 Janri Geldenhuys1,2
Janri Geldenhuys1,2 Mathys J. Redelinghuys1,2
Mathys J. Redelinghuys1,2 Hendrik A. Lombaard3
Hendrik A. Lombaard3 Marthie M. Ehlers1,2,4
Marthie M. Ehlers1,2,4 Don Cowan2
Don Cowan2 Marleen M. Kock1,2,4*
Marleen M. Kock1,2,4*- 1Department of Medical Microbiology, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
- 2Centre for Microbial Ecology and Genomics, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa
- 3Obstetrics and Gynecology, Rahima Moosa Mother and Child Hospital, Wits Obstetrics and Gynecology Clinical Research Division, School of Clinical Medicine, Faculty of Health Sciences, University of Witwatersrand, Johannesburg, South Africa
- 4Department of Medical Microbiology, Tshwane Academic Division, National Health Laboratory Service, Pretoria, South Africa
Background: Changes in microbial communities are a known characteristic of various inflammatory diseases and have been linked to adverse pregnancy outcomes, such as preterm birth. However, there is a paucity of information regarding the taxonomic composition and/or diversity of microbial communities in pre-eclampsia. The aim of this study was to determine the diversity of the gut, vaginal and oral microbiome in a cohort of South African pregnant women with and without pre-eclampsia. The diversity of the gut, vaginal and oral microbiome was determined by targeted next generation sequencing (NGS) of the V3 and V4 region of the 16S rRNA gene on the Illumina MiSeq platform.
Results: In this study population, pre-eclampsia was associated with a significantly higher alpha diversity (P = 0.0472; indicated by the Shannon index) in the vaginal microbiome accompanied with a significant reduction in Lactobacillus spp. (P = 0.0275), compared to normotensive pregnant women. Lactobacillus iners was identified as the predominant species of the vaginal microbiome in both cohorts. High inter-individual variation in alpha diversity was observed in the gut and oral microbiome in both cohorts. Although differences in the relative abundance of bacteria at all phylogenetic levels were observed, overall microbial composition of the gut, oral and vaginal microbiome was not significantly different in the pre-eclampsia cohort compared to the normotensive cohort.
Conclusion: Collectively, a reduction of Lactobacillus spp., and predominance of L. iners in pregnant women with pre-eclampsia could suggest an unstable vaginal microbiome that might predispose pregnant women to develop pre-eclampsia. The lack of significant structural changes in the gut, oral and vaginal microbiome does not suggest that the characterized communities play a role in pre-eclampsia, but could indicate a characteristic unique to the study population. The current study provided novel information on the diversity of the gut, oral and vaginal microbiome among pregnant women in South Africa with and without pre-eclampsia. The current study provides a baseline for further investigations on the potential role of microbial communities in pre-eclampsia.
Background
Pre-eclampsia is a multisystem and multifactorial disorder unique to pregnancy, diagnosed by new-onset hypertension during pregnancy and damage to one or more organ systems featuring proteinuria, hemolysis, elevated liver enzymes and low platelet count, neurological or visual symptoms, abnormal Doppler ultrasound and fetal growth restriction (1–3). This disorder is further characterized by two subtypes known as the maternal subtype, with metabolic and immunological involvement, and the placental subtype characterized by an ischemic placenta followed by maternal systemic inflammation (4, 5). Pre-eclampsia has a complex etiology for which diagnostic biomarkers and approved therapies are not yet available. The exact cause of this condition remains unknown; however, its association with exaggerated systemic inflammation and certain risk factors, such as diabetes and obesity, has suggested the microbiome may play a role in disease pathogenesis (6–8). The maternal microbiome is considered an important factor that is affected by and which influences the physiological processes in pregnancy including immune, metabolic and hormonal changes (9). Microbial communities in the placenta, vagina, the distal gut and the oral cavity have previously been implicated in maternal health and potentially also in the pathogenesis of pre-eclampsia (10–12). Since Amarasekara et al. (13) described the placental microbiome in a group of pregnant women with pre-eclampsia, conflicting information on the presence and potential role of a placental microbiome in pre-eclampsia have been reported (14–18). However, the presence of commensal microorganisms originating from the oral cavity, gut and vagina in the placenta of pre-eclampsia and hypertensive pregnancies has refocused the role of the placental microbiome by means of bacterial translocation (12, 13, 19).
Dysbiosis of the gut microbiome may contribute to the pathogenesis of hypertension and affect maternal adaptation in pregnancy including placental function (20, 21). The contributing role of a disrupted gut microbiome in metabolic disease, blood pressure (BP) regulation, chronic inflammatory diseases and complicated pregnancies point to a possible role in the development of pre-eclampsia (19, 22, 23). Several studies investigated the composition and stability of the vaginal microbiome in reproductive-age women, uncomplicated pregnancy and in pregnant women with spontaneous preterm delivery (24–26). The vaginal microbiome in uncomplicated pregnancy is characterized by lower richness and diversity with an increase in abundance of the Lactobacillus species, to obtain stability and resilience during pregnancy (27). However, there is uncertainty on the composition and contributing role of the vaginal microbiome in preterm birth. Although a reduction in Lactobacillus species has been suggested as a risk factor for preterm delivery, different findings have also been reported (25, 26, 28–30).
The oral microbiome is known as one of the most diverse microbiomes in the human body with a strong impact on systemic health (31–33). Periodontal disease can cause systemic illness including atherosclerotic cardiovascular disease, rheumatoid arthritis, diabetes and adverse pregnancy outcomes (34–39). Bacterial species associated with oral infections have the ability to translocate and colonize extra-oral sites such as the placenta, and this phenomenon has led to the establishment of a link between oral infections and adverse systemic conditions (40–42). In addition, maternal periodontal disease has previously been found to be associated with an increased risk of preterm birth and pre-eclampsia (43–45).
Currently available next generation sequencing (NGS) techniques, including 16S targeted rRNA sequencing and metagenomics enable the characterization of microbial communities associated with pregnancy and pregnancy complications, such as pre-eclampsia. The characterization of microbial communities in pre-eclampsia may reveal whether structural changes in microbial communities are associated with pre-eclampsia and may also provide indicative biomarkers for the development of future preventative and therapeutic strategies. The aim of this study was to determine the diversity of the gut, vaginal and oral microbiome in a cohort of pre-eclampsia and normotensive pregnant women.
Methods
Sample collection
All participants in this study were recruited from a provincial hospital in Johannesburg, South Africa. Ten primiparous pregnant women, ≥18 years and with a gestational age ≥28 gestational weeks (3rd trimester), were included in either one of two cohorts (N = normal/normotensive and P = pre-eclampsia) following the rule of ten (46). Participants were selected according to the study criteria after a medical examination was done by a healthcare practitioner or using hospital records when available. Participants who had symptomatic vaginal infections including known viral infections, such as human papilloma virus (HPV), antibiotic usage in the 4 weeks prior to sample collection, previous miscarriages and a gestational age ≤ 28 weeks were excluded from the study. Informed consent was obtained prior to sample collection.
Three samples were collected from each enrolled participant to characterize the gut, vaginal and oral microbiome, respectively. A rectal swab as an alternative to a stool sample was collected by an obstetrician by inserting a flocked dry swab (FLOQSwabs 552C, Copan, CA) into the anal canal (±3 cm) beyond the anal verge (47). A midvaginal swab was collected by the insertion of a dry flocked swab into the vaginal canal (±2 cm) to absorb vaginal fluid. Saliva was obtained by the use of a sterile flocked swab in a stable position under the tongue for 2 min. Fresh collected samples were labeled, kept and transported on ice to the laboratory of the Department of Medical Microbiology, University of Pretoria, Pretoria and stored at −20°C until processing. The sample labeling used in this study refers to the site of collection, i.e., R = rectal/gut, V = vaginal, O = oral sample in both cohorts.
Bacterial genomic DNA isolation
The extraction methods used in this study were adapted with the use of different enzymes for bacterial lysis and sufficient isolation of all bacterial organisms present in the gut, oral cavity and the vagina (24, 48, 49). The stored dry swabs (−20°C; F700-SAEV-TSC; Thermo Scientific, Waltham, MA) were thawed on ice for 30 min prior to genomic DNA isolation. A volume of 2 mL sterile 1X phosphate buffered saline (PBS) (Life Technologies Corporation, Carlsbad, CA, USA) was added to thawed samples. Rectal and vaginal swabs were incubated at room temperature (25°C ± 5°C) for 2–3 h and mixed in a VX-100 vortex mixer (Labnet International, Edison, NJ) at maximum speed for 5 min to efficiently suspend all cellular material (24). Oral swabs were incubated overnight at 4°C and ere mixed vigorously at maximum speed for 5 min to release all cellular material into the PBS solution (24). Total bacterial genomic DNA (gDNA) was isolated using the Bioline ISOLATE II Genomic DNA kit (Bioline) following the manufacturer's instructions with minor modifications (see Supplementary material). The concentration of the isolated gDNA was determined by using the NanoDropR ND-1000 Spectrophotometer (Thermo Scientific) and the purity was determined by the ratio of absorbance at 260 and 280 nm (A260/280). The isolated DNA was normalized to a final concentration of 10 ng/μL in a total volume of 25 μL. The normalized DNA was used for 16S rRNA amplification and sequencing.
Targeted amplification of the 16S rRNA variable domains (V3 and V4)
A genome sequence spanning the V3 and V4 regions of the 16S rRNA gene were amplified using the Illumina ultramer oligonucleotides (Integrated DNA Technologies, Coralville, IA) that include Illumina adapter overhang nucleotide sequences (50). Polymerase chain reaction (PCR) conditions and reagent volumes are available as Supplementary Information. A negative control from each gDNA isolation step was included in amplification steps to determine the possibility of carry-over contamination between samples and reagents during these steps. Amplification products were purified using the ISOLATE II PCR and Gel Kit (Bioline) according to the manufacturer's instructions with minor adjustments (see Supplementary material).
Library preparation and 16S targeted NGS
The purified amplified products were submitted to the ARC Biotechnology Platform, Onderstepoort, Pretoria, South Africa for library preparation and NGS on the Illumina MiSeq platform. Sequencing libraries were created according to the Illumina 16S metagenomic library preparation protocol available online (see Supplementary material). The 16S rRNA gene was sequenced on a MiSeq sequencer creating 300 bp paired-end reads with the V3 600 cycles kit (Illumina, San Diego, CA). Raw paired-end FASTQ files of each sample were received for subsequent analysis.
Sequence and statistical analysis
The targeted metagenomic sequence data were analyzed using the Quantitative Insights Into Microbial Ecology (QIIME) pipeline, version 9.1 (51). Matching paired-end sequences were merged using the fastq-join command in the ea-utils software package (52). Chimeric sequences were identified from merged sequences and removed using the USEARCH database version 6.1 (53). The open reference method was used to assign the sequences to 97% identity (ID) operational taxonomic units (OTUs). Taxonomy was assigned when the representative sequence of each OTU was aligned against the GreenGenes database 12_10, using the Basic Local Alignment Search Tool (BLAST) program incorporated into the QIIME software (54).
The resulting unrarefied OTU table and phylogenetic tree generated in QIIME were used for diversity and statistical analysis done using Rstudio version 1.0.136 (55). Statistical analysis and the generation of graphs using ggplots (integrated in the package phyloseq version 1.19.1 and the package tidyverse version 1.1.1) were used as implemented in Rstudio. These packages were also used to generate stacked bar plots for visualization of the taxonomic composition in each sample. To select only sequences representing bacteria, sequences representing mitochondria, Archaea and chloroplast were removed from the OTU table and phylogenetic tree in Rstudio. Samples that contained fewer than the rarefaction depth of 1,021 OTUs were removed for alpha and beta diversity analysis. The resulting dataset included a total of fourteen samples in the gut microbiome, ten samples in the vaginal microbiome and eleven samples in the oral microbiome. The data of each microbiome were extracted from the rarefied OTU table to infer statistical analysis of each microbiome individually. Due to the small sample size, it was assumed that the data do not follow a normal distribution pattern. Therefore, the non-parametric Kruskal-Wallis rank chi-squared test was used to evaluate differences (P-value < 0.05) in alpha diversity and the relative percent abundances of taxa between cohorts with a 95% confidence interval (56). Alpha diversity was measured by the Chao1 index, an indirect measure of richness and Shannon's diversity index, an indirect measure of evenness (56).
In beta diversity analysis, principal coordinate analysis (PCoA) plots were generated with weighted and unweighted unifrac distance matrices to vizualize the differences between cohorts (57). The statistical significance in the differences in microbial communities between the cohorts was calculated using a permanova test, Adonis (Analysis of Dissimilarity), as implemented in the R package vegan version 2.4–3.0 (58). This method included 999 permutations and the weighted and unweighted unifrac distance matrices.
Results
Samples were obtained from 21 participants who provided informed consent and who met the inclusion criteria. Eleven participants were recruited in the control group and ten participants were recruited to the study group. All participants were in an age group between 18 and 35 years with a gestational age between 32 and 40 weeks. DNA extracted from each sample was quantified, followed by amplification and then used for subsequent analysis. In the sequence analysis, the selection of OTUs in QIIME resulted in a dataset of 545,677 sequences and 1,751 identified taxa between the 63 samples. An average of 9,094 OTUs were formed with a minimum of 73 and a maximum of 64,247 OTUs between 63 samples. Rarefaction prior to statistical analysis allowed normalization of sequencing data and identification of samples with insufficient sequencing data; insufficient sequencing data could affect subsequent analysis due to inaccurate representation of microbial communities. After rarefaction, the OTU table including the gut, vaginal and oral microbiome consisted of 884 taxa and 35 samples in total (reduced from an initial 63 samples). The rarefied cohort consisted of a total of 14 samples (normotensive, N = 8; pre-eclampsia, P = 6) for analysis of the gut microbiome, 10 samples (N = 5; P = 5) of the vaginal microbiome and 11 samples (N = 5; P = 6) of the oral microbiome. The 35 samples on which all analyses were carried out come from a pool of 14 participants in the final cohort (i.e., three and four participants were excluded from the normotensive and pre-eclampsia cohorts, respectively, as none of their samples were included after rarefaction). All participants were of black South African ethnicity except one of the participants who was of white South African ethnicity.
Alpha and beta diversity analysis
A higher average alpha diversity was found in the gut, oral and vaginal microbiome in pregnant women with pre-eclampsia in comparison to normotensive pregnant women (Table 1). The differences in alpha diversity observed between cohorts in the gut microbiome was not significant for both the Chao1 index (P = 0.1213) and Shannon index (P = 0.09329). Differences were also not significant for both the Chao1 (P = 1) and the Shannon index (P = 0.715) in the oral microbiome. In the vaginal microbiome, a significant difference in diversity was observed between the two groups (P = 0.0472) for the Shannon index, but not the Chao1 index (P = 0.3472) (Figure 1).
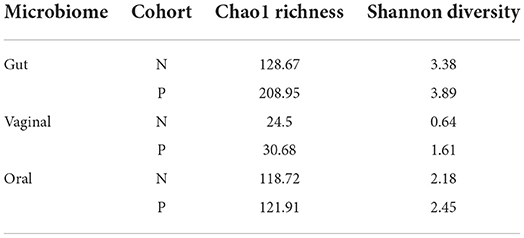
Table 1. The average alpha diversity measures of the gut, vaginal and oral microbiome as assessed by the Chao1 richness Index and the Shannon Index.
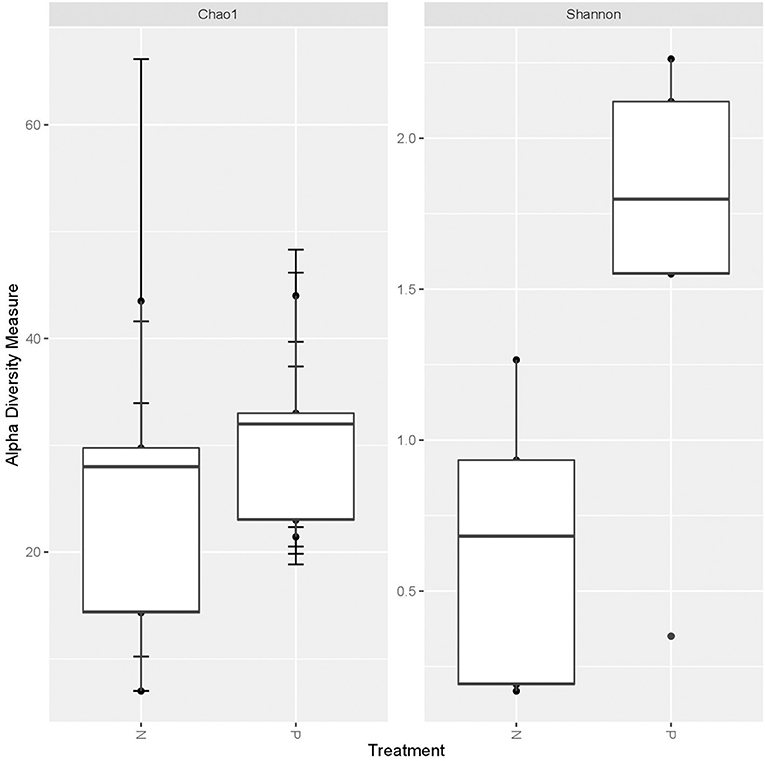
Figure 1. Box-plot illustrating alpha diversity indices (Chao1 and Shannon diversity index) of the vaginal microbiome comparing the normotensive (n = 5) and pre-eclampsia (p = 5) cohort.
Principal coordinate analysis based on the UniFrac distances using phylogenetic information revealed no distinct separation of microbial communities of the gut, vaginal and oral microbiome amongst both cohorts (Figures 2A–F). The dissimilarity test, Adonis, indicated that the gut microbiome in the pre-eclampsia group is not significantly different compared to that of the normotensive cohort using both the UniFrac weighted (R2 = 0.02266; P = 0.987) and unweighted (R2 = 0.07072; P = 0.568) distance matrices. Similarly, the oral microbiome of the pre-eclampsia group was also not significantly different to that of the normotensive cohort using the UniFrac weighted (R2 = 0.10288, P =0.354) and unweighted (R2 = 0.12881, P = 0.185) distance matrices. In the vaginal microbiome, a clear separation of the pre-eclampsia and normotensive cohort was visible on the PCoA plot with the UniFrac weighted distance matrix; however, the finding was not statistically significant with both the weighted (R2 = 0.30017, P = 0.05) and unweighted distance matrix (R2 = 0.2344, P = 0.068) (Figure 2C).
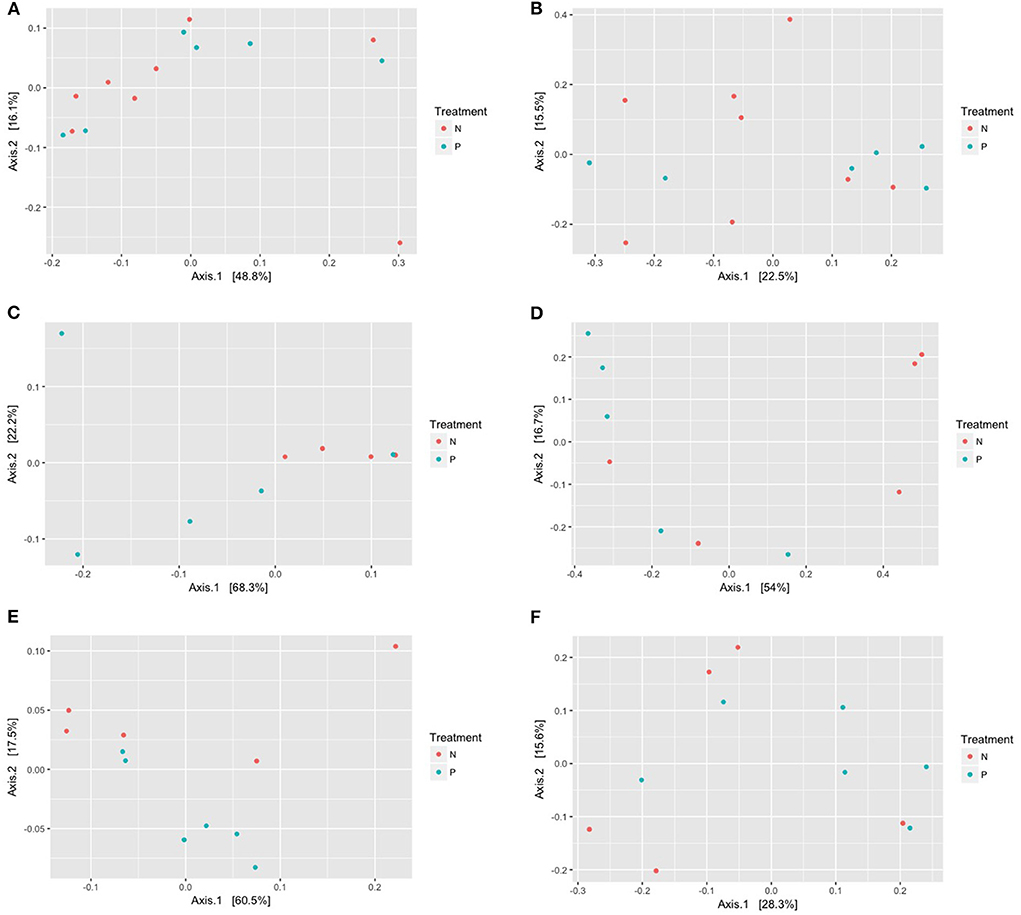
Figure 2. (A–F) Principal coordinate analysis (PCoA) of the gut, vaginal, and oral microbiome displaying the dissimilarities among the gut microbiome in the normotensive group compared to the pre-eclampsia group using the weighted (A,C,E) and the unweighted (B,D,F) UniFrac distance metrices. Each point indicates the microbiome of each sample in the N = normotensive cohort and P = pre-eclampsia cohort.
Comparison of the gut microbiome between cohorts
The gut microbiome of both groups was analyzed at different taxonomic levels including at phylum, genus and species level. Although the Firmicutes was the most abundant phylum, the relative abundance did not differ significantly between cohorts (Firmicutes; P = 0.6985, Bacteroidetes; P = 0.8973, Fusobacteria; P = 0.08117, Proteobacteria; P = 0.7026, Actinobacteria; P = 0.9485). Significant differences of relative abundance were not observed on genus level (P values > 0.05) between cohorts; however, changes in the relative abundance of several genera were observed. The pre-eclampsia group had a higher relative abundance of Bacteroides (6.41% as opposed to 4.73%), Faecalibacterium (12.35% as opposed to 10.61%) and Blautia (7.61% as opposed to 3.67%). In contrast, the normotensive group had a higher relative abundance of Anaerococcus (7.56% as opposed to 3.59%), Clostridioides (5.64% as opposed to 3.21%), Finegoldia (4.78% as opposed to 2.9%) and Prevotella (15.17% as opposed to 12.30%).
On species level, a higher relative abundance of Peptostreptococcus anaerobius (average relative abundance of 23.36%) was observed in the pre-eclampsia group compared to the normotensive group (7.8%), although the difference was not significant (P = 0.1824). Faecalibacterium prausnitzii (average relative abundance of 42.69 and 35.79%, respectively) and Lactobacillus iners (average relative abundance of 36.49 and 21.67%, respectively) were more abundant in the pre-eclampsia group compared to the normotensive group.
Comparison of the vaginal microbiome between cohorts
The Firmicutes was the predominant phylum in the vaginal microbiome with an average relative abundance of 95.2 and 74.28% in normotensive and pre-eclampsia cohort, respectively. Figure 3 indicates the overall microbial composition at genus level. Only genera with a relative abundance of more than 30% in each sample is indicated on the plot. The genus Lactobacillus was significantly lower (P = 0.0275) in the pre-eclampsia cohort (61.5%) compared to the normotensive cohort (92.6%). Higher relative abundances of the genera Prevotella, Peptoniphilus and Anaerococcus and an increase in Dialister were also observed in the pre-eclampsia cohort. The genera Sneathia, Parvimonas, Clostridium, Megasphaera and Peptostreptococcus were uniquely detected in only the pre-eclampsia cohort (Figure 3).
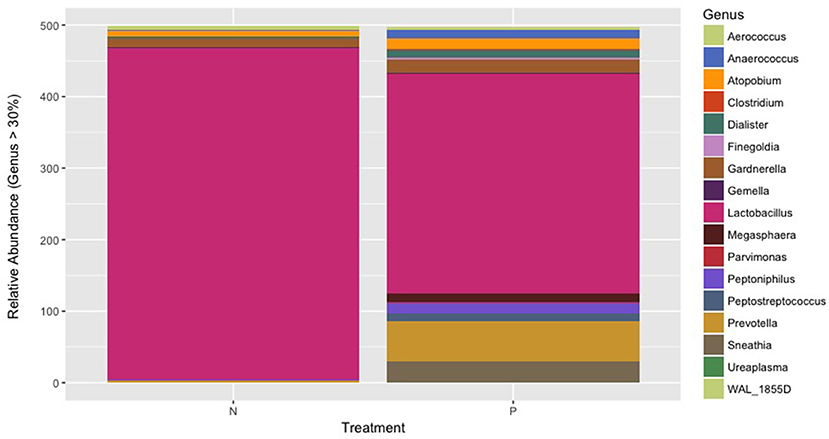
Figure 3. The overall microbial composition of the vaginal microbiome present at genus level showing the genera present in more than 30% of each sample in both the normotensive cohort and the pre-eclampsia cohort.
Present in low levels of abundance, Atopobium spp. were present in equal relative abundance in both groups (3.93 and 3.89%), whereas a slight increase in Aerococcus spp. (average relative abundance of 2.94% compared to 1.92%) and Gardnerella spp. (5.84% average relative abundance compared to 4.83%) was detected in the normotensive group. Lactobacillus iners was identified as the predominant species in the vaginal microbiome of both the normotensive (97.71%) and pre-eclampsia cohort (86.14%). A higher relative abundance of Atopobium vaginae (average relative abundance of 6.56% compared to 4.51%) was observed in the pre-eclampsia cohort. The presence of Prevotella melaninogenica and Peptostreptococcus anaerobius were also uniquely detected in the pre-eclampsia cohort.
Comparison of the oral microbiome between cohorts
The PCoA of the oral microbiome illustrated in Figures 2E,F indicated no clear separation of the oral microbial communities between normotensive pregnant women and women with pre-eclampsia. However, this plot also displayed the similarity and high variation within cohorts, which was confirmed by the relative abundance of oral bacteria on genus and species level. The Firmicutes was the predominant phylum in the oral microbiome among the normotensive (70.98%) and pre-eclampsia cohort (67.76%) respectively. On genus level, Streptococcus was the predominant bacterial genus in both cohorts followed by order of decreasing abundance of Haemophilus, Neisseria, Prevotella and Veillonella. Higher relative abundance values of Streptococcus, Granulicatella and Veilonella were observed in the normotensive group. Neisseria (4.46 and 4.27%) and Porphyromonas (2.49 and 2.68%) were detected in similar relative abundance in both cohorts. Prevotella (5.16 and 4.13%) and Haemophilus (13.29% compared to 8.4%) were more abundant in the pre-eclampsia cohort. Genera such as Rothia, Sneathia, Treponema, Parvimonas, Prevotella, Pseudomonas and Gemella were detected in low abundance.
The pre-eclampsia cohort had higher average relative abundance of the species Prevotella melaninogenica (23.57%) and Neisseria subflava (6.96%) compared to the normotensive cohort (17.84%, 3.93% respectively). Neisseria cinerea and N. Oralis were detected in both cohorts. The relative abundance of the species Rothia dentocariosa was significantly higher (P = 0.02535) in the normotensive cohort compared to the pre-eclampsia cohort.
Discussion
To our knowledge, this is the first study characterizing and determining the diversity of the gut, vaginal and oral microbiomes among pregnant women in South Africa with pre-eclampsia. Pre-eclampsia is a complication of hypertension in pregnancy and is one of the main contributions to the high maternal/fetal morbidity and mortality rates globally as well as in South Africa (59, 60). Research to uncover the complicated pathology involved in pre-eclampsia is ongoing in order to develop preventative and therapeutic strategies. In the current study, pregnant women with pre-eclampsia displayed a greater phylogenetic diversity in terms of richness (the number of taxa present) and evenness (abundance of many microbial constituents) compared to normotensive pregnant women. High inter-individual variation was evident in the characterization of the gut and oral microbiome and with a lesser extent in the vaginal microbiome and significant structural changes of microbial communities were not found. The high inter-individual variation in this study is supported by the observations of the Human Microbiome Project Consortium, which reported that individuals differ with the carriage of specific microbes and that inter-individual variation in the microbiome is specific, functionally relevant and personalized (61). Regardless of inter-individual variation and small sample size, a significant increase in alpha diversity accompanied with a reduction in Lactobacillus species was found in the vaginal microbiome of this cohort of pregnant women with pre-eclampsia, in comparison to normotensive pregnant women.
A decrease in gut microbial diversity is a known suggestive indicator of microbial imbalance associated with human disease (62). In contrast to the findings of the current study (19), found a decrease in alpha diversity in pregnant women with pre-eclampsia. Another study found no differences in diversity between pregnant women with and without pre-eclampsia (63). Alpha diversity analysis of the current study is therefore not suggestive of an imbalanced or disrupted gut microbiome. As indicated by Frost et al. (62), an increase in alpha diversity could rather contribute to greater stability and resilience of the gut microbiome. Taxonomic characterization of the gut microbiome revealed a mixed species microbiome with the exception of a few samples having a single dominant bacterial species, including Bifidobacterium longum, Moryella indoligenes and Lactobacillus iners. Diversity and composition of gut microbiome is versatile and is influenced by various external and internal factors such as diet and host genetics. Species dominance could be specific to an individual at a specific time with presence relating to a species specific function (64). Several species with anti-inflammatory properties such as Faecalibacterium prausnitzii was detected in the pre-eclampsia cohort, indicated a balanced gut microbiome with no significant structural changes to play a possible role in the development of pre-eclampsia (65–69).
Similarly to the gut microbiome, a diverse oral microbiome was characterized in women with and without pre-eclampsia. Diversity of the oral microbiome has been found to be stable during pregnancy and has also not been found to change in oral disease (70). Changes in alpha diversity is therefore not suspected to play a role in pre-eclampsia. Microbial characterization indicated dominance of Streptococcus spp., that has been reported in healthy oral microbiomes (71, 72). Other species, such as Rothia, Haemophilus and Neisseria spp., have been observed in healthy individual and therefore it is unknown whether these species could have contributed to pre-eclampsia in the current study (73–75). The presence of these species indicates a diverse oral microbiome present in pregnant women with and without pre-eclampsia. In addition, no pathogenic species associated with periodontitis were detected in the current study to hypothesize any link with pre-eclampsia (76).
Characterization of the vaginal microbiome in both cohorts has provided more information on the structure of vaginal microbial communities of South African pregnant women. Racial variation has a strong influence on community structure; for example, Aerococcus spp., Peptoniphilus spp., Dialister spp., Atopobium spp., Sneathia spp. and Gardnerella spp., have been detected more among healthy African American and African women (77, 78). In addition, a less acidic vaginal environment described in black and Hispanic women could also influence microbial diversity (24). Results within the South African population have similarly reported dominance of L. iners in black South Africa women (79). Lactobacillus iners has also been reported to co-exist with other BV-related bacteria (24, 78, 80–83).
Taxonomic observations in the pre-eclampsia cohort showed similarities to a BV-associated vaginal microbiome, which is characterized by higher bacterial diversity, lower abundances of Lactobacillus spp. and higher abundances of Atopobium spp., Dialister spp., Gardnerella spp., Prevotella spp. and Sneathia spp. (84). Low abundances of Lactobacillus spp., may have significant health consequences such as increase vaginal inflammation and susceptibility to pathogenic infection (79). A decrease in relative abundance of Lactobacillus spp., together with dominance of L. iners could further contribute to an unstable vaginal microbiome in women with pre-eclampsia. Petricevic et al. (85) described a vaginal microbiome dominated by a single vaginal Lactobacillus species, specifically L. iners in the late first trimester of pregnancy that might have been associated with preterm birth. Another study by Kindlinger et al. (86) identified the predominance of L. iners as a risk factor for preterm birth, while the predominance of L. crispatus was highly predictive of a term birth. Other studies found L. iners to represent a transitional phase of the vaginal microbiome between healthy and dysbiotic states that may promote the recurrence of BV (80, 87–89).
Regardless of low sample numbers, the results of the current study align with previous studies that found an increase in bacterial diversity accompanied by a reduction in Lactobacillus spp., in complicated pregnancies (11, 86, 90–92). Diversity analysis by the weighted UniFrac distance matrix suggested that the relative abundance of bacterial taxa may play a more important role in comparative analysis than the absence or presence of taxa between the two groups. The co-existence of L. iners and anaerobic taxa in both cohorts could indicate an unstable and transitional vaginal microbiome in South African pregnant women (78, 93–95). However, these findings concurrently with a significant reduction of Lactobacillus spp., found in pregnant women with pre-eclampsia could discriminate a vaginal microbiome unique to a study population to a vaginal microbiome that could increase the risk of developing pre-eclampsia.
The strengths of this study were the identification of microbial taxa on all phylogenetic levels, especially on species level in low percentage abundances, which is needed to unlock the functional complexity of a microbiome in health and disease. Analysis at species level is critical as different species may have different functional capacities, which is needed to link changes in the human microbiome to health or disease. The sample collection strategy used in this study accounted for as much variability as possible by including a homogenous group of pregnant women in their third trimester of pregnancy, of the same ethnic group, age and HIV status. Other strengths of the study are the inclusion of the normotensive group to draw comparisons to a matched control cohort and simultaneous characterization of microbiomes in three distinct body sites.
A limitation of this study includes the small sample size used for statistical analysis due to the exclusion of samples with insufficient sequencing data. To control for inter-individual variability, samples needed to be as homogenous as possible. However, by excluding for women who presented with symptomatic infection (vaginal and oral infection), it could possibly have excluded some species that could be implicated in the pathogenesis of pre-eclampsia. The absence of asymptomatic BV in the participants of both cohorts was not confirmed using laboratory methods and should be taken into account with the results reported in this study. A limitation of the study is therefore that the exclusion or diagnosis of BV was not confirmed amongst participants of both cohorts and therefore it is not known whether BV had a contributing role to the observed vaginal microbiome in the pre-eclampsia cohort and weather BV could have increased the risk of these women to develop pre-eclampsia. Another limitation was the lack of the captured medical information of the enlisted participants, such as body mass index (BMI), white blood cell (WBC) counts and birth outcomes.
Conclusion
The current study provided a diversity analysis of the gut, vaginal and oral microbiomes in a cohort of South African pregnant women with and without pre-eclampsia. Pregnant women with a vaginal microbiome characterized by a significant reduction in Lactobacillus spp., and dominance by L. iners could have an instable vaginal microbiome and that may increase the risk of developing pre-eclampsia. Diversity and compositional analysis have revealed important data that are otherwise scanty on the gut, oral and vaginal microbiomes in pregnant women (predominantly black African) with pre-eclampsia in South Africa. In future research, larger cohorts with functional and longitudinal analysis of the gut, vaginal and oral microbiomes may reveal additional changes across these microbial communities in pregnant women with pre-eclampsia.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA database, accession number PRJNA798597 (BioProjectID).
Ethics statement
The studies involving human participants were reviewed and approved by Research Ethics Committee of the Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa (116/2016) and the Human Research Ethics Committee of the University of the Witwatersrand, Johannesburg, South Africa (M160573). The patients/participants provided their written informed consent to participate in this study.
Author contributions
JG is the project leader and was involved in the concept design, sample collection, laboratory work, data analysis, and writing of the manuscript. MK is the principal investigator and grant holder. MK, MR, and ME were involved in the conceptual design of the study as well as the review of the manuscript. HL was involved in the conceptual design of the study, assisted in sample collection, and contributed toward the clinical aspect of the study. DC was involved by providing support for data analysis. All authors read and approved the final manuscript.
Funding
Financial support was provided by the NHLS Research trust.
Acknowledgments
The authors would like to thank the University of Pretoria and the National Health Laboratory Service (NHLS) for the financial assistance received as well as the Rahima Moosa Mother and Child Hospital for the sample collection and the Center of Microbial Ecology and Genomics (CMEG) for their computer facilities and assistance in the data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgwh.2022.810673/full#supplementary-material
References
1. Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, Saito S, et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension. (2018) 72:24–43. doi: 10.1161/HYPERTENSIONAHA.117.10803
2. Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. (2005) 365:785–99. doi: 10.1016/S0140-6736(05)71003-5
3. Spradley FT, Palei AC, Granger JP. Immune mechanisms linking obesity and preeclampsia. Biomolecules. (2015) 4:3142–76. doi: 10.3390/biom5043142
4. Phipps E, Prasanna D, Brima W, Jim B. Preeclampsia: updates in pathogenesis, definitions, and guidelines. Clin J Am Soc Nephrol. (2016) 6:1102–13. doi: 10.2215/CJN.12081115
5. Geldenhuys J, Rossouw TM, Lombaard HA, Ehlers MM, Kock MM. Pre-eclampsia, a pregnancy disorder associated with disruption in the regulation of immune responses. Front Immunol. (2011) 9:1659. doi: 10.3389/fimmu.2018.01659
6. Bell MJ. A historical overview of preeclampsia-eclampsia. J Obstet Gynecol Neonatal Nurs. (2010) 39:510–8. doi: 10.1111/j.1552-6909.2010.01172.x
7. Crusell MK, Hansen TH, Nielsen T, Allin KH, Rühlemann MC, Damm P, et al. Gestational diabetes is associated with change in the gut microbiota composition in third trimester of pregnancy and postpartum. Microbiome. (2018) 6:1–9. doi: 10.1186/s40168-018-0472-x
8. Crusell MK, Brink LR, Nielsen T, Allin KH, Hansen T, Damm P, et al. Gestational diabetes and the human salivary microbiota: a longitudinal study during pregnancy and postpartum. BMC Preg Childbirth. (2020) 20:1–2. doi: 10.1186/s12884-020-2764-y
9. Nuriel-Ohayon M, Neuman H, Koren O. Microbial changes during pregnancy, birth, and infancy. Front Microbiol. (2016) 7:1031. doi: 10.3389/fmicb.2016.01031
10. Charbonneau MR, Blanton LV, Digiulio DB, Relman DA, Lebrilla B, Mills DA, et al. Human developmental biology viewed from a microbial perspective. Nature. (2016) 535:48–55. doi: 10.1038/nature18845
11. DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci USA. (2015) 112:11060–5. doi: 10.1073/pnas.1502875112
12. Ishimwe JA. Maternal microbiome in preeclampsia pathophysiology and implications on offspring health. Physiol Rep. (2021) 10:e14875. doi: 10.14814/phy2.14875
13. Amarasekara R, Jayasekara RW, Senanayake H, Dissanayake VHW. Microbiome of the placenta in pre-eclampsia supports the role of bacteria in the multifactorial cause of pre-eclampsia. J Obst Gynaecol Res. (2015) 41:662–9. doi: 10.1111/jog.12619
14. Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. (2016) 4:1–1. doi: 10.1186/s40168-016-0172-3
15. Leiby JS, McCormick K, Sherrill-Mix S, Clarke EL, Kessler LR, Taylor LJ, et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome. (2018) 6:1–1. doi: 10.1186/s40168-018-0575-4
16. de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, et al. Human placenta has no microbiome but can contain potential pathogens. Nature. (2019) 572:329–34. doi: 10.1038/s41586-019-1451-5
17. Theis KR, Romero R, Greenberg JM, Winters AD, Garcia-Flores V, Motomura K, et al. No consistent evidence for microbiota in murine placental and fetal tissues. Msphere. (2020) 1:e00933–19. doi: 10.1128/mSphere.00933-19
18. Olaniyi KS, Moodley J, Mahabeer Y, Mackraj I. Placental microbial colonization and its association with pre-eclampsia. Front Cell Infect Microbiol. (2020) 10:413. doi: 10.3389/fcimb.2020.00413
19. Chen X, Li P, Liu M, Zheng H, He Y, Chen MX, et al. Gut dysbiosis induces the development of pre-eclampsia through bacterial translocation. Gut. (2020) 3:513–22. doi: 10.1136/gutjnl-2019-319101
20. Gohir W, Ratcliffe EM, Sloboda DM, Sciences B. Of the bugs that shape us: maternal obesity, the gut microbiome, and long-term disease risk. Pediatric Res. (2014) 77:196–204. doi: 10.1038/pr.2014.169
21. Sanz Y, Olivares M, Moya-Pérez Á, Agostoni C. Understanding the role of gut microbiome in metabolic disease risk. Pediatric Res. (2015) 77:236-44. doi: 10.1038/pr.2014.170
22. Liu J, Yang H, Yin Z, Jiang X, Zhong H, Qiu D, et al. Remodeling of the gut microbiota and structural shifts in preeclampsia patients in South China. Eur J Clin Microbiol Infect Dis. (2016) 36:713–9. doi: 10.1007/s10096-016-2853-z
23. Lv WJ, Wu XL, Chen WQ, Li YF, Zhang GF, Chao LM, et al. The gut microbiome modulates the changes in liver metabolism and in inflammatory processes in the brain of chronic unpredictable mild stress rats. Oxidat Med Cell Long. (2019) 24:2019. doi: 10.1155/2019/7902874
24. Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. (2011) 108:4680–7. doi: 10.1073/pnas.1002611107
25. Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Bieda J, et al. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome. (2014) 2:18. doi: 10.1186/2049-2618-2-18
26. Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Nikita L, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. (2014) 2:4. doi: 10.1186/2049-2618-2-10
27. Macintyre DA, Chandiramani M, Lee YS, Kindinger L, Smith A, Angelopoulos N, et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci Rep. (2015) 5:8988. doi: 10.1038/srep08988
28. Cao B, Stout MJ, Lee I, Mysorekar IU. Placental microbiome and its role in preterm birth. NeoReviews. (2014) 15:e537–45. doi: 10.1542/neo.15-12-e537
29. Hyman RW, Fukushima M, Jiang H, Fung E, Rand L, Johnson B, et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod Sci. (2014) 21:32–40. doi: 10.1177/1933719113488838
30. Kindinger LM, MacIntyre DA, Lee YS, Marchesi JR, Smith A, McDonald JAK, et al. Relationship between vaginal microbial dysbiosis, inflammation, and pregnancy outcomes in cervical cerclage. Sci Transl Med. (2016) 8:350ra102. doi: 10.1126/scitranslmed.aag1026
31. Fujiwara N, Tsuruda K, Iwamoto Y, Kato F, Odaki T, Yamane N, et al. Significant increase of oral bacteria in the early pregnancy period in Japanese women. J Invest Clin Dent. (2015) 8:e12189. doi: 10.1111/jicd.12189
32. Lazarevic V, Whiteson K, Huse S, Hernandez D, Farinelli L, Osterås M, et al. Metagenomic study of the oral microbiota by illumina high-throughput sequencing. J Microbiol Meth. (2009) 79:266–71. doi: 10.1016/j.mimet.2009.09.012
33. Wade WG. The oral microbiome in health and disease. Pharmacol Res. (2013) 69:137–43. doi: 10.1016/j.phrs.2012.11.006
34. Alchalabi HA, Al Habashneh R, Jabali OA, Khader YS. Association between periodontal disease and adverse pregnancy outcomes in a cohort of pregnant women in Jordan. Clin Exp Obst Gynecol. (2013) 40:399–402. doi: 10.4314/ahs.v18i3.18
35. Blaizot A, Vergnes J-N, Nuwwareh S, Amar J, Sixou M. Periodontal diseases and cardiovascular events: meta-analysis of observational studies. Int Dental J. (2009) 59:197–209. doi: 10.1922/IDJ_2114Sixou13
36. Detert J, Pischon N, Burmester GR, Buttgereit F. The association between rheumatoid arthritis and periodontal disease. Arthr Res Therapy. (2010) 12:218. doi: 10.1186/ar3106
37. Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, et al. Periodontitis and diabetes: a two-way relationship. Diabetologia. (2012) 55:21–31. doi: 10.1007/s00125-011-2342-y
38. Wei B-J, Chen Y-J, Yu L, Wu B. Periodontal disease and risk of preeclampsia: a meta-analysis of observational studies. PLoS ONE. (2013) 8:e70901. doi: 10.1371/journal.pone.0070901
39. Xiong X, Buekens P, Vastardis S, Yu SM. Periodontal disease and pregnancy outcomes: state-of-the-science. Obst Gynecol Survey. (2007) 62:605–15. doi: 10.1097/01.ogx.0000279292.63435.40
40. Barak S, Oettinger-Barak O, Machtei EE, Sprecher H, Ohel G. Evidence of periopathogenic microorganisms in placentas of women with preeclampsia. J Periodontol. (2007) 78:670–6. doi: 10.1902/jop.2007.060362
41. Ha YW, Wang X. Mobile microbiome: Oral bacteria in extra-oral infections and inflammation. J Dent Res. (2013) 92:485–91. doi: 10.1177/0022034513487559
42. Kumar PS, Mason MR. Mouthguards: does the indigenous microbiome play a role in maintaining oral health? Front Cell Infect Microbiol. (2015) 5:1–9. doi: 10.3389/fcimb.2015.00035
43. Contreras A, Herrera JA, Soto JE, Arce RM, Jaramillo A, Botero JE. Periodontitis is associated with preeclampsia in pregnant women. J Periodontol. (2006) 77:182–8. doi: 10.1902/jop.2006.050020
44. Offenbacher S, Boggess KA, Murtha AP, Jared HL, Lieff S, McKaig RG, et al. Progressive periodontal disease and risk of very preterm delivery. Obst Gynecol. (2006) 107:29–36. doi: 10.1097/01.AOG.0000190212.87012.96
45. Huang X, Wang J, Liu J, Hua L, Zhang D, Hu T, et al. Maternal periodontal disease and risk of preeclampsia: a meta-analysis. J Huazhong Univ Sci Technol Med Sci. (2014) 34:729–35.
46. Gotelli NJ, Ellison AM. A Primer of Ecological Statistics. Sunderland, MA: Sinauer and Associates (2004).
47. Budding AE, Grasman ME, Eck A, Bogaards JA, Vandenbroucke-Grauls CMJE, et al. Rectal swabs for analysis of the intestinal microbiota. PLoS ONE. (2014) 9:5–12. doi: 10.1371/journal.pone.0101344
48. Bai G, Gajer P, Nandy M, Ma B, Yang H, Sakamoto J, et al. Comparison of storage conditions for human vaginal microbiome studies. PLoS ONE. (2012) 7:1–10. doi: 10.1371/journal.pone.0036934
49. Yuan S, Cohen DB, Ravel J, Abdo Z, Forney LJ. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE. (2012) 7:e33865. doi: 10.1371/journal.pone.0033865
50. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl Acids Res. (2013) 41:e1. doi: 10.1093/nar/gks808
51. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Meth. (2011) 7:335–6. doi: 10.1038/nmeth.f.303
52. Giloteaux L, Goodrich JK, Walters WA, Levine SM, Ley RE, Hanson MR. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome. (2016) 4:30. doi: 10.1186/s40168-016-0171-4
53. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. (2010) 26:2460–1. doi: 10.1093/bioinformatics/btq461
54. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with AR. Appl Environ Microbiol. (2006) 72:5069–72. doi: 10.1128/AEM.03006-05
55. Team RC. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2017).
56. McMurtry VE, Gupta RW, Tran L, Iv EEB, Penn D, Taylor CM, et al. Bacterial diversity and clostridia abundance decrease with increasing severity of necrotizing enterocolitis. Microbiome. (2015) 3:11. doi: 10.1186/s40168-015-0075-8
57. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. (2005) 71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005
58. Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, et al. Vegan: Community Ecology Package. R Package Version 2.2-0. (2014). Available online at: http://CRAN.Rproject.org/package=vegan (accessed March 29, 2017).
59. Nathan HL, Seed PT, Hezelgrave NL, De Greeff A, Lawley E, Conti-Ramsden F, et al. Maternal and perinatal adverse outcomes in women with pre-eclampsia cared for at facility-level in South Africa: a prospective cohort study. J Glob Health. (2018) 8:020401. doi: 10.7189/jogh.08-020401
60. Say L, Chou D, Gemmill A, Tunçalp Ö, Moller AB, Daniels J, et al. Global causes of maternal death: a WHO systematic analysis. Lancet Glob Health. (2014) 6:e323–33. doi: 10.1016/S2214-109X(14)70227-X
61. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
62. Frost F, Kacprowski T, Rühlemann M, Pietzner M, Bang C, Franke A, et al. Long-term instability of the intestinal microbiome is associated with metabolic liver disease, low microbiota diversity, diabetes mellitus and impaired exocrine pancreatic function. Gut. (2021) 3:522–30. doi: 10.1136/gutjnl-2020-322753
63. Wang J, Shi ZH, Yang J, Wei Y, Wang XY, Zhao YY. Gut microbiota dysbiosis in preeclampsia patients in the second and third trimesters. Chin Med J. (2020) 9:1057. doi: 10.1097/CM9.0000000000000734
64. O'Callaghan A, van Sinderen D. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol. (2016) 7:925. doi: 10.3389/fmicb.2016.00925
65. Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol. (2000) 66:1654–61. doi: 10.1128/AEM.66.4.1654-1661.2000
66. Carlier J-P, K'ouas G, Han XY. Moryella indoligenes gen. nov., sp. nov., an anaerobic bacterium isolated from clinical specimens. Int J Syst Evol Microbiol. (2007) 57:725–9. doi: 10.1099/ijs.0.64705-0
67. Chen X, Fruehauf J, Goldsmith JD, Xu H, Katchar KK, Koon H, et al. Saccharomyces boulardii inhibits EGF receptor signaling and intestinal tumor growth in Apc(Min) mice. Gastroenterology. (2009) 137:914–23. doi: 10.1053/j.gastro.2009.05.050
68. Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med. (2017) 1:73. doi: 10.1186/s12967-017-1175-y
69. Venturi A, Gionchetti P, Rizzello F, Johansson R, Zucconi E, Brigidi P, et al. Impact on the composition of the faecal flora by a new probiotic preparation: preliminary data on maintenance treatment of patients with ulcerative colitis. Alim Pharmacol Therap. (1999) 13:1103–8. doi: 10.1046/j.1365-2036.1999.00560.x
70. Jang H, Patoine A, Wu TT, Castillo DA, Xiao J. Oral microflora and pregnancy: a systematic review and meta-analysis. Sci Rep. (2021) 1:1–31. doi: 10.1038/s41598-021-96495-1
71. Marcotte H, Lavoie MC. Oral microbial ecology and the role of salivary immunoglobulin microbiology A. Mol Biol Rev. (1998) 62:71–109. doi: 10.1128/MMBR.62.1.71-109.1998
72. Palmer RJ Jr. Composition and development of oral bacterial communities. Periodontology 2000. (2014) 64:20–39. doi: 10.1111/j.1600-0757.2012.00453.x
73. Adams SE, Arnold D, Murphy B, Carroll P, Green AK, Smith AM, et al. A randomised clinical study to determine the effect of a toothpaste containing enzymes and proteins on plaque oral microbiome ecology. Sci Rep. (2017) 7:43344. doi: 10.1038/srep43344
74. Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. (2012) 6:1176–85. doi: 10.1038/ismej.2011.191
75. Zamakhchari M, Wei G, Dewhirst F, Lee J, Schuppan D, Oppenheim FG, et al. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PLoS ONE. (2011) 6:e24455. doi: 10.1371/journal.pone.0024455
76. Duran-Pinedo AE, Frias-Lopez J. Beyond microbial community composition: functional activities of the oral microbiome in health and disease. Microbes Infect. (2015) 17:505–16. doi: 10.1016/j.micinf.2015.03.014
77. Fettweis JM, Brooks JP, Serrano MG, Sheth NU, Girerd PH, Edwards DJ, et al. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology. (2014) 160:2272–82. doi: 10.1099/mic.0.081034-0
78. Juliana NC, Peters RP, Al-Nasiry S, Budding AE, Morré SA, Ambrosino E. Composition of the vaginal microbiota during pregnancy in women living in sub-Saharan Africa: a PRISMA-compliant review. BMC Preg Childbirth. (2021) 1:1–5. doi: 10.1186/s12884-021-04072-1
79. Gosmann C, Anahtar MN, Handley SA, Farcasanu M, Abu-Ali G, Bowman BA, et al. Lactobacillus-deficient cervicovaginal bacterial communities are associated with increased HIV acquisition in young South African women. Immunity. (2017) 46:29–37. doi: 10.1016/j.immuni.2016.12.013
80. Jespers V, Menten J, Smet H, Poradosú S, Abdellati S, et al. Quantification of bacterial species of the vaginal microbiome in different groups of women, using nucleic acid amplification tests. BMC Microbiol. (2012) 12:83. doi: 10.1186/1471-2180-12-83
81. Mitchell C, Balkus JE, Fredricks D, Liu C, McKernan-Mullin J, Frenkel LM, et al. Interaction between lactobacilli, bacterial vaginosis-associated bacteria, and HIV type 1 RNA and DNA genital shedding in U.S. and Kenyan women. AIDS Res Human Retrov. (2013) 29:13–9. doi: 10.1089/aid.2012.0187
82. Petrova MI, Reid G, Vaneechoutte M, Lebeer S. Lactobacillus iners: friend or foe? Trends Microbiol. (2017) 25:182–91. doi: 10.1016/j.tim.2016.11.007
83. Srinivasan S, Hoffman NG, Morgan MT, Matsen FA, Fiedler TL, Hall RW, et al. Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS ONE. (2012) 7:e37818. doi: 10.1371/journal.pone.0037818
84. Liu M, Xu S, He Y, Deng G, Sheng H, Huang X. Diverse vaginal microbiomes in reproductive-age women with vulvovaginal candidiasis. PLoS ONE. (2013) 8:e79812. doi: 10.1371/journal.pone.0079812
85. Petricevic L, Domig KJ, Nierscher FJ, Sandhofer MJ, Fidesser M, Krondorfer I, et al. Characterisation of the vaginal lactobacillus microbiota associated with preterm delivery. Sci Rep. (2014) 4:5136. doi: 10.1038/srep05136
86. Kindinger LM, Bennett PR, Lee YS, Marchesi JR, Smith A, Cacciatore S, et al. The interaction between vaginal microbiota, cervical length, and vaginal progesterone treatment for preterm birth risk. Microbiome. (2017) 5:6. doi: 10.1186/s40168-016-0223-9
87. Brooks JP, Buck GA, Chen G, Diao L, Edwards DJ, Fettweis JM, et al. Changes in vaginal community state types reflect major shifts in the microbiome. Micro Ecol Health Dis. (2017) 1:1303265. doi: 10.1080/16512235.2017.1303265
88. Lopes dos Santos Santiago G, Tency I, Verstraelen H, Verhelst R, Trog M, Temmerman M, et al. Longitudinal qPCR study of the dynamics of crispatus L, L. iners, vaginae A, (sialidase positive) G. vaginalis, and P. bivia in the vagina. PLoS ONE. (2012) 7:e45281. doi: 10.1371/journal.pone.0045281
89. Macklaim JM, Gloor GB, Anukam KC, Cribby S, Reid G. At the crossroads of vaginal health and disease, the genome sequence of lactobacillus iners AB-1. Proc Natl Acad Sci USA. (2011) 108:4688–95. doi: 10.1073/pnas.1000086107
90. Borgdorff H, Armstrong SD, Tytgat HLP, Xia D, Ndayisaba GF, Wastling JM, et al. Unique insights in the cervicovaginal lactobacillus iners and L. crispatus proteomes and their associations with microbiota dysbiosis. PLoS ONE. (2016) 11:e0150767. doi: 10.1371/journal.pone.0150767
91. Freitas AC, Bocking A, Hill JE, Money DM. Increased richness and diversity of the vaginal microbiota and spontaneous preterm birth. Microbiome. (2018) 1:117. doi: 10.1186/s40168-018-0502-8
92. Leitich H, Kiss H. A symptomatic bacterial vaginosis and intermediate flora as risk factors for adverse pregnancy outcome. Best Pract Res Clin Obst Gynaecol. (2007) 21:375–90. doi: 10.1016/j.bpobgyn.2006.12.005
93. Fredricks DN. Molecular methods to describe the spectrum and dynamics of the vaginal microbiota. Anaerobe. (2011) 17:191–5. doi: 10.1016/j.anaerobe.2011.01.001
94. Fredricks DN, Fiedler TL, Marrazzo JM. Molecular identification of bacteria associated with bacterial vaginosis. N Engl J Med. (2005) 353:1899–911. doi: 10.1056/NEJMoa043802
Keywords: gut microbiome, vaginal microbiome, oral microbiome, pre-eclampsia, pregnancy, 16S rRNA sequence, diversity
Citation: Geldenhuys J, Redelinghuys MJ, Lombaard HA, Ehlers MM, Cowan D and Kock MM (2022) Diversity of the gut, vaginal and oral microbiome among pregnant women in South Africa with and without pre-eclampsia. Front. Glob. Womens Health 3:810673. doi: 10.3389/fgwh.2022.810673
Received: 07 November 2021; Accepted: 29 July 2022;
Published: 16 September 2022.
Edited by:
Marta Cohen, Sheffield Children's Hospital, United KingdomReviewed by:
Antonio Simone Laganà, University of Palermo, ItalyKingsley C. Anukam, Nnamdi Azikiwe University, Nigeria
Copyright © 2022 Geldenhuys, Redelinghuys, Lombaard, Ehlers, Cowan and Kock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marleen M. Kock, bWFybGVlbi5rb2NrQHVwLmFjLnph