 Morgane Canonne
Morgane Canonne Fabienne George2
Fabienne George2- 1Research Laboratory, CHU UCL Namur-Godinne, Yvoir, Belgium
- 2Biobank, CHU UCL Namur-Godinne, Yvoir, Belgium
- 3Department of Hematology, CHU UCL Namur-Godinne, Yvoir, Belgium
Mature B cell malignancies constitute a wide range of biologically and clinically heterogeneous hematological diseases. Despite an increasingly thorough understanding of the pathophysiology of these pathologies and significant improvements in therapies, a dismal outcome still affects a large number of patients. Therefore, further investigations into new treatment perspectives are highly needed and they depend entirely on the ex vivo culture of patient cells. Primary cells usually demand superior culture models, as they are notoriously difficult to cultivate. The literature is not devoid of approaches ranging from two- to three-dimensional systems for culturing mature malignant primary B cells. However, they display substantial protocol inter-variation. This imposes a high risk of failures, repeats, and inconsistent results, which are neither compatible with the rare value of primary cells nor the efficiency of the drug discovery process. In this review, we provide a thorough overview of the different approaches that have been implemented in the literature for the culture of mature malignant primary B cells, and we discuss associated considerations and limitations to assist researchers in determining a fit-for-purpose culture system, thereby attempting to reduce the number of trials and errors as well as associated biomaterial expenditure.
Introduction
Mature B neoplasms represent a heterogeneous group of pathologically and clinically divergent malignancies originating from developed B cells. They comprise Chronic Lymphocytic Leukemia (CLL) (1), Follicular Lymphoma (FL) (2), Marginal Zone Lymphoma (MZL) (3), Hairy Cell Leukemia (HCL) (4), Mantle Cell Lymphoma (MCL) (5), Diffuse Large B-Cell Lymphoma (DLBCL) (6), Burkitt Lymphoma (BL) (7), and Multiple Myeloma (MM) (8). Despite significant progress in therapeutic options and regimen efficacy, clinical outcomes generally remain unsatisfactory due to disease complexity and intra/inter-heterogeneity. Therefore, there is a germane need to further decipher the mechanisms underlying not only disease etiopathology but also drug efficacy and resistance. This implies the extensive use of pre-clinical models, such as ex vivo culture systems.
For this review, we perused the literature to present an overview of the strategies ranging from two-dimensional (2D) to three-dimensional (3D) approaches that have been implemented to maintain mature malignant primary B cells in an ex vivo culture environment. A 2D culture consists of culturing cells as monocultures on flat and hard surfaces. In 2.5D culture platforms, one or several other relevant cell types are cocultured with the cells of interest to better recapitulate cellular interactions occurring in the natural microenvironment. Finally, 3D culture approaches strive to increase the fidelity of the cellular and molecular Tumor MicroEnvironment (TME) in terms of composition, structure, and dynamics. Scaffold-based 3D models (9), organoids (10), on-chip technology (11), bioprinted constructs (12), and bioreactors (13) are all 3D culture approaches that can assist in emulating the complexity of the TME.
Our goal is to provide researchers with a comprehensive summary of the approaches that have been implemented in the literature for the ex vivo maintenance of mature malignant B cells to dispense them from time-consuming literature searches and to minimize the laborious technical optimization performed on precious and limited primary material. To that end, we describe the growth outcomes resulting from the different culture methods published in the scientific literature while pinpointing associated technical and biological pitfalls.
Normal B cell dynamics and associated malignancies
Natural B cell development
The efficient generation of a diverse immune B cell repertoire that protect against (re)infection results from finely tuned and remarkably engineered genetic and molecular processes originating in primary lymphoid tissue (bone marrow) and progressing in secondary lymphoid organs (such as lymph nodes and the spleen) (14).
Early maturation of B cells occurs in the bone marrow, the seat of hematopoiesis, wherein stepwise gene rearrangement-based events lead to the development of a primary virgin collection of B cells (i.e., unchallenged by antigens) that features B Cell Receptors (BCRs) with sundry variable regions of surface immunoglobulins (14). Following bone marrow egress and blood- or lymph-mediated immune challenges, the final maturation and selection of B cells occur in transient, dynamic, and polarized microstructures called Germinal Centers (GCs) that are located in the B cell follicles of secondary lymphoid organs (15, 16). Specifically, once antigen-engaged, activated B cells migrate to the follicle border zone enriched in CD4+ helper T cells (Th) (hence termed the T zone) under the influence of chemokines, such as C-C motif Ligand 21 (CCL21) (17). This homing molecule is secreted by resident stromal Fibroblastic Reticular Cells (FRCs), which support the structure and function of the T zone (18).
At the follicle-T zone interface, B cells present Th with cognate antigens and subsequently undergo an initial in loco proliferation (15, 16). At this stage, after Th-mediated isotype switching, a fraction of the pre-GC early-activated B cells (seemingly presenting the highest initial BCR affinity) differentiates into short-lived plasma or memory cells (15, 16). Another preferred portion will enter the B follicle and coalesce into intensively proliferating and tight cell clusters, thereby forming the GC Dark Zone (DZ), together with a supportive network of stromal C-X-C motif chemokine Ligand 12 (CXCL12)-expressing Reticular Cells (CRCs) and some follicular CD4+ T helper cells (Tfh) (15, 16). There, GC DZ B cells (also called centroblasts) undergo active proliferation and substantial intraclonal diversification via Somatic HyperMutation (SHM) of the exon coding for the surface immunoglobulin variable region (15, 16).
Centroblasts then transition to the most distal zone of the GC, the Light Zone (LZ) (where they are referred to as centrocytes), to enable the selection of the most affine variants. These variants can undergo repetitive bidirectional travel between the DZ and the LZ in order to further increase antigen affinity (15, 16). In parallel, dysfunctional BCR-bearing centrocytes endure apoptosis and are eliminated by tingible body macrophages (19). Aside from macrophages and centrocytes, numerous auxiliary cell populations also reside in the GC LZ. These include naïve follicular B cells, CD8+ T cells, and Follicular Dendritic Cells (FDCs), which drive BCR affinity testing by presenting antigens and supporting selected cell survival/proliferation in concert with Tfh (15, 16). Selected high-affinity B cells can commit to either plasma or memory cell differentiation, although the discriminative factor(s) driving each cell fate decision remains to be fully elucidated (15, 16).
The inner mantle zone and the outer marginal zone both constitute GC circumscribing compartments. While the former is the result of the hyperactive GC-mediated displacement of primary resident cells (20), the latter, best appreciated in the spleen, mostly harbors sentinel memory B cells that are strategically positioned to ensure a swift frontline response during pathogenic provocation (21).
Mature B lymphoma/leukemia ontogeny and treatment strategies
Although intricately efficient, the overall natural history of B cells remains an error-prone process that can give rise to a large spectrum of malignancies resulting from distinct stages of B cell maturation (22, 23) (Figure 1). It is noteworthy that the cell of origin of each B neoplasm subtype has been under continuous debate. Recent investigations have compelled us to discard the compartmented view relative to the follicle reaction since they point out gradual phenotypical transitions in the GC, thereby defining multiple subpopulations and further complicating the physiological and pathological picture (27).
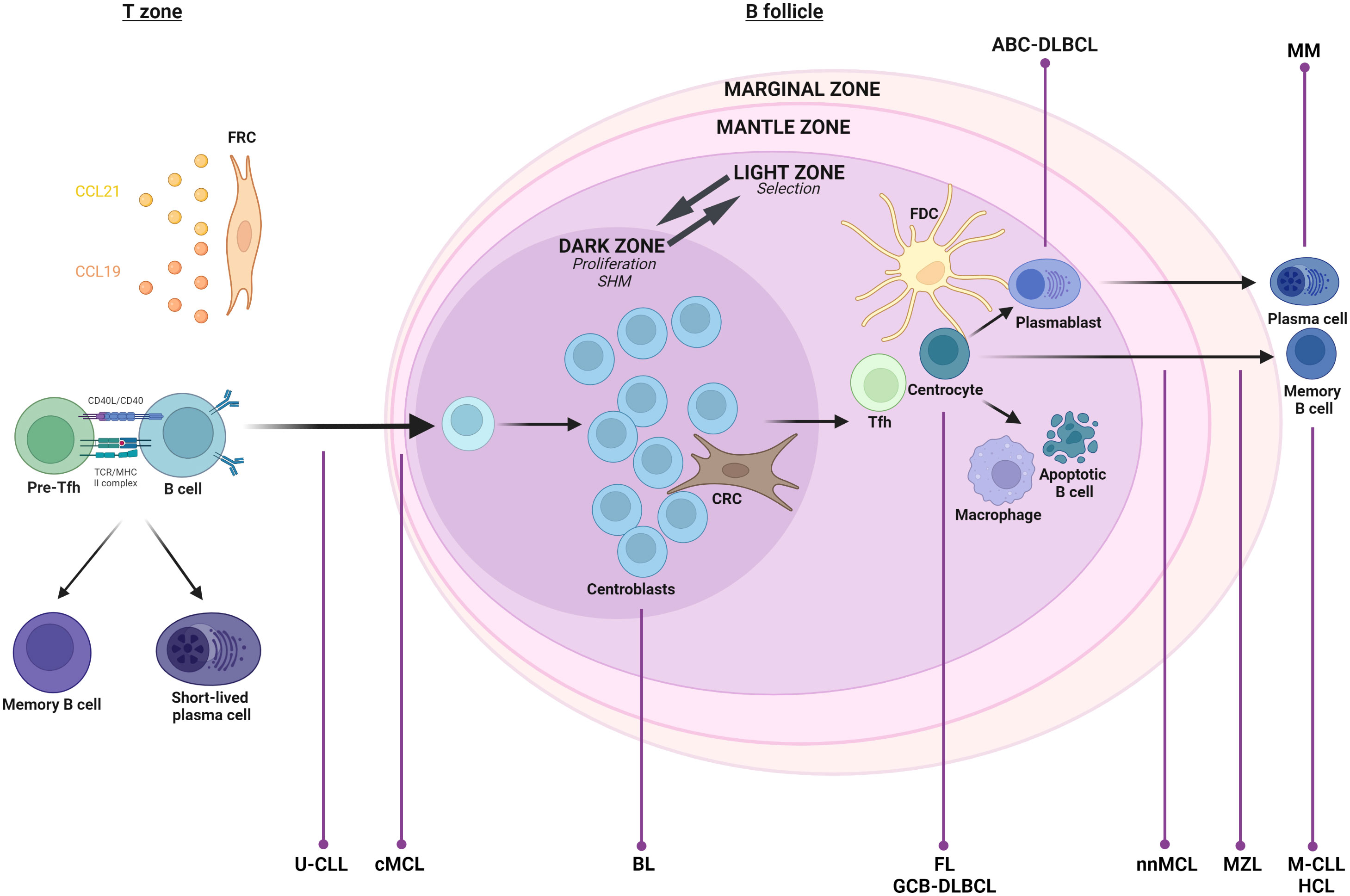
Figure 1 Germinal Center (GC) reaction. Upon cognate antigen B Cell Receptor (BCR) engagement, naive B cells migrate to secondary lymphoid organs, where a prime interaction with activated helper T cells in the T zone initiates three possible cell fates: cell differentiation into short-term plasma cells, maturation into memory B cells, or commitment to the GC response (15, 16). In the last case, B cells undergo iterative cycles of extensive proliferation, Somatic HyperMutation (SHM), and affinity-based selection in functionally and spatially segregated GC compartments, namely the Dark Zone (DZ) and Light Zone (LZ) (15, 16). Eventually, while B cells presenting a crippled BCR experience apoptosis, selected B cells undergo terminal differentiation into long-lived progeny, either plasma cells or memory cells (15, 16). All these processes are coordinated/supported by diverse and zone-specific cell populations, such as Fibroblastic Reticular Cells (FRCs) (T zone support and function; C-C motif Ligand 19 and 21 (CCL19 and CCL21)- mediated homing of B and T lymphocytes), stromal C-X-C motif chemokine Ligand 12 (CXCL12)-expressing Reticular Cells (CRCs) (DZ migration and support), and Follicular Dendritic Cells (FDCs) (selection through antigen display; B cell survival), as well as other immune cells, such as follicular CD4+ T helper cells (Tfh) (B cell survival and differentiation) and macrophages (apoptotic B cell clearance) (15, 16, 24). The GC is surrounded by the mantle zone composed notably of resting B cells and the marginal zone inhabited by guarding B cells involved in the innate-like head immune response (20, 21). Although essential for host protection against pathogens, the B-mediated immune response inherently possesses a detrimental downside, specifically the potential for lymphomagenesis at various stages of the B cell’s transition through its maturation process. Thus, normal counterparts of Follicular Lymphoma (FL), Germinal Center B cell-like (GCB) Diffuse Large B-Cell Lymphoma (GCB-DLBCL), Activated B Cell-DLBCL (ABC-DLBCL), and Burkitt Lymphoma (BL) all originate from the GC. While the two former types seem related to LZ residing cells, ABC-DLBCL offers signs of plasmablast engagement, and BL coincides with DZ residing cells (22, 23). Marginal Zone Lymphoma (MZL) is thought to be derived from an antigen-activated cell originating from the marginal zone (21). Chronic Lymphocytic Leukemia (CLL) is divided into two molecular subgroups according to the mutational status of the ImmunoGlobulin Heavy-chain Variable region (IGHV). Cells presenting unmutated IGHV (U-CLL) are likely GC-virgin, while mutated ones (M-CLL) would correspond to GC-experienced cells (25). Likewise, Mantle Cell Lymphoma (MCL) cells can present a naïve-like (conventional MCL: cMCL) or memory-like (non-nodal MCL: nnMCL) phenotype (26). Finally, Hairy Cell Leukemia (HCL) presents features of late-activated memory B cells (4), and Multiple Myeloma (MM) is characterized by abnormal plasma cells (8). Created with BioRender.com.
Therapeutic recommendations for B lymphoma are regularly updated, and they depend on the patient’s phenotypic and genetic profiles. However, the frontline treatment is usually based on an immunochemo approach, consisting of variations around the classic rituximab plus cyclophosphamide, vincristine, doxorubicin and prednisone (R-CHOP) treatment (28–32).
While substantial progress has been made regarding B lymphoma/leukemia treatment strategies, there is still a general unmet need experienced by ineligible, relapsed, or refractory patients who face bleak outcomes. Intensification of treatments with high doses of chemotherapy followed by autologous stem cell transplant is the gold standard for responders to salvage regimens, but this strategy is restricted to patients who are fit and still chemo-sensitive (28–32). Although the development of new technologies, such as Bi-specific T-cell Engagers (BiTE) and Chimeric Antigen Receptor T (CAR-T) cells, have yielded hope for the aforementioned patient subsets, therapy success is often thwarted by treatment-related toxicities (33, 34). Therefore, there is an urgent need to improve not only salvage treatments but, more importantly, first-line approaches because the best way to treat relapse is to avoid it.
In an era of emerging personalized medicine where the implemented treatment is tailored to a patient’s cell-specific make-up, it has become more necessary than ever to establish ex vivo culture models to further decipher disease pathophysiology and support and expedite drug testing and discovery.
2D culture
Partial supplementation outcomes from the literature
Inevitably, the dynamic and zone-specific microenvironment is the main player in B cell physiological development. In line with this, B cell neoplasms tend to be paragons of microenvironment-dependent tumors, as reflected by the fact that they swiftly undergo apoptosis when cultured ex vivo (35, 36). Hence, attempts at 2D monoculture supplementation have stemmed from the inherent readiness of malignant cells to exploit and skew cellular and molecular crosstalk between their normal counterparts and in vivo niches (37). As such, despite a lack of standardization in the literature, governing strategies include B cell activation mediated by the main receptors BCR, CD40, and/or Toll-Like Receptor (TLR).
While the former is critical to overall normal B cell maturation (38), CD40 engagement by a productive immune synapse, with notably CD40L-expressing Tfh cells, is a sine qua non to the proper induction, progression, and completion of the aforementioned B follicle-mediated humoral response (39). TLRs are pattern recognition receptors that physiologically provide B cells with an innate response capacity (40). Numerous lines of evidence support the key pathogenic role of these actors in mature B cell malignancies. The prime implication of BCR in B cell-related malignancies notably pertains to the presence of stereotyped BCR, especially in CLL (41) but also in MCL and MZL (42), which argues more for genuine antigen pressure than sheer serendipity (43). This is further demonstrated by the therapeutic success of Bruton’s tyrosine kinase inhibitors, mostly in the clinical management of CLL (44), but also MCL and MZL (45). Several observations have also positioned CD40 as a disease driver. For example, CCL22-expressing CLL cells chemo-attract CD40L-expressing CD4+ T cells, which then seemingly favor CLL cell proliferation, since CD40L+ T cells colocalize with the cluster of proliferative malignant cells in lymph node and bone marrow pseudo-follicles (46). The relevance of TLR in B lymphoma/leukemia ontogeny can be speculated following the observation that its pathology can be associated with viral or bacterial infections, as evidenced by MALT lymphomas and Helicobacter pylori (47).
Generally speaking, the most prominent results in terms of the ex vivo maintenance of mature malignant primary B cells are elicited from combined supplementations. This is best explained by the fact that crosstalk and intersecting nodes exist between the different signaling pathways (48) and because it is now well established that no single mechanism ever constitutes the sole pathological driver in a complex hijacked microenvironment. These co-inputs usually consist of the activation of one or several of the abovementioned B cell receptors in concert with stimulatory growth factors (e.g., cytokines) expressed by adjuvant cell populations co-residing in vivo with (malignant) B cells in secondary lymphoid organs, blood, or bone marrow (Figure 2). In vitro, BCR activation is technically achieved following exogenous anti-IgM antibody binding. CD40 stimulation is implemented using either CD40 Ligands (CD40Ls) or other CD40 agonists (i.e., anti-CD40 antibodies), delivered exogenously in a soluble free form or cell-associated in a coculture system. In the latter case, murine fibroblasts are either stably transfected with CD40L (CD40LT cells) (58) or artificially express FcγRII (CDw32 L cells), thereby allowing the crosslinking of anti-CD40 antibodies (59). Finally, the stimulation of TLR9, located on the endoplasmic reticulum of resting cells, is mediated by endocytosed synthetic unmethylated Cytosine-phosphate-Guanine OligoDeoxyNucleotides (CpG ODN) (60).
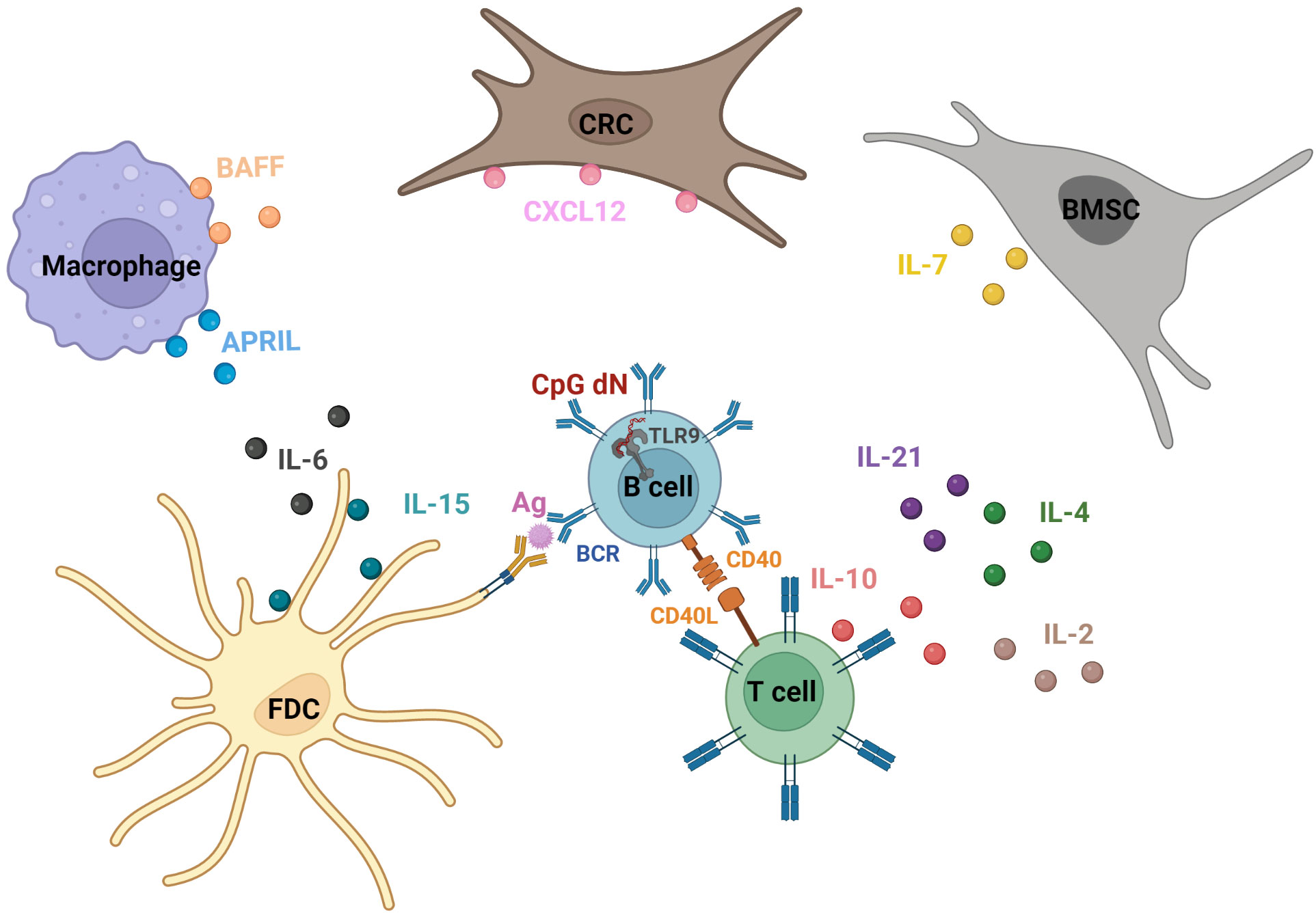
Figure 2 Example of actors exploited to drive the ex vivo survival/proliferation of mature malignant B cells. The implemented strategies for the ex vivo maintenance of malignant B cells are based on the pro-survival, pro-proliferative, and/or pro-migratory physiological signaling pirated by malignant B cells. These cues encountered in vivo are in part mediated by the activation of B cell conventional receptors, namely B Cell Receptor (BCR) (38), CD40 (39), and the Toll-Like Receptor 9 (TLR9) (40), respectively, via antigen presentation (notably by Follicular Dendritic Cells (FDCs) in Germinal Centers (GCs) during antigen-affinity selection) (15, 16), interaction with CD40L (notably displayed by follicular helper T cells (Tfh) in the GC) (39), and encounter with microbial unmethylated Cytosine-phosphate-Guanine diNucleotides (CpG dN) (40). Moreover, a cytokine/chemokine-based regulation is orchestrated by diverse ancillary cell types co-populating the B cell niches (i.e., the lymphoid organs, the blood and the bone marrow). For instance, InterLeukin-7 (IL-7), expressed notably by Bone Marrow Stromal Cells (BMSCs), is a crucial actor in the survival, proliferation, and maturation of B cell progenitors (49). Furthermore, B cell interactions in lymphoid organs with Tfh support proper progression through the follicular humoral immunity process, notably through CD40L-, IL-21-, IL-4- and IL-10-mediated signals (50, 51). In addition, C-X-C motif chemokine Ligand 12 (CXCL12)-expressing Reticular Cells (CRCs) produce the chemokine CXCL12, presumably in a proteoglycan-adsorbed manner on the cell membrane (52), thereby allowing the confinement of B cells in the dark zone of the GC (53). FDCs produce IL-15 and IL-6. While the presentation of the former as a membrane-bound form positively affects B cell proliferation (54), the latter drives proper GC reaction (55). Monocyte-derived cells, such as macrophages, produce B cell Activating Factor (BAFF) and A PRoliferation Inducing Ligand (APRIL), two transmembrane proteins that can be processed into soluble forms and are implicated in B cell maintenance and survival (56). IL-2, produced notably by activated T cells, controls B cell proliferation and differentiation (57). Created with BioRender.com.
Figure 3 summarizes the growth outcomes (detailed in depth hereafter) obtained from mature malignant primary B cells following diverse partial ex vivo supplementations. Specifically, while sole BCR activation has been shown to favor CLL cell proliferation (61) and survival (62), a single treatment with TLR9 Ligands (TLR9Ls) has been associated with increased CLL (63–65), SLL (65), FL (65), and MZL (65, 66) cell proliferation. CD40 activation induces FL (67, 68) and CLL (61, 63, 69) cell proliferation and promotes FL (70, 71), CLL (72–75), and MCL (76) cell survival. Notably, TLR9 stimulation potentiates CD40-activated CLL cell proliferation (77).
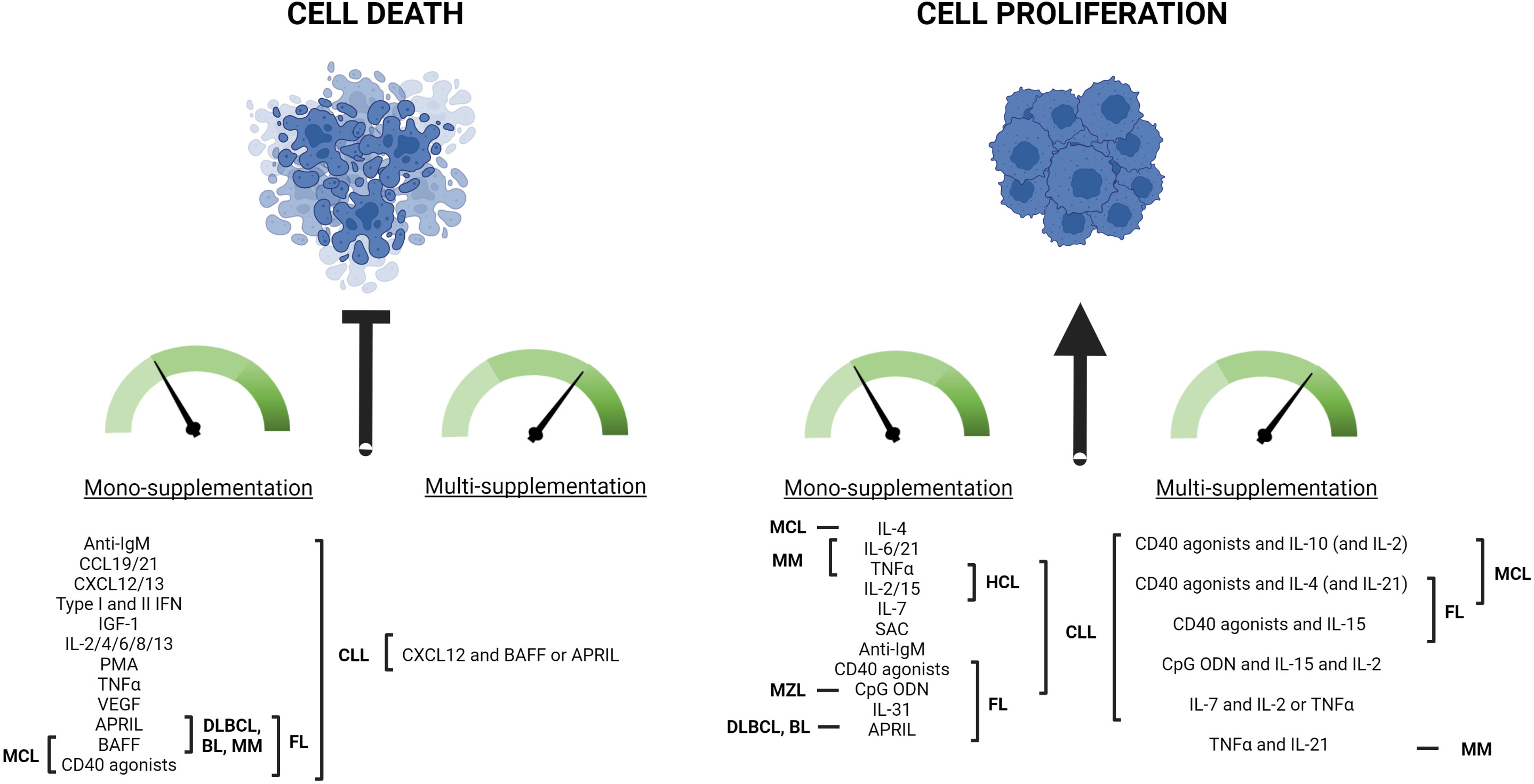
Figure 3 Summary from the scientific literature on the ex vivo 2D culture outcomes obtained with various exogenous partial supplementations of mature malignant primary B cells. Various molecules have been shown to inhibit malignant B cell death and/or to promote their proliferation when monocultured in vitro. They include B cell main receptor activators, such as Cytosine-phosphate-Guanine OligoDeoxyNucleotides (CpG ODN), anti-IgM antibodies (anti-IgM), and CD40 agonists, as well as many InterLeukins (IL-). The B malignancy subtype(s), for which each molecule has proven effective for ex vivo cell growth, is indicated in the figure. For more details and associated references, the reader is invited to refer to the main text in Partial supplementation outcomes from the literature. Combined supplementation generally potentiates the growth outcomes already observed with each actor due to synergy/additional effects. Chronic Lymphocytic Leukemia (CLL), Small Lymphocytic Lymphoma (SLL), Follicular Lymphoma (FL), Marginal Zone Lymphoma (MZL), Hairy Cell Leukemia (HCL), Mantle Cell Lymphoma (MCL), Diffuse Large B-Cell Lymphoma (DLBCL), Burkitt Lymphoma (BL), Multiple Myeloma (MM), Tumor Necrosis Factor α (TNFα), Insulin Growth Factor-1 (IGF-1), Vascular Endothelial Growth Factor (VEGF), InterFeroN (IFN), Staphylococcus Aureus Cowan strain I (SAC), Phorbol 12-Myristate 1-Acetate (PMA), B cell Activating Factor (BAFF), A PRoliferation Inducing Ligand (APRIL), C-X-C motif chemokine Ligand 12 (CXCL12), C-C motif Ligand 19 and 21 (CCL19 and CCL21). Created with BioRender.com.
CD40 stimulation is often combined with InterLeukin-4 (IL-4) supplementation. On its own, IL-4 enhances MCL cell proliferation (78) and CLL (73, 74, 79–85) cell viability; however, it seems to yield no success in terms of proliferation for this latter subtype (79, 83, 85, 86). Importantly, dual CD40/IL-4 stimulation has been shown to improve/potentiate the abovementioned pro-survival/proliferative outcomes (61, 78, 83, 86–92). The extra presence of IL-21 in this duo further heightens the proliferative response of CLL cells (86, 93). Conversely, BCR activation in combination with IL-4 does not seem to efficiently promote CLL cell proliferation (61, 83, 86, 94).
CD40 activation synergizes with IL-10 to promote CLL (69) and MCL (95) cell proliferation. The addition of IL-2 to this cocktail further dramatically enhances the mitogenic effect in the former subtype, since IL-10 upregulates the α chain of the IL-2 receptor (69). Sole IL-2 promotes CLL cell proliferation (83, 96, 97) and survival (98). Pro-mitogenic IL-2 also enhances the (IL-15-driven)-proliferation of primary CLL and HCL cells (96). IL-2 also synergizes with TLR9 activation to promote primary CLL cell proliferation (63, 99) and so does IL-15 (100) because both interleukin receptors share a β subunit that is upregulated by TLR9L (101). In line with this, this trio displays a pro-proliferative effect on CLL cells (64). Similarly, IL-2 positively drives CLL cell proliferation in synergy with BCR (83, 94, 102) or CD40 (69) activation as well as co-stimulation mediated by protein kinase C activator Phorbol 12-Myristate 1-Acetate (PMA) (96) or Staphylococcus Aureus Cowan strain I (SAC) (102), which already display, on their own, pro-survival (62, 80, 81) and pro-proliferative (61) effects, respectively. Enhanced proliferative effects were also observed in CLL (103) and FL (67) cells when IL-15 was combined with CD40 activation.
The B cell Activating Factor (BAFF) and A PRoliferation Inducing Ligand (APRIL) enhance primary CLL (80, 85, 104–107), FL, BL, DLBCL (108), MCL (109) and MM (110) cell survival. APRIL also enhances FL, BL, and DLBCL cell proliferation (108) while BAFF promotes FL cell proliferation in association with BCR activation (87). IL-7 increases CLL cell proliferation, even more in the presence of IL-2 or Tumor Necrosis Factor α (TNFα) (97). IL-8 (111, 112) and IL-13 (83) have been shown to favor CLL cell survival. However, IL-13 also inhibits IL-2-mediated CLL cell proliferation, as does IL-4 (83, 94).
Insulin Growth Factor-1 (IGF-1) (113), CXCL12 (52, 62, 107) and CXCL13 (62), CCL19 and CCL21 (62), Vascular Endothelial Growth Factor (VEGF) (85, 114, 115), type I and II InterFeroN (IFN) (116, 117), and TNFα (118) are also implicated in CLL cell survival, as well as CLL (97, 119) and HCL (120) cell proliferation for the lastly mentioned actor.
In contrast, IL-5 augments CLL cell apoptosis, an effect partially counteracted by IL-4 (82). IL-31 (68) and IL-21 (121) increase FL and MM cell proliferation (especially in the presence of TNF for the latter subtype), respectively. IL-6 increases MM cell proliferation and survival (121) as well as CLL cell viability (111, 122).
Intrinsic considerations
Although recurrent tendencies can be outlined (Figure 3), one ought to keep in mind that ex vivo culture outcomes remain highly dependent on the patient, regardless of the implemented supplementation approach. In congruity with the well-established fact that patients suffering from the same pathology can display variable/opposite treatment responses (as exemplified by CD40 agonism-based DLBCL therapy (123)), one can easily anticipate that malignant primary cells will display a breadth of behaviors under the same stimuli due to their inherent inter-heterogeneity.
Hence, patient cells can be divided into CD40 (95, 124) and BCR (125) responders and non-responders. In line with this, the added value of the various supplementations discussed above is not straightforward, and culture behaviors can be highly heterogeneous in amplitude response (65, 69, 81, 83, 86, 93, 97, 102, 111, 119, 121, 126–128). Dual/opposite effects resulting from a specific cocktail can even be observed (61, 69, 98) as exemplified by the occasional inhibitory action of IL-4 on malignant cell proliferation (88, 95), in accordance with its described counter-proliferative function regarding IL-2 (94, 129).
Researchers have increasingly succeeded in establishing genetic/phenotypic correlations to explain these multifarious responses to stimulation. For instance, while IL-4 induces proliferation in GCB-DLBCL cell lines, it displays the opposite effect on ABC-DLBCL ones (130). Additionally, primary GCB-DLBCL specifically featuring CD10+, Bcl2-, and extra nodal lesions are more favorably prone to long-term ex vivo maintenance when co-stimulated with IL-4 and CD40L than other subsets (131). In addition, Wang et al. reported that IL-21-driven cell growth and survival seem specific only to type III latency Epstein-Barr Virus (EBV)+-DLBCL and that the opposite effect can be expected for EBV–DLBCL (132). IL-21 was also found to show a dual effect on BL and t (14, 18)-featuring DLBCL and FL cell lines, with an increase in proliferation among the former and apoptosis induction among the others (133).
Relative to CLL mutational status, Unmutated (U) CLL cells seem more prone to ex vivo apoptosis than their Mutated (M) counterparts when cultured in a simple growth medium (74, 134) although contrasting evidence has also been reported (135). Along those lines, while TLR9 activation generally leads to the proliferation and survival of primary U-CLL cells, it induces apoptosis in most of the M-subtype samples (100, 136), although antagonizing evidence also exists (137). Notably, this last study showed that IL-21 amplifies both differential TLR9L-induced growth behaviors. Moreover, while CLL samples featuring 13q deletion as a unique aberration are particularly sensitive to TLR9L-induced apoptosis (138), those bearing the additional ataxia-telangiectasia mutated protein anomaly are prone to display greater viability under the TLR9L+IL-15 supplementation regimen (100). Furthermore, cells bearing only Trisomy 12 as a genetic abnormality show superior proliferative proclivity under the same stimulatory conditions, although both U-CLL and M-CLL cells are positive responders to this cocktail (100). Combined CD40/TLR9 activation seems to more prominently drive U-CLL cell proliferation (139). Similarly, only the CD38+ CLL subset was found to undergo apoptosis in response to crosslinked IgM stimulation. However, the physiological outcome was the opposite when stimulated with crosslinked IgD (140).
Technical considerations
Attributes aside from intrinsic cell make-up can influence cell response to external stimuli, such as the sequential and combinatory nature of the supplementation system. For instance, although IL-10 can potentiate CD40-activated CLL cell proliferation (69), it acts deleteriously when used singly (141). This proapoptotic effect, however, seems restricted to CLL, as other types of mature B neoplasms do not display such an IL-10 response (141). Furthermore, while the sole (63, 103, 142) or post-CD40 activation (143) addition of IL-21 can result in some proapoptotic activity on CLL cells, it can also yield a pro-proliferative/survival effect when concomitant to CD40 activation (63) or following IL-4 priming (86, 93). However, a single IL-21 treatment did not consistently lead to CLL cell apoptosis (93), suggesting that alternative parameters could be the reason for the observed discrepancies.
Indeed, one should also keep in mind that reagent specificities can account for the divergences observed in the literature (144). For instance, IL-6 antiapoptotic activity on CLL cells appears to be strictly restricted to the dimer form and is modulated by the origin of the cytokine, possibly because of variations in stability (122). Molecule dosage also determines cell responsiveness (102, 119), as receptor/ligand interactions cannot be restrictively defined as “on-off” operations (145).
Importantly, the specifics (e.g., valency) of the different anti-CD40 antibodies modulate the level of B cell activation (145). Moreover, multimerized soluble or membrane-bound forms of CD40L and crosslinked anti-CD40 antibodies have been shown to facilitate CD40 activation compared to free monomeric ligands or antibodies, as they better allow receptor clustering and downstream signaling cascades (145, 146). Furthermore, depending on the membrane-based model used, the CD40 agonist density on the cell membrane can vary, thereby leading to differential signal modulation (145). Similarly, immobilized anti-IgM antibodies induce more potent BCR activation in CLL cells compared to the free entities (147), or they even induce growth behavior in opposition to the one triggered by the untethered counterparts, possibly due to the sustained vs. transient nature of the signal (125).
Of note, the full influence of CD40 activation can be tricky to assess in systems utilizing CD40L- or FcγRII-expressing cell lines, as it can be misconstrued or outweighed by the cytoprotective impact putatively already provided by the feeder layer itself. Notably, while murine fibroblasts expressing CD40L were found to provide death protection for CLL cells, their untransfected counterparts did not (72, 90). However, the addition of anti-CD40 antibodies to the CDw32 L coculture system did not add value to FL cell survival (148).
Biological considerations
One major caveat of 2D monoculture is its inability to exhaustively recapitulate, quantitatively and qualitatively, but also spatially and temporally, the intricate in vivo microenvironment, which is characterized by a dynamic and finely tuned interplay of abundant regulatory molecules. Hence, attempts to improve cell survival and proliferation through partial exogenous supplementation carry the risk of inducing subpopulation selection and/or phenotypic shifts that are exaggerated and/or unrepresentative compared to the in vivo situation.
For instance, aside from cell survival and proliferation, specific medium supplementation can influence other cellular outcomes, such as cell differentiation. Although one may assume that this effect would be negligible considering the supposed “maturation arrest” state characteristic of tumor cells, CLL cells have been demonstrated to possess not only an inherent propensity to undergo plasmacytic differentiation but also Class Switch Recombination (CSR) and SHM. This provocative plasticity, and blockage thereof, is entirely dependent on environmental cues (149). Hence, ex vivo, the combination of CD40 activation with IL-10 induces CLL cell immunoglobulin secretion (69, 150). This phenotype shift into Immunoglobulin-Secreting Cells (ISCs) is potentialized by the addition of IL-2 (69, 151), although CD40 activation alone (61, 69) or in the presence of sole IL-2 (61, 69) or IL-4 (61, 88, 151), or single IL-4 (85) treatment, has seemingly failed to significantly induce ISCs. On the other hand, IL-4, in concert with CD40 activation (150) or hydrocortisone (152) is linked to CLL IgE switching, although quite inconsistently (61). The additional presence of IL-21 in the CD40L and IL-4 cocktail does not seem to trigger the plasmacytic phenotype (93), although sole IL-21 stimulation of primary CLL cells yields various results in terms of the induction of the differentiation marker Blimp1 (153). These discrepancies can be explained by the fact that the differentiation potential of CLL cells is inversely correlated with their anergic state (153). Of note, sole IL-21 treatment increases Blimp1 abundance in DLBCL cells (132).
While TLR9L-, IL-2-, and IL-15-mediated stimulation of CLL cells first induces robust cell proliferation, it eventually leads to cell differentiation into ISCs (64). Furthermore, IL-2, in association with SAC (61) or crosslinked-IgD (140), induces CLL cell differentiation into ISCs. PMA is a strong inducer of ISCs (154–156), and this effect is amplified in the presence of residual T lymphocytes (156) or associated cytokines IL-2 (157) or IL-4 (158). Using a two-step culture system consisting of the sequential use of pro-proliferation and pro-differentiation multi-cytokine cocktails, Ghamlouch et al. concluded, after exhaustive assessment of immunophenotypic, morphologic, and molecular features, that CLL cells undergo plasmacytic differentiation in the presence of PMA, or TLR9L and CD40L, but not in CD40L-only (159, 160). Of interest, BAFF and APRIL induce differentiation into ISCs as well as CSR in CLL cells (85).
Although it can bear a significant analytical downstream impact, the progression of malignant B cells through the maturation process has seldom been investigated in studies using a stimulatory cocktail primarily aimed at maintaining primary cells ex vivo. However, CLL cell plasmacytic differentiation has been associated with poorer cell survival capacity and decreased expression of actors driving malignant programming (64, 161). Thus, while this observation justifies the impetus for “differentiation therapy” (162), it can easily lead to skewed/overemphasized in vitro drug sensitivity when exogenous molecules are used naively for sole cell survival/proliferation purposes.
As medium supplementation modulates spontaneous cell death, cell proliferation, and cell differentiation, it is also expected to mitigate the drug response profile of mature malignant primary B cells. Therefore, partial supplementation can, albeit in its own limited way, recreate the protective haven encountered in vivo (70, 115, 163–165) or even reveal drug-permissive synergies that are worth exploring (65, 163, 166).
Additional considerations
As bystander cell-secreted molecules seem crucial to improving malignant mature B cell survival and proliferation, culturing malignant B cells as unfractionated/unpurified portions among autologous cells is a well-grounded practice whenever allowed by downstream analysis. Hence, autologous T cells have been shown to increase CLL cell survival (74, 111), although not consistently (35). Interestingly, CCL2 and CXCL2 pro-survival effects are restricted to unpurified CLL samples, highlighting the inter-dependence of cell populations in terms of survival (111).
The medium change frequency significantly affects cell survival, as it can deplete molecular talks. Hence, Burgess et al. found that enhanced cell survival was obtained with only a weekly or biweekly medium refresh (111). Higher cell densities that enable closer cell-to-cell contact also greatly impact cell survival, and while some authors have shown that a density of 2.5 million cells per mL already displays remarkable added value compared to a lower density (167), others have found that only densities from 40 million cells per mL induced marked enhanced survival (111).
Commercial serum-free AIM-V and Hybridoma media (Thermo Fisher Scientific) can enhance CLL cell survival due to undetermined factors (168, 169). Aside from quelling ethical concerns, removing the need for serum from the culture medium decreases biological interference, improves result consistency, and facilitates analysis, thanks to a refined medium composition (170). Of importance, autologous patient serum promotes better CLL cell survival compared to heterologous serum or a complete medium (80), albeit inconsistently (35).
Although inconsistently beneficial (168), the exogenous addition of glutathione or its precursor N-acetylcysteine in the culture medium can promote CLL cell survival, as it thwarts the limited ability of malignant cells to uptake cystine for cysteine production, which would subsequently impair the glutathione-dependent redox balance (171). The presence of the reducing agent β-mercaptoethanol in a cystine-containing culture medium also provides similar protective effect (171).
It may be important to assess the relevancy of IL-9 (172) and IL-27A (173) as culture supplements for the growth of primary DLBCL cells, as they both promote DLBCL-derived cell line proliferation, counter drug-induced apoptosis, and seem prognostically meaningful with regard to patient serum levels. Similarly, IL-22 supports the growth of MCL cell lines (174).
Overall, the biological limitations of 2D monocultures are proportional to the technical advantages offered. Incorporating a relevant secondary cell type into the culture is a technically accessible way to enhance cell survival and/or proliferation while better mitigating putative exogenous supplementation-induced phenotypical shifts.
2.5D culture
2D coculture (2.5D) models from the literature
2.5D models implemented for the ex vivo culture of diverse mature B neoplasms are presented in Table 1.
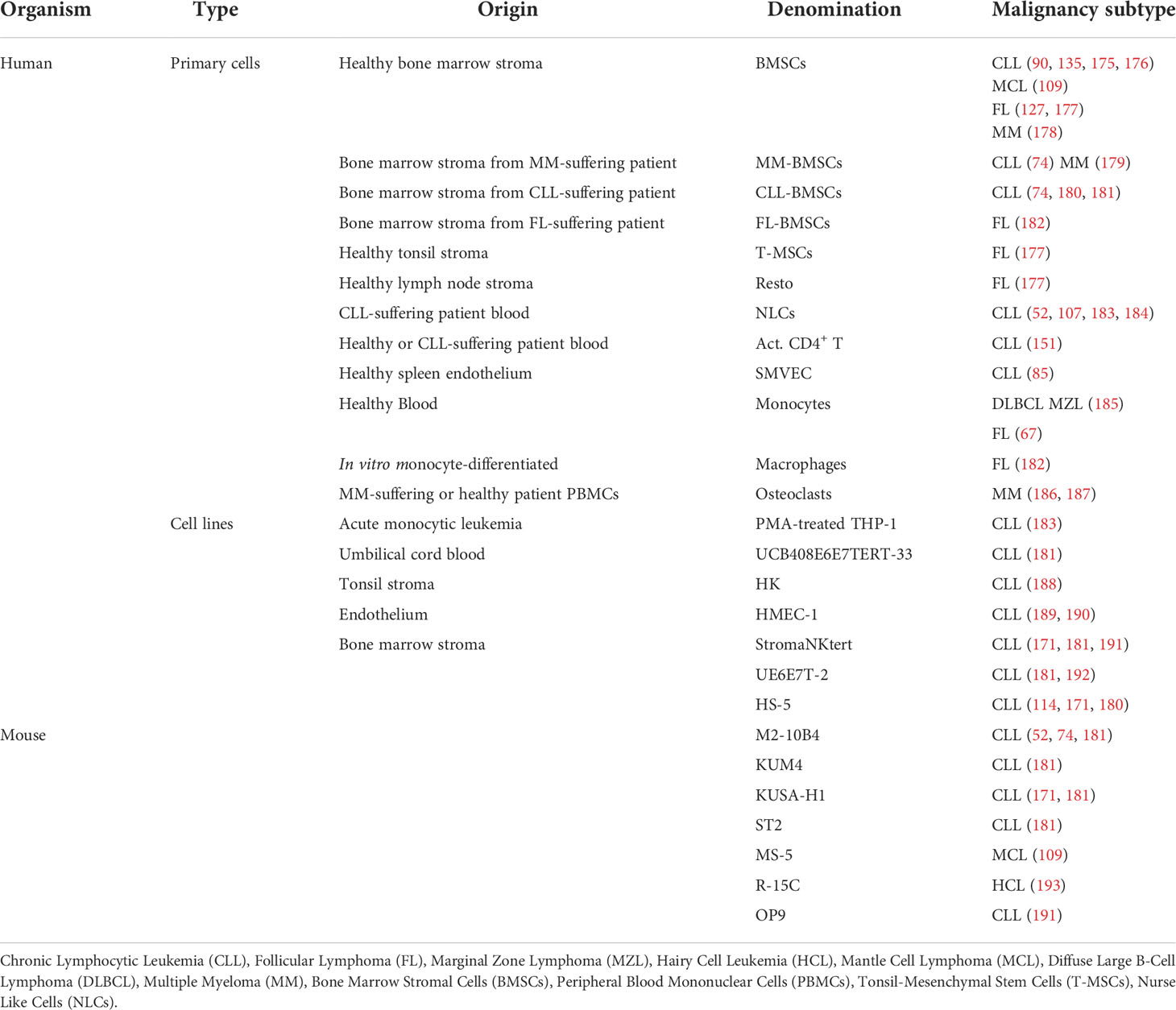
Table 1 Summary of the scientific literature on the 2.5D culture models implemented for the ex vivo maintenance of mature malignant primary B cells.
As demonstrated using IL-21 (63), attempts at partial supplementations can induce unsought cell growth behaviors otherwise not observed in the additional presence of (in vivo) ancillary co-stimulations. Hence, although CLL cells display decreased survival in response to a single IL-10 treatment (141), they exhibit enhanced survival when directly cocultured with IL-10-secreting Bone Marrow Stromal Cells (BMSCs) (175). The overall cytoprotective effect of lymphoid tissue-derived stromal cells on diverse malignant B cells has been reported with either primary cells or cell lines of human or murine origin (Table 1). Notably, primary BMSCs with a pathological origin display increased efficacy in maintaining malignant B cells ex vivo (74, 180, 182). Furthermore, supplementing the BMSC-based coculture model with additional co-stimulators further enhances cell proliferation (127, 135, 192). FL cell survival is supported by Resto cells (lymph node-derived FRCs that can be differentiated in vitro from tonsil and bone marrow mesenchymal stem cells) (177). Similarly, the human tonsil dendritic follicular HK cell line supports CLL cell survival (188).
Monocytes also constitute crucial growth assistants, as coculture sometimes appears indispensable to DLBCL and MZL cells’ ex vivo persistence otherwise lacking in plain medium or in the simple presence of BAFF and IL-2, which, however, further potentiate the monocyte-driven effect (185). Moreover, monocytes favor FL cell growth, especially through IL-15 trans-presentation and concomitant CD40 activation (67). Along those lines, CLL cell survival is sustained by patient blood-derived cells called nurse-like cells, which differentiate in vitro from CD14+ mononuclear cells in a CLL cell-driven manner and are considered tumor associated macrophages (52, 107, 183, 184, 194). This cytoprotective effect is partially mediated by CXCL12 (52), BAFF, and APRIL (107). On the same note, primary macrophages promote FL cells’ ex vivo maintenance (182), and the (M2) macrophage-differentiated leukemic cell line THP-1 supports CLL cell survival (183).
CLL and CD3-activated CD4+ T cells direct coculture drastically enhances CLL cell proliferative capacity, which is further heightened by IL-2 (151). Coculture with endothelial cells also guards CLL cells against spontaneous apoptosis (85, 189, 190). Finally, primary MM cell survival is supported by osteoclasts (186, 187).
Biological and technical considerations
Although coculture systems are no exception to the “patient-dependent outcome” rule (127), they generally benefit from a higher potential for long-term ex vivo cell maintenance thanks to the incorporation of cellular support, which provides both biological and physical assistance. On that account, cocultured CLL cells spontaneously migrate beneath the sessile BMSC feeder layer, a phenomenon termed “pseudoemperipolesis” (195). This adherence to the nurturing layer also occurs with cocultured MCL cells and manifests itself in cobblestone areas seemingly enriched in tumor-initiating cells (196). In harmony with this idea, direct cell contact between malignant cells and the feeder layer either dictates (52, 109, 151, 175, 176, 181, 188) or enhances (85, 90, 180) the pro-survival effect, highlighting the extracellular matrix and/or cell membrane as crucial interactive structures. Indeed, concerning the latter, some aforementioned molecular actors present functionally relevant membrane-bound forms, as exemplified by BAFF (197) and CXCL12 (198). As overall upgraded biomimetic models that better recapitulate the protective effect of the in vivo niche, coculture systems appear more suitable for drug efficacy assessment (165, 171, 181, 183, 184, 199).
The choice of a feeder layer depends realistically on financial, technical, and logistic factors. Indeed, while unmodified primary feeder cells inevitably recapitulate the in vivo niche more genuinely than cell lines, they offer a less consistent and reproducible set-up, and their use can be hampered by a lack of a regular supply and/or production tediousness (183). However, inter-lab standardization of coculture systems resorting to characterized cell lines can also be hindered by a selective lack of access. For instance, the ATCC cell line HS-5 is no longer available in European countries due to more rigid regulations.
Because cell lines often lack contact inhibition, the feeder layer must be disabled in terms of proliferation to prevent overgrowth-induced (patient) cell death and medium growth factor depletion during long-term cocultures. The two major approaches used to do so are γ-irradiation or cytotoxic drugs, such as mitomycin C. While the former induces DNA breakage, the latter acts as a DNA crosslinker (200). However, both have downsides, as they have been associated with metabolic disturbances (200, 201). Moreover, γ-irradiation necessitates expensive ad hoc equipment. Mitomycin C can be a more accessible and budget-effective approach, but dose optimization according to the cell line used, as well as thorough subsequent washing to prevent the deleterious effect of mitomycin C leftover on the patient cells, are required (202).
3D culture
Although they bolster efforts to reconstitute the tumor-residing microenvironment, 2.5D cultures are still far from being capable of offering accurate representations of the complexity and authenticity of a niche. In a bid to better decipher spatiotemporal aspects of lymphoma/leukemia pathophysiology, and to obtain improved concordance between pre-clinical and clinical studies, 3D models have increasingly drawn the interest of researchers, as they help to better capture heterogeneous multicellular interactions (and overall biological and chemical cues). Furthermore, they are also more capable of reconstructing structural, mechanical, and temporal organization/constraints that greatly influence cell behavior, as well as drug response and delivery (203–205). Although they have been increasingly researched, 3D culture models are still difficult to construct because the degree of sophistication/complexity of the platform is proportional to its cost, labor intensity, and applicability in terms of throughput series. Nevertheless, biologically relevant 3D platforms focus on mechanical/structural, cellular, and dynamic aspects that together shape a disease in vivo, enabling it to thrive into a hijacked pro-tumor sanctuary (Figure 4).
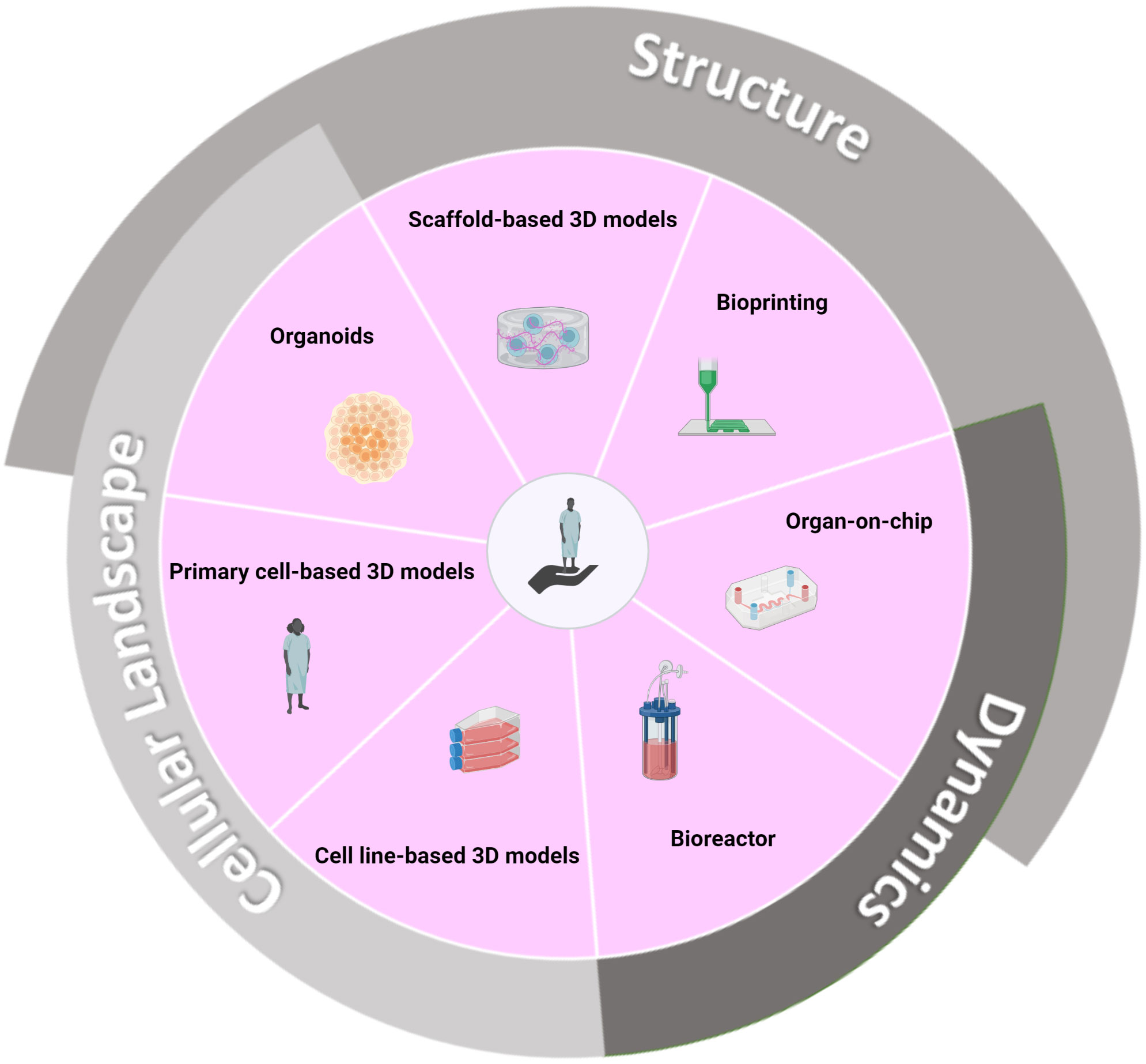
Figure 4 3D culture strategies that biomimicry crucial in vivo microenvironment features and ultimately enable improved predictive value for clinical success. Pathophysiologically relevant 3D models demand a faithful representation of the tumor architecture and microenvironment. Tumor structural support and organization are encouraged by the biomimetic reconstitution of the ExtraCellular Matrix (ECM) using natural or synthetic scaffolds (206). At a larger scale, tissue rigidity/plasticity and associated compressive forces in the tumor-residing organ are reproduced using ad hoc material, such as hard porous scaffolds with high compressive strength for the bone (207). Further spatial refinements are enabled by on-chip 3D platforms that can recreate tissue/organ intra-compartmentalization (208). Additionally, a dynamic condition is achieved thanks to the perfused nature of the devices (209). 3D bioprinting is a computer-aided technology that allows a defined object geometry and precise cell spatial distribution, thanks to a layer-wise deposition approach (12). Bioreactors can account for the circulating nature of a malignancy and allow the study of cell homing and retention in tissues (13). Finally, yet importantly, the tumor microenvironment comprises an intricate ecosystem of normal cells that can be simulated using several cell lines (210), primary cells (179), or, as exemplified with the bone marrow, recreated via organoid technology (10). This last one also allows for the partial recreation of cellular structural units, such as the vasculature (10). Created with BioRender.com.
Mechanical/structural component
The ExtraCellular Matrix (ECM) is an extensive non-cellular network composed of fibrous proteins (e.g., collagen), glycoproteins (e.g., laminin), and glycosaminoglycans (e.g., heparan sulfate). It provides both structural and functional cell support (206). Indeed, (malignant) cell interactions with the ECM directly drive cell organization, survival, proliferation, and migration (211). Scaffold-based 3D models imitate the ECM biostructure using either biological (e.g., collagen) or synthetic (e.g., polyethylene glycol) materials. While the latter type offers cost-efficiency and reproducibility, the former is intrinsically bioactive and is a natural reservoir for various growth-related actors (206).
Moreover, as exemplified by the work of De la Puente et al. who developed 3D tissue-engineered bone marrow supporting the proliferation of primary MM cells, ECM-anchored 3D models mimic physiological gradients (e.g., oxygen and drug), and therefore permit relevant investigations of drug delivery and response (9). Following this idea, ECM stiffness in 3D models has been shown to affect drug diffusion (36, 204).
Tumors evolve in a mechanically constrained setting, where tensile forces are determined not only by an organ’s external borders but also by the internal meshwork of proliferating cells and dynamic ECM. Since mechano-sensing affects biological processes (212), it is relevant to focus efforts on generating platforms possessing mechanical properties, as exemplified by the work of Lamaison et al. who generated elastic spheroids using alginate-based microfluidic-generated encapsulation. They could reproduce lymph node/tumor-mediated compressive forces and possibly associated cues. In this model, co-encapsulation with primary tonsil stromal cells maintained FL patient cell viability (204).
At the microscale, on-chip 3D devices can assist in mirroring an organ microarchitecture, such as its internal multi-compartmentalization, as exemplified by lymph nodes-on-chip models (208). At the nanoscale, 3D bioprinting technology allows for the precisely controlled spatial deposition of cellular and molecular constituents in a computer-generated construct. While constituting an under-explored avenue in hematological malignancies, this technique is permissive to primary CLL cells’ long-term maintenance (12).
Cellular component
The TME also comprises a heterogeneous population of normal cells, such as immune and stromal cells, that are not mere bystanders but rather key effectors in tumor persistence and therapy resistance (213). 3D models attempting to reconstruct the initial cellular diversity display different degrees of complexity (and therefore translational values) with the concomitant use of one or two additional cell types originating from cell lines or primary cells. For instance, thanks to a conical agarose microwell allowing both the formation of cancer cell aggregates and the diffusion of biomolecules originating from a bone marrow stromal cell line, Waldschmidt et al. were able to propagate primary MM cells in a cytoprotective environment (210). Furthermore, Foxall et al. developed collagen-scaffolded spheroids comprising fibroblasts (differentiated from commercial adipose-derived stem cells) and primary monocyte-derived macrophages, ultimately creating a culture system that altogether favors primary DLBCL cell survival. This model also seems to exert opposing action against rituximab-induced antibody-dependent cell phagocytosis, therefore putatively partially mimicking in vivo drug resistance (36). Jakubikova et al. created a 3D hydrogel model wherein patient-derived BMSCs induced niche-like structures that enabled primary MM cell proliferation. The 3D platform showed potential to serve as a testbed for the study of niche-mediated MM survival/drug resistance (179).
While the above-mentioned systems are conducive to the ex vivo maintenance of malignant B cells, they remain minimalistic views of the gamut of cell lineages (and associated structures) harbored in the initial ecosystem. In particular, the presence of a bone-like niche is relevant in 3D models investigating MM, since osteolytic disease is a hallmark of this pathology that researchers have attempted to phenocopy, notably by exploiting 3D mineralized bone models (207). Moreover, although the use of primary (bone marrow) mesenchymal stem/stromal cells rather than cell lines can more accurately model the original TME, their current shaky definition and the variations in associated isolation and culture methods seem antagonistic to the development of a defined and reproducible 3D model (214, 215). The generation of (bone marrow) organoids, which are self-organizing and self-renewing microstructures, is a way to partially address these issues (216). However, the literature on bone marrow organoids is scarce due to the technical and technological challenges associated with reproducing the fully mature, diverse, and functional units of the bone marrow (e.g., the bone, and hemopoietic, and vascular niches), originating from a multidirectional differentiation of stem cells (215). Although still imperfect, the self-generation of bone marrow organoids presenting a vessel-like network and able to support the engraftment of leukemic blasts is a step forward toward the generation of promising organoid systems amenable to hematological preclinical applications (10).
Another prime consideration regarding the accuracy of the cellular landscape in 3D models is the representativeness of all the malignant subclones, notably cancer stem cells, that drive disease relapse and resistance (217). Kirshner et al. established a patient-derived, self-stratified bone marrow 3D model, allowing not only the proliferation of MM cells but also the persistence of putative dormant stem-like malignant cells, thereby permitting the preclinical assessment of the MM clone as a whole (218).
Dynamic component
One of the shortcomings of most actual 3D models is their static nature. Nonetheless, authors are increasingly striving to reproduce the dynamic flows that occur physiologically through the vascular network and regulate nutrient supply and waste disposal. Hence, although it has not yet been assessed in the context of B lymphoma/leukemia studies, Chou et al. developed an on-chip model comprising a dynamic vascular channel that supports the generation of a multi-lineage bone marrow-like proxy. This model allows the incorporation of a relevant pharmacokinetic aspect and the study of the post-treatment recovery of the generated tissue-like structure (209). In a more accessible and economical set-up, Mannino et al. generated a DLBCL-on-chip model where the blood vessel directly traverses the artificial tumor niche, closely resembling what occurs in vivo (11).
Moreover, mature B malignancies are dynamic neoplasms, as exemplified by CLL, for which lymph nodes constitute active proliferation sites (in areas called pseudofollicles) that feed the quiescent circulating compartment (44). Therefore, peripheral blood, lymph node, and bone marrow-originating malignant B cells are expected to display differential signaling/behavioral signatures due, in most part, to the influence of these distinct specialized microenvironments (13). Hence, the addition of lymph node-enriched IL-4 restores the BCR component balance of primary CLL cells by upregulating the surface expressions of IgM and CD79b, thus enhancing BCR responsiveness, a feature that is not shared by resting (anergic) CLL cells in peripheral blood (219, 220). To account for the circulating nature of the disease, a bioreactor model has been customized for the study of CLL cell retention and egress into/from a reconstructed bone marrow niche (13). Moreover, Walsby et al. developed a circulating model system allowing for the study of primary CLL cell behavior and signature under shear forces reflective of what they (along with endothelial cells) are subjected to in the peripheral vasculature, thereby further clarifying what occurs during transendothelial cell migration (221).
Discussion: literature-to-bench translation
The choice of an ex vivo culture model (Figure 5) is realistically contingent on both the research objectives and financial/technical feasibility. Although 3D ex vivo culture models are undeniably best suited for preclinical investigations, considering their elaborate design and subsequent upgraded translational value (223), the development of 3D platforms for the ex vivo maintenance of mature malignant B cells is still in its infancy and remains challenging and expensive, both in its elaboration and application. This is especially the case as models become progressively more complex to enhance their ability to recapitulate the native niche.
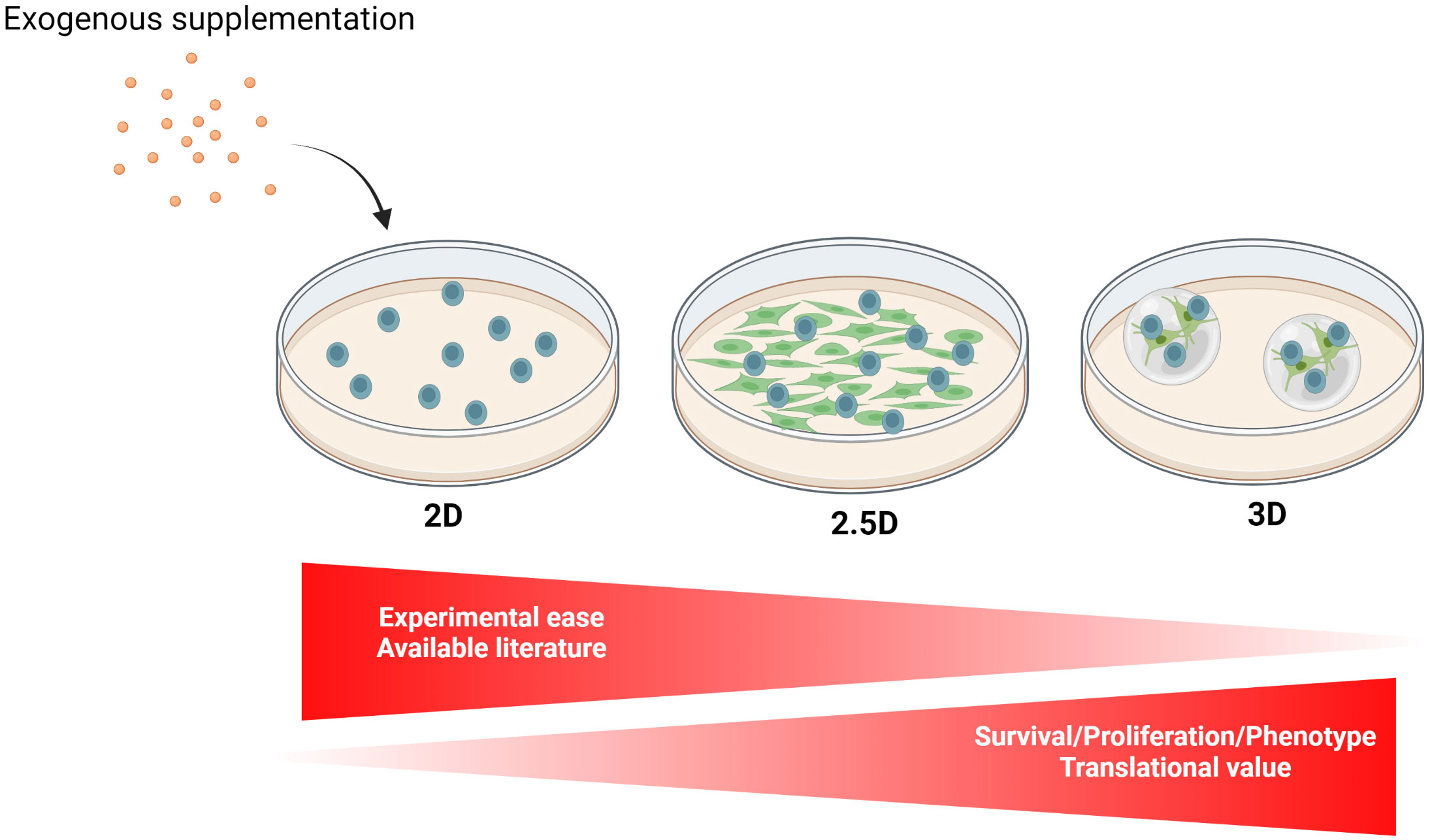
Figure 5 Different culture models implemented for the ex vivo propagation of mature malignant B cells. The ex vivo culture of mature B neoplasm-derived primary cells can be implemented as a 2D monoculture, 2D coculture (2.5D), or 3D culture which, in this order, offers an increase in cell survival/proliferation, as well as a higher translational value to the experimental results. Although they are characterized by different degrees of investigation/development by researchers, none of these models have been experimentally standardized. 2D monocultures, which are more adapted for short-term experiments, are often partially supplemented with exogenous growth factors, most commonly CD40 agonists and/or InterLeukin (IL)-4, allowing improved cell survival and proliferation. Furthermore, Toll-Like Receptor 9 activation combined with IL-2 treatment is widely used to trigger B cell proliferation for subsequent cytogenetic analysis (222). 2D cocultures (2.5D) of malignant B cells in direct contact with various ancillary cell populations, such as stromal cells derived from lymphoid organs, improve ex vivo cell maintenance and can be achieved with primary cells or cell lines. Finally, interest is growing in the development of 3D models for hematological malignancies, ranging from scaffolded spheroids to more intricate bioreactors and microfluidic devices (213). Created with BioRender.com.
As a second-best option, 2.5D coculture system allow for the integration of interpopulation interactions while enabling higher throughput experiments. Despite the absence of standardization, successful attempts to coculture malignant primary B cells have been achieved with stromal cells of relevant origins and with various immune cells, preferably in direct cell–cell contact. Notably, the conceptual significance of cocultures is not just to improve cancer cell survival/proliferation but also to better adjust culture techniques to develop therapeutic strategies that are more and more microenvironment-centric, rather than focused on the cancer cells themselves (224).
Although completely incapable of replicating the in vivo complexity, 2D monoculture remains an easy, fast, and low-cost alternative offering reproducible and easy-to-interpret results, especially for experiments that do not require prolonged cultures. Experimental ease, however, is hindered by patient-dependent cell behaviors in response to partial supplementation. While the effort to standardize and improve cost/time efficiency are salient considerations to perform achievable science, we advocate that researchers, whenever feasible, address the culture needs specific to a pathological subtype and subset more congruously. Along those lines, we suggest that the research community systematically shares the differential growth behaviors of the primary samples used in their studies for the implemented culture conditions, along with, if known, the patient’s genetic/molecular characteristics to help in establishing the interconnections as those mentioned earlier. We feel that seemingly experimental failures that constitute the most invaluable data are often not exploited and/or communicated enough.
On a side-but-related note, the limitations encountered by researchers regarding the ex vivo culture of patient cells induce an initial selection bias in favor of patient samples displaying a high percentage of cancer cells and, in the context of retrospective use, toward patients for whom significant biomaterial was retrieved and cryopreserved, thereby enabling both culture condition optimization and actual experimental replicates. Biobanks can indeed be challenged with scant biomaterial to process, as the accelerating in-depth characterization of patient samples implies the decreased availability of biomaterial, which is dispatched between diverse diagnostic services and clinical studies. This is especially true for the biomaterial retrieved from bone marrow biopsies, whose invasive and painful nature precludes ad libitum supply. Although not devoid of liabilities, an approach worth considering involves the transitory use of Patient Derived Xenografts (PDXs), which enable the expansion of valuable primary cells for high-throughput studies, such as drug screening (225).
To conclude, we feel, like many others, that 3D culture models have a bright future for studies related to drug development and disease mechanisms. Technology-based attempts to recreate multifaceted microenvironments are sharpening and striving to incorporate additional critical physiological aspects. Accumulating evidence seems to indicate that researchers should focus on the establishment of a customizable/personalized set-up, as patient-derived material (e.g., ECM) unsurprisingly offers added value in terms of primary cell maintenance (9). Although increasing the arduousness of the task, the use of autologous TME would allow the faithful recreation of the in vivo niche, a process mostly undertaken using cell lines or allogenic cells from healthy individuals. This approach, however, neglects a prime characteristic of the TME co-inhabiting with malignant cells, which is the fact that it is re-educated (FL), effaced (BL), or recruited. For example, cells resembling FRCs and FDCs have been spotted in FL-infiltrated bone marrow, although they are not detected under healthy conditions. Whether they originate from MSCs or are recruited to the bone marrow is still unclear (226). Furthermore non-malignant stromal cells display specific gene signatures among DLBCL patients, which are predictive of treatment outcomes (227). Overall, such an approach is in congruity with the growing trend of personalized medicine.
We anticipate that the increasing efforts invested by the research community will lead to the establishment of sophisticated but accessible models that attempt a difficult balance between disease/patient specifics, standardization, and translational value. Therefore, we propose this review as a modest contribution to the collective effort.
Author contributions
MC wrote the manuscript and designed the figures and tables. FG and CG contributed to the writing and intellectually/critically revised the paper. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG, et al. Chronic lymphocytic leukemia. Nat Rev Dis Prim (2017) 3(1):16096. doi: 10.1038/nrdp.2016.96
2. Freedman A, Jacobsen E. Follicular lymphoma: 2020 update on diagnosis and management. Am J Hematol (2020) 95(3):316–27. doi: 10.1002/ajh.25696
3. Cheah CY, Zucca E, Rossi D, Habermann TM. Marginal zone lymphoma: present status and future perspectives. Haematologica (2022) 107(1):35–43. doi: 10.3324/haematol.2021.278755
4. Maitre E, Cornet E, Troussard X. Hairy cell leukemia: 2020 update on diagnosis, risk stratification, and treatment. Am J Hematol (2019) 94(12):1413–22. doi: 10.1002/ajh.25653
5. Maddocks K. Update on mantle cell lymphoma. Blood (2018) 132(16):1647–56. doi: 10.1182/blood-2018-03-791392
6. Susanibar-Adaniya S, Barta SK. Update on diffuse large b cell lymphoma: A review of current data and potential applications on risk stratification and management. Am J Hematol (2021) 96(5):617–29. doi: 10.1002/ajh.26151
7. Kalisz K, Alessandrino F, Beck R, Smith D, Kikano E, Ramaiya NH, et al. An update on burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging (2019) 10(1):56. doi: 10.1186/s13244-019-0733-7
8. Cowan AJ, Green DJ, Kwok M, Lee S, Coffey DG, Holmberg LA, et al. Diagnosis and management of multiple myeloma: A review. JAMA (2022) 327(5):464–77. doi: 10.1001/jama.2022.0003
9. de la Puente P, Muz B, Gilson RC, Azab F, Luderer M, King J, et al. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials (2015) 73:70–84. doi: 10.1016/j.biomaterials.2015.09.017
10. Giger S, Hofer M, Miljkovic-Licina M, Hoehnel S, Brandenberg N, Guiet R, et al. Microarrayed human bone marrow organoids for modeling blood stem cell dynamics. APL Bioeng (2022) 6(3):036101. doi: 10.1063/5.0092860
11. Mannino RG, Santiago-Miranda AN, Pradhan P, Qiu Y, Mejias JC, Neelapu SS, et al. 3D microvascular model recapitulates the diffuse large b-cell lymphoma tumor microenvironment in vitro. Lab Chip (2017) 17(3):407–14. doi: 10.1039/c6lc01204c
12. Sbrana FV, Pinos R, Barbaglio F, Ribezzi D, Scagnoli F, Scarfò L, et al. 3D bioprinting allows the establishment of long-term 3D culture model for chronic lymphocytic leukemia cells. Front Immunol (2021) 12:639572. doi: 10.3389/fimmu.2021.639572
13. Barbaglio F, Belloni D, Scarfò L, Sbrana FV, Ponzoni M, Bongiovanni L, et al. Three-dimensional co-culture model of chronic lymphocytic leukemia bone marrow microenvironment predicts patient-specific response to mobilizing agents. Haematologica (2021) 106(9):2334–44. doi: 10.3324/haematol.2020.248112
14. Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology (2020) 160(3):233–47. doi: 10.1111/imm.13176
15. Mesin L, Ersching J, Victora GD. GERMINAL CENTER b CELL DYNAMICS. Immunity (2016) 45(3):471–82. doi: 10.1016/j.immuni.2016.09.001
16. Young C, Brink R. The unique biology of germinal center b cells. Immunity (2021) 54(8):1652–64. doi: 10.1016/j.immuni.2021.07.015
17. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged b cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PloS Biol (2005) 3(6):e150. doi: 10.1371/journal.pbio.0030150
18. Masters AR, Jellison ER, Puddington L, Khanna KM, Haynes L. Attrition of T cell zone fibroblastic reticular cell number and function in aged spleens. ImmunoHorizons (2018) 2(5):155–63. doi: 10.4049/immunohorizons.1700062
19. Gotur S, Wadhwan V. Tingible body macrophages. J Oral Maxillofac Pathol (2020) 24(3):418–20. doi: 10.4103/jomfp.JOMFP_314_20
20. Sagaert X, Sprangers B, De Wolf-Peeters C. The dynamics of the b follicle: Understanding the normal counterpart of b-cell-derived malignancies. Leukemia (2007) 21(7):1378–86. doi: 10.1038/sj.leu.2404737
21. Marinkovic D, Marinkovic T. Putative role of marginal zone b cells in pathophysiological processes. Scand J Immunol (2020) 92(3):e12920. doi: 10.1111/sji.12920
22. Basso K, Dalla-Favera R. Germinal centres and b cell lymphomagenesis. Nat Rev Immunol (2015) 15(3):172–84. doi: 10.1038/nri3814
23. Mlynarczyk C, Fontán L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev (2019) 288(1):214–39. doi: 10.1111/imr.12755
24. Cinti I, Denton AE. Lymphoid stromal cells–more than just a highway to humoral immunity. Oxford Open Immunol (2021) 2(1):iqab011. doi: 10.1093/oxfimm/iqab011
25. Puente XS, Jares P, Campo E. Chronic lymphocytic leukemia and mantle cell lymphoma: crossroads of genetic and microenvironment interactions. Blood (2018) 131(21):2283–96. doi: 10.1182/blood-2017-10-764373
26. Veloza L, Ribera-Cortada I, Campo E. Mantle cell lymphoma pathology update in the 2016 WHO classification. Ann Lymphoma (2019) 3:3. doi: 10.21037/aol.2019.03.01
27. Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-cell analysis of germinal-center b cells informs on lymphoma cell of origin and outcome. J Exp Med (2020) 217(10):e20200483. doi: 10.1084/jem.20200483
28. Poletto S, Novo M, Paruzzo L, Frascione PMM, Vitolo U. Treatment strategies for patients with diffuse large b-cell lymphoma. Cancer Treat Rev (2022) 110:102443. doi: 10.1016/j.ctrv.2022.102443
29. Dreyling M, Campo E, Hermine O, Jerkeman M, Le Gouill S, Rule S, et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2017) 28:iv62–71. doi: 10.1093/annonc/mdx223
30. Bonnet C, Janssens A, Wu K, Schroyens W, Van Hende V, Heimann P, et al. BHS guidelines for the treatment of burkitt’s lymphoma. Belgian J. Hematol (2015) 6(2):61–9.
31. Dreyling M, Ghielmini M, Rule S, Salles G, Ladetto M, Tonino SH, et al. Newly diagnosed and relapsed follicular lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2021) 32(3):298–308. doi: 10.1016/j.annonc.2020.11.008
32. Zucca E, Arcaini L, Buske C, Johnson PW, Ponzoni M, Raderer M, et al. Marginal zone lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2020) 31(1):17–29. doi: 10.1016/j.annonc.2019.10.010
33. Abramson JS, Ghosh N, Smith SM. ADCs, BiTEs, CARs, and small molecules: A new era of targeted therapy in non-Hodgkin lymphoma. Am. Soc Clin Oncol Educ book Am Soc Clin Oncol Annu Meet (2020) 40(40):302–13. doi: 10.1200/EDBK_279043
34. Hallek M, Al-Sawaf O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am J Hematol (2021) 96(12):1679–705. doi: 10.1002/ajh.26367
35. Collins RJ, Verschuer LA, Harmon BV, Prentice RL, Pope JH, Kerr JFR. Spontaneous programmed death (apoptosis) of b-chronic lymphocytic leukaemia cells following their culture in vitro. Br J Haematol (1989) 71(3):343–50. doi: 10.1111/j.1365-2141.1989.tb04290.x
36. Foxall R, Narang P, Glaysher B, Hub E, Teal E, Coles MC, et al. Developing a 3D b cell lymphoma culture system to model antibody therapy. Front Immunol (2021) 11:605231. doi: 10.3389/fimmu.2020.605231
37. van Attekum MHA, Eldering E, Kater AP. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica (2017) 102(9):1469–76. doi: 10.3324/haematol.2016.142679
38. Yam-Puc JC, Toellner KM, Zhang L, Zhang Y. Role of b-cell receptors for b-cell development and antigen-induced differentiation. F1000Research (2018) 7:429. doi: 10.12688/f1000research.13567.1
39. Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev (2009) 229(1):152–72. doi: 10.1111/j.1600-065X.2009.00782.x
40. Hua Z, Hou B. TLR signaling in b-cell development and activation. Cell Mol Immunol (2013) 10(2):103–6. doi: 10.1038/cmi.2012.61
41. Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med (2004) 200(4):519–25. doi: 10.1084/jem.20040544
42. Agathangelidis A, Psomopoulos F, Stamatopoulos K. Stereotyped b cell receptor immunoglobulins in b cell lymphomas. Methods Mol Biol (2019) 1956:139–55. doi: 10.1007/978-1-4939-9151-8_7
43. Gemenetzi K, Agathangelidis A, Zaragoza-Infante L, Sofou E, Papaioannou M, Chatzidimitriou A, et al. B cell receptor immunogenetics in b cell lymphomas: Immunoglobulin genes as key to ontogeny and clinical decision making. Front Oncol (2020) 10:67. doi: 10.3389/fonc.2020.00067
44. Haselager MV, Kater AP, Eldering E. Proliferative signals in chronic lymphocytic leukemia; what are we missing? Front Oncol (2020) 10:592205. doi: 10.3389/fonc.2020.592205
45. Burger JA. BTK inhibitors: present and future. Cancer J (2019) 25(6):386–93. doi: 10.1097/PPO.0000000000000412
46. Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, et al. Chronic lymphocytic leukemia b cells are endowed with the capacity to attract CD4 + , CD40L + T cells by producing CCL22. Eur J Immunol 32(5):1403–13. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y
47. Isaza-Correa JM, Liang Z, Van Den Berg A, Diepstra A, Visser L. Toll-like receptors in the pathogenesis of human b cell malignancies. J Hematol Oncol (2014) 7(1):57. doi: 10.1186/s13045-014-0057-5
48. Wen Y, Jing Y, Yang L, Kang D, Jiang P, Li N, et al. The regulators of BCR signaling during b cell activation. Blood Sci (2019) 1(2):119–29. doi: 10.1097/BS9.0000000000000026
49. Corfe SA, Paige CJ. The many roles of IL-7 in b cell development; mediator of survival, proliferation and differentiation. Semin Immunol (2012) 24(3):198–208. doi: 10.1016/j.smim.2012.02.001
50. Crotty ST. Follicular helper cell biology: A decade of discovery and diseases. Immunity (2019) 50(5):1132–48. doi: 10.1016/j.immuni.2019.04.011
51. Jandl C, King C. Cytokines in the germinal center niche. Antibodies (2016) 5(1):5. doi: 10.3390/antib5010005
52. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia b cells from spontaneous apoptosis through stromal cell–derived factor-1. Blood (2000) 96(8):2655–63. doi: 10.1182/blood.V96.8.2655
53. Rodda LB, Bannard O, Ludewig B, Nagasawa T, Cyster JG. Phenotypic and morphological properties of germinal center dark zone Cxcl12-expressing reticular cells (CRCs). J Immunol (2015) 195(10):4781–91. doi: 10.4049/jimmunol.1501191
54. Park C-S, Yoon S-O, Armitage RJ, Choi YS. Follicular dendritic cells produce IL-15 that enhances germinal center b cell proliferation in membrane-bound form. J Immunol (2004) 173(11):6676–83. doi: 10.4049/jimmunol.173.11.6676
55. Kopf M, Herren S, Wiles MV, Pepys MB, Kosco-Vilbois MH. Interleukin 6 influences germinal center development and antibody production via a contribution of C3 complement component. J Exp Med (1998) 188(10):1895–906. doi: 10.1084/jem.188.10.1895
56. Nakayamada S, Tanaka Y. BAFF- and APRIL-targeted therapy in systemic autoimmune diseases. Inflammation Regener (2016) 36(1):1–6. doi: 10.1186/s41232-016-0015-4
57. Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 requirement for human plasma cell generation: Coupling differentiation and proliferation by enhancing MAPK–ERK signaling. J Immunol (2012) 189(1):161–73. doi: 10.4049/jimmunol.1200301
58. Urashima M, Chauhan D, Uchiyama H, Freeman GJ, Anderson KC. CD40 ligand triggered interleukin-6 secretion in multiple myeloma. Blood (1995) 85(7):1903–12. doi: 10.1182/blood.V85.7.1903.bloodjournal8571903
59. Banchereau J, De Paoli P, Vallé A, Garcia E, Rousset F. Long-term human b cell lines dependent on interleukin-4 and antibody to CD40. Science (1991) 251(4989):70–2. doi: 10.1126/science.1702555
60. Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol (2004) 173(2):1179–83. doi: 10.4049/jimmunol.173.2.1179
61. Fluckiger AC, Rossi JF, Bussel A, Bryon P, Banchereau J, Defrance T. Responsiveness of chronic lymphocytic leukemia b cells activated Via surface igs or CD40 to b-cell tropic factors. Blood (1992) 80(12):3173–81. doi: 10.1182/blood.V80.12.3173.3173
62. Ticchioni M, Essafi M, Jeandel PY, Davi F, Cassuto JP, Deckert M, et al. Homeostatic chemokines increase survival of b-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene (2007) 26(50):7081–91. doi: 10.1038/sj.onc.1210519
63. Pascutti MF, Jak M, Tromp JM, Derks IAM, Remmerswaal EBM, Thijssen R, et al. IL-21 and CD40L signals from autologous T cells can induce antigen-independent proliferation of CLL cells. Blood (2013) 122(17):3010–9. doi: 10.1182/blood-2012-11-467670
64. Gutierrez A, Arendt BK, Tschumper RC, Kay NE, Zent CS, Jelinek DF. Differentiation of chronic lymphocytic leukemia b cells into immunoglobulin secreting cells decreases LEF-1 expression. PloS One (2011) 6(10):e26056. doi: 10.1371/journal.pone.0026056
65. Jahrsdorfer B, Mühlenhoff L, Blackwell SE, Wagner M, Poeck H, Hartmann E, et al. B-cell lymphomas differ in their responsiveness to CpG oligodeoxynucleotides. Clin Cancer Res (2005) 11(4):1490–9. doi: 10.1158/1078-0432.CCR-04-1890
66. Fonte E, Agathangelidis A, Reverberi D, Ntoufa S, Scarfò L, Ranghetti P, et al. Toll-like receptor stimulation in splenic marginal zone lymphoma can modulate cell signaling, activation and proliferation. Haematologica (2015) 100(11):1460–8. doi: 10.3324/haematol.2014.119933
67. Epron G, Ame-Thomas P, Le Priol J, Pangault C, Dulong J, Lamy T, et al. Monocytes and T cells cooperate to favor normal and follicular lymphoma b-cell growth: role of IL-15 and CD40L signaling. Leukemia (2012) 26(1):139–48. doi: 10.1038/leu.2011.179
68. Ferretti E, Tripodo C, Pagnan G, Guarnotta C, Marimpietri D, Corrias MV, et al. The interleukin (IL)-31/IL-31R axis contributes to tumor growth in human follicular lymphoma. Leuk 2015 (2014) 29(4):958–67. doi: 10.1038/leu.2014.291
69. Fluckiger AC, Garrone P, Durand I, Galizzi JP, Banchereau J. Interleukin 10 (IL-10) upregulates functional high affinity IL-2 receptors on normal and leukemic b lymphocytes. J Exp Med (1993) 178(5):1473–81. doi: 10.1084/jem.178.5.1473
70. Travert M, Ame-Thomas P, Pangault C, Morizot A, Micheau O, Semana G, et al. CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-κB activation and up-regulation of c-FLIP and bcl-xL. J Immunol (2008) 181(2):1001–11. doi: 10.4049/jimmunol.181.2.1001
71. Ghia P, Boussiotis VA, Schultze JL, Cardoso AA, Dorfman DM, Gribben JG, et al. Unbalanced expression of bcl-2 family proteins in follicular lymphoma: Contribution of CD40 signaling in promoting survival. Blood (1998) 91(1):244–51. doi: 10.1182/blood.V91.1.244
72. Kater AP, Evers LM, Remmerswaal EBM, Jaspers A, Oosterwijk MF, Van Lier RAW, et al. CD40 stimulation of b-cell chronic lymphocytic leukaemia cells enhances the anti-apoptotic profile, but also bid expression and cells remain susceptible to autologous cytotoxic T-lymphocyte attack. Br J Haematol (2004) 127(4):404–15. doi: 10.1111/j.1365-2141.2004.05225.x
73. Hewamana S, Lin TT, Jenkins C, Burnett AK, Jordan CT, Fegan C, et al. The novel nuclear factor-κB inhibitor LC-1 is equipotent in poor prognostic subsets of chronic lymphocytic leukemia and shows strong synergy with fludarabine. Clin Cancer Res (2008) 14(24):8102–11. doi: 10.1158/1078-0432.CCR-08-1673
74. Coscia M, Pantaleoni F, Riganti C, Vitale C, Rigoni M, Peola S, et al. IGHV unmutated CLL b cells are more prone to spontaneous apoptosis and subject to environmental prosurvival signals than mutated CLL b cells. Leuk (2011) 25(5):828–37. doi: 10.1038/leu.2011.12
75. Kitada S, Zapata JM, Andreff M, Reed JC. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in b-cell chronic lymphocytic leukaemia. Br J Haematol (1999) 106(4):995–1004. doi: 10.1046/j.1365-2141.1999.01642.x
76. Clodi K, Snell V, Zhao S, Cabanillas F, Andreeff M, Younes A. Unbalanced expression of fas and CD40 in mantle cell lymphoma. Br J Haematol (1998) 103(1):217–9. doi: 10.1046/j.1365-2141.1998.00958.x
77. Decker T, Schneller F, Sparwasser T, Tretter T, Lipford GB, Wagner H, et al. Immunostimulatory CpG-oligonucleotides cause proliferation, cytokine production, and an immunogenic phenotype in chronic lymphocytic leukemia b cells. Blood (2000) 95(3):999–1006. doi: 10.1182/blood.V95.3.999.003k10_999_1006
78. Castillo R, Mascarenhas J, Telford W, Chadburn A, Friedman SM, Schattner EJ. Proliferative response of mantle cell lymphoma cells stimulated by CD40 ligation and IL-4. Leuk (2000) 14(2):292–8. doi: 10.1038/sj.leu.2401664
79. Dancescu M, Rubio-Trujillo M, Biron G, Bron D, Delespesse G, Sarfati M. Interleukin 4 protects chronic lymphocytic leukemic b cells from death by apoptosis and upregulates bcl-2 expression. J Exp Med (1992) 176(5):1319–26. doi: 10.1084/jem.176.5.1319
80. Ghamlouch H, Ouled-Haddou H, Damaj G, Royer B, Gubler B, Marolleau JP. A combination of cytokines rescues highly purified leukemic CLL b-cells from spontaneous apoptosis In vitro. PloS One (2013) 8(3):e60370. doi: 10.1371/journal.pone.0060370
81. Barragán M, Bellosillo B, Campàs C, Colomer D, Pons G, Gil J. Involvement of protein kinase c and phosphatidylinositol 3–kinase pathways in the survival of b-cell chronic lymphocytic leukemia cells. Blood (2002) 99(8):2969–76. doi: 10.1182/blood.V99.8.2969
82. Mainou-Fowler T, Craig V, Copplestone J, Hamon M, Prentice A. Interleukin-5 (IL-5) increases spontaneous apoptosis of b-cell chronic lymphocytic leukemia cells In vitro independently of bcl-2 expression, and is inhibited by IL-4. Blood (1994) 84(7):2297–304. doi: 10.1182/blood.V84.7.2297.2297
83. Chaouchi N, Wallon C, Goujard C, Tertian G, Rudent A, Caput D, et al. Interleukin-13 inhibits Interleukin-2–induced proliferation and protects chronic lymphocytic leukemia b cells from In vitro apoptosis. Blood (1996) 87(3):1022–9. doi: 10.1182/blood.V87.3.1022.bloodjournal8731022
84. Panayiotidis P, Ganeshaguru K, Jabbar SAB, Hoffbrand AV. Interleukin-4 inhibits apoptotic cell death and loss of the bcl-2 protein in b-chronic lymphocytic leukaemia cells in vitro. Br J Haematol (1993) 85(3):439–45. doi: 10.1111/j.1365-2141.1993.tb03330.x
85. Cols M, Barra CM, He B, Puga I, Xu W, Chiu A, et al. Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the TNF-related factors BAFF, APRIL and CD40L. J Immunol (2012) 188(12):6071–83. doi: 10.4049/jimmunol.1102066
86. Schleiss C, Ilias W, Tahar O, Güler Y, Miguet L, Mayeur-Rousse C, et al. BCR-associated factors driving chronic lymphocytic leukemia cells proliferation ex vivo. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-018-36853-8
87. Briones J, Timmerman JM, Hilbert DM, Levy R. BLyS and BLyS receptor expression in non-hodgkin’s lymphoma. Exp Hematol (2002) 30(2):135–41. doi: 10.1016/S0301-472X(01)00774-3
88. Crawford DH, Catovsky D. In vitro activation of leukaemic b cells by interleukin-4 and antibodies to CD40. Immunology (1993) 80(1):40–4.
89. Johnson PWM, Watt SM, Betts DR, Davies D, Jordan S, Norton AJ, et al. Isolated follicular lymphoma cells are resistant to apoptosis and can be grown In vitro in the CD40/Stromal cell system. Blood (1993) 82(6):1848–57. doi: 10.1182/blood.V82.6.1848.1848
90. Willimott S, Baou M, Huf S, Wagner SD. Separate cell culture conditions to promote proliferation or quiescent cell survival in chronic lymphocytic leukemia. Leuk Lymphoma (2007) 48(8):1647–50. doi: 10.1080/10428190701447353
91. Schmitter D, Koss MV, Niederer E, Stahel R, Pichert G. T-Cell derived cytokines co-stimulate proliferation of CD40-activated germinal centre as well as follicular lymphoma cells. Hematol Oncol (1997) 15(4):197–207. doi: 10.1002/(SICI)1099-1069(199711)15:4<197::AID-HON614>3.0.CO;2-V
92. Guidoboni M, Zancai P, Cariati R, Rizzo S, Dal Col J, Pavan A, et al. Retinoic acid inhibits the proliferative response induced by CD40 activation and interleukin-4 in mantle cell lymphoma. Cancer Res (2005) 65(2):587–95. doi: 10.1158/0008-5472.587.65.2
93. Ahearne MJ, Willimott S, Piñon L, Kennedy DB, Miall F, Dyer MJS, et al. Enhancement of CD154/IL4 proliferation by the T follicular helper (Tfh) cytokine, IL21 and increased numbers of circulating cells resembling tfh cells in chronic lymphocytic leukaemia. Br J Haematol (2013) 162(3):360–70. doi: 10.1111/bjh.12401
94. Karray S, Defrance T, Merle-Beral H, Banchereau J, Debré P, Galanaud P. Interleukin 4 counteracts the interleukin 2-induced proliferation of monoclonal b cells. J Exp Med (1988) 168(1):85–94. doi: 10.1084/jem.168.1.85
95. Visser HPJ, Tewis M, Willemze R, Kluin-Nelemans JC. Mantle cell lymphoma proliferates upon IL-10 in the CD40 system. Leuk (2000) 14(8):1483–9. doi: 10.1038/sj.leu.2401829
96. Trentin L, Cerutti A, Zambello R, Sancetta R, Tassinari C, Facco M, et al. Interleukin-15 promotes the growth of leukemic cells of patients with b-cell chronic lymphoproliferative disorders. Blood (1996) 87(8):3327–35. doi: 10.1182/blood.V87.8.3327.bloodjournal8783327
97. Digel W, Schmid M, Heil G, Conrad P, Gillis S, Porzsolt F. Human interleukin-7 induces proliferation of neoplastic cells from chronic lymphocytic leukemia and acute leukemias. Blood (1991) 78(3):753–9. doi: 10.1182/blood.V78.3.753.753
98. Decker T, Bogner C, Oelsner M, Peschel C, Ringshausen I. Antiapoptotic effect of interleukin-2 (IL-2) in b-CLL cells with low and high affinity IL-2 receptors. Ann Hematol. [Internet] (2010) 89(11):1125–32. doi: 10.1007/s00277-010-0994-1
99. Decker T, Schneller F, Hipp S, Miething C, Jahn T, Duyster J, et al. Cell cycle progression of chronic lymphocytic leukemia cells is controlled by cyclin D2, cyclin D3, cyclin-dependent kinase (cdk) 4 and the cdk inhibitor p27. Leuk (2002) 16(3):327–34. doi: 10.1038/sj.leu.2402389
100. Mongini PKA, Gupta R, Boyle E, Nieto J, Lee H, Stein J, et al. TLR-9 and IL-15 synergy promotes the In vitro clonal expansion of chronic lymphocytic leukemia b cells. J Immunol (2015) 195(3):901–23. doi: 10.4049/jimmunol.1403189
101. Gupta R, Yan XJ, Barrientos J, Kolitz JE, Allen SL, Rai K, et al. Mechanistic insights into CpG DNA and IL-15 synergy in promoting b cell chronic lymphocytic leukemia clonal expansion. J Immunol (2018) 201(5):1570–85. doi: 10.4049/jimmunol.1800591
102. Lantz O, Grillot-Courvalin C, Schmitt C, Fermand JP, Brouet JC. Interleukin 2-induced proliferation of leukemic human b cells. J Exp Med (1985) 161(5):1225–30. doi: 10.1084/jem.161.5.1225
103. De Totero D, Meazza R, Capaia M, Fabbi M, Azzarone B, Balleari E, et al. The opposite effects of IL-15 and IL-21 on CLL b cells correlate with differential activation of the JAK/STAT and ERK1/2 pathways. undefined (2008) 111(2):517–24. doi: 10.1182/blood-2007-04-087882
104. Novak AJ, Bram RJ, Kay NE, Jelinek DF. Aberrant expression of b-lymphocyte stimulator by b chronic lymphocytic leukemia cells: a mechanism for survival. Blood (2002) 100(8):2973–9. doi: 10.1182/blood-2002-02-0558
105. Endo T, Nishio M, Enzler T, Cottam HB, Fukuda T, James DF, et al. BAFF and APRIL support chronic lymphocytic leukemia b-cell survival through activation of the canonical NF-kappaB pathway. Blood (2007) 109(2):703–10. doi: 10.1182/blood-2006-06-027755
106. Kern C, Cornuel JF, Billard C, Tang R, Rouillard D, Stenou V, et al. Involvement of BAFF and APRIL in the resistance to apoptosis of b-CLL through an autocrine pathway. Blood (2004) 103(2):679–88. doi: 10.1182/blood-2003-02-0540
107. Nishio M, Endo T, Tsukada N, Ohata J, Kitada S, Reed JC, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1α. Blood (2005) 106(3):1012–20. doi: 10.1182/blood-2004-03-0889
108. He B, Chadburn A, Jou E, Schattner EJ, Knowles DM, Cerutti A. Lymphoma b cells evade apoptosis through the TNF family members BAFF/BLyS and APRIL. J Immunol (2004) 172(5):3268–79. doi: 10.4049/jimmunol.172.5.3268
109. Medina DJ, Goodell L, Glod J, Gélinas C, Rabson AB, Strair RK. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of b-cell activating factor and activation of the canonical and non-canonical nuclear factor κB pathways. Haematologica (2012) 97(8):1255–63. doi: 10.3324/haematol.2011.040659
110. Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood (2004) 103(8):3148–57. doi: 10.1182/blood-2003-06-1984
111. Burgess M, Cheung C, Chambers L, Ravindranath K, Minhas G, Knop L, et al. CCL2 and CXCL2 enhance survival of primary chronic lymphocytic leukemia cells in vitro. Leuk Lymphoma (2012) 53(10):1988–98. doi: 10.3109/10428194.2012.672735
112. Francia Di Celle P, Mariani S, Riera L, Stacchini A, Reato G, Foa R. Intereukin-8 induces the accumulation of b-cell chronic lymphocytic leukemia cells by prolonging survival in an autocrine fashion. Blood (1996) 87(10):4382–9. doi: 10.1182/blood.V87.10.4382.bloodjournal87104382
113. Schillaci R, Galeano A, Becu-Villalobos D, Spinelli O, Sapia S, Bezares RF. Autocrine/paracrine involvement of insulin-like growth factor-I and its receptor in chronic lymphocytic leukaemia. Br J Haematol (2005) 130(1):58–66. doi: 10.1111/j.1365-2141.2005.05579.x
114. Gehrke I, Gandhirajan RK, Poll-Wolbeck SJ, Hallek M, Kreuzer KA. Bone marrow stromal cell-derived vascular endothelial growth factor (VEGF) rather than chronic lymphocytic leukemia (CLL) cell-derived VEGF is essential for the apoptotic resistance of cultured CLL cells. Mol. Med (2011) 17(7–8):619–27. doi: 10.2119/molmed.2010.00210
115. Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in b-cell chronic lymphocytic leukemia. Blood (2004) 104(3):788–94. doi: 10.1182/blood-2003-08-2763
116. Bauvois B, Pramil E, Jondreville L, Quiney C, Nguyen-Khac F, Susin SA. Activation of interferon signaling in chronic lymphocytic leukemia cells contributes to apoptosis resistance via a JAK-Src/STAT3/Mcl-1 signaling pathway. Biomedicines (2021) 9(2):1–15. doi: 10.3390/biomedicines9020188
117. Buschle M, Campana D, Carding SR, Richard C, Victor Hoffbrand A, Brenner MK. Interferon gamma inhibits apoptotic cell death in b cell chronic lymphocytic leukemia. J Exp Med (1993) 177(1):213–8. doi: 10.1084/jem.177.1.213
118. Dürr C, Hanna BS, Schulz A, Lucas F, Zucknick M, Benner A, et al. Tumor necrosis factor receptor signaling is a driver of chronic lymphocytic leukemia that can be therapeutically targeted by the flavonoid wogonin. Haematologica (2018) 103(4):688–97. doi: 10.3324/haematol.2017.177808
119. Digel W, Stefanic M, Schoniger W, Buck C, Raghavachar A, Frickhofen N, et al. Tumor necrosis factor induces proliferation of neoplastic b cells from chronic lymphocytic leukemia. Blood (1989) 73(5):1242–6. doi: 10.1182/blood.V73.5.1242.1242
120. Barut B, Chauhan D, Uchiyama H, Anderson KC. Interleukin-6 functions as an intracellular growth factor in hairy cell leukemia in vitro. J Clin Invest (1993) 92(5):2346–52. doi: 10.1172/JCI116839
121. Brenne AT, Ro TB, Waage A, Sundan A, Borset M, Hjorth-Hansen H. Interleukin-21 is a growth and survival factor for human myeloma cells. Blood (2002) 99(10):3756–62. doi: 10.1182/blood.V99.10.3756
122. Moreno A, Villar ML, Cámara C, Luque R, Cespón C, González-Porqué P, et al. Interleukin-6 dimers produced by endothelial cells inhibit apoptosis of b-chronic lymphocytic leukemia cells. Blood (2001) 97(1):242–9. doi: 10.1182/blood.V97.1.242
123. Shi X, Dornan D. To respond or not to respond to CD40 agonism: That is the prediction. Oncoimmunology (2012) 1(1):83–5. doi: 10.4161/onci.1.1.17827
124. Granziero L, Ghia P, Circosta P, Gottardi D, Strola G, Geuna M, et al. Survivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in b-cell chronic lymphocytic leukemia. Blood (2001) 97(9):2777–83. doi: 10.1182/blood.V97.9.2777
125. Deglesne PA, Chevallier N, Letestu R, Baran-Marszak F, Beitar T, Salanoubat C, et al. Survival response to b-cell receptor ligation is restricted to progressive chronic lymphocytic leukemia cells irrespective of Zap70 expression. Cancer Res (2006) 66(14):7158–66. doi: 10.1158/0008-5472.CAN-06-0085
126. Touw I, Dorssers L, Lowenberg B. The proliferative response of b cell chronic lymphocytic leukemia to interleukin 2: Functional characterization of the interleukin 2 membrane receptors. Blood (1987) 69(6):1667–73. doi: 10.1182/blood.V69.6.1667.1667
127. Hajjami HMEl, Amé-Thomas P, Pangault C, Tribut O, DeVos J, Jean R, et al. Functional alteration of the lymphoma stromal cell niche by the cytokine context: Role of indoleamine-2,3 dioxygenase. Cancer Res (2009) 69(7):3228–37. doi: 10.1158/0008-5472.CAN-08-3000
128. Hivroz C, Grillot-Courvalin C, Brouet J -C, Seligmann M. Heterogeneity of responsiveness of chronic lymphocytic leukemic b cells to b cell growth factor or interleukin 2. Eur J Immunol (1986) 16(8):1001–4. doi: 10.1002/eji.1830160821
129. Luo H, Rubio M, Biron G, Delespesse G, Sarfati M. Antiproliferative effect of interleukin-4 in b chronic lymphocytic leukemia. J Immunother (1991) 10(6):418–25. doi: 10.1097/00002371-199112000-00005
130. Lu X, Nechushtan H, Ding F, Rosado MF, Singal R, Alizadeh AA, et al. Distinct IL-4-induced gene expression, proliferation, and intracellular signaling in germinal center b-cell-like and activated b-cell-like diffuse large-cell lymphomas. Blood (2005) 105(7):2924–32. doi: 10.1182/blood-2004-10-3820
131. Kato H, Kagami Y, Nakagawa M, Karnan S, Yatabe Y, Nakamura S, et al. IL-4 / CD 40 l Co-stimulation induces long-term proliferation for CD 10-positive germinal center b cell-like diffuse Large b-cell lymphoma. The Open Leukemia Journal (2010) 3:60–8. doi: 10.2174/1876816401003010060
132. Wang Y, Wang C, Cai X, Mou C, Cui X, Zhang Y, et al. IL-21 stimulates the expression and activation of cell cycle regulators and promotes cell proliferation in EBV-positive diffuse large b cell lymphoma. Sci Rep (2020) 10(1):12326. doi: 10.1038/s41598-020-69227-0
133. Akamatsu N, Yamada Y, Hasegawa H, Makabe K, Asano R, Kumagai I, et al. High IL-21 receptor expression and apoptosis induction by IL-21 in follicular lymphoma. Cancer Lett (2007) 256(2):196–206. doi: 10.1016/j.canlet.2007.06.001
134. Damle RN, Temburni S, Banapour T, Paul S, Mongini PKA, Allen SL, et al. T-Cell independent, b-cell receptor-mediated induction of telomerase activity differs among IGHV mutation-based subgroups of chronic lymphocytic leukemia patients. Blood (2012) 120(12):2438–49. doi: 10.1182/blood-2012-02-409110
135. Plander M, Seegers S, Ugocsai P, Diermeier-Daucher S, Iványi J, Schmitz G, et al. Different proliferative and survival capacity of CLL-cells in a newly established in vitro model for pseudofollicles. Leuk (2009) 23(11):2118–28. doi: 10.1038/leu.2009.145
136. Wagner M, Oelsner M, Moore A, Götte F, Kuhn PH, Haferlach T, et al. Integration of innate into adaptive immune responses in ZAP-70–positive chronic lymphocytic leukemia. Blood (2016) 127(4):436–48. doi: 10.1182/blood-2015-05-646935
137. Ghalamfarsa G, Jadidi-Niaragh F, Hojjat-Farsangi M, Asgarian-Omran H, Yousefi M, Tahmasebi F, et al. Differential regulation of b-cell proliferation by IL21 in different subsets of chronic lymphocytic leukemia. Cytokine (2013) 62(3):439–45. doi: 10.1016/j.cyto.2013.03.023
138. Jahrsdörfer B, Wooldridge JE, Blackwell SE, Taylor CM, Griffith TS, Link BK, et al. Immunostimulatory oligodeoxynucleotides induce apoptosis of b cell chronic lymphocytic leukemia cells. J Leukoc Biol (2005) 77(3):378–87. doi: 10.1189/jlb.0604373
139. Tromp JM, Tonino SH, Elias JA, Jaspers A, Luijks DM, Kater AP, et al. Dichotomy in NF-κB signaling and chemoresistance in immunoglobulin variable heavy-chain-mutated versus unmutated CLL cells upon CD40/TLR9 triggering. Oncogene (2010) 29(36):5071–82. doi: 10.1038/onc.2010.248
140. Zupo S, Massara R, Dono M, Rossi E, Malavasi F, Cosulich ME, et al. Apoptosis or plasma cell differentiation of CD38-positive b-chronic lymphocytic leukemia cells induced by cross-linking of surface IgM or IgD. Blood (2000) 95(4):1199–206. doi: 10.1182/blood.V95.4.1199.004k21_1199_1206
141. Fluckiger AC, Durand I, Banchereau J. Interleukin 10 induces apoptotic cell death of b-chronic lymphocytic leukemia cells. J Exp Med (1994) 179(1):91–9. doi: 10.1084/jem.179.1.91
142. Gowda A, Roda J, Hussain SRA, Ramanunni A, Joshi T, Schmidt S, et al. IL-21 mediates apoptosis through up-regulation of the BH3 family member BIM and enhances both direct and antibody-dependent cellular cytotoxicity in primary chronic lymphocytic leukemia cells in vitro. Blood (2008) 111(9):4723–30. doi: 10.1182/blood-2007-07-099531
143. De Totero D, Meazza R, Zupo S, Cutrona G, Matis S, Colombo M, et al. Interleukin-21 receptor (IL-21R) is up-regulated by CD40 triggering and mediates proapoptotic signals in chronic lymphocytic leukemia b cells. Blood (2006) 107(9):3708–15. doi: 10.1182/blood-2005-09-3535
144. Zupo S, Cutrona G, Mangiola M, Ferrarini M, Schattner E, Bernal A. Role of surface IgM and IgD on survival of the cells from b-cell chronic lymphocytic leukemia. Blood (2002) 99(6):2277–8. doi: 10.1182/blood-2001-11-0126
145. Néron S, Nadeau PJ, Darveau A, Leblanc JF. Tuning of CD40-CD154 interactions in human b-lymphocyte activation: A broad array of in vitro models for a complex in vivo situation. Arch Immunol Ther Exp (Warsz) (2011) 59(1):25–40. doi: 10.1007/s00005-010-0108-8
146. Delgado R, Kielbassa K, Ter Burg J, Klein C, Trumpfheller C, de Heer K, et al. Co-Stimulatory versus cell death aspects of agonistic cd40 monoclonal antibody selicrelumab in chronic lymphocytic leukemia. Cancers (Basel) (2021) 13(12):3084. doi: 10.3390/cancers13123084
147. Rombout A, Lust S, Offner F, Naessens E, Verhasselt B, Philippé J. Mimicking the tumour microenvironment of chronic lymphocytic leukaemia in vitro critically depends on the type of b-cell receptor stimulation. Br J Cancer (2016) 114(6):704–12. doi: 10.1038/bjc.2016.35
148. Buske C, Twiling A, Gogowski G, Schreiber K, Feuring-Buske M, Wulf CG, et al. In vitro activation of low-grade non-hodgkin’s lymphoma by murine fibroblasts, IL-4, anti-CD40 antibodies and the soluble CD40 ligand. Leuk (1997) 11(11):1862–7. doi: 10.1038/sj.leu.2400822
149. Patten PEM, Ferrer G, Chen S-S, Simone R, Marsilio S, Yan X-J, et al. Chronic lymphocytic leukemia cells diversify and differentiate in vivo via a nonclassical Th1-dependent, Bcl-6–deficient process. JCI Insight (2016) 1(4):e86288. doi: 10.1172/jci.insight.86288
150. Malisan F, Fluckiger AC, Ho S, Guret C, Banchereau J, Martinez-Valdez H. B-chronic lymphocytic leukemias can undergo isotype switching In vivo and can be induced to differentiate and switch In vitro. Blood (1996) 87(2):717–24. doi: 10.1182/blood.V87.2.717.bloodjournal872717
151. Tretter T, Schuler M, Schneller F, Brass U, Esswein M, Aman MJ, et al. Direct cellular interaction with activated CD4+T cells overcomes hyporesponsiveness of b-cell chronic lymphocytic leukemiain vitro. Cell Immunol (1998) 189(1):41–50. doi: 10.1006/cimm.1998.1360
152. Sarfati M, Luo H, Delespesse G. IgE synthesis by chronic lymphocytic leukemia cells. J Exp Med (1989) 170(5):1775–80. doi: 10.1084/jem.170.5.1775
153. Duckworth A, Glenn M, Slupsky JR, Packham G, Kalakonda N. Variable induction of PRDM1 and differentiation in chronic lymphocytic leukemia is associated with anergy. Blood (2014) 123(21):3277–85. doi: 10.1182/blood-2013-11-539049
154. Cossman J, Neckers LM, Braziel RM, Trepel JB, Korsmeyer SJ, Bakhshi A. In vitro enhancement of immunoglobulin gene expression in chronic lymphocytic leukemia. J Clin Invest (1984) 73(2):587–92. doi: 10.1172/JCI111247
155. Carlsson M, Totterman T, Matsson P, Nilsson K. Cell cycle progression of b-chronic lymphocytic leukemia cells induced to differentiate by TPA. Blood (1988) 71(2):415–21. doi: 10.1182/blood.V71.2.415.415
156. TANGYE SG, WESTON KM, RAISON RL. Phorbol ester activates CD5+ leukaemic b cells via a T cell-independent mechanism. Immunol Cell Biol (1995) 73(1):44–51. doi: 10.1038/icb.1995.7
157. Kabelitz D, Pfeffer K, von Steldern D, Bartmann P, Brudler O, Nerl C, et al. In vitro maturation of b cells in chronic lymphocytic leukemia. i. synergistic action of phorbol ester and interleukin 2 in the induction of tac antigen expression and interleukin 2 responsiveness in leukemic b cells. J Immunol (1985) 135(4):2876–81.
158. van Kooten C, Rensink I, Aarden L, van Oers R. Effect of IL-4 and IL-6 on the proliferation and differentiation of b-chronic lymphocytic leukemia cells. Leukemia (1993) 7(4):618–24
159. Ghamlouch H, Ouled-Haddou H, Guyart A, Regnier A, Trudel S, Claisse JF, et al. Phorbol myristate acetate, but not CD40L, induces the differentiation of CLL b cells into ab-secreting cells. Immunol Cell Biol (2014) 92(7):591–604. doi: 10.1038/icb.2014.37
160. Ghamlouch H, Ouled-Haddou H, Guyart A, Regnier A, Trudel S, Claisse JF, et al. TLR9 ligand (CpG oligodeoxynucleotide) induces CLL b-cells to differentiate into CD20+ antibody-secreting cells. Front Immunol (2014) 5:292. doi: 10.3389/fimmu.2014.00292
161. Ghamlouch H, Darwiche W, Hodroge A, Ouled-Haddou H, Dupont S, Singh AR, et al. Factors involved in CLL pathogenesis and cell survival are disrupted by differentiation of CLL b-cells into antibody-secreting cells. Oncotarget (2015) 6(21):18484–503. doi: 10.18632/oncotarget.3941
162. Degos L, Wang ZY. All trans retinoic acid in acute promyelocytic leukemia. Oncogene (2001) 20(49):7140–5. doi: 10.1038/sj.onc.1204763
163. Jak M, Van Bochove GGW, Van Lier RAW, Eldering E, Van Oers MHJ. CD40 stimulation sensitizes CLL cells to rituximab-induced cell death. Leuk (2011) 25(6):968–78. doi: 10.1038/leu.2011.39
164. Hallaert DYH, Jaspers A, Van Noesel CJ, Van Oers MHJ, Kater AP, Eldering E. C-abl kinase inhibitors overcome CD40-mediated drug resistance in CLL: implications for therapeutic targeting of chemoresistant niches. Blood (2008) 112(13):5141–9. doi: 10.1182/blood-2008-03-146704
165. Lu K, Fang XS, Feng LL, Jiang YJ, Zhou XX, Liu X, et al. The STAT3 inhibitor WP1066 reverses the resistance of chronic lymphocytic leukemia cells to histone deacetylase inhibitors induced by interleukin-6. Cancer Lett (2015) 359(2):250–8. doi: 10.1016/j.canlet.2015.01.021
166. Mankaï A, Buhé V, Hammadi M, Youinou P, Ghedira I, Berthou C, et al. Improvement of rituximab efficiency in chronic lymphocytic leukemia by CpG-mediated upregulation of CD20 expression independently of PU.1. Ann N Y Acad Sci (2009) 1173(1):721–8. doi: 10.1111/j.1749-6632.2009.04614.x
167. Schulz A, Toedt G, Zenz T, Stilgenbauer S, Lichter P, Seiffert M. Inflammatory cytokines and signaling pathways are associated with survival of primary chronic lymphocytic leukemia cells in vitro: a dominant role of CCL2. Haematologica (2011) 96(3):408–16. doi: 10.3324/haematol.2010.031377
168. Levesque MC, O’Loughlin CW, Weinberg JB. Use of serum-free media to minimize apoptosis of chronic lymphocytic leukemia cells during in vitro culture. Leuk (2001) 15(8):1305–7. doi: 10.1038/sj.leu.2402192
169. Zent CS, Chen JB, Kurten RC, Kaushal GP, Lacy HM, Schichman SA. Alemtuzumab (CAMPATH 1H) does not kill chronic lymphocytic leukemia cells in serum free medium. Leuk Res (2004) 28(5):495–507. doi: 10.1016/j.leukres.2003.09.011
171. Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukemia. Nat Cell Biol (2012) 14(3):276–86. doi: 10.1038/ncb2432
172. Lv X, Feng L, Ge X, Lu K, Wang X. Interleukin-9 promotes cell survival and drug resistance in diffuse large b-cell lymphoma. J Exp Clin Cancer Res (2016) 35(1):1–9. doi: 10.1186/s13046-016-0374-3
173. Zhong W, Xu X, Zhu Z, Yang L, Du H, Xia Z, et al. Increased interleukin-17A levels promote rituximab resistance by suppressing p53 expression and predict an unfavorable prognosis in patients with diffuse large b cell lymphoma. Int J Oncol (2018) 52(5):1528–38. doi: 10.3892/ijo.2018.4299
174. Gelebart P, Zak Z, Dien-Bard J, Anand M, Lai R. Interleukin 22 signaling promotes cell growth in mantle cell lymphoma. Transl Oncol (2011) 4(1):9–19. doi: 10.1593/tlo.10172
175. Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic b cells but not normal b cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood (1998) 91(7):2387–96. doi: 10.1182/blood.V91.7.2387
176. Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand AV. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol (1996) 92(1):97–103. doi: 10.1046/j.1365-2141.1996.00305.x
177. Amé-Thomas P, Hajjami HMEl, Monvoisin C, Jean R, Monnier D, Caulet-Maugendre S, et al. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor b-cell growth: role of stromal cells in follicular lymphoma pathogenesis. Blood (2007) 109(2):693–702. doi: 10.1182/blood-2006-05-020800
178. Zlei M, Egert S, Wider D, Ihorst G, Wäsch R, Engelhardt M. Characterization of in vitro growth of multiple myeloma cells. Exp Hematol (2007) 35(10):1550–61. doi: 10.1016/j.exphem.2007.06.016
179. Jakubikova J, Cholujova D, Hideshima T, Gronesova P, Soltysova A, Harada T, et al. A novel 3D mesenchymal stem cell model of the multiple myeloma bone marrow niche: biologic and clinical applications. Oncotarget (2016) 7(47):77326–41. doi: 10.18632/oncotarget.12643
180. Trimarco V, Ave E, Facco M, Chiodin G, Frezzato F, Martini V, et al. Cross-talk between chronic lymphocytic leukemia (CLL) tumor b cells and mesenchymal stromal cells (MSCs): implications for neoplastic cell survival. Oncotarget (2015) 6(39):42130–49. doi: 10.18632/oncotarget.6239
181. Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood (2009) 114(20):4441–50. doi: 10.1182/blood-2009-07-233718
182. Guilloton F, Caron G, Ménard C, Pangault C, Amé-Thomas P, Dulong J, et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2-dependent recruitment and polarization of monocytes. Blood (2012) 119(11):2556–67. doi: 10.1182/blood-2011-08-370908
183. Mohammad IS, Lin K, Oates M, Khan UT, Burger J, Pettitt AR, et al. Development of a cell-line model to mimic the pro-survival effect of nurse-like cells in chronic lymphocytic leukemia. Leuk Lymphoma (2021) 62(1):45–57. doi: 10.1080/10428194.2020.1811274
184. Boissard F, Fournié JJ, Quillet-Mary A, Ysebaert L, Poupot M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J (2015) 5(10):e355. doi: 10.1038/bcj.2015.74
185. Mueller CG, Boix C, Kwan W-H, Daussy C, Fournier E, Fridman WH, et al. Critical role of monocytes to support normal b cell and diffuse large b cell lymphoma survival and proliferation. J Leukoc Biol (2007) 82(3):567–75. doi: 10.1189/jlb.0706481
186. Abe M, Hiura K, Wilde J, Shioyasono A, Moriyama K, Hashimoto T, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood (2004) 104(8):2484–91. doi: 10.1182/blood-2003-11-3839
187. Yaccoby S, Wezeman MJ, Henderson A, Cottler-Fox M, Yi Q, Barlogie B, et al. Cancer and the microenvironment. Cancer Res (2004) 64(6):2016–23. doi: 10.1158/0008-5472.CAN-03-1131
188. Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, et al. Protection of CLL b cells by a follicular dendritic cell line is dependent on induction of mcl-1. Blood (2002) 100(5):1795–801. doi: 10.1182/blood.V100.5.1795.h81702001795_1795_1801
189. Buggins AGS, Pepper C, Patten PEM, Hewamana S, Gohil S, Moorhead J, et al. Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF-κB activation and de novo gene transcription. Cancer Res (2010) 70(19):7523–33. doi: 10.1158/0008-5472.CAN-10-1634
190. Hamilton E, Pearce L, Morgan L, Robinson S, Ware V, Brennan P, et al. Mimicking the tumour microenvironment: three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br J Haematol (2012) 158(5):589–99. doi: 10.1111/j.1365-2141.2012.09191.x
191. Hacken ET, Valentin R, Regis FFD, Sun J, Yin S, Werner L, et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight (2018) 3(19):e121438. doi: 10.1172/jci.insight.121438
192. Purroy N, Abrisqueta P, Carabia J, Carpio C, Palacio C, Bosch F, et al. Co-Culture of primary CLL cells with bone marrow mesenchymal cells, CD40 ligand and CpG ODN promotes proliferation of chemoresistant CLL cells phenotypically comparable to those proliferating in vivo. Oncotarget (2015) 6(10):7632–43. doi: 10.18632/oncotarget.2939
193. Sivina M, Kreitman RJ, Peled A, Ravandi F, Burger JA. Adhesion of hairy cells leukemia (HCL) cells to stromal cells can be inhibited by blocking VLA-4 integrins and CXCR4 chemokine receptors. Blood (2011) 118(21):1760. doi: 10.1182/blood.V118.21.1760.1760
194. Domagala M, Ysebaert L, Ligat L, Lopez F, Fournié JJ, Laurent C, et al. IL-10 rescues CLL survival through repolarization of inflammatory nurse-like cells. Cancers (Basel) (2022) 14(1):16. doi: 10.3390/cancers14010016
195. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia b cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood (1999) 94(11):3658–67. doi: 10.1182/blood.V94.11.3658
196. Medina DJ, Abass-Shereef J, Walton K, Goodell L, Aviv H, Strair RK, et al. Cobblestone-area forming cells derived from patients with mantle cell lymphoma are enriched for CD133+ tumor-initiating cells. PloS One (2014) 9(4):e91042. doi: 10.1371/journal.pone.0091042
197. Kowalczyk-Quintas C, Chevalley D, Willen L, Jandus C, Vigolo M, Schneider P. Inhibition of membrane-bound BAFF by the anti-BAFF antibody belimumab. Front Immunol (2018) 9:2698. doi: 10.3389/fimmu.2018.02698
198. Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, Lolis E. Structural and functional basis of CXCL12 (Stromal cell-derived factor-1α) binding to heparin. J Biol Chem (2007) 282(13):10018–27. doi: 10.1074/jbc.M608796200
199. Pettirossi V, Santi A, Imperi E, Russo G, Pucciarini A, Bigerna B, et al. Lymphoid neoplasia: BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood (2015) 125(8):1207–16. doi: 10.1182/blood-2014-10-603100
200. Almenario FM, Asi RJL, Jacinto SD, Mazahery ARF. Reducing mitomycin-c-induced ROS levels in mouse feeder cells improves induced pluripotent stem cell colony growth. Biotechniques (2020) 68(5):270–4. doi: 10.2144/btn-2019-0118
201. Kwon YK, Ha IJ, Bae HW, Jang WG, Yun HJ, Kim SR, et al. Dose-dependent metabolic alterations in human cells exposed to gamma irradiation. PloS One (2014) 9(11):e113573. doi: 10.1371/journal.pone.0113573
202. Ponchio L, Duma L, Oliviero B, Gibelli N, Pedrazzoli P, Della Cuna GR. Mitomycin c as an alternative to irradiation to inhibit the feeder layer growth in long-term culture assays. Cytotherapy (2000) 2(4):281–6. doi: 10.1080/146532400539215
203. Decaup E, Jean C, Laurent C, Gravelle P, Fruchon S, Capilla F, et al. Anti-tumor activity of obinutuzumab and rituximab in a follicular lymphoma 3D model. Blood Cancer J (2013) 3(8):e131. doi: 10.1038/bcj.2013.32
204. Lamaison C, Latour S, Hélaine N, Le Morvan V, Saint-Vanne J, Mahouche I, et al. A novel 3D culture model recapitulates primary FL b-cell features and promotes their survival. Blood Adv (2021) 5(23):5372–86. doi: 10.1182/bloodadvances.2020003949
205. Decaup E, Rossi C, Gravelle P, Laurent C, Bordenave J, Tosolini M, et al. A tridimensional model for NK cell-mediated ADCC of follicular lymphoma. Front Immunol (2019) 10:1943. doi: 10.3389/fimmu.2019.01943
206. Law AMK, Rodriguez de la Fuente L, Grundy TJ, Fang G, Valdes-Mora F, Gallego-Ortega D. Advancements in 3D cell culture systems for personalizing anti-cancer therapies. Front Oncol (2021) 11:782766. doi: 10.3389/fonc.2021.782766
207. Reagan MR, Mishima Y, Glavey SV, Zhang Y, Manier S, Lu ZN, et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood (2014) 124(22):3250–9. doi: 10.1182/blood-2014-02-558007
208. Shanti A, Hallfors N, Petroianu GA, Planelles L, Stefanini C. Lymph nodes-On-Chip: Promising immune platforms for pharmacological and toxicological applications. Front Pharmacol (2021) 12:711307. doi: 10.3389/fphar.2021.711307
209. Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat BioMed Eng (2020) 4(4):394–406. doi: 10.1038/s41551-019-0495-z
210. Waldschmidt JM, Fruttiger SJ, Wider D, Jung J, Thomsen AR, Hartmann TN, et al. Ex vivo propagation in a novel 3D high-throughput co-culture system for multiple myeloma. J Cancer Res Clin Oncol (2022) 148(5):1045–55. doi: 10.1007/s00432-021-03854-6
211. Hervás-Salcedo R, Martín-Antonio B. A journey through the inter-cellular interactions in the bone marrow in multiple myeloma: Implications for the next generation of treatments. Cancers (2022) 14(15):3796. doi: 10.3390/cancers14153796
212. Amer M, Shi L, Wolfenson H. The ‘Yin and yang’ of cancer cell growth and mechanosensing. Cancers (Basel) (2021) 13(19):4754. doi: 10.3390/cancers13194754
213. Scielzo C, Ghia P. Modeling the leukemia microenviroment in vitro. Front Oncol (2020) 10:607608. doi: 10.3389/fonc.2020.607608
214. Mancini SJC, Balabanian K, Corre I, Gavard J, Lazennec G, Le Bousse-Kerdilès MC, et al. Deciphering tumor niches: Lessons from solid and hematological malignancies. Front Immunol (2021) 12:766275. doi: 10.3389/fimmu.2021.766275
215. Calvi LM, Link DC. Cellular complexity of the bone marrow hematopoietic stem cell niche. Calcif Tissue Int (2014) 94(1):112–24. doi: 10.1007/s00223-013-9805-8
216. Xu H, Lyu X, Yi M, Zhao W, Song Y, Wu K. Organoid technology and applications in cancer research. J Hematol Oncol (2018) 11(1):1–15. doi: 10.1186/s13045-018-0662-9
217. Ghosh N, Matsui W. Cancer stem cells in multiple myeloma. Cancer Lett (2009) 277(1):1–7. doi: 10.1016/j.canlet.2008.08.005
218. Kirshner J, Thulien KJ, Martin LD, Marun CD, Reiman T, Belch AR, et al. A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood (2008) 112(7):2935–45. doi: 10.1182/blood-2008-02-142430
219. Guo B, Zhang L, Chiorazzi N, Rothstein TL. IL-4 rescues surface IgM expression in chronic lymphocytic leukemia. Blood (2016) 128(4):553–62. doi: 10.1182/blood-2015-11-682997
220. Efremov DG. IL-4 regulates b-cell receptor signaling in CLL. Blood (2016) 128(4):468–9. doi: 10.1182/blood-2016-06-719872
221. Walsby E, Buggins A, Devereux S, Jones C, Pratt G, Brennan P, et al. Development and characterization of a physiologically relevant model of lymphocyte migration in chronic lymphocytic leukemia. Blood (2014) 123(23):3607–17. doi: 10.1182/blood-2013-12-544569
222. Put N, Konings P, Rack K, Jamar M, Van Roy N, Libouton JM, et al. Improved detection of chromosomal abnormalities in chronic lymphocytic leukemia by conventional cytogenetics using CpG oligonucleotide and interleukin-2 stimulation: A Belgian multicentric study. Genes Chromosom Cancer (2009) 48(10):843–53. doi: 10.1002/gcc.20691
223. Jensen C, Teng Y. Is it time to start transitioning from 2D to 3D cell culture? Front Mol Biosci (2020) 7:33. doi: 10.3389/fmolb.2020.00033
224. Forte D, Krause DS, Andreeff M, Bonnet D, Méndez-Ferrer S. Updates on the hematologic tumor microenvironment and its therapeutic targeting. Haematologica (2019) 104(10):1928–34. doi: 10.3324/haematol.2018.195396
225. Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol (2017) 10(1):106. doi: 10.1186/s13045-017-0470-7
226. Scott DW, Gascoyne RD. The tumour microenvironment in b cell lymphomas. Nat Rev Cancer (2014) 14(8):517–34. doi: 10.1038/nrc3774
Keywords: B lymphoma cell, primary cell culture, 2D cell culture, 3D cell culture, coculture (co-culture), B leukemic cells
Citation: Canonne M, George F and Graux C (2022) Ex vivo culture of malignant primary B cells. Front. Hematol. 1:1004717. doi: 10.3389/frhem.2022.1004717
Received: 27 July 2022; Accepted: 30 September 2022;
Published: 19 October 2022.
Edited by:
Wei Ding, Mayo Clinic, United StatesReviewed by:
Abdullah Khan, University of Birmingham, United KingdomRyan Wilcox, University of Michigan, United States
Copyright © 2022 Canonne, George and Graux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Morgane Canonne, bW9yZ2FuZS5jYW5vbm5lQGNodXVjbG5hbXVyLnVjbG91dmFpbi5iZQ==