 Monica S. Chung
Monica S. Chung Sang Jun Han
Sang Jun Han- 1Division of Reproductive Endocrinology and Infertility, Department of Ob/Gyn, Baylor College of Medicine, Houston, TX, United States
- 2Laboratory of Dan L. Duncan Cancer Center and Reproductive Medicine, Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, United States
Endometriosis is a known estrogen-dependent inflammatory disease affecting reproductive-aged women. Common symptoms include pelvic pain, dysmenorrhea, dyspareunia, heavy menstrual bleeding, and infertility. The exact etiology of endometriosis is largely unknown, and, thus, the diagnosis and treatment of endometriosis are challenging. A complex interplay of many molecular mechanisms is thought to aid in the progression of endometriosis, most notably angiogenesis. This mini-review examines our current knowledge of the molecular etiology of endometriosis-associated angiogenesis and discusses anti-angiogenic therapy, in the blockade of endometriosis-associated angiogenesis, as potential non-hormonal therapy for the treatment of endometriosis.
Introduction
Endometriosis is an estrogen-dependent inflammatory disease (1), defined as the presence of endometrial glands and stroma outside the uterine cavity. Endometriosis affects up to 5–10% of reproductive-aged women (2, 3) and has a higher prevalence in infertile women (4). The prevalence, however, may be underestimated because of diagnostic difficulty (3) and variation in clinical presentation. Early age at menarche, short menstrual cycle length, lean body size, and decreased parity are characteristically associated with a greater risk of endometriosis (3). Common symptoms that are predictive of the diagnosis of endometriosis include abdominopelvic pain, severe dysmenorrhea, dyspareunia, heavy menstrual bleeding, infertility, and a prior diagnosis of irritable bowel syndrome or pelvic inflammatory disease (5, 6). Clinicians should also suspect endometriosis in women of reproductive age with cyclical dyschezia, dysuria, or hematuria (6).
Diagnosis and treatment of endometriosis are challenging, given the large knowledge gap in specific cellular and molecular pathways. Surgical visualization and sampling with histologic review are necessary to confirm the diagnosis of endometriosis, which makes diagnosis more difficult. In addition, clinical presentation, treatment response, or prognosis do not frequently correlate with classification and staging (7, 8).
The exact etiology of endometriosis is largely unknown, but a few theories have been proposed and include retrograde menstruation, coelomic metaplasia, and lymphatic and vascular metastasis (9). Retrograde menstruation is a widely accepted proposed mechanism that refers to the overflow of menstrual debris, containing endometrial tissue, through the fallopian tubes and into the pelvic peritoneal cavity (10). Additional factors are necessary to explain retrograde menstruation however, since retrograde menstruation occurs in most reproductive-aged women, but only 10% of reproductive-aged women have a diagnosis of endometriosis. It is suggested that endometrial stromal cells provide adhesive ability through integrin and localized inflammatory responses compared to normal endometrial stromal cells (9).
Current medical treatment focuses on hormonal manipulation to induce a hypoestrogenic state in women. Common medications include combined oral contraceptive pills, progestins, and gonadotropin-releasing hormone (GnRH) agonists and antagonists (11, 12). However, their ineffectiveness, side effects, and recurrence after discontinuation often limit these options. In addition, adverse side effects, including hot flashes, memory loss, and insomnia are often associated with GnRH agonists (13). Surgical management for excision of endometriosis is an option for patients who desire relief of symptoms, but recurrence is commonly encountered postoperatively. Overall, safe, effective, non-hormonal targeted therapies are limited/inadequate for patients with endometriosis. This article aims to review our current knowledge of endometriosis-associated angiogenesis and discuss anti-angiogenic treatment as non-hormonal therapy for the treatment of endometriosis (14).
Angiogenesis and Endometriosis
The precise pathways in the pathogenesis of endometriosis are complex and largely unknown. Various theories have been proposed as potential sources of endometriotic lesions (9), but the maintenance and progression of endometriosis entail a complex interplay of many molecular mechanisms. Immune dysregulation with localized inflammation, hyperproliferation, anti-apoptosis, and enhanced angiogenesis have a critical role in the progression of this disease. For instance, in endometriotic endometrial cells, TNFα-induced apoptosis signaling is effectively suppressed by steroid receptor coactivator-1 (SRC-1) isoform/ERβ axis (15–17). ERβ also causes inflammasome-mediated hyperproliferation of endometriotic lesions for the progression of endometriosis (16).
Angiogenesis is a physiologic process that provides fundamental vasculature for the overall growth and repair of organisms' systemic and local tissue needs. Highly regulated angiogenesis is essential for normal reproduction and plays a crucial role in follicular maturation, development of a functional corpus luteum, and endometrial growth (18). Angiogenic dysregulation, or the excessive growth of new blood vessels, can contribute to the establishment and progression of many diseases. In this frame of reference, angiogenesis has a critical role in the pathogenesis of endometriosis, because the growth of new blood vessels from pre-existing vessels is necessary for the survival and progression of ectopic endometrial implants. Limited information describing the precise mechanism of endometriosis-associated angiogenesis is available, however. Neovascularization from a complex system of cytokines, growth factors, steroids, and eucasanoids in the peritoneal environment, is thought to aid in recruiting new capillaries for the progression of ectopic lesions (18). Furthermore, mobilization and recruitment of bone marrow-derived endothelial progenitor cells (EPCs) to areas of hypoxic tissue, or “vasculogenesis,” also provides de novo formation of microvessels in endometriosis (19, 20). Vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 were found to stimulate the mobilization of EPCs from bone marrow (19). Moreover, the dynamics of hypoxia with endothelial injury, inflammation, and ERα expression aid in the employment of EPCs for the growth of endometriotic implants (21, 22).
Factors for Endometriosis-Associated Angiogenesis
The distinct interplay between cytokines, growth factors, and angiogenic factors aid in establishing and progressing endometriotic implants.
Cytokine
The immune system, particularly Interleukin (IL)-1β, the dominant interleukin-1 secreted by activated peritoneal macrophages, stimulates stromal cells to produce angiogenic molecules (14). Interleukin (IL)-6, produced by endometriotic stromal cells in the presence of (IL)-1β, also increases angiogenic factors in neutrophils to stimulate endometriosis-associated angiogenesis (17, 23). IL-8, a pro-angiogenic factor, may potentiate neovascularization of ectopic implants, as elevation of IL-8 is observed in endometriosis patients (24, 25). IL-17A, in human endometriotic lesions, significantly increases angiogenic (VEGF, IL-8, IL-6, and IL-1β) and chemotactic cytokines (G-CSF, CXCL12, CXCL1, and CX3CL1) in endometrial cells (26). VEGF is a very potent and highly responsive angiogenic factor. Numerous factors aid in VEGF modulation, including Activin A (27) and IL-1β (14). VEGF protein expression is present in normal endometrial stromal cells, with levels increasing in response to estrogen and progesterone (17, 28). Cyclic VEGF expression is observed throughout the menstrual cycle and has the most significant expression during the secretory phase (28). Compared to women without endometriosis, increased VEGF levels are found in both peritoneal fluid of women with endometriosis and ectopic endometriotic tissue and contribute to the angiogenic microenvironment in endometriosis (29–31). In addition to endometriotic lesions, the VEGFR1/VEGF signaling in macrophages and fibroblasts enhance the growth of endometriotic lesions by activating lymphangiogenesis (32).
Transcription Factors
Hypoxia-inducible factor enhances expression of pro-angiogenic factors, such as VEGF, in vascular endothelial cells to enhance hypoxia-induced angiogenesis (33, 34). Ovarian endometriomas have a higher level of HIF-1α compared to normal endometrium (35). In the presence of HIF-1α, VEGF mRNA expression levels increase in response to hypoxia; moreover, HIF-1α is required for oxygen-regulated transcriptional activation of genes encoding VEGF to enhance hypoxia-induced angiogenesis (36, 37). Thus, the HIF-1α/VEGF axis is critical in endometriosis-associated angiogenesis. In addition to angiogenesis, HIF-1α also promotes endometriotic stroma cell migration and invasion by up-regulating autophagy in endometriosis (38).
Since endometriosis is an estrogen-dependent disease, estrogen and estrogen receptors (ERs) are critical for the progression of endometriosis (39). Additionally, endometriotic tissue has higher local estradiol concentrations than normal endometrium by increasing steroidogenic factor I (SF-I) and aromatase (2, 10). In a preclinical model of endometriosis, targeting ERs, chloroindazole (CLI) for ERα and oxabicycloheptene sulfonate (OBHS) for ERβ, effectively suppresses endometriosis progression by inhibiting ER-dependent inflammatory activity (40). Additionally, PHTPP, a selective ERβ antagonist, effectively suppresses endometriosis progression in mice with endometriosis (16).
Estrogen is a pro-angiogenic hormone, widely known for its effects of neovascularization and angiogenesis in the uterus and endometrium through proliferation and migration of endothelial cells and formation of new matrices around vessels. Estrogen-mediated angiogenesis, however, is also necessary in non-reproductive tissue for wound healing, reestablishment of blood supply to ischemic tissue, tumor growth, and repair of damaged organs (41–43). Furthermore, endothelial progenitor cells, which play an important role in angiogenesis, are hormonally regulated (44).
What is the correlation between estrogen and angiogenesis for endometriosis progression? The 17β-Estradiol (E2) up-regulates VEGF expression in human primary endometrial stromal cells by activating the Wnt/β-catenin axis through ERs and thus enhances their ability to establish a new blood supply to the human exfoliated endometrium (Figure 1) (45, 49). Estrogen and selective agonist for each subtype of ER, such as ERα agonist, 4,4′,4″-(4-propyl-(1H)-pyrazole-1,3,5-tryl) triphenol and ERβ agonist 2,3-bis(4-hydroxy-phenyl)-propionitrile [DPN]) regulates the axonal guidance molecules of the SLIT/ROBO signaling that have a critical role in neuroangiogenesis occurring in endometriosis lesions found on the peritoneal wall (50). Furthermore, ERβ directly regulates the expression of genes involved in hypoxia-induced angiogenesis, such as HIF1α, VEGF, and Angiotensin (Ang)1 in ectopic lesions of mice with endometriosis to stimulate endometriosis progression (51). Therefore, E2/ERs axis has a critical role in regulating genes involved in endometriosis-associated angiogenesis in endometriotic lesions to promote endometriosis progression. In addition to E2, exposure of bisphenol A elevates ERβ in mouse endometrium, promoting endometriosis progression by activating ERβ-regulated endometriosis cellular pathways involving angiogenesis (52). Besides ERs, G Protein-Coupled Estrogen Receptor (GPER) levels are significantly elevated in endometriotic lesions and endometriosis-associated macrophages by stress-related hormones and inflammation (53, 54). Therefore, rapid estrogen effects mediated by GPER also have a critical role in the hormonal regulation of endometriosis.
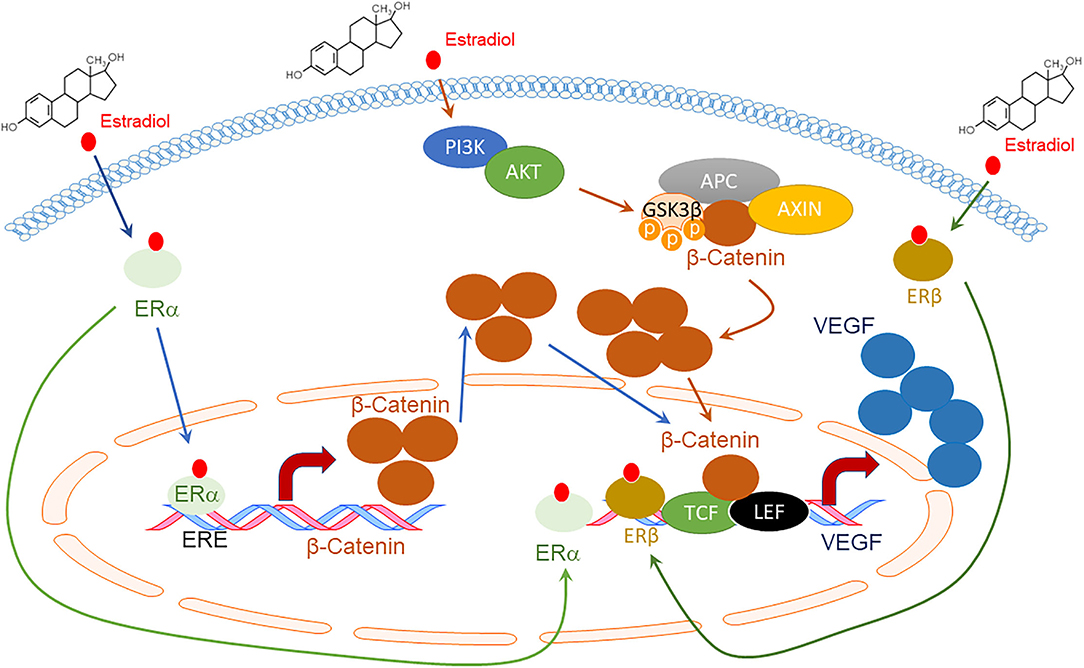
Figure 1. Schematic diagram of VEGF expression by Estradiol (E2) and Estrogen Receptors (ERs) through the Wnt/β-catenin signaling pathway. E2 promotes the direct binding of ERα to the Estrogen Response Element (ERE) site of the β-catenin promotor, enhancing its expression (45). E2 also activates the Phosphoinositide 3-kinases (PI3Ks)/AKT serine-threonine protein kinase (AKT) axis, which inactivates Glycogen synthase kinase (GSK) 3β through phosphorylation. The inhibited β-catenin destruction complex, which consists of APC regulator of WNT signaling pathways (APC) and Axis Inhibition Protein (AXIN), decreases β-catenin degradation. Accumulated β-Catenin enters the nucleus to bind to transcription factor 3/lymphoid enhancing binding factor 1 (TCF3/LEF1), enhancing VEGF expression (45). ERα and ERβ also directly bind to the VEGF promoter region and increase VEGF expression upon E2 activation (46–48).
Peptide hormones regulate angiogenesis by stimulation or inhibition to promote or prevent the growth of target tissue. Proteolysis converts the original hormone to either pro- or anti-angiogenic peptides. For example, growth hormone, prolactin, and placental lactogen family are structurally and functionally related, all released from the anterior pituitary. These hormones are pro-angiogenic when released, but upon proteolysis, they display angiogenic inhibitory properties. Endothelin, gonadotropins, insulin-like growth factor I (IGF-I), parathyroid hormone, and thyroid-stimulating hormone exhibit pro-angiogenic properties, whereas angiotensin, somatostatin, and natriuretic peptides demonstrate angiogenic inhibitory properties (55). Endothelin and IGF-1 may also play a role in endometriosis-associated angiogenesis as their levels were found to be significantly higher in women with endometriosis compared to controls (56, 57).
Although the exact molecular etiology of thyroid hormone and pro-angiogenesis has yet to be discovered, thyroid hormone has been implicated in both physiologic and pathologic angiogenesis in experimental models. Thyroid hormone-induced cardiac hypertrophy and ischemia models have demonstrated sustained angiogenesis and coronary blood flow. The hormone may also induce the expression of transcription factors that play a role in coronary artery collateralization in hypoxia (58). Furthermore, larger endometriotic implants were found with increased thyroid hormone levels, and increased chronic pelvic pain and disease score were noted in endometriotic patients with thyroid disorder (59). Given these findings, thyroid hormone may contribute to endometriosis-associated angiogenesis.
Angiogenesis in Tumor Progression
Tumor angiogenesis differs significantly from physiologic angiogenesis. However, distinctions between angiogenesis in tumors vs. endometriosis are mainly unknown. Therefore, understanding tumor angiogenesis is essential for developing and advancing anti-angiogenic therapy, which has been employed as a potential treatment option for endometriosis.
Angiogenesis is constitutively activated during tumor progression to promote cancer cell progression by activating neovascularization (60). Physiologic angiogenesis is more tightly regulated and stabilizes once new vessels are formed. However, angiogenic tumor vessels are dilated and tortuous, and vascular density and blood vessel diameter are not uniform (61). Moreover, the tumor microenvironment favors hypoxic conditions. Therefore, the cancer-associated hypoxic conditions up-regulate Hypoxia-Inducible Factor (HIF)-1α transcription factor to increase its target genes, which include vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), placental growth factor (PlGF), and hepatocyte growth factor (HFG) in tumors to generate angiogenic tumor vessels (62–65).
The hypoxia /HIF1-α axis is required for normal endometrial repair during menstruation (66). In addition to normal endometrial function, endometriosis is associated with local angiogenic and hypoxic mechanisms, similar to cancer angiogenesis. For example, ovarian endometriomas express high levels of HIF-1/2α, and VEGF-A expression compared to endometrium of women without endometriosis (35). HIF-1α levels are elevated in ectopic endometrial lesions, and hypoxia plays a critical role in the survival of retrograde reflux of endometrial fragments during angiogenesis in early implanted ectopic endometrial lesions (67). Therefore, HIF1-α-mediated angiogenesis has a critical role in endometriosis-like cancer progression.
Non-Angiogenic Pathways in Tumor Progression
Anti-angiogenesis therapy has demonstrated only modest success in cancer patients, and, thus, further efforts have been geared toward examining non-angiogenic modalities for tumor growth. Formation of blood vessels in cancer cells through non-angiogenic modalities that rely on alternative vascularization methods include Vascular mimicry, Vascular co-option of vessels, and Intussusceptive microvascular growth, or IMG (68).
Vasculogenic mimicry creates microvascular channels in tumor cells without the presence of endometrial cells, providing a network of fluid-conducting channels (69).
Vascular co-option of vessels is a non-angiogenic means for cancer cells to obtain a blood supply by hijacking pre-existing blood vessels in the surrounding tissue to support tumor growth and metastasis (70).
Vessel intussusception, or intussusceptive microvascular growth (IMG), generates new vascular structures by extending the capillary wall into the lumen of pre-existing vessels. Compared to angiogenesis, IMG is typically rapid as it doesn't rely on the proliferation of endothelial cells but rather on the remodeling of existing vascular structures (71). However, it has not been reported whether non-angiogenic pathways are also involved in endometriosis-associated angiogenesis.
Anti-Angiogenic Therapy for Endometriosis Treatment
Angiogenesis has a critical role in endometriosis progression. Therefore, various angiogenic blockers have been employed as non-hormonal therapy for endometriosis (Table 1). For example, VEGF blockers and inhibitors have demonstrated promising results in mice, decreasing the number of endometriotic implants, reducing vascular density, increasing apoptosis, and reducing VEGF levels in peritoneal fluid (81, 121, 122). Similar results were achieved in a rat model without compromising ovarian reserve (120). Furthermore, in a human clinical trial, a patient was treated for severe endometriosis with Bevacizumab (Avastin, a monoclonal antibody directed against VEGF) and reported complete disappearance of her therapy-refractory dysmenorrhea. Diffuse fibrosis of her endometriosis lesions was also observed at second-look laparoscopy (123).
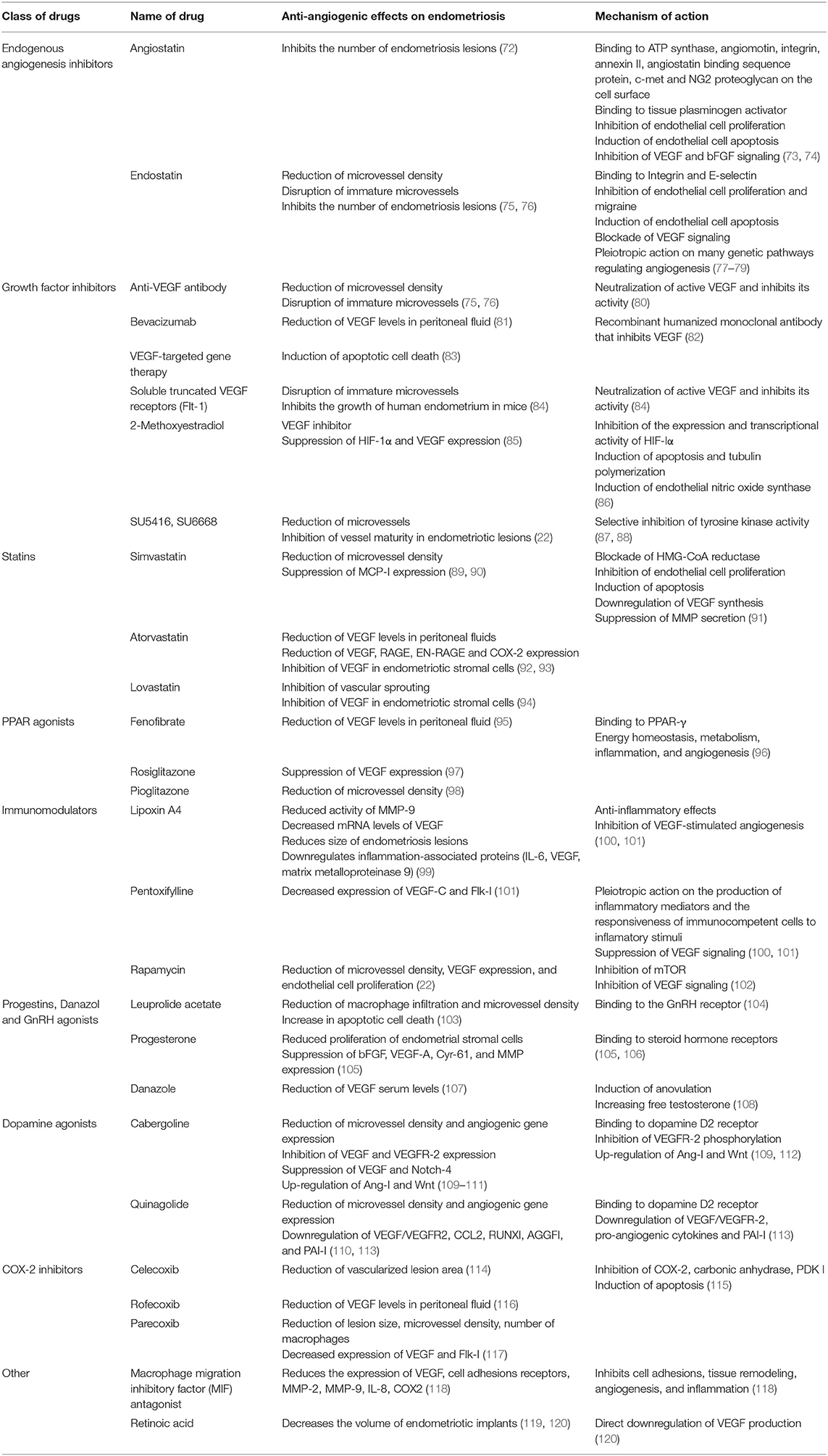
Table 1. Anti-angiogenic therapy and their mechanisms of action.
Other assuring anti-angiogenic therapies have been investigated. For example, macrophage migration inhibitory factor (MIF) demonstrates the development of endometriosis in vivo and demonstrated pro-apoptosis activity (118, 121). Retinoic acid has known anti-angiogenic characteristics, suppressing the growth of endometriotic lesions and inhibiting peritoneal cytokine secretion in an immunocompetent mouse model (119, 121). Statins inhibit inflammation and angiogenic genes, cyclooxygenase-2 and VEGF, in endometriotic stromal cells (92, 121). Cabergoline, a dopamine agonist, inhibits the development of endometriosis by inhibiting VEGF and VEGFR-2 (109, 121) and may prove an effective therapy for women with chronic pain due to endometriosis (124).
Furthermore, women with endometriosis display significantly elevated levels of ERβ in ovarian endometrioma when compared to normal endometrium (125). ERβ overexpression could potentiate infertility in women with endometriosis because ERβ overexpression impairs decidualization in the stroma of their endometrium in mice (16). ERβ also has a critical role in the progression of endometriotic lesions, including angiogenesis (51). Therefore, targeting ERβ could benefit both regression of ectopic implants by inhibiting proliferation and angiogenesis of endometriotic lesions and optimizing endometrial receptivity in patients with endometriosis. The 2-methoxyestradiol is a natural metabolite of estradiol and binds to GPER, but not ERs (126). The 2-methoxyestradiol suppresses the growth of ectopic lesions in mice model of endometriosis (85) by inhibiting angiogenesis of endometriosis progression because 2-methoxyestradiol downregulates angiotensin AT1 receptor (127). Also, 2-methoxyestradiol suppressed HIF-1α expression in vivo, results in a decreased expression of HIF-1α target genes, such as VEGF, phosphoglycerate kinase, and glucose transporter-1 (85). Therefore, targeting GPER can be employed as anti-angiogenic therapy for endometriosis treatment.
Modes of Resistance to Anti-Angiogenic Therapies
Although angiogenesis is a critical regulator in the progression of malignant tumors, individuals may respond differently to anti-angiogenic therapy. These anti-angiogenic agents are not equally active across all tumor types. Few clinical studies support the theory that anti-angiogenic treatments can prevent activation of the angiogenic switch in the progression and metastasis of tumors (128). Furthermore, anti-angiogenic biomarkers do not adequately represent a specific response to these therapeutic agents (129).
Anti-angiogenic therapy has resulted in transient improvements in cancer treatment with tumor stability or shrinkage and increased survival. Over time, however, the tumor re-establishes its growth and progression, challenging the notion that angiogenesis is necessary for the advancement of tumors. Adaptive, or evasive, resistance and intrinsic, or pre-existing, non-responsiveness have been suggested as distinct pathways in evading anti-angiogenic therapy. Adaptive resistance theorizes that angiogenic tumors can adapt to the presence of an anti-angiogenic agent and obtain mechanisms to escape the inhibition of angiogenesis. Comparably, intrinsic resistance likely encompasses a similar mechanism, but the tumor may have underlying characteristics (type of tumor, stage of progression, treatment history, individual genotype) that place an individual at greater risk. In addition, those with pre-existing non-responsiveness typically see no clinical benefit with angiogenesis inhibitors, whereas those with evasive non-responsiveness demonstrate transient benefit (130).
Although resistance to anti-angiogenic therapy has also been noted, interestingly, in some cases, acquired resistance may be a transient occurrence. Sequential therapy with a similar, but not identical, anti-angiogenic drug is a strategy to delay the onset of acquired resistance or treat cancers that have progressed through anti-angiogenic therapy (131).
Discussion
Non-hormonal targeted therapy for endometriosis is critical for caring for women with endometriosis to circumvent the adverse effects of current hormonal treatment. Our present knowledge of endometriosis-associated angiogenesis suggests that anti-angiogenesis therapy can aid in targeted treatment for these patients. The proposed pathogenic pathways discussed in this article may assist in developing targeted therapies through adequately powered studies and a multidisciplinary approach.
Anti-angiogenic therapy may adversely impact normal physiologic angiogenesis, such as ovulation and wound healing (132), leading to adverse effects on reproductive function and teratogenicity during the treatment of endometriosis in reproductive-aged women. Thus, it is crucial to define the distinct angiogenic pathways specific for endometriosis to mitigate these potential side effects. ERβ and GPER are highly elevated in endometriotic tissue, compared to normal tissue, and could serve as new molecular therapeutic targets to suppress endometriosis-specific angiogenesis.
Smaller anti-angiogenic molecules, derived from natural products, have significant advantages over synthetic inhibitors. For instance, polyphenols, polysaccharides, alkaloids, terpenoids, and saponins containing natural products target tumor angiogenesis (133). Furthermore, smaller anti-angiogenic natural products can be more easily manufactured, confer lower cost, provide higher efficacy, and have little to no known toxicity (134). Ultimately, it is imperative to conduct further studies to explore natural angiogenesis inhibitors and evaluate its anti-angiogenic efficacy as a non-hormonal therapy for endometriosis treatment in the future.
Author Contributions
MC drafted the manuscript. SH revised the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by grant funding from NICHD (5R01HD098059).
Author Disclaimer
This manuscript review does not necessarily reflect the opinions or views of Baylor College of Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kitawaki J, Kado N, Ishihara H, Koshiba H, Kitaoka Y, Honjo H. Endometriosis: the pathophysiology as an estrogen-dependent disease. J Steroid Biochem Mol Biol. (2002) 83:149e155. doi: 10.1016/S0960-0760(02)00260-1
3. Shafrir AL, Farland LV, Shah DK, Harris HR, Kvaskoff M, Zondervan K, et al. Risk for and consequences of endometriosis: a critical epidemiologic review. Best Pract Res Clin Obstet Gynaecol. (2018) 51:1–15. doi: 10.1016/j.bpobgyn.2018.06.001
4. Ozkan S, Murk W, Arici A. Endometriosis and infertility. Epidemiology and evidence based treatments. Ann N Y Acad Sci. (2008) 1127:92e100. doi: 10.1196/annals.1434.007
5. Ballard KD, Seaman HE, de Vries CS, Wright JT. Can symptomatology help in the diagnosis of endometriosis? Findings from a national case-control study—part 1. BJOG. (2008) 115:1382–91. doi: 10.1111/j.1471-0528.2008.01878.x
6. Dunselman GA, Vermeulen N, Becker C, Calhaz-Jorge C, D'Hooghe T, De Bie B, et al. ESHRE guideline: management of women with endometriosis. Hum Reprod. (2014) 29:400–12. doi: 10.1093/humrep/det457
7. Schliep KC, Mumford SL, Peterson CM. Pain typology and incident endometriosis. Hum Reprod. (2015) 30:2427–38. doi: 10.1093/humrep/dev147
8. Johnson NP, Hummelshoj L, Adamson GD. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. (2017) 32:315–24. doi: 10.1093/humrep/dew293
9. Zondervan KT, Becker CM, Missmer SA. Endometriosis. N Engl J Med. (2020) 382:1244–56. doi: 10.1056/NEJMra1810764
10. Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol. (1927) 14:422–69. doi: 10.1016/S0002-9378(15)30003-X
11. Ozkan S, Arici A. Advances in treatment options of endometriosis. Gynecol Obstet Invest. (2009) 67:81–91. doi: 10.1159/000163071
12. Vercellini P, Buggio L, Frattaruolo MP, Borghi A, Dridi D, Somigliana E. Medical treatment of endometriosis-related pain. Best Pract Res Clin Obstet Gynaecol. (2018) 51:68–91. doi: 10.1016/j.bpobgyn.2018.01.015
13. Gallagher JS, Missmer SA, Hornstein MD, Laufer MR, Gordon CM, DiVasta AD. Long-term effects of gonadotropin-releasing hormone agonists and add-back in adolescent endometriosis. J Pediatr Adolesc Gynecol. (2018) 31:376–81. doi: 10.1016/j.jpag.2018.03.004
14. Lebovic DI, Bentzien F, Chao VA, Garrett EN, Meng YG, Taylor RN. Induction of an angiogenic phenotype in endometriotic stromal cell cultures by interleukin-1beta. Mol Hum Reprod. (2000) 6:269–75. doi: 10.1093/molehr/6.3.269
15. Han SJ, Hawkins SM, Begum K, Jung SY, Kovanci E, Qin J, et al. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat Med. (2012) 18:1102–11. doi: 10.1038/nm.2826
16. Han SJ, Jung SY, Wu S-P. Estrogen receptor β modulates apoptosis complexes and the inflammasome to drive the pathogenesis of endometriosis. Cell. (2015) 163:960–74. doi: 10.1016/j.cell.2015.10.034
17. Lebovic DI, Shifren JL, Ryan IP, Mueller MD, Korn AP, Darney PD, et al. Ovarian steroid and cytokine modulation of human endometrial angiogenesis. Hum Reprod. (2000) 15(Suppl.3):67–77. doi: 10.1093/humrep/15.suppl_3.67
18. Taylor RN, Lebovic DI, Mueller MD. Angiogenic factors in endometriosis. Ann N Y Acad Sci. (2002) 955:89–100. doi: 10.1111/j.1749-6632.2002.tb02769.x
19. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. (2002) 29(6 Suppl.16):15–8. doi: 10.1053/sonc.2002.37263
20. Taylor HS. Bone marrow in the pathophysiology of endometriosis. Fertility Sterility. (2020) 113:942. doi: 10.1016/j.fertnstert.2020.03.012
21. Laschke MW, Elitzsch A, Vollmar B, Menger MD. In vivo analysis of angiogenesis in endometriosis-like lesions by intravital fluorescence microscopy. Fertil Steril. (2005) 84(Suppl.2):1199–209. doi: 10.1016/j.fertnstert.2005.05.010
22. Laschke MW, Elitzsch A, Vollmar B, Vajkoczy P, Menger MD. Combined inhibition of vascular endothelial growth factor (VEGF), fibroblast growth factor and platelet-derived growth factor, but not inhibition of VEGF alone, effectively suppresses angiogenesis and vessel maturation in endometriotic lesions. Hum Reprod. (2006) 21:262–8. doi: 10.1093/humrep/dei308
23. Guo F, He Y, Fan Y, Du Z, Sun H, Feng Z, et al. G-CSF and IL-6 may be involved in formation of endometriosis lesions by increasing the expression of angiogenic factors in neutrophils. Mol Hum Reprod. (2021) 27:gaab064. doi: 10.1093/molehr/gaab064
24. Barcz E, Rózewska ES, Kaminski P, Demkow U, Bobrowska K, Marianowski L. Angiogenic activity and IL-8 concentrations in peritoneal fluid and sera in endometriosis. Int J Gynaecol Obstet. (2002) 79:229–35. doi: 10.1016/S0020-7292(02)00308-9
25. Conroy S, Kruyt FAE, Wagemakers M, Bhat KPL, den Dunnen WFA. IL-8 associates with a pro-angiogenic and mesenchymal subtype in glioblastoma. Oncotarget. (2018) 9:21. doi: 10.18632/oncotarget.24595
26. Ahn SH, Edwards AK, Singh SS, Young SL, Lessey BA, Tayade C. IL-17A contributes to the pathogenesis of endometriosis by triggering proinflammatory cytokines and angiogenic growth factors. J Immunol. (2015) 195:2591–600. doi: 10.4049/jimmunol.1501138
27. Ervolino De Oliveira C, Dourado MR, Sawazaki-Calone Í, Costa De Medeiros M, Rossa Júnior C, De Karla Cervigne N, et al. Activin A triggers angiogenesis via regulation of VEGFA and its overexpression is associated with poor prognosis of oral squamous cell carcinoma. Int J Oncol. (2020) 57:364–76. doi: 10.3892/ijo.2020.5058
28. Shifren JL, Tseng JF, Zaloudek CJ, Ryan IP, Meng YG, Ferrara N, et al. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: implications for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis. J Clin Endocrinol Metab. (1996) 81:3112–8. doi: 10.1210/jcem.81.8.8768883
29. Donnez J, Smoes P, Gillerot S, Casanas-Roux F, Nisolle M. Vascular endothelial growth factor (VEGF) in endometriosis. Hum Reprod. (1998) 13:1686–90. doi: 10.1093/humrep/13.6.1686
30. Bourlev V, Volkov N, Pavlovitch S, Lets N, Larsson A, Olovsson M. The relationship between microvesseldensity, proliferative activity and expression of vascular endothelial growth factor-A and its receptors in eutopic endometrium and endometriotic lesions. Reproduction. (2006) 132:501–9. doi: 10.1530/rep.1.01110
31. McLaren J. Vascular endothelial growth factor and endometriotic angiogenesis. Hum Reprod Update. (2000) 6:45–55. doi: 10.1093/humupd/6.1.45
32. Hattori K, Ito Y, Honda M, Sekiguchi K, Hosono K, Shibuya M, et al. Lymphangiogenesis induced by vascular endothelial growth factor receptor 1 signaling contributes to the progression of endometriosis in mice. J Pharmacol Sci. (2020) 143:255–63. doi: 10.1016/j.jphs.2020.05.003
33. Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5' enhancer. Circ Res. (1995) 77:638–43. doi: 10.1161/01.RES.77.3.638
34. Hashimoto T, Shibasaki F. Hypoxia-inducible factor as an angiogenic master switch. Front Pediatr. (2015) 3:33. doi: 10.3389/fped.2015.00033
35. Filippi I, Carrarelli P, Luisi S, Batteux F, Chapron C, Naldini A, et al. Different expression of hypoxic and angiogenic factors in human endometriotic lesions. Reprod Sci. (2016) 23:492–7. doi: 10.1177/1933719115607978
36. Li J, Li SX, Gao XH, Zhao LF, Du J, Wang TY, et al. HIF1A and VEGF regulate each other by competing endogenous RNA mechanism and involve in the pathogenesis of peritoneal fibrosis. Pathol Res Practice. (2019) 215:644–52. doi: 10.1016/j.prp.2018.12.022
37. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 12:149–62. doi: 10.1101/gad.12.2.149
38. Liu H, Zhang Z, Xiong W, Zhang L, Xiong Y, Li N, et al. Hypoxia-inducible factor-1α promotes endometrial stromal cells migration and invasion by up-regulating autophagy in endometriosis. Reproduction. (2017) 153:809–20. doi: 10.1530/REP-16-0643
39. Burns KA, Rodriguez KF, Hewitt SC, Janardhan KS, Young SL, Korach KS. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology. (2012) 153:3960–71. doi: 10.1210/en.2012-1294
40. Zhao Y, Gong P, Chen Y, Nwachukwu JC, Srinivasan S, Ko C, et al. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Scie Transl Med. (2015) 7:271ra9. doi: 10.1126/scitranslmed.3010626
41. Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev. (2008) 60:210–41. doi: 10.1124/pr.107.08002
42. Cid MC, Schnaper HW, Kleinman HK. Estrogens and the vascular endothelium. Ann N Y Acad Sci. (2002) 966:143–57. doi: 10.1111/j.1749-6632.2002.tb04211.x
43. Rubanyi GM, Johns A, Kauser K. Effect of estrogen on endothelial function and angiogenesis. Vascul Pharmacol. (2002) 38:89–98. doi: 10.1016/S0306-3623(02)00131-3
44. Sieveking DP, Chow RW, Ng MK. Androgens, angiogenesis and cardiovascular regeneration. Curr Opin Endocrinol Diabetes Obes. (2010) 17:277–83. doi: 10.1097/MED.0b013e3283394e20
45. Zhang L, Xiong W, Xiong Y, Liu H, Liu Y. 17 β-Estradiol promotes vascular endothelial growth factor expression via the Wnt/β-catenin pathway during the pathogenesis of endometriosis. Mol Hum Reprod. (2016) 22:526–35. doi: 10.1093/molehr/gaw025
46. Mueller MD, Vigne JL, Minchenko A, Lebovic DI, Leitman DC, Taylor RN. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors alpha and beta. Proc Natl Acad Sci USA. (2000) 97:10972–7. doi: 10.1073/pnas.200377097
47. Dadiani M, Seger D, Kreizman T, Badikhi D, Margalit R, Eilam R, et al. Estrogen regulation of vascular endothelial growth factor in breast cancer in vitro and in vivo: the role of estrogen receptor alpha and c-Myc. Endocr Relat Cancer. (2009) 16:819–34. doi: 10.1677/ERC-08-0249
48. Chen L, Yang Y, Zhang L, Li C, Coffie JW, Geng X, et al. Aucubin promotes angiogenesis via estrogen receptor beta in a mouse model of hindlimb ischemia. J Steroid Biochem Mol Biol. (2017) 172:149–59. doi: 10.1016/j.jsbmb.2017.07.007
49. Liu S, Fan W, Gao X, Huang K, Ding C, Ma G, et al. Estrogen receptor alpha regulates the Wnt/β-catenin signaling pathway in colon cancer by targeting the NOD-like receptors. Cell Signal. (2019) 61:86–92. doi: 10.1016/j.cellsig.2019.05.009
50. Greaves E, Collins F, Esnal-Zufiaurre A, Giakoumelou S, Horne AW, Saunders PT. Estrogen receptor (ER) agonists differentially regulate neuroangiogenesis in peritoneal endometriosis via the repellent factor SLIT3. Endocrinology. (2014) 155:4015–426. doi: 10.1210/en.2014-1086
51. Han SJ, Lee JE, Cho YJ, Park MJ, O'Malley BW. Genomic function of estrogen receptor β in endometriosis. Endocrinology. (2019) 160:2495–516. doi: 10.1210/en.2019-00442
52. Xue W, Yao X, Ting G, Ling J, Huimin L, Yuan Q, et al. BPA modulates the WDR5/TET2 complex to regulate ERβ expression in eutopic endometrium and drives the development of endometriosis. Environ Poll. (2021) 268:115748. doi: 10.1016/j.envpol.2020.115748
53. Samartzis N, Samartzis EP, Noske A, Fedier A, Dedes KJ, Caduff R, et al. Expression of the G protein-coupled estrogen receptor (GPER) in endometriosis: a tissue microarray study. Reprod Biol Endocrinol. (2012) 10:30. doi: 10.1186/1477-7827-10-30
54. Heublein S, Vrekoussis T, Kuhn C, Friese K, Makrigiannakis A, Mayr D, et al. Inducers of G-protein coupled estrogen receptor (GPER) in endometriosis: potential implications for macrophages and follicle maturation. J Reprod Immunol. (2013) 97:95–103. doi: 10.1016/j.jri.2012.10.013
55. Clapp C, Thebault S, Jeziorski MC, Martínez De La Escalera G. Peptide hormone regulation of angiogenesis. Physiol Rev. (2009) 89:1177–215. doi: 10.1152/physrev.00024.2009
56. Abaé M, Glassberg M, Majercik MH, Yoshida H, Vestal R, Puett D. Immunoreactive endothelin-1 concentrations in follicular fluid of women with and without endometriosis undergoing in vitro fertilization-embryo transfer. Fertil Steril. (1994) 61:1083–7. doi: 10.1016/S0015-0282(16)56760-0
57. Heidari S, Kolahdouz-Mohammadi R, Khodaverdi S, Tajik N, Delbandi AA. Expression levels of MCP-1, HGF, IGF-1 in endometriotic patients compared with non endometriotic controls. BMC Womens Health. (2021) 21:422. doi: 10.1186/s12905-021-01560-6
58. Luidens MK, Mousa SA, Davis FB, Lin HY, Davis PJ. Thyroid hormone and angiogenesis. Vascul Pharmacol. (2010) 52:142–5. doi: 10.1016/j.vph.2009.10.007
59. Peyneau M, Kavian N, Chouzenoux S, Nicco C, Jeljeli M, Toullec L, et al. Role of thyroid dysimmunity and thyroid hormones in endometriosis. Proc Natl Acad Sci USA. (2019) 116:11894–9. doi: 10.1073/pnas.1820469116
60. Katayama Y, Uchino J, Chihara Y, Tamiya N, Kaneko Y, Yamada T, et al. Tumor neovascularization and developments in therapeutics. Cancers. (2019) 11:316. doi: 10.3390/cancers11030316
61. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. (2003) 3:401–10. doi: 10.1038/nrc1093
62. Bos R, Van Diest PJ, De Jong JS, Van Der Groep P, Van Der Valk P, Van Der Wall E. Hypoxia-inducible factor-1alpha is associated with angiogenesis, and expression of bFGF, PDGF-BB, EGFR in invasive breast cancer. Histopathology. (2005) 46:31–6. doi: 10.1111/j.1365-2559.2005.02045.x
63. Razban V, Lotfi AS, Soleimani M, Ahmadi H, Massumi M, Khajeh S, et al. HIF-1α overexpression induces angiogenesis in mesenchymal stem cells. Biores Open Access. (2012) 1:174–83. doi: 10.1089/biores.2012.9905
64. Tudisco L, Orlandi A, Tarallo V, De Falco S. Hypoxia activates placental growth factor expression in lymphatic endothelial cells. Oncotarget. (2017) 8:32873–83. doi: 10.18632/oncotarget.15861
65. Zhu H, Zhang S. Hypoxia inducible factor-1α/vascular endothelial growth factor signaling activation correlates with response to radiotherapy and its inhibition reduces hypoxia-induced angiogenesis in lung cancer. J Cell Biochem. (2018) 119:7707–18. doi: 10.1002/jcb.27120
66. Maybin JA, Murray AA, Saunders PTK, Hirani N, Carmeliet P, Critchley HOD. Hypoxia and hypoxia inducible factor-1α are required for normal endometrial repair during menstruation. Nat Commun. (2018) 9:295. doi: 10.1038/s41467-017-02375-6
67. Wu MH, Chen KF, Lin SC, Lgu CW, Tsai SJ. Aberrant expression of leptin in human endometriotic stromal cells is induced by elevated levels of hypoxia inducible factor 1alpha. Am J Pathol. (2007) 170:590–8. doi: 10.2353/ajpath.2007.060477
68. El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, et al. A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain. (2010) 133:973–82. doi: 10.1093/brain/awq044
69. Fernández-Cortés M, Delgado-Bellido D, Oliver FJ. Vasculogenic mimicry: become an endothelial cell “but not so much”. Front Oncol. (2019) 9:803. doi: 10.3389/fonc.2019.00803
70. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel co-option in cancer. Nat Rev Clin Oncol. (2019) 16:469–3. doi: 10.1038/s41571-019-0181-9
71. Zuazo-Gaztelu I, Casanovas O. Unraveling the role of angiogenesis in cancer ecosystems. Front Oncol. (2018) 8:248. doi: 10.3389/fonc.2018.00248
72. Dabrosin C, Gyorffy S, Margetts P, Ross C, Gauldie J. Therapeutic effect of angiostatin gene transfer in a murine model of endometriosis. Am J Pathol. (2002) 161:909–18. doi: 10.1016/S0002-9440(10)64251-4
73. Sim BK, MacDonald NJ, Gubish ER. Angiostatin and endostatin: endogenous inhibitors of tumor growth. Cancer Metastasis Rev. (2000) 19:181–90. doi: 10.1023/a:1026551202548
74. Wahl ML, Kenan DJ, Gonzalez-Gronow M, Pizzo SV. Angiostatin's molecular mechanism: aspects of specificity and regulation elucidated. J Cell Biochem. (2005) 96:242–61. doi: 10.1002/jcb.20480
75. Nap AW, Griffioen AW, Dunselman GA, Bouma-Ter Steege JC, Thijssen VL, Evers JL, et al. Antiangiogenesis therapy for endometriosis. J Clin Endocrinol Metab. (2004) 89:1089–195. doi: 10.1210/jc.2003-031406
76. Nap AW, Dunselman GA, Griffioen AW, Mayo KH, Evers JL, Groothuis PG. Angiostatic agents prevent the development of endometriosis-like lesions in the chicken chorioallantoic membrane. Fertil Steril. (2005) 83:793–75. doi: 10.1016/j.fertnstert.2004.06.080
77. Kim YM, Jang JW, Lee OH, Yeon J, Choi EY, Kim KW, et al. Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res. (2000) 60:5410–543.
78. Folkman J. Antiangiogenesis in cancer therapy–endostatin and its mechanisms of action. Exp Cell Res. (2006) 312:594–607. doi: 10.1016/j.yexcr.2005.11.015
79. Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, et al. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. (2002) 277:27872–9. doi: 10.1074/jbc.M202771200
80. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. (2005) 333:328–35. doi: 10.1016/j.bbrc.2005.05.132
81. Ricci AG, Olivares CN, Bilotas MA, Meresman GF, Barañao RI. Effect of vascular endothelial growth factor inhibition on endometrial implant development in a murine model of endometriosis. Reprod Sci. (2011) 18:614–22. doi: 10.1177/1933719110395406
82. Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. (2020) 86:102017. doi: 10.1016/j.ctrv.2020.102017
83. Rein DT, Schmidt T, Bauerschmitz G, Hampl M, Beyer IM, Paupoo AA, et al. Treatment of endometriosis with a VEGF-targeted conditionally replicative adenovirus. Fertil Steril. (2010) 93:2687–94. doi: 10.1016/j.fertnstert.2009.04.042
84. Hull ML, Charnock-Jones DS, Chan CL, Bruner-Tran KL, Osteen KG, Tom BD, et al. Anti-angiogenic agents are effective inhibitors of endometriosis. J Clin Endocrinol Metab. (2003) 88:2889–99. doi: 10.1210/jc.2002-021912
85. Becker CM, Rohwer N, Funakoshi T, Cramer T, Bernhardt W, Birsner A, et al. 2-methoxyestradiol inhibits hypoxia-inducible factor-1α and suppresses growth of lesions in a mouse model of endometriosis. Am J Pathol. (2008) 172:534–44. doi: 10.2353/ajpath.2008.061244
86. Verenich S, Gerk PM. Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges. Mol Pharm. (2010) 7:2030–9. doi: 10.1021/mp100190f
87. Mendel DB, Laird AD, Smolich BD, Blake RA, Liang C, Hannah AL, et al. Development of SU5416, a selective small molecule inhibitor of VEGF receptor tyrosine kinase activity, as an anti-angiogenesis agent. Anticancer Drug Des. (2000) 15:29–41.
89. Bruner-Tran KL, Osteen KG, Duleba AJ. Simvastatin protects against the development of endometriosis in a nude mouse model. J Clin Endocrinol Metab. (2009) 94:2489–94. doi: 10.1210/jc.2008-2802
90. Cakmak H, Basar M, Seval-Celik Y, Osteen KG, Duleba AJ, Taylor HS, et al. Statins inhibit monocyte chemotactic protein 1 expression in endometriosis. Reprod Sci. (2012) 19:572–9. doi: 10.1177/1933719111430998
91. Dulak J, Józkowicz A. Anti-angiogenic and anti-inflammatory effects of statins: relevance to anti-cancer therapy. Curr Cancer Drug Targets. (2005) 5:579–94. doi: 10.2174/156800905774932824
92. Sharma I, Dhawan V, Mahajan N, Saha SC, Dhaliwal LK. In vitro effects of atorvastatin on lipopolysaccharide-induced gene expression in endometriotic stromal cells. Fertil Steril. (2010) 94:1639–46.e1. doi: 10.1016/j.fertnstert.2009.10.003
93. Oktem M, Esinler I, Eroglu D, Haberal N, Bayraktar N, Zeyneloglu HB. High-dose atorvastatin causes regression of endometriotic implants: a rat model. Hum Reprod. (2007) 22:1474–80. doi: 10.1093/humrep/del505
94. Esfandiari N, Khazaei M, Ai J, Bielecki R, Gotlieb L, Ryan E, et al. Effect of a statin on an in vitro model of endometriosis. Fertil Steril. (2007) 87:257–62. doi: 10.1016/j.fertnstert.2006.06.040
95. Onalan G, Zeyneloglu HB, Bayraktar N. Fenofibrate causes regression of endometriotic implants: a rat model. Fertil Steril. (2009) 92:2100–2. doi: 10.1016/j.fertnstert.2009.05.065
96. Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. (2011) 2:236–40. doi: 10.4103/2231-4040.90879
97. Peeters LL, Vigne JL, Tee MK, Zhao D, Waite LL, Taylor RN. PPAR gamma represses VEGF expression in human endometrial cells: implications for uterine angiogenesis. Angiogenesis. (2005) 8:373–9. doi: 10.1007/s10456-005-9027-4
98. Herington JL, Crispens MA, Carvalho-Macedo AC, Camargos AF, Lebovic DI, Bruner-Tran KL, et al. Development and prevention of postsurgical adhesions in a chimeric mouse model of experimental endometriosis. Fertil Steril. (2011) 95:1295–301.e1. doi: 10.1016/j.fertnstert.2010.09.017
99. Xu Z, Zhao F, Lin F, Chen J, Huang Y. Lipoxin A4 inhibits the development of endometriosis in mice: the role of anti-inflammation and anti-angiogenesis. Am J Reprod Immunol. (2012) 67:491–7. doi: 10.1111/j.1600-0897.2011.01101.x
100. Olive DL, Lindheim SR, Pritts EA. New medical treatments for endometriosis. Best Pract Res Clin Obstet Gynaecol. (2004) 18:319–28. doi: 10.1016/j.bpobgyn.2004.03.005
101. Vlahos NF, Gregoriou O, Deliveliotou A, Perrea D, Vlachos A, Zhao Y, et al. Effect of pentoxifylline on vascular endothelial growth factor C and flk-1 expression on endometrial implants in the rat endometriosis model. Fertil Steril. (2010) 93:1316–23. doi: 10.1016/j.fertnstert.2008.10.056
102. Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. (2002) 8:128–35. doi: 10.1038/nm0202-128
103. Khan KN, Kitajima M, Hiraki K, Fujishita A, Sekine I, Ishimaru T, et al. Changes in tissue inflammation, angiogenesis and apoptosis in endometriosis, adenomyosis and uterine myoma after GnRH agonist therapy. Hum Reprod. (2010) 25:642–53. doi: 10.1093/humrep/dep437
104. Wilson AC, Meethal SV, Bowen RL, Atwood CS. Leuprolide acetate: a drug of diverse clinical applications. Expert Opin Investig Drugs. (2007) 16:1851–163. doi: 10.1517/13543784.16.11.1851
105. Mönckedieck V, Sannecke C, Husen B, Kumbartski M, Kimmig R, Tötsch M, et al. Progestins inhibit expression of MMPs and of angiogenic factors in human ectopic endometrial lesions in a mouse model. Mol Hum Reprod. (2009) 15:633–43. doi: 10.1093/molehr/gap063
106. Sitruk-Ware R. New progestagens for contraceptive use. Hum Reprod Update. (2006) 12:169–78. doi: 10.1093/humupd/dmi046
107. Matalliotakis IM, Goumenou AG, Koumantakis GE, Neonaki MA, Koumantakis EE, Dionyssopoulou E, et al. Serum concentrations of growth factors in women with and without endometriosis: the action of anti-endometriosis medicines. Int Immunopharmacol. (2003) 3:81–9. doi: 10.1016/S1567-5769(02)00216-3
108. Canavan TP, Radosh L. Managing endometriosis. Strategies to minimize pain and damage. Postgrad Med. (2000) 107:213–6. doi: 10.3810/pgm.2000.03.951
109. Novella-Maestre E, Carda C, Noguera I, Ruiz-Saur, í A, García-Velasco JA, Simón C, Pellicer A. Dopamine agonist administration causes a reduction in endometrial implants through modulation of angiogenesis in experimentally induced endometriosis. Hum Reprod. (2009) 24:1025–35. doi: 10.1093/humrep/den499
110. Delgado-Rosas F, Gómez R, Ferrero H, Gaytan F, Garcia-Velasco J, Simón C, et al. The effects of ergot and non-ergot-derived dopamine agonists in an experimental mouse model of endometriosis. Reproduction. (2011) 142:745–55. doi: 10.1530/REP-11-0223
111. Novella-Maestre E, Carda C, Ruiz-Sauri A, Garcia-Velasco JA, Simon C, Pellicer A. Identification and quantification of dopamine receptor 2 in human eutopic and ectopic endometrium: a novel molecular target for endometriosis therapy. Biol Reprod. (2010) 83:866–73. doi: 10.1095/biolreprod.110.084392
112. Gomez R, Gonzalez-Izquierdo M, Zimmermann RC, Novella-Maestre E, Alonso-Muriel I, Sanchez-Criado J, et al. Low-dose dopamine agonist administration blocks vascular endothelial growth factor (VEGF)-mediated vascular hyperpermeability without altering VEGF receptor 2-dependent luteal angiogenesis in a rat ovarian hyperstimulation model. Endocrinology. (2006) 147:5400–11. doi: 10.1210/en.2006-0657
113. Gómez R, Abad A, Delgado F, Tamarit S, Simón C, Pellicer A. Effects of hyperprolactinemia treatment with the dopamine agonist quinagolide on endometriotic lesions in patients with endometriosis-associated hyperprolactinemia. Fertil Steril. (2011) 95:882–8.e1. doi: 10.1016/j.fertnstert.2010.10.024
114. Olivares C, Ricci A, Bilotas M, Barañao RI, Meresman G. The inhibitory effect of celecoxib and rosiglitazone on experimental endometriosis. Fertil Steril. (2011) 96:428–33. doi: 10.1016/j.fertnstert.2011.05.063
115. Schönthal AH. Direct non-cyclooxygenase-2 targets of celecoxib and their potential relevance for cancer therapy. Br J Cancer. (2007) 97:1465–8. doi: 10.1038/sj.bjc.6604049
116. Dogan E, Saygili U, Posaci C, Tuna B, Caliskan S, Altunyurt S, et al. Regression of endometrial explants in rats treated with the cyclooxygenase-2 inhibitor rofecoxib. Fertil Steril. (2004) 82(Suppl.3):1115–120. doi: 10.1016/j.fertnstert.2004.06.033
117. Machado DE, Berardo PT, Landgraf RG, Fernandes PD, Palmero C, Alves LM, et al. A selective cyclooxygenase-2 inhibitor suppresses the growth of endometriosis with an anti-angiogenic effect in a rat model. Fertil Steril. (2010) 93:2674–9. doi: 10.1016/j.fertnstert.2009.11.037
118. Khoufache K, Bazin S, Girard K, Guillemette J, Roy MC, Verreault JP, et al. Macrophage migration inhibitory factor antagonist blocks the development of endometriosis in vivo. PLoS ONE. (2012) 7:e37264. doi: 10.1371/journal.pone.0037264
119. Wieser F, Wu J, Shen Z, Taylor RN, Sidell N. Retinoic acid suppresses growth of lesions, inhibits peritoneal cytokine secretion, and promotes macrophage differentiation in an immunocompetent mouse model of endometriosis. Fertil Steril. (2012) 97:1430–7. doi: 10.1016/j.fertnstert.2012.03.004
120. Ozer H, Boztosun A, Açmaz G, Atilgan R, Akkar OB, Kosar MI. The efficacy of bevacizumab, sorafenib, and retinoic acid on rat endometriosis model. Reprod Sci. (2013) 20:26–32. doi: 10.1177/1933719112452941
121. Rocha AL, Reis FM, Taylor RN. Angiogenesis and endometriosis. Obstet Gynecol Int. (2013) 2013:859619. doi: 10.1155/2013/859619
122. Laschke MW, Menger MD. Anti-angiogenic treatment strategies for the therapy of endometriosis. Hum Reprod Update. (2012) 18:682–702. doi: 10.1093/humupd/dms026
123. Bouquet de., Joliniere J, Fruscalzo A, Khomsi F, Stochino Loi E, Cherbanyk F, Ayoubi JM, et al. Antiangiogenic therapy as a new strategy in the treatment of endometriosis? the first case report. Front Surg. (2021) 8:791686. doi: 10.3389/fsurg.2021.791686
124. DiVasta AD, Stamoulis C, Gallagher JS, Laufer MR, Anchan R, Hornstein MD. Non-hormonal therapy for endometriosis: a randomized, placebo-controlled, pilot study of cabergoline versus norethindrone acetate. F S Rep. (2021) 2:454–461. doi: 10.1016/j.xfre.2021.07.003
125. Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, Attar E, et al. Role of estrogen receptor-β in endometriosis. Semin Reprod Med. (2012) 30:39–45. doi: 10.1055/s-0031-1299596
126. Thekkumkara T, Snyder R, Karamyan VT. Competitive binding assay for the G-protein-coupled receptor 30 (GPR30) or G-protein-coupled estrogen receptor (GPER). Methods Mol Biol. (2016) 1366:11–7. doi: 10.1007/978-1-4939-3127-9_2
127. Koganti S, Snyder R, Gumaste U, Karamyan VT, Thekkumkara T. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur J Pharmacol. (2014) 723:131–40. doi: 10.1016/j.ejphar.2013.10.064
128. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. (1971) 285:1182–6. doi: 10.1056/NEJM197111182852108
129. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. (2014) 17:471–94. doi: 10.1007/s10456-014-9420-y
130. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. (2008) 8:592–603. doi: 10.1038/nrc2442
131. Tang TC, Man S, Xu P, Francia G, Hashimoto K, Emmenegger U, et al. Development of a resistance-like phenotype to sorafenib by human hepatocellular carcinoma cells is reversible and can be delayed by metronomic UFT chemotherapy. Neoplasia. (2010) 12:928–40. doi: 10.1593/neo.10804
132. Klauber N, Rohan RM, Flynn E, D'Amato RJ. Critical components of the female reproductive pathway are suppressed by the angiogenesis inhibitor AGM-1470. Nat Med. (1997) 3:443–46. doi: 10.1038/nm0497-443
133. Li R, Song X, Guo Y, Song P, Duan D, Chen ZS. Natural products: a promising therapeutics for targeting tumor angiogenesis. Front Oncol. (2021) 11:772915. doi: 10.3389/fonc.2021.772915
Keywords: endometriosis, angiogenesis, estrogen receptors, vascular endothelial growth factor, anti-angiogenic therapy
Citation: Chung MS and Han SJ (2022) Endometriosis-Associated Angiogenesis and Anti-angiogenic Therapy for Endometriosis. Front. Glob. Womens Health 3:856316. doi: 10.3389/fgwh.2022.856316
Received: 17 January 2022; Accepted: 24 February 2022;
Published: 05 April 2022.
Edited by:
Thomas G. Gray, Norfolk and Norwich University Hospital, United KingdomReviewed by:
Domenico Ribatti, University of Bari Aldo Moro, ItalyMegha Mistry, Norfolk and Norwich University Hospital, United Kingdom
Copyright © 2022 Chung and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sang Jun Han, c2poYW5AYmNtLmVkdQ==