarized in Figure 2B and the Supplemental Table. Although observed at low frequency, mutations in start/stop codons (M1T, M1I, and X414R) result in similar changes. One insertion of five amino acid residues is also included in this group.
We identified many frameshift and terminating mutations, which are shown by fs and asterisks (*) in the figure, respectively. These mutations appear to accumulate specifically in the region encoding the N-terminal transactivation domain of GATA1, which corresponds to the upstream region of the second ATG codon on exon 3 (see Figure 1). These mutations strongly influence the production of GATA1fl from the canonical transcript, while the production of GATA1s from the noncanonical transcript is predicted to be unaffected. Rather, many of these frameshift and termination mutations reside in the N-terminal transactivation domain, leading to the exclusive expression of GATA1s.
The pathological significance of the exclusive expression of GATA1s was first reported as a causative mutation of acute megakaryocytic leukemia (AMKL) in patients with trisomy 21, also known as Down syndrome (64). Subsequently, Hollanda, L.M. et al. (65) identified the first family carrying germline mutations causing exclusive GATA1 expression. Affected patients suffered from severe anemia accompanied by dyserythropoiesis, while the megakaryocytic lineage phenotype was subtle, as were the abnormalities in the platelet aggregation test. Accumulating lines of evidence support the notion that mutations causing GATA1s provoke severe erythroid dysplasia resembling Diamond-Blackfan anemia (DBA), an infant-onset disease involving red cell hypoplasia or aplasia (66, 67). Both hemizygous male and heterozygous female patients who express GATA1s exclusively also have a high potential for developing AMKL by somatic acquisition of trisomy 21, in addition to DBA-like symptoms (68, 69). The phenotypes in erythroid and megakaryocyte lineages are probably diversified due to the characteristic feature of GATA1s that inhibits an erythroid transcriptional program and induces a megakaryocytic program (70).
According to the DBA International Clinical Care Consensus Document in 2008 (71), the diagnosis of classical DBA is made when all four criteria are met, i.e., “age less than 1 year”, “macrocytic anemia with no other significant cytopenia”, “reticulocytopenia”, and “normal marrow cellularity with a paucity of erythroid precursors”. To date, a diverse set of causative genes for DBA has been identified, and almost half of DBA patients have a single mutation in a gene encoding ribosomal protein or a coupling factor with a ribosomal protein (72), suggesting that ribosome dysfunction is involved in the pathogenesis of DBA. Mutations in the causative genes are used as supporting criteria for the diagnosis of DBA (71). In addition to ribosomal genes, genes encoding GATA1, erythropoietin, and adenosine deaminase 2 have also been implicated in DBA-like diseases (69). However, DBA caused by the GATA1 mutation shows distinct characteristics from those caused by ribosomal gene mutations (69).
If in-frame stop codons mark the translation termination upstream of the alternative initiation codon on exon 3 (Figure 2C upper panel), posttermination ribosomes can reinitiate translation at codon 84 (the second ATG) of the canonical transcript and produce GATA1s utilizing the alternative initiation codon in exon 3. Concomitantly, GATA1s is also translated from the noncanonical transcript, albeit at low levels. Consequently, substantial levels of GATA1s are translated from the canonical and noncanonical transcripts and accumulate.
In contrast, if a premature stop codon does not appear upstream of the alternative initiation codon, the frameshift is maintained, and neither GATA1fl nor GATA1s is produced from the canonical transcript (Figure 2C lower panel). In this case, only a small amount of GATA1s is produced from the noncanonical transcript. These results thus indicate that the level of GATA1s produced varies by case depending on the mutation type, and the quantitative difference in GATA1s levels modifies the characteristics of the erythroid progenitors carrying the GATA1s mutation. Splicing mutations that result in exon 2 skipping produce high levels of GATA1s (Figure 2D).
In addition to the frameshift and splicing mutations producing GATA1s, germline variants that are predicted to produce neither GATA1fl nor GATA1s have been identified (Figure 2B). Two studies demonstrated that a 5-amino acid insertion following Q290 caused severe CDA in boys (73, 74). It has been reported that adding 42 extra amino acids to the C-terminus of the GATA1fl protein by removing the terminal codon leads to the rare blood group Lu(a-b-) rather than disease accompanying the mild platelet disorder in boys (75). It is predicted that the reduction of EKLF, a target gene of GATA1, causes the Lu(a-b-) phenotype, since haploinsufficiency for EKLF gene is causative in a family with the rare blood group Lu(a-b-) (76). Finally, although the S251fs, N272fs and Y364* variants are predicted to change the structure of GATA1, no reports in the literature have described the pathological significance of these variants. Further studies are expected to deepen our understanding of the molecular pathogenesis related to GATA1 dysfunction.
Conclusions
Accumulating lines of evidence support the notion that multiple transcription factors and their modifiers contribute to homeostasis of the hematopoietic system. GATA1 is a representative transcription factor that plays central roles in erythropoiesis and megakaryopoiesis. Since GATA1 is an X-chromosome gene, and since GATA1 has two functional isoforms that may exert different consequences on hematopoietic homeostasis, the impacts of GATA1 gene mutations vary in different situations. However, the mechanisms by which GATA1 mutations contribute to pathogenesis are not yet fully understood. More comprehensive analyses of GATA1 function are awaited to clarify the pathogenesis of GATA1-related hematopoietic diseases.
Author contributions
RS preformed literature search, and RS and MY wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by MEXT/JSPS KAKENHI (19H03555 to RS, 22K19450 to MY) and BINDS/AMED (JP22ama121038 to MY).
Acknowledgments
The authors acknowledge the support from the Tohoku University Library in assisting with the literature search.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2023.1181216/full#supplementary-material
References
1. Hirasawa R, Shimizu R, Takahashi S, Osawa M, Takayanagi S, Kato Y, et al. Essential and instructive roles of GATA factors in eosinophil development. J Exp Med (2002) 195(11):1379–86. doi: 10.1084/jem.20020170
2. Migliaccio AR, Rana RA, Sanchez M, Lorenzini R, Centurione L, Bianchi L, et al. GATA-1 as a regulator of mast cell differentiation revealed by the phenotype of the GATA-1low mouse mutant. J Exp Med (2003) 197(3):281–96. doi: 10.1084/jem.20021149
3. Nei Y, Obata-Ninomiya K, Tsutsui H, Ishiwata K, Miyasaka M, Matsumoto K, et al. GATA-1 regulates the generation and function of basophils. Proc Natl Acad Sci USA (2013) 110(46):18620–5. doi: 10.1073/pnas.1311668110
4. Shimizu R, Yamamoto M. GATA-related hematologic disorders. Exp Hematol (2016) 44(8):696–705. doi: 10.1016/j.exphem.2016.05.01
5. Shimizu R, Yamamoto M. Gene expression regulation and domain function of hematopoietic GATA factors. Semin Cell Dev Biol (2005) 16(1):129–36. doi: 10.1016/j.semcdb.2004.11.001
6. Suzuki M, Shimizu R, Yamamoto M. Transcriptional regulation by GATA1 and GATA2 during erythropoiesis. Int J Hematol (2011) 93(2):150–5. doi: 10.1007/s12185-011-0770-6
7. Suzuki N, Ohneda O, Minegishi N, Nishikawa M, Ohta T, Takahashi S, et al. Combinatorial Gata2 and Sca1 expression defines hematopoietic stem cells in the bone marrow niche. Proc Natl Acad Sci USA (2006) 103(7):2202–7. doi: 10.1073/pnas.0508928103
8. Takai J, Moriguchi T, Suzuki M, Yu L, Ohneda K, Yamamoto M. The Gata1 5' region harbors distinct cis-regulatory modules that direct gene activation in erythroid cells and gene inactivation in HSCs. Blood (2013) 122(20):3450–60. doi: 10.1182/blood-2013-01-476911
9. Ohneda K, Yamamoto M. Roles of hematopoietic transcription factors GATA-1 and GATA-2 in the development of red blood cell lineage. Acta Haematol (2002) 108(4):237–45. doi: 10.1159/00006566
10. Kaneko H, Shimizu R, Yamamoto M. GATA factor switching during erythroid differentiation. Curr Opin Hematol (2010) 17(3):163–8. doi: 10.1097/MOH.0b013e32833800b8
11. Yu L, Takai J, Otsuki A, Katsuoka F, Suzuki M, Katayama S, et al. Derepression of the DNA methylation machinery of the Gata1 gene triggers the differentiation cue for erythropoiesis. Mol Cell Biol (2017) 37(8):e00592–16. doi: 10.1128/MCB.00592-16
12. Suzuki N, Suwabe N, Ohneda O, Obara N, Imagawa S, Pan X, et al. Identification and characterization of 2 types of erythroid progenitors that express GATA-1 at distinct levels. Blood (2003) 102(10):3575–83. doi: 10.1182/blood-2003-04-1154
13. Whyatt D, Lindeboom F, Karis A, Ferreira R, Milot E, Hendriks R, et al. An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature (2000) 406(6795):519–24. doi: 10.1038/35020086
14. Shimizu R, Hasegawa A, Ottolenghi S, Ronchi A, Yamamoto M. Verification of the in vivo activity of three distinct cis-acting elements within the Gata1 gene promoter-proximal enhancer in mice. Genes Cells (2013) 18(11):1032–41. doi: 10.1111/gtc.12096
15. Tusi BK, Wolock SL, Weinreb C, Hwang Y, Hidalgo D, Zilionis R, et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature (2018) 555(7694):54–60. doi: 10.1038/nature25741
16. Nicolis S, Bertini C, Ronchi A, Crotta S, Lanfranco L, Moroni E, et al. An erythroid specific enhancer upstream to the gene encoding the cell-type specific transcription factor GATA-1. Nucleic Acids Res (1991) 19(19):5285–91. doi: 10.1093/nar/19.19.5285
17. Tsai SF, Strauss E, Orkin SH. Functional analysis and in vivo footprinting implicate the erythroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes Dev (1991) 5(6):919–31. doi: 10.1101/gad.5.6.919
18. Ito E, Toki T, Ishihara H, Ohtani H, Gu L, Yokoyama M, et al. Erythroid transcription factor GATA-1 is abundantly transcribed in mouse testis. Nature (1993) 362(6419):466–8. doi: 10.1038/362466a0
19. Onodera K, Yomogida K, Suwabe N, Takahashi S, Muraosa Y, Hayashi N, et al. Conserved structure, regulatory elements, and transcriptional regulation from the GATA-gene testis promoter. J Biochem (1997) 121(2):251–63. doi: 10.1093/oxfordjournals.jbchem.a021581
20. Dyer KD, Czapiga M, Foster B, Foster PS, Kang EM, Lappas CM, et al. Eosinophils from lineage-ablated delta dblGATA bone marrow progenitors: the dblGATA enhancer in the promoter of GATA-1 is not essential for differentiation ex vivo. J Immunol (2007) 179(3):1693–9. doi: 10.4049/jimmunol.179.3.1693
21. Kaneko H, Kobayashi E, Yamamoto M, Shimizu R. N- and c-terminal transactivation domains of GATA1 protein coordinate hematopoietic program. J Biol Chem (2012) 287(25):21439–49. doi: 10.1074/jbc.M112.370437
22. Hasegawa A, Kaneko H, Ishihara D, Nakamura M, Watanabe A, Yamamoto M, et al. GATA1 binding kinetics on conformation-specific binding sites elicit differential transcriptional regulation. Mol Cell Biol (2016) 36(16):2151–67. doi: 10.1128/MCB.00017-16
23. Tsang AP, Visvader JE, Turner CA, Fujiwara Y, Yu C, Weiss MJ, et al. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell (1997) 90(1):109–19. doi: 10.1016/s0092-8674(00)80318-9
24. Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature (1998) 396(6711):594–8. doi: 10.1038/25166
25. Blobel GA, Nakajima T, Eckner R, Montminy M, Orkin SH. CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci USA (1998) 95(5):2061–6. doi: 10.1073/pnas.95.5.2061
26. Stumpf M, Waskow C, Krotschel M, van Essen D, Rodriguez P, Zhang X, et al. The mediator complex functions as a coactivator for GATA-1 in erythropoiesis via subunit Med1/TRAP220. Proc Natl Acad Sci USA (2006) 103(49):18504–9. doi: 10.1073/pnas.0604494103
27. Shimizu R, Takahashi S, Ohneda K, Engel JD, Yamamoto M. In vivo requirements for GATA-1 functional domains during primitive and definitive erythropoiesis. EMBO J (2001) 20(18):5250–60. doi: 10.1093/emboj/20.18.5250
28. Rainis L, Bercovich D, Strehl S, Teigler-Schlegel A, Stark B, Trka J, et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood (2003) 102(3):981–6. doi: 10.1182/blood-2002-11-3599
29. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci USA (1996) 93(22):12355–8. doi: 10.1073/pnas.93.22.12355
30. Takahashi S, Shimizu R, Suwabe N, Kuroha T, Yoh K, Ohta J, et al. GATA factor transgenes under GATA-1 locus control rescue germline GATA-1 mutant deficiencies. Blood (2000) 96(3):910–6. doi: 10.1182/blood.V96.3.910
31. Takahashi S, Onodera K, Motohashi H, Suwabe N, Hayashi N, Yanai N, et al. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J Biol Chem (1997) 272(19):12611–5. doi: 10.1074/jbc.272.19.12611
32. McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H, Orkin SH. A "knockdown" mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc Natl Acad Sci USA (1997) 94(13):6781–5. doi: 10.1073/pnas.94.13.6781
33. Vannucchi AM, Bianchi L, Cellai C, Paoletti F, Rana RA, Lorenzini R, et al. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood (2002) 100(4):1123–32. doi: 10.1182/blood-2002-06-1913
34. Martelli F, Ghinassi B, Panetta B, Alfani E, Gatta V, Pancrazzi A, et al. Variegation of the phenotype induced by the Gata1low mutation in mice of different genetic backgrounds. Blood (2005) 106(13):4102–13. doi: 10.1182/blood-2005-03-1060
35. Gutierrez L, Tsukamoto S, Suzuki M, Yamamoto-Mukai H, Yamamoto M, Philipsen S, et al. Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood (2008) 111(8):4375–85. doi: 10.1182/blood-2007-09-115121
36. Kobayashi E, Shimizu R, Kikuchi Y, Takahashi S, Yamamoto M. Loss of the Gata1 gene IE exon leads to variant transcript expression and the production of a GATA1 protein lacking the n-terminal domain. J Biol Chem (2010) 285(1):773–83. doi: 10.1074/jbc.M109.030726
37. Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet (2000) 24(3):266–70. doi: 10.1038/73480
38. Svidnicki M, Filho MAF, Brandao MM, Dos Santos M, de Oliveira Dias R, Tavares RS, et al. New germline GATA1 variant in females with anemia and thrombocytopenia. Blood Cells Mol Dis (2021) 88:102545. doi: 10.1016/j.bcmd.2021.102545
39. Shimizu R, Kuroha T, Ohneda O, Pan X, Ohneda K, Takahashi S, et al. Leukemogenesis caused by incapacitated GATA-1 function. Mol Cell Biol (2004) 24(24):10814–25. doi: 10.1128/MCB.24.24.10814-10825.2004
40. Wang W, Wang SA, Medeiros LJ, Khoury JD. Pure erythroid leukemia. Am J Hematol (2017) 92(3):292–6. doi: 10.1002/ajh.24626
41. Pan X, Ohneda O, Ohneda K, Lindeboom F, Iwata F, Shimizu R, et al. Graded levels of GATA-1 expression modulate survival, proliferation, and differentiation of erythroid progenitors. J Biol Chem (2005) 280(23):22385–94. doi: 10.1074/jbc.M500081200
42. Abe K, Shimizu R, Pan X, Hamada H, Yoshikawa H, Yamamoto M. Stem cells of GATA1-related leukemia undergo pernicious changes after 5-fluorouracil treatment. Exp Hematol (2009) 37(4):435–45.e1. doi: 10.1016/j.exphem.2008.12.004
43. Turro E, Astle WJ, Megy K, Graf S, Greene D, Shamardina O, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature (2020) 583(7814):96–102. doi: 10.1038/s41586-020-2434-2
44. Tadaka S, Hishinuma E, Komaki S, Motoike IN, Kawashima J, Saigusa D, et al. jMorp updates in 2020: large enhancement of multi-omics data resources on the general Japanese population. Nucleic Acids Res (2021) 49(D1):D536–D44. doi: 10.1093/nar/gkaa1034
45. Kuriyama S, Yaegashi N, Nagami F, Arai T, Kawaguchi Y, Osumi N, et al. The tohoku medical megabank project: design and mission. J Epidemiol (2016) 26(9):493–511. doi: 10.2188/jea.JE20150268
46. Wilkinson-White L, Gamsjaeger R, Dastmalchi S, Wienert B, Stokes PH, Crossley M, et al. Structural basis of simultaneous recruitment of the transcriptional regulators LMO2 and FOG1/ZFPM1 by the transcription factor GATA1. Proc Natl Acad Sci USA (2011) 108(35):14443–8. doi: 10.1073/pnas.1105898108
47. Trainor CD, Omichinski JG, Vandergon TL, Gronenborn AM, Clore GM, Felsenfeld G. A palindromic regulatory site within vertebrate GATA-1 promoters requires both zinc fingers of the GATA-1 DNA-binding domain for high-affinity interaction. Mol Cell Biol (1996) 16(5):2238–47. doi: 10.1128/MCB.16.5.2238
48. Trainor CD, Ghirlando R, Simpson MA. GATA zinc finger interactions modulate DNA binding and transactivation. J Biol Chem (2000) 275(36):28157–66. doi: 10.1074/jbc.M000020200
49. Newton A, Mackay J, Crossley M. The n-terminal zinc finger of the erythroid transcription factor GATA-1 binds GATC motifs in DNA. J Biol Chem (2001) 276(38):35794–801. doi: 10.1074/jbc.M106256200
50. Mehaffey MG, Newton AL, Gandhi MJ, Crossley M, Drachman JG. X-Linked thrombocytopenia caused by a novel mutation of GATA-1. Blood (2001) 98(9):2681–8. doi: 10.1182/blood.v98.9.2681
51. Freson K, Matthijs G, Thys C, Marien P, Hoylaerts MF, Vermylen J, et al. Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum Mol Genet (2002) 11(2):147–52. doi: 10.1093/hmg/11.2.147
52. Del Vecchio GC, Giordani L, De Santis A, De Mattia D. Dyserythropoietic anemia and thrombocytopenia due to a novel mutation in GATA-1. Acta Haematol (2005) 114(2):113–6. doi: 10.1159/000086586
53. Tubman VN, Levine JE, Campagna DR, Monahan-Earley R, Dvorak AM, Neufeld EJ, et al. X-Linked gray platelet syndrome due to a GATA1 Arg216Gln mutation. Blood (2007) 109(8):3297–9. doi: 10.1182/blood-2006-02-004101
54. Hermans C, De Waele L, Van Geet C, Freson K. Novel GATA1 mutation in residue D218 leads to macrothrombocytopenia and clinical bleeding problems. Platelets (2014) 25(4):305–7. doi: 10.3109/09537104.2013.815339
55. Di Pierro E, Russo R, Karakas Z, Brancaleoni V, Gambale A, Kurt I, et al. Congenital erythropoietic porphyria linked to GATA1-R216W mutation: challenges for diagnosis. Eur J Haematol (2015) 94(6):491–7. doi: 10.1111/ejh.12452
56. Bastida JM, Malvestiti S, Boeckelmann D, Palma-Barqueros V, Wolter M, Lozano ML, et al. A novel GATA1 variant in the c-terminal zinc finger compared with the platelet phenotype of patients with a likely pathogenic variant in the n-terminal zinc finger. Cells (2022) 11(20):3223. doi: 10.3390/cells11203223
57. Campbell AE, Wilkinson-White L, Mackay JP, Matthews JM, Blobel GA. Analysis of disease-causing GATA1 mutations in murine gene complementation systems. Blood (2013) 121:5218–27. doi: 10.1182/blood-2013-03-488080
58. Saultier P, Cabantous S, Puceat M, Peiretti F, Bigot T, Saut N, et al. GATA1 pathogenic variants disrupt MYH10 silencing during megakaryopoiesis. J Thromb Haemost (2021) 19(9):2287–301. doi: 10.1111/jth.15412
59. Jurk K, Adenaeuer A, Sollfrank S, Gross K, Hauser F, Czwalinna A, et al. Novel GATA1 variant causing a bleeding phenotype associated with combined platelet alpha-/delta-Storage pool deficiency and mild dyserythropoiesis modified by a SLC4A1 variant. Cells (2022) 11(19):3071. doi: 10.3390/cells11193071
60. Pereira J, Bento C, Manco L, Gonzalez A, Vagace J, Ribeiro ML. Congenital dyserythropoietic anemia associated to a GATA1 mutation aggravated by pyruvate kinase deficiency. Ann Hematol (2016) 95(9):1551–3. doi: 10.1007/s00277-016-2720-0
61. Hetzer B, Meryk A, Kropshofer G, Bargehr C, Jimenez-Heredia R, Boztug K, et al. An R307H substitution in GATA1 that prevents Ser310 phosphorylation causes severe fetal anemia. Blood Adv (2022) 6(14):4330–4. doi: 10.1182/bloodadvances.2021006347
62. Ludwig LS, Lareau CA, Bao EL, Liu N, Utsugisawa T, Tseng AM, et al. Congenital anemia reveals distinct targeting mechanisms for master transcription factor GATA1. Blood (2022) 139(16):2534–46. doi: 10.1182/blood.2021013753
63. Bianchi P, Fermo E. Molecular heterogeneity of pyruvate kinase deficiency. Haematologica (2020) 105(9):2218–28. doi: 10.3324/haematol.2019.241141
64. Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of down syndrome. Nat Genet (2002) 32(1):148–52. doi: 10.1038/ng955
65. Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet (2006) 38(7):807–12. doi: 10.1038/ng1825
66. Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, et al. Exome sequencing identifies GATA1 mutations resulting in diamond-blackfan anemia. J Clin Invest (2012) 122(7):2439–43. doi: 10.1172/JCI63597
67. van Dooijeweert B, Kia SK, Dahl N, Fenneteau O, Leguit R, Nieuwenhuis E, et al. GATA-1 defects in diamond-blackfan anemia: phenotypic characterization points to a specific subset of disease. Genes (Basel) (2022) 13(3):447. doi: 10.3390/genes13030447
68. Camargo R, Sahoo SS, Cordoba JC, Magalhaes IQ. Germline GATA1 exon 2 mutation associated with chronic cytopenia and a non-down syndrome transient abnormal myelopoiesis with clonal trisomy 21. Leukemia (2022) 36(9):2347–50. doi: 10.1038/s41375-022-01638-6
69. Hasle H, Kline RM, Kjeldsen E, Nik-Abdul-Rashid NF, Bhojwani D, Verboon JM, et al. Germline GATA1s-generating mutations predispose to leukemia with acquired trisomy 21 and down syndrome-like phenotype. Blood (2022) 139(21):3159–65. doi: 10.1182/blood.2021011463
70. Byrska-Bishop M, VanDorn D, Campbell AE, Betensky M, Arca A, Yao Y. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 n-terminus. J Clin Invest (2015) 125:993–1005. doi: 10.1172/JCI75714
71. Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, et al. Diagnosing and treating diamond blackfan anaemia: results of an international clinical consensus conference. Br J Haematol (2008) 142(6):859–76. doi: 10.1111/j.1365-2141.2008.07269.x
72. Da Costa L, Leblanc T, Mohandas N. Diamond-blackfan anemia. Blood (2020) 136(11):1262–73. doi: 10.1182/blood.2019000947
73. Abdulhay NJ, Fiorini C, Verboon JM, Ludwig LS, Ulirsch JC, Zieger B, et al. Impaired human hematopoiesis due to a cryptic intronic GATA1 splicing mutation. J Exp Med (2019) 216(5):1050–60. doi: 10.1084/jem.20181625
74. Kobayashi A, Ohtaka R, Toki T, Hara J, Muramatsu H, Kanezaki R, et al. Dyserythropoietic anaemia with an intronic GATA1 splicing mutation in patients suspected to have diamond-blackfan anaemia. EJHaem (2022) 3(1):163–7. doi: 10.1002/jha2.374
75. Singleton BK, Roxby DJ, Stirling JW, Spring FA, Wilson C, Poole J, et al. A novel GATA1 mutation (Stop414Arg) in a family with the rare X-linked blood group lu(a-b-) phenotype and mild macrothrombocytic thrombocytopenia. Br J Haematol (2013) 161(1):139–42. doi: 10.1111/bjh.12184
Keywords: GATA1, erythropoiesis, leukemia, structural change, expression level change, X-chromosome inactivation
Citation: Shimizu R and Yamamoto M (2023) Recent progress in analyses of GATA1 in hematopoietic disorders: a mini-review. Front. Hematol. 2:1181216. doi: 10.3389/frhem.2023.1181216
Received: 07 March 2023; Accepted: 24 April 2023;
Published: 03 May 2023.
Edited by:
Anna Rita Migliaccio, Campus Bio-Medico University, ItalyReviewed by:
John Strouboulis, King’s College London, United KingdomAnnarita Miccio, INSERM U1163 Institut Imagine, France
Copyright © 2023 Shimizu and Yamamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ritsuko Shimizu, cnNoaW1penVAbWVkLnRvaG9rdS5hYy5qcA==