 Isabel Rada
Isabel Rada Juan Francisco Calderón
Juan Francisco Calderón Gonzalo Martínez2
Gonzalo Martínez2 Paula Muñoz Venturelli
Paula Muñoz Venturelli- 1Instituto de Ciencias e Innovación en Medicina, Facultad de Medicina Clínica Alemana Universidad del Desarrollo, Santiago, Chile
- 2División de Enfermedades Cardiovasculares, Pontificia Universidad Católica de Chile, Santiago, Chile
- 3The George Institute for Global Health, Faculty of Medicine, University of New South Wales, Sydney, NSW, Australia
Objectives: Spontaneous cervical artery dissections (SCeAD) and coronary artery dissections (SCoAD) are major causes of neurovascular and cardiovascular morbidity in young adults. Although multiple aspects of their etiology are still unknown, most consensuses are focused on the presence of constitutional genetic aspects and environmental triggers. Since recent evidence of genetic contribution points to a possible overlap between these conditions, we aimed to describe current information on SCeAD and SCoAD genetics and their potential shared pathological aspects.
Materials and methods: A narrative review is presented. Publications in English and Spanish were queried using database search. The articles were evaluated by one team member in terms of inclusion criteria. After collecting, the articles were categorized based on scientific content.
Results: Given that patients with SCeAD and SCoAD rarely present connective tissue disorders, other genetic loci are probably responsible for the increased susceptibility in some individuals. The common variant rs9349379 at PHACTR1 gene is associated with predisposition to pathologies of the arterial wall, likely mediated by variations in Endothelin-1 (ET-1) levels. The risk of arterial dissection may be increased for those who carry the rs9349379(A) allele, associated with lower expression levels of ET-1; however, the local effect of this vasomotor imbalance remains unclear. Sex differences seen in SCeAD and SCoAD support a role for sex hormones that could modulate risk, tilting the delicate balance and forcing vasodilator actions to prevail over vasoconstriction due to a reduction in ET-1 expression.
Conclusions: New evidence points to a common gene variation that could explain dissection in both the cervical and coronary vasculatures. To further confirm the risk conferred by the rs9349379 variant, genome wide association studies are warranted, hopefully in larger and ethnically diverse populations.
Introduction
Arterial dissections are characterized by the separation of the arterial wall layers, creating an intramural hematoma either between vessel intima and media or media and adventitia. This can impair blood flow and adequate tissue perfusion, leading to ischemia and organ failure, and eventually cause death (1). Recently, there has been a renewed interest in spontaneous cervical artery dissections (SCeAD) and coronary artery dissections (SCoAD), due to improved diagnosis and the recognition that ischemic events derived from them can have severe clinical consequences (2, 3). Both types of artery dissections affect young populations and are particularly important in women since they occur more frequently during pregnancy and postpartum (4).
The pathological mechanisms behind SCeAD and SCoAD have still to be clarified. Although their presentation is mostly sporadic (5, 6), consensus exists regarding the presence in these patients of a constitutional genetically determined weakness of the vessel wall that, combined with triggering environmental factors, such as acute infection, minor trauma or physical stress, results in vessel injury (7–9). An underlying vasculopathy is suggested by the fact that patients with SCeAD and SCoAD commonly present with concomitant arterial anomalies, such as fibromuscular dysplasia (FMD) (1, 10). Classically considered distinct entities, recent evidence describes possible genetic associations for both types of dissections (11, 12). Our aim is to describe the current literature on SCeAD and SCoAD genetics and the potential shared mechanisms underpinning these two conditions.
Methods
The studies included in this narrative review (Supplementary Table eS1) were identified by searching in the electronic databases PubMed and ScienceDirect. Keywords were related to the pathological conditions and genomic/genetic terms “cervical artery dissection”; “carotid artery dissection”; “coronary artery dissection”; “mRNA”; “single nucleotide variant”; “single nucleotide polymorphism”; “polymorphism”; “genetic”; “genomic” using Boolean connectors AND/OR respectively. The search was restricted to manuscripts published from 1950 to present published in English. The selection criteria were as follows: (i) type of study: case studies, case-control studies, cohort studies; (ii) population type: diagnosis of SCeAD or SCoAD, without age or comorbidities restrictions; (iii) type of measurements: biological confirmation through genetic testing; (iv) type of outcomes: genetic/genomics reports, e.g.,: copy number variants (CNV), single nucleotide variants (SNV) and microRNA.
Results
Epidemiology and clinical implications of spontaneous cervical and coronary dissections
While SCeAD has an annual incidence rate of 2.6–2.9 cases per 100,000 people (13), official epidemiological data from SCoAD is not so well documented (14). There is a sex predominance for SCoAD, affecting more women (81%–92%) (15). Conversely, it appears that SCeAD has a slight tendency for higher cases in males (53%–57%), with the caveat that this estimated predominance can be biased because SCeAD patients with local symptoms might go under-recognized (13, 16). The overall population affected by these dissections is classically young-middle aged adults with scarce cardiovascular risk factors. The mean age of onset for SCeAD is 45 years (13); similarly, SCoAD reports a mean age ranging between 42 and 53 years (15, 17).
Both dissections can lead to severe clinical consequences. SCeAD accounts for 10%–25% of all stroke events in young patients (13) with increased risk within the first 2 weeks after dissection (18). Likewise, SCoAD can lead to myocardial injury and infarct, causing 1%–4% of all acute coronary syndromes and it has been associated to 0.5% of sudden cardiac deaths, particularly in young individuals (7, 19).
The occurrence of each condition is related to different environmental triggers. While SCeAD associates with minor cervical trauma (20), seasonal peak during winter (21), recent acute infection and migraine (11), SCoAD has been related to physiological and physical stress (e.g., strenuous exercise), leaving aside traumatic, iatrogenic or atherosclerotic etiologies (7–9). Nonetheless, SCeAD and SCoAD share common risk factors, including connective tissue disorders (CTD), FMD, and pregnancy/postpartum (8, 20, 22, 23), as it will be discussed later.
Regarding sex differences, the clear predominance of SCoAD in women is particularly important since this entity represents the major cause of myocardial infarction in pregnant women, particularly during the third trimester or early postpartum (14). Likewise, a series of SCeAD cases have been reported involving postpartum women, not only related to the stress of vaginal delivery but also after cesarean section (4). Estrogen, a steroid hormone whose most active metabolite is 17β-estradiol, increases its levels among adolescence, pre-menopause women and fluctuates and drops at older ages (24). Estrogens have vasodilatory effects throughout the activation of its receptors, promoting the release of nitric oxide in endothelial cells by activation of the PI3-kinase/AKT pathway (25); as well as endothelial prostacyclin (PGI2) production via arachidonic acid derivatives (26, 27). Additionally, the action of estrogens on the inhibition of the vasoconstrictor role of the renin-angiotensin-aldosterone system has been shown, favoring vasodilatation by increasing natriuretic peptide (28, 29). Also, estradiol has an inhibitory effect on endothelin 1 (ET-1) synthesis, further favoring a vasodilatory phenotype (30–32). Also, preclinical studies have suggested that pregnancy can increase the expression of oxytocin receptors in vascular tissue and can influence the risk of dissection. Previous research in murine models with CTDs showed that oxytocin levels are involved in extracellular signal-regulated kinase (ERK) pathway activation inducing aneurysm progression. This was confirmed by dissection reduction after suppression of lactation stimulus, ERK inhibition and oxytocin antagonist treatment (33, 34).
Thanks to the improved recognition and more precise diagnosis of these conditions by clinicians during the last couple of decades, a gradually more robust data have been accumulating that hopefully will help to better describe the role of modulating factors and environmental triggers, which end up resulting in arterial dissection.
Fibromuscular dysplasia, spontaneous cervical and coronary dissection
FMD is defined as “an idiopathic, segmental, non-atheromatous disease of the musculature of arterial walls, leading to stenosis of small and medium-sized arteries” (35). The presence of FMD has been described in series of both SCeAD and SCoAD patients, ranging between 5%–31% and 45%–86%, respectively (1, 36–40). Several epidemiological reports of US and European origin point to a preponderance of females in FMD cases, mostly diagnosed at age 50–55 years (41, 42), highlighting epidemiological similarities with SCoAD, where FMD is believed to be the underlying arteriopathy in the majority of cases (43). Considering the possible association between FMD, SCeAD and SCoAD, it has been suggested that these disorders comprise clinical manifestations of a common pathological entity (44). Beyond its association to vessel dissection, FMD in the renal arteries can lead to arterial hypertension, further increasing cardiovascular risk.
Recently, systematic recognition of FMD has been performed in large prospective cohort studies. For example, among 750 SCoAD patients, the overall presence of FMD was identified in 31.1% (39). Specifically, 56.7% (233/411) of patients with complete screening had FMD (39). Meanwhile, the screening of 1,283 SCeAD resulted in 8.0% of FMD diagnosis (103/1,283). Of note, the cerebrovascular FMD was detected as a potential marker of recurrence (40). However, a delay in FMD diagnosis and incomplete screening in SCeAD and SCoAD patients might favor the underestimation of the shared prevalence between these conditions.
FMD appears to be much more prevalent than previously thought. For example, silent FMD has been recognized in up to 6% of potential kidney donors (45, 46). While renal and cervico-cephalic arteries are the most commonly affected vascular beds, almost half of patients can have multivessel compromise, including visceral, ilio-femoral and coronary arteries (47). Furthermore, a familial presentation of FMD has been previously described (48–50), however, this association is now believed to be uncommon. In the ARCADIA study, familial FMD was present in 2.9% of cases (47); thus, most cases are sporadic. Nevertheless, as genetic background is highly probable, potential gene variants have been proposed. Using exome sequencing, the presence of multifocal FMD was associated to myosin light chain kinase (MYLK – also involved in thoracic aneurysms), dynein cytoplasmic heavy chain 1 (DYNC2H1), sarcomeric protein obscuring (OBSCN), and RNF213 (involved in Moyamoya disease) genes (42). Additionally, (51) Kiando et al. 2017 found that allele A of a genetic variant (rs9349379) of the phosphate and actin regulator 1 gene (PHACTR1) was associated to a 40% increase in the relative risk of FMD (51). As it will be discussed later, this gene is also associated with vascular dissections (Figure 1).
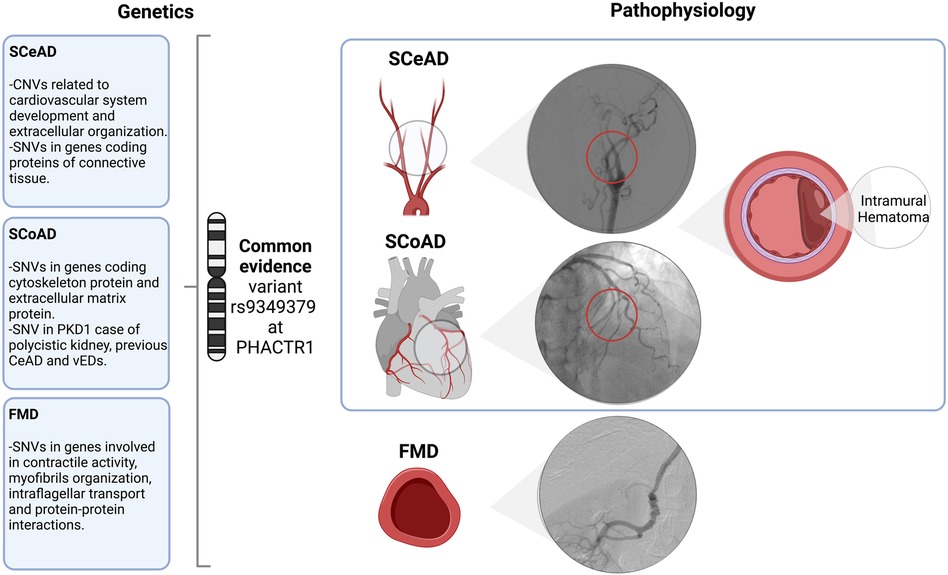
Figure 1. Overview and associated arteriopathy of spontaneous cervical and coronary dissection and fibromuscular dysplasia. SCeAD, SCoAD and FMD have been previously associated as clinical manifestations of a common pathological entity. Though, literature reported specific genetic contribution and its functional correlation for each condition, the common variant rs9349379 at PHACTR1 might represent a potential shared underlying mechanism. Specifically, SCeAD and SCoAD results from the separation of arterial wall layers producing an intramural hematoma with or without intimal tear. A low frequency of SCeAD, SCoAD and FMD cases are related to connective tissue disorders. SCeAD, spontaneous cervical artery dissection; SCoAD, spontaneous coronary artery dissection; FMD, fibromuscular dysplasia; CNVs, copy number variants; SNVs, single nucleotide variants; PDK1, polycystin-1; vEDs, vascular Ehlers-Danlos syndrome.
Spontaneous cervical artery dissection genetic studies
The CTDs monogenic disorders have been explored in regard to the predisposition to SCeAD. Autosomal dominant conditions such as Marfan syndrome (MFS) caused by variants in FBN1 gene (52), vascular Ehlers-Danlos syndrome (vEDS) caused by variants in COL3A1 (53) and Loeys-Dietz syndrome (LDS) caused by variants in TGFBR1, TGFBR2 and SMAD3 (54, 55), among other genes, have been considered as potential disorders favoring SCeAD (3, 56). However, a comprehensive systematic review in 2009 reported low frequency of these heritable cases, showing that vEDS accounted for only 2% of all SCeAD cases, while in MFS the overall reported cases were less than 1% (10). Although these studies may have resulted in an underestimation due to the lack of a systematic diagnostic protocol and disease recognition mainly derived from clinical features without molecular confirmation, clinical registry data seem to support these findings, as an association with CTDs is low (56, 57). The systematic review also reported predominantly negative findings from 15 genetic association studies, although the candidate genes coding for intercellular adhesion molecule 1 (ICAM-1), collagen type III a-1 (COL3A1) and methylenetetrahydrofolate reductase (MTHFR) involved in regulation of extracellular matrix (ECM) were associated in five studies (58–62). Moreover, a meta-analysis based on evidence of MTHFR 677TT genotype yielded significant association with SCeAD; however, a subsequent case-control study elicited opposite evidence, where the genotypic frequencies for this variant and others linked to SCeAD, thrombosis, coronary artery disease, hypertension and lipoprotein levels did not differ with their control counterparts (63). Furthermore, analyses of other gene variants involved in ECM homeostasis, (promoter region of matrix metalloproteinases or MMPs), lacked statistical differences between individuals with SCeAD and controls (64).
Regarding the identification of causative copy number variants (CNVs) in SCeAD, a case-control study from patients with or without dermal tissue alterations and its comparison with control samples and earlier published data, retrieved 34 CNVs associated with SCeAD (65). Those affected with dermal tissue alterations showed higher density of CNVs and ontology analyses revealed enrichment for genes related to ECM and collagen fibril organization. Although authors found novel rare CNVs in SCeAD population (duplication MYH11/ABCC6 and deletion SGCZ) that have also been described for aortic dissection, none of these were located in any other dissected participant (65). In line with exploration of CNVs, Grond-Grinsbach et al. used a Single Nucleotide Polymorphism (SNPs) array and genotyped a large set of SNPs in 883 patients. CNVs of SCeAD carriers covered 433 protein-coding genes associated to muscle organ development and cell differentiation of cardiovascular system; interestingly, the altered MYH11 encoding for smooth muscle myosin heavy chain and ABCC6 involved in membrane transportation were also found in SCeAD cases (3 patients) (66). Continuing with SNPs examination, the association between variants and arterial connective disorders was tested in SCeAD cases from 9 families with 2 affected members. The SNPs genotyping yielded 1,242 variants in the sample from which 142 were nonsense or missense substitutions. Among them, 9 were non-benign variants identified in 4 out of the 9 families. Specifically, these variants were found in COL3A1, FBN1, COL4A1 and TGFBR2 genes each in one of these 4 families. Although an increased susceptibility is suggested due to familial SCeAD, genetic heterogeneity makes difficult to establish a single causal variant, thus favoring a polygenic model (67).
More recently, an inverse association of a variant located in PHACTR1 with SCeAD was recognized in a large collection of cases from European origin recruited in European and US centers (11). Specifically, a genome wide association analysis revealed an association of rs9349379 variant in PHACTR1 with SCeAD during discovery and follow-up phases with the rs9349379(G) allele conferring lower risk of dissection (11) (Figure 2). This variant has been previously associated with myocardial infarction and coronary calcifications in various ancestry groups (68–71), with effects in the opposite direction of that in SCeAD. Conversely, a comparison between African American (AA) and European American (EA) population with stroke and SCeAD from the 1,000 genome project data, showed a higher allele frequency for rs9349379(A) in the AA group with SCeAD, but, paradoxically, AA had less SCeAD prevalence compared to EA (72). Of note, those from AA group with allele A also presented a greater rate of cardiovascular risk factors. These conflicting results could be explained by the existence of intermediate modulators of the effect of this variant, but also to bias of a single center database, where SCeAD can be underdiagnosed in low cardiovascular risk populations (72).
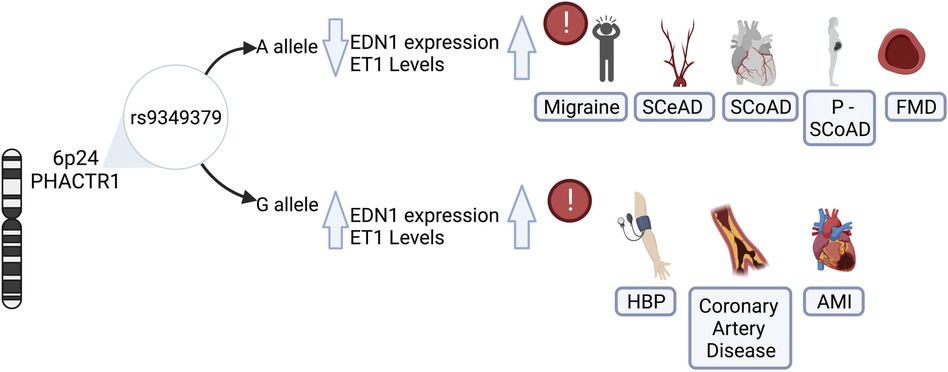
Figure 2. Role of rs9349379 genotypes on endothelin-1 levels and its risk associated with vascular diseases. The genetic variant rs9349379 in PHACTR1 gene regulars EDN1 gene expression, influencing protein levels and inducing its imbalance. The rs9349379 major allele (A) down regulate EDN1 gene expression, lowering ET1 secretion and favoring vasodilatation. Those carrying A allele are at risk for SCeAD, SCoAD migraine and FMD predominantly in the female population. Contrary to the effect G allele which increase ET1 levels that favors vasoconstriction and is associated with HBP, CAS, and AMI, mainly affecting the male population. EDN1, endothelin-1 gene; ET1, endothelin-1 protein; SCeAD, cervical artery dissections; SCoAD, spontaneous coronary artery dissections; P-SCoAD, pregnancy-associated SCoAD; FMD, fibromuscular dysplasia; HBP, high blood pressure; CAS, coronary artery disease; AMI, acute myocardial infarction.
Spontaneous coronary artery dissection genetic studies
Similarly to SCeAD, CTDs have historically represented the main focus of SCoAD genetic research, yet it has been identified as the causative disorder in less than 5% of these cases (73). Few case studies have reported the presence of vEDS-related variants in COL3A1 gene, mainly in patients with multivessel events and/or SCoAD recurrence (74–77). Interestingly, two of these cases were pregnancy-associated SCoAD (74, 76). Studies referring patients to genetic analysis from larger SCoAD case series have led to the detection of a small proportion of participants with disease-causing variants. Henkin et al. (78), used diverse panels of genes associated with CTDs to test SCoAD-affected individuals, in which only 3 out of 59 - the total sample who underwent genetic testing - had variants either in FBN1 or COL3A1 (78). Similarly, a prospective cohort confirmed the presence of COL3A1 variants in 3 patients and SMAD3 in one case from subjects who agreed to the genetic evaluation. Noteworthy, one patient with variants in polycystin-1 (PKD1) - who was diagnosed with polycystic kidney disease - had a previous SCeAD, followed by a SCoAD four years after the first dissection event (79). The autosomal dominant polycystic kidney disease (ADPKD) might produce arterial wall weakening and increase susceptibility to vascular events. Evidence has documented that half of deaths among patients with ADPKD were produced by myocardial ischemia (80). Moreover, studies looking beyond traditional CTDs have reported the detection of rare variants in the gene encoding cytoskeletal protein Talin 1 (TLN1) in a familial SCoAD case composed of 5 members from which 3 had dissections. This variant was subsequently found in 10 cases of a large SCoAD cohort, thus, it is believed to have detrimental vascular effects, since TLN1 alteration has been also found in aortic dissection (81).
The association of SCoAD with the variant rs9349379 in PHACTR1 has been recently recognized. This variant was previously described for coronary diseases such as atherosclerosis and acute myocardial infarction (82–84). Adlam et al. (12) analyzed SCoAD cases and controls studies, along with pregnancy-associated SCoAD cases from the United States, United Kingdom, Australia and France with confirmed European Ancestry. In accordance with epidemiological data of SCoAD, the study revealed a female predominance (87%–96%), with pregnancy-associated SCoAD accounting for 10% of cases. Besides sex influence, based on more than 1,000 cases, it was estimated that those who carried the rs9349379-A allele had 70% greater risk of SCoAD. This was associated with lower plasma levels of Endothelin-1 (ET-1) according to measurements from a subset of SCoAD patients from the United Kingdom with similar characteristics (Figure 2) (12). Similarly, a meta-analysis of genome wide SCoAD discovery and replication reported an association with the variant rs9349379 in PHACTR1 (85). In addition, the genome wide approach in a prospective large cohort of SCoAD patients revealed an association of the variant rs12740679 in ADAMTSL4 gene chromosome 1q21.2, which encode an extracellular matrix protein involved in microfibril formation (85). Although the pathophysiology of SCoAD is not yet well understood, these latest findings give new insights for further research focused on elucidating the role of the common rs9349379 variant.
Potential common underlying mechanisms in spontaneous cervical and coronary dissection
As previously mentioned, the common variant rs9349379 at PHACTR1 locus on chromosome 6p24 has been proposed as a potential causative variant for pathologies of vascular phenotype, mostly described for chronic coronary diseases (84, 86, 87). However, the emerging evidence of other vascular disorders such as FMD (51), SCeAD (11, 72, 88) and SCoAD (12, 85) have raised the attention to explore the potential underlying mechanisms of this genetic locus. Gupta et al., reported a common intergenic area between rs9349379 at PHACTR1 locus and the promoter of endothelin-1 gene (EDN1), with putative regulatory function on the expression of these genes (89). Specifically, PHACTR1 encodes for a protein of phosphatase and actin regulator family related to regulation of the actin cytoskeleton and, indirectly, on the endothelial cell survival (90). In turn, this could have deleterious effects in several vascular phenotypes such as formation of atherosclerotic plaques (91), neo-angiogenesis (92), cell proliferation and migration in cerebral microvasculature (93). Several experiments in stem cell lines reprogramed with CRISPR/Cas9 validated the influence of the genotype at rs9349379 on EDN1 levels, in which the minor allele in homozygosity (G/G) resulted in higher EDN1 expression, contrary to the reduced levels observed in association with homozygous major allele (A/A) (89). The association was confirmed in plasma samples from healthy individuals: those who carry the minor allele expressed higher levels of the precursor Big- ET-1 that is subsequently converted to the active form ET-1. The authors proposed a model where rs9349379 (G) confers risk for coronary artery disease/myocardial infarct, whereas it reduces susceptibility to SCeAD, FMD and migraine (58, 89). Given these findings, the major allele rs9349379 (A) may impose the risk genotype for both dissections, along with reduced ET-1 levels as described in a large sample of SCoAD patients (12). ET-1 is a peptide with potent vasoconstrictor action synthesized by different cell types, predominantly vascular endothelium. ET-1 activates receptors ETA y ETB2 generating a vasoconstrictor effect mediated by calcium, while ETB1 promotes the release of vasodilator agents as nitric oxide and prostacyclin (94). Although the response of increased levels of ET-1 have been widely characterized as endothelial damaging (95), the vascular susceptibility that could result from reduced expression of EDN1 has not been explored.
The findings of the present study may have relevant clinical implications for the prediction of dissection, based on genetic contribution described in the evidence. Particularly, populations carrying the major allele rs9349379 (A) at PHACTR1 might have down regulated expression of EDN1 and lower ET-1 levels. This imbalance favoring vasodilation and affecting vascular integrity, could also interplay with sex hormones. This can be critical for specific interest groups such as women at all stages of their life, particularly during pregnancy. Therefore, the study of the hormonal influence on arterial wall vulnerability could improve the understanding of female predominance in SCoAD and pregnancy-associated dissections.
Conclusion
Emerging evidence has related vascular disorders such as SCeAD, SCoAD and FMD to the common variant rs9349379 at PHACTR1, likely mediated by ET-1 levels. The risk of arterial dissections may increase for those carrying the rs9349379(A) variant and thus express lower ET-1 levels; however, local effect or vascular remodeling resulting from this vasomotor imbalance remains unclear. Sex differences support a role for sex hormones on risk modulation, by which vasodilator actions might likewise prevail over-active vasoconstriction. Further research is needed to confirm the risk conferred by the rs9349379 variant, including genome wide association studies in larger and ethnically diverse SCeAD, SCoAD and FMD patients.
Author contributions
IR: performed the review and drafted the manuscript. JFC, GM, PMV: critically reviewed the manuscript and wrote the final version. All authors contributed to the article and approved the submitted version.
Funding
ANID Fondecyt Regular 1181238 (PMV), ANID Fondecyt Iniciación 11170353 (JFC), ACT210012 (JFC), EQM190110 (JFC).
Conflict of interest
JFC is coauthor of a patent application for pharmacological treatment of vascular Ehlers-Danlos Syndrome. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The handling editor CC declared a shared affiliation with the author PMV at the time of review.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgwh.2023.1007795/full#supplementary-material.
References
1. Saw J, Ricci D, Starovoytov A, Fox R, Buller CE. Spontaneous coronary artery dissection: prevalence of predisposing conditions including fibromuscular dysplasia in a tertiary center cohort. JACC Cardiovasc Interv. (2013) 6(1):44–52. doi: 10.1016/j.jcin.2012.08.017
2. Lewey J, El Hajj SC, Hayes SN. Spontaneous coronary artery dissection: new insights into this not-so-rare condition. Annu Rev Med. (2022) 73:339–54. doi: 10.1146/annurev-med-052819-023826
3. Marciniec M, Sapko K, Kulczynski M, Popek-Marciniec S, Szczepanska-Szerej A, Rejdak K. Non-traumatic cervical artery dissection and ischemic stroke: a narrative review of recent research. Clin Neurol Neurosurg. (2019) 187:105561. doi: 10.1016/j.clineuro.2019.105561
4. Urrutia F, Mazzon E, Brunser A, Diaz V, Calderon JF, Stecher X, et al. Cervical artery dissection in postpartum women after cesarean and vaginal delivery. J Stroke Cerebrovasc Dis. (2022) 31(8):106572. doi: 10.1016/j.jstrokecerebrovasdis.2022.106572
5. Goel K, Tweet M, Olson TM, Maleszewski JJ, Gulati R, Hayes SN. Familial spontaneous coronary artery dissection: evidence for genetic susceptibility. JAMA Intern Med. (2015) 175(5):821–6. doi: 10.1001/jamainternmed.2014.8307
6. Grond-Ginsbach C, De Freitas GR, Campos CR, Thie A, Caso V, Machetanz J, et al. Familial occurrence of cervical artery dissection–coincidence or sign of familial predisposition? Cerebrovasc Dis. (2012) 33(5):466–70. doi: 10.1159/000337035
7. Macaya F, Salinas P, Gonzalo N, Fernandez-Ortiz A, Macaya C, Escaned J. Spontaneous coronary artery dissection: contemporary aspects of diagnosis and patient management. Open Heart. (2018) 5(2):e000884. doi: 10.1136/openhrt-2018-000884
8. Saw J, Mancini GBJ, Humphries KH. Contemporary review on spontaneous coronary artery dissection. J Am Coll Cardiol. (2016) 68(3):297–312. doi: 10.1016/j.jacc.2016.05.034
9. Tweet MS, Gulati R, Hayes SN. Spontaneous coronary artery dissection. Curr Cardiol Rep. (2016) 18(7):60. doi: 10.1007/s11886-016-0737-6
10. Debette S, Markus HS. The genetics of cervical artery dissection: a systematic review. Stroke. (2009) 40(6):e459–66. doi: 10.1161/STROKEAHA.108.534669
11. Debette S, Kamatani Y, Metso TM, Kloss M, Chauhan G, Engelter ST, et al. Common variation in Phactr1 is associated with susceptibility to cervical artery dissection. Nat Genet. (2015) 47(1):78–83. doi: 10.1038/ng.3154
12. Adlam D, Olson TM, Combaret N, Kovacic JC, Iismaa SE, Al-Hussaini A, et al. Association of the Phactr1/Edn1 genetic locus with spontaneous coronary artery dissection. J Am Coll Cardiol. (2019) 73(1):58–66. doi: 10.1016/j.jacc.2018.09.085
13. Blum CA, Yaghi S. Cervical artery dissection: a review of the epidemiology, pathophysiology, treatment, and outcome. Arch Neurosci. (2015) 2(4):e26670. doi: 10.5812/archneurosci.26670
14. Hayes SN, Kim ESH, Saw J, Adlam D, Arslanian-Engoren C, Economy KE, et al. Spontaneous coronary artery dissection: current state of the science: a scientific statement from the American heart association. Circulation. (2018) 137(19):e523–57. doi: 10.1161/CIR.0000000000000564
15. Tweet MS, Kok SN, Hayes SN. Spontaneous coronary artery dissection in women: what is known and what is yet to be understood. Clin Cardiol. (2018) 41(2):203–10. doi: 10.1002/clc.22909
16. Mazzon E, Rocha D, Brunser A, De la Barra C, Stecher X, Bernstein T, et al. Cervical artery dissections with and without stroke, risk factors and prognosis: a Chilean prospective cohort. J Stroke Cerebrovasc Dis. (2020) 29(8):104992. doi: 10.1016/j.jstrokecerebrovasdis.2020.104992
17. Garcia-Guimaraes M, Bastante T, Macaya F, Roura G, Sanz R, Barahona Alvarado JC, et al. Spontaneous coronary artery dissection in Spain: clinical and angiographic characteristics, management, and in-hospital events. Rev Esp Cardiol. (2021) 74(1):15–23. doi: 10.1016/j.rec.2020.04.002
18. Morris NA, Merkler AE, Gialdini G, Kamel H. Timing of incident stroke risk after cervical artery dissection presenting without ischemia. Stroke. (2017) 48(3):551–5. doi: 10.1161/STROKEAHA.116.015185
19. Hill SF, Sheppard MN. Non-atherosclerotic coronary artery disease associated with sudden cardiac death. Heart. (2010) 96(14):1119–25. doi: 10.1136/hrt.2009.185157
20. Robertson JJ, Koyfman A. Cervical artery dissections: a review. J Emerg Med. (2016) 51(5):508–18. doi: 10.1016/j.jemermed.2015.10.044
21. Paciaroni M, Georgiadis D, Arnold M, Gandjour J, Keseru B, Fahrni G, et al. Seasonal variability in spontaneous cervical artery dissection. J Neurol Neurosurg Psychiatry. (2006) 77(5):677–9. doi: 10.1136/jnnp.2005.077073
22. Yip A, Saw J. Spontaneous coronary artery dissection-a review. Cardiovasc Diagn Ther. (2015) 5(1):37–48. doi: 10.3978/j.issn.2223-3652.2015.01.08
23. Saw J, Humphries K, Aymong E, Sedlak T, Prakash R, Starovoytov A, et al. Spontaneous coronary artery dissection: clinical outcomes and risk of recurrence. J Am Coll Cardiol. (2017) 70(9):1148–58. doi: 10.1016/j.jacc.2017.06.053
24. Knowlton AA, Lee AR. Estrogen and the cardiovascular system. Pharmacol Ther. (2012) 135(1):54–70. doi: 10.1016/j.pharmthera.2012.03.007
25. Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res. (2016) 118(6):994–1007. doi: 10.1161/CIRCRESAHA.115.305376
26. Sobrino A, Oviedo PJ, Novella S, Laguna-Fernandez A, Bueno C, García-Pérez MA, et al. Estradiol selectively stimulates endothelial prostacyclin production through estrogen receptor-Α. J Mol Endocrinol. (2010) 44(4):237–46. doi: 10.1677/JME-09-0112
27. O’Sullivan MG, Goodrich JA, Adams MR. Increased prostacyclin synthesis by atherosclerotic arteries from estrogen-treated monkeys. Life Sci. (2001) 69(4):395–401. doi: 10.1016/S0024-3205(01)01131-6
28. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ. (2017) 8(1):1–16. doi: 10.1186/s13293-017-0152-8
29. O’Donnell E, Floras JS, Harvey PJ. Estrogen status and the renin angiotensin aldosterone system. Am J Physiol Regul Integr Comp Physiol. (2014) 307(5):R498–R500. doi: 10.1152/ajpregu.00182.2014
30. Dubey RK, Jackson EK, Keller PJ, Imthurn B, Rosselli M. Estradiol metabolites inhibit endothelin synthesis by an estrogen receptor-independent mechanism. Hypertension. (2001) 37(2):640–4. doi: 10.1161/01.HYP.37.2.640
31. Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev. (2008) 60(2):210–41. doi: 10.1124/pr.107.08002
32. Bilsel AS, Moini H, Tetik E, Aksungar F, Kaynak B, Özer A. 17β-estradiol modulates endothelin-1 expression and release in human endothelial cells. Cardiovasc Res. (2000) 46(3):579–84. doi: 10.1016/S0008-6363(00)00046-8
33. Habashi JP, MacFarlane EG, Bagirzadeh R, Bowen C, Huso N, Chen Y, et al. Oxytocin antagonism prevents pregnancy-associated aortic dissection in a mouse model of marfan syndrome. Sci Transl Med. (2019) 11(490):eaat4822. doi: 10.1126/scitranslmed.aat4822
34. Bowen CJ, Calderon Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, et al. Targetable cellular signaling events mediate vascular pathology in vascular ehlers-danlos syndrome. J Clin Invest. (2020) 130(2):686–98. doi: 10.1172/JCI130730
35. Persu A, Giavarini A, Touze E, Januszewicz A, Sapoval M, Azizi M, et al. European consensus on the diagnosis and management of fibromuscular dysplasia. J Hypertens. (2014) 32(7):1367–78. doi: 10.1097/HJH.0000000000000213
36. Kadian-Dodov D, Gornik HL, Gu X, Froehlich J, Bacharach JM, Chi Y-W, et al. Dissection and aneurysm in patients with fibromuscular dysplasia: findings from the Us registry for Fmd. J Am Coll Cardiol. (2016) 68(2):176–85. doi: 10.1016/j.jacc.2016.04.044
37. Saw J, Poulter R, Fung A, Wood D, Hamburger J, Buller CE. Spontaneous coronary artery dissection in patients with fibromuscular dysplasia: a case series. Circ Cardiovasc Interv. (2012) 5(1):134–7. doi: 10.1161/CIRCINTERVENTIONS.111.966630
38. Bejot Y, Aboa-Eboule C, Debette S, Pezzini A, Tatlisumak T, Engelter S, et al. Characteristics and outcomes of patients with multiple cervical artery dissection. Stroke. (2014) 45(1):37–41. doi: 10.1161/STROKEAHA.113.001654
39. Saw J, Starovoytov A, Humphries K, Sheth T, So D, Minhas K, et al. Canadian spontaneous coronary artery dissection cohort study: in-hospital and 30-day outcomes. Eur Heart J. (2019) 40(15):1188–97. doi: 10.1093/eurheartj/ehz007
40. Bonacina S, Grassi M, Zedde M, Zini A, Bersano A, Gandolfo C, et al. Clinical features of patients with cervical artery dissection and fibromuscular dysplasia. Stroke. (2021) 52(3):821–9. doi: 10.1161/STROKEAHA.120.031579
41. Hayes SN, Tweet MS, Adlam D, Kim ESH, Gulati R, Price JE, et al. Spontaneous coronary artery dissection: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 76(8):961–84. doi: 10.1016/j.jacc.2020.05.084
42. Di Monaco S, Georges A, Lengele JP, Vikkula M, Persu A. Genomics of fibromuscular dysplasia. Int J Mol Sci. (2018) 19(5):1526. doi: 10.3390/ijms19051526
43. Gilhofer TS, Saw J. Spontaneous coronary artery dissection: a review of complications and management strategies. Expert Rev Cardiovasc Ther. (2019) 17(4):275–91. doi: 10.1080/14779072.2019.1598261
44. Maas A, Bouatia-Naji N, Persu A, Adlam D. Spontaneous coronary artery dissections and fibromuscular dysplasia: current insights on pathophysiology, sex and gender. Int J Cardiol. (2019) 286:220–5. doi: 10.1016/j.ijcard.2018.11.023
45. Cragg AH, Smith TP, Thompson BH, Maroney TP, Stanson AW, Shaw GT, et al. Incidental fibromuscular dysplasia in potential renal donors: long-term clinical follow-up. Radiology. (1989) 172(1):145–7. doi: 10.1148/radiology.172.1.2662248
46. Blondin D, Lanzman R, Schellhammer F, Oels M, Grotemeyer D, Baldus SE, et al. Fibromuscular dysplasia in living renal donors: still a challenge to computed tomographic angiography. Eur J Radiol. (2010) 75(1):67–71. doi: 10.1016/j.ejrad.2009.03.014
47. Plouin PF, Baguet JP, Thony F, Ormezzano O, Azarine A, Silhol F, et al. High prevalence of multiple arterial bed lesions in patients with fibromuscular dysplasia: the arcadia registry (assessment of renal and cervical artery dysplasia). Hypertension. (2017) 70(3):652–8. doi: 10.1161/HYPERTENSIONAHA.117.09539
48. Halpern MM, Sanford HS, Viamonte M. Renal-artery abnormalities in three hypertensive sisters: probable familial fibromuscular hyperplasia. JAMA. (1965) 194(5):512–3. doi: 10.1001/jama.1965.03090180036007
49. Hansen J, Holten C, Thorborg JV. Hypertension in two sisters caused by so-called fibromuscular hyperplasia of the renal arteries. Acta Med Scand. (1965) 178(4):461–74. doi: 10.1111/j.0954-6820.1965.tb04291.x
50. Plagnol P, Gillet JM, Cambuzat JM, Broussin J. Familial reno-vascular hypertension (author’s transl). J Radiol Electrol Med Nucl. (1975) 56(2):173–4. Journal code: 7610508. ISSN: 0368-3966. L-ISSN: 0368-39661151915
51. Kiando SR, Tucker NR, Castro-Vega L-J, Katz A, D’Escamard V, Tréard C, et al. Phactr1 is a genetic susceptibility locus for fibromuscular dysplasia supporting its Complex genetic pattern of inheritance. PLoS Genet. (2016) 12(10):e1006367. doi: 10.1371/journal.pgen.1006367
52. Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. (1991) 352(6333):337–9. doi: 10.1038/352337a0
53. Germain DP. Ehlers-Danlos syndrome type iv. Orphanet J Rare Dis. (2007) 2(1):32. doi: 10.1186/1750-1172-2-32
54. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in Tgfbr1 or Tgfbr2. Nat Genet. (2005) 37(3):275–81. doi: 10.1038/ng1511
55. Van Hemelrijk C, Renard M, Loeys B. The loeys-dietz syndrome: an update for the clinician. Curr Opin Cardiol. (2010) 25(6):546–51. doi: 10.1097/HCO.0b013e32833f0220
56. Grond-Ginsbach C, Debette S. The association of connective tissue disorders with cervical artery dissections. Curr Mol Med. (2009) 9(2):210–4. doi: 10.2174/156652409787581547
57. Debette S, Goeggel Simonetti B, Schilling S, Martin JJ, Kloss M, Sarikaya H, et al. Familial occurrence and heritable connective tissue disorders in cervical artery dissection. Neurology. (2014) 83(22):2023–31. doi: 10.1212/WNL.0000000000001027
58. Pezzini A, Del Zotto E, Archetti S, Negrini R, Bani P, Albertini A, et al. Plasma homocysteine concentration, C677t Mthfr genotype, and 844ins68bp Cbs genotype in young adults with spontaneous cervical artery dissection and atherothrombotic stroke. Stroke. (2002) 33(3):664–9. doi: 10.1161/hs0302.103625
59. Arauz A, Hoyos L, Cantú C, Jara A, Martínez L, García I, et al. Mild hyperhomocysteinemia and low folate concentrations as risk factors for cervical arterial dissection. Cerebrovasc Dis. (2007) 24(2-3):210–4. doi: 10.1159/000104479
60. Longoni M, Grond-Ginsbach C, Grau A, Genius J, Debette S, Schwaninger M, et al. The Icam-1 E469k gene polymorphism is a risk factor for spontaneous cervical artery dissection. Neurology. (2006) 66(8):1273–5. doi: 10.1212/01.wnl.0000208411.01172.0b
61. von Pein F, Valkkila M, Schwarz R, Morcher M, Klima B, Grau A, et al. Analysis of the Col3a1 gene in patients with spontaneous cervical artery dissections. J Neurol. (2002) 249(7):862–6. doi: 10.1007/s00415-002-0745-x
62. Pezzini A, Grassi M, Del Zotto E, Giossi A, Monastero R, Dalla Volta G, et al. Migraine mediates the influence of C677t Mthfr genotypes on ischemic stroke risk with a stroke-subtype effect. Stroke. (2007) 38(12):3145–51. doi: 10.1161/STROKEAHA.107.491506
63. Jara-Prado A, Alonso M, Ruano LM, Camacho JG, Leyva A, Lopez M, et al. Mthfr C677t, Fii G20210a, Fv Leiden G1691a, Nos3 Intron 4 Vntr, and Apoe Ε4 gene polymorphisms are not associated with spontaneous cervical artery dissection. Int J Stroke. (2010) 5(2):80–5. doi: 10.1111/j.1747-4949.2010.00412.x
64. Buss A, Pech K, Roelver S, Bloemeke B, Klotzsch C, Breuer S. Functional polymorphisms in matrix metalloproteinases −1, −3, −9 and −12 in relation to cervical artery dissection. BMC Neurol. (2009) 9(1):40. doi: 10.1186/1471-2377-9-40
65. Grond-Ginsbach C, Chen B, Pjontek R, Wiest T, Jiang Y, Burwinkel B, et al. Copy number variation in patients with cervical artery dissection. Eur J Hum Genet. (2012) 20(12):1295–9. doi: 10.1038/ejhg.2012.82
66. Grond-Ginsbach C, Chen B, Krawczak M, Pjontek R, Ginsbach P, Jiang Y, et al. Genetic imbalance in patients with cervical artery dissection. Curr Genomics. (2017) 18(2):206–13. doi: 10.2174/1389202917666160805152627
67. Grond-Ginsbach C, Brandt T, Kloss M, Aksay SS, Lyrer P, Traenka C, et al. Next generation sequencing analysis of patients with familial cervical artery dissection. Eur Stroke J. (2017) 2(2):137–43. doi: 10.1177/2396987317693402
68. Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. (2009) 41(3):334–41. doi: 10.1038/ng.327
69. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. (2013) 45(1):25–33. doi: 10.1038/ng.2480
70. Hager J, Kamatani Y, Cazier J-B, Youhanna S, Ghassibe-Sabbagh M, Platt DE, et al. Genome-wide association study in a Lebanese cohort confirms Phactr1 as a major determinant of coronary artery stenosis. PLoS One. (2012) 7(6):e38663. doi: 10.1371/journal.pone.0038663
71. Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in europeans and south asians identifies five new loci for coronary artery disease. Nat Genet. (2011) 43(4):339–44. doi: 10.1038/ng.782
72. Green IE, Rahman SA, Owens DL, Sale MM, Currie LJ, Keene KL, et al. Cervical artery dissection in patients of African ancestry. Cerebrovasc Dis. (2018) 46(5-6):218–22. doi: 10.1159/000494704
73. Hayes S, Kim E, Saw J, Adlam D, Arslanian-Engoren C, Economy K, et al. Council on clinical cardiology; council on cardiovascular and stroke nursing; council on genomic and precision medicine; and stroke council. Spontaneous coronary artery dissection: current state of the science: a scientific statement from the American heart association. Circulation. (2018) 137(19):e523–57. doi: 10.1161/CIR.0000000000000564
74. Nakamura M, Yajima J, Oikawa Y, Ogasawara K, Uejima T, Abe K, et al. Vascular ehlers-danlos syndrome:—all three coronary artery spontaneous dissections. J Cardiol. (2009) 53(3):458–62. doi: 10.1016/j.jjcc.2008.09.007
75. Hampole CV, Philip F, Shafii A, Pettersson G, Anesi GL, Patel JB, et al. Spontaneous coronary artery dissection in ehlers-danlos syndrome. Ann Thorac Surg. (2011) 92(5):1883–4. doi: 10.1016/j.athoracsur.2011.03.136
76. Reinhold SM, Lima B, Khalid A, Gonzalez-Stawinski GV, Stoler RC, Hall SA, et al. editors. Heart transplantation in the ehlers-danlos syndrome. Baylor university medical center proceedings (2015). Taylor & Francis.
77. Ades LC, Waltham RD, Chiodo AA, Bateman JF. Myocardial infarction resulting from coronary artery dissection in an adolescent with ehlers-danlos syndrome type iv due to a type iii collagen mutation. Heart. (1995) 74(2):112–6. doi: 10.1136/hrt.74.2.112
78. Henkin S, Negrotto SM, Tweet MS, Kirmani S, Deyle DR, Gulati R, et al. Spontaneous coronary artery dissection and its association with heritable connective tissue disorders. Heart. (2016) 102(11):876–81. doi: 10.1136/heartjnl-2015-308645
79. Kaadan MI, MacDonald C, Ponzini F, Duran J, Newell K, Pitler L, et al. Prospective cardiovascular genetics evaluation in spontaneous coronary artery dissection. Circ Genom Precis Med. (2018) 11(4):e001933. doi: 10.1161/CIRCGENETICS.117.001933
80. Neves JB, Rodrigues FB, Lopes JA. Autosomal dominant polycystic kidney disease and coronary artery dissection or aneurysm: a systematic review. Ren Fail. (2016) 38(4):493–502. doi: 10.3109/0886022X.2016.1144209
81. Turley TN, Theis JL, Sundsbak RS, Evans JM, O'Byrne MM, Gulati R, et al. Rare missense variants in Tln1 are associated with familial and sporadic spontaneous coronary artery dissection. Circ Genom Precis Med. (2019) 12(4):e002437. doi: 10.1161/CIRCGEN.118.002437
82. Zhao Q, Wei H, Liu D, Shi B, Li L, Yan M, et al. Phactr1 and Slc22a3 gene polymorphisms are associated with reduced coronary artery disease risk in the male Chinese han population. Oncotarget. (2017) 8(1):658–63. doi: 10.18632/oncotarget.13506
83. Pérez-Hernández N, Vargas-Alarcón G, Posadas-Sánchez R, Martínez-Rodríguez N, Tovilla-Zárate CA, Rodríguez-Cortés AA, et al. Phactr1 gene polymorphism is associated with increased risk of developing premature coronary artery disease in Mexican population. Int J Environ Res Public Health. (2016) 13(8):803. doi: 10.3390/ijerph13080803
84. Beaudoin M, Gupta RM, Won H-H, Lo KS, Do R, Henderson CA, et al. Myocardial infarction–associated Snp at 6p24 interferes with Mef2 binding and associates with Phactr1 expression levels in human coronary arteries. Arterioscler Thromb Vasc Biol. (2015) 35(6):1472–9. doi: 10.1161/ATVBAHA.115.305534
85. Saw J, Yang ML, Trinder M, Tcheandjieu C, Xu C, Starovoytov A, et al. Chromosome 1q21.2 and additional loci influence risk of spontaneous coronary artery dissection and myocardial infarction. Nat Commun. (2020) 11(1):4432. doi: 10.1038/s41467-020-17558-x
86. Reschen ME, Lin D, Chalisey A, Soilleux EJ, O’Callaghan CA. Genetic and environmental risk factors for atherosclerosis regulate transcription of phosphatase and actin regulating gene Phactr1. Atherosclerosis. (2016) 250:95–105. doi: 10.1016/j.atherosclerosis.2016.04.025
87. Vargas JD, Manichaikul A, Wang XQ, Rich SS, Rotter JI, Post WS, et al. Common genetic variants and subclinical atherosclerosis: the multi-ethnic study of atherosclerosis (mesa). Atherosclerosis. (2016) 245:230–6. doi: 10.1016/j.atherosclerosis.2015.11.034
88. Abe A, Nito C, Sakamoto Y, Nogami A, Hokama H, Takahashi S, et al. Spontaneous bilateral cervical internal carotid and vertebral artery dissection in a Japanese patient without collagen vascular disease with special reference to single-nucleotide polymorphisms. J Stroke Cerebrovasc Dis. (2016) 25(8):e114–7. doi: 10.1016/j.jstrokecerebrovasdis.2016.04.027
89. Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. (2017) 170(3):522–33.e15. doi: 10.1016/j.cell.2017.06.049
90. Allen PB, Greenfield AT, Svenningsson P, Haspeslagh DC, Greengard P. Phactrs 1–4: a family of protein phosphatase 1 and actin regulatory proteins. Proc Natl Acad Sci U S A. (2004) 101(18):7187–92. doi: 10.1073/pnas.0401673101
91. Kasikara C, Schilperoort M, Gerlach B, Xue C, Wang X, Zheng Z, et al. Deficiency of macrophage Phactr1 impairs efferocytosis and promotes atherosclerotic plaque necrosis. J Clin Invest. (2021) 131(8):e145275. doi: 10.1172/JCI145275
92. Jarray R, Allain B, Borriello L, Biard D, Loukaci A, Larghero J, et al. Depletion of the novel protein Phactr-1 from human endothelial cells abolishes tube formation and induces cell death receptor apoptosis. Biochimie. (2011) 93(10):1668–75. doi: 10.1016/j.biochi.2011.07.010
93. Jing Y, Zhang L, Xu Z, Chen H, Ju S, Ding J, et al. Phosphatase actin regulator-1 (Phactr-1) knockdown suppresses cell proliferation and migration and promotes cell apoptosis in the bend.3 mouse brain capillary endothelial cell line. Med Sci Monit. (2019) 25:1291–300. doi: 10.12659/MSM.912586
94. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev. (2016) 68(2):357–418. doi: 10.1124/pr.115.011833
Keywords: genetics, genetic variation, cervical artery dissection, coronary artery dissection, vascular disease in women, genetic association, genetics of artery dissections
Citation: Rada I, Calderón JF, Martínez G and Muñoz Venturelli P (2023) Genetics of spontaneous cervical and coronary artery dissections. Front. Glob. Womens Health 4:1007795. doi: 10.3389/fgwh.2023.1007795
Received: 30 July 2022; Accepted: 14 April 2023;
Published: 5 May 2023.
Edited by:
Cheryl Carcel, University of New South Wales, AustraliaReviewed by:
Yuki Sakamoto, Nippon Medical School, Japan© 2023 Rada, Calderón, Martínez and Muñoz Venturelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paula Muñoz Venturelli cGF1bXVub3pAdWRkLmNs