
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genome Ed., 10 November 2022
Sec. Genome Editing in Blood Disorders
Volume 4 - 2022 | https://doi.org/10.3389/fgeed.2022.1050507
This article is part of the Research TopicTherapeutic Gene Correction Strategies Based on CRISPR Systems or Other Engineered Site-specific NucleasesView all 12 articles
 Beeke Wienert1
Beeke Wienert1 M. Kyle Cromer2,3,4*
M. Kyle Cromer2,3,4*The discovery of CRISPR has allowed site-specific genomic modification to become a reality and this technology is now being applied in a number of human clinical trials. While this technology has demonstrated impressive efficacy in the clinic to date, there remains the potential for unintended on- and off-target effects of CRISPR nuclease activity. A variety of in silico-based prediction tools and empirically derived experimental methods have been developed to identify the most common unintended effect—small insertions and deletions at genomic sites with homology to the guide RNA. However, large-scale aberrations have recently been reported such as translocations, inversions, deletions, and even chromothripsis. These are more difficult to detect using current workflows indicating a major unmet need in the field. In this review we summarize potential sequencing-based solutions that may be able to detect these large-scale effects even at low frequencies of occurrence. In addition, many of the current clinical trials using CRISPR involve ex vivo isolation of a patient’s own stem cells, modification, and re-transplantation. However, there is growing interest in direct, in vivo delivery of genome editing tools. While this strategy has the potential to address disease in cell types that are not amenable to ex vivo manipulation, in vivo editing has only one desired outcome—on-target editing in the cell type of interest. CRISPR activity in unintended cell types (both on- and off-target) is therefore a major safety as well as ethical concern in tissues that could enable germline transmission. In this review, we have summarized the strengths and weaknesses of current editing and delivery tools and potential improvements to off-target and off-tissue CRISPR activity detection. We have also outlined potential mitigation strategies that will ensure that the safety of CRISPR keeps pace with efficacy, a necessary requirement if this technology is to realize its full translational potential.
Gene therapy to correct, add, or modify genes holds great promise for many genetic disorders, including hemoglobinopathies, immunodeficiencies, and lysosomal storage disorders. Historically, gene therapy referred to viral-mediated gene addition, however this has the potential to disrupt essential genes or activate oncogenes due to semi-random genomic integration (Hacein-Bey-Abina et al., 2008). Gene editing tools such as CRISPR-Cas, TALENs, mega nucleases, or zinc finger nucleases have thus emerged as exciting alternatives due to the ability to target them to specific sites in the genome. Among these, the more straightforward and modular design of CRISPR guide RNAs (gRNA), which direct the Cas protein to a complementary site in the genome, has made them the preferred tool for both research and clinical applications. Ongoing clinical trials using CRISPR-modified cells have published results without any adverse events for both genome editing in T cells (Lu et al., 2020; Stadtmauer et al., 2020) and hematopoietic stem and progenitor cells (HSPCs) (Frangoul et al., 2021). In addition, the first clinical trial using CRISPR-Cas9 to treat transthyretin amyloidosis by editing hepatocytes in vivo has reported disease phenotype improvements in a small group of patients (Gillmore et al., 2021). These early clinical trials highlight the immense potential of CRISPR-Cas9 to treat disease, albeit lacking long-term follow-up data to support safety in humans.
One concern of clinical genome editing is the potential to cause unintended DNA alterations that may have a detrimental effect on cellular function (Figure 1). These undesired consequences can stem from on-target or off-target edits causing unwanted insertions and deletions (indels) or larger rearrangements (structural variants (SVs)) such as translocations, inversions, and duplications. The field has made great progress in developing methods to detect undesired editing events in silico, in cell-free DNA in vitro, and in live cells ex vivo (Blattner et al., 2020), but often it is challenging to link genomic alterations to their impact on cellular health and function. For example, off-target indels occurring in a gene desert may have no phenotypic effect, while some indels at the on-target site may lead to aberrant mRNA and protein products (Tuladhar et al., 2019) that significantly impact cell function (Lindeboom et al., 2019). As CRISPR-Cas9 genome editing moves towards in vivo therapeutic applications, making this link becomes even more critical as rare events could be detrimental if occurring in an oncogenic context. In addition, in vivo applications carry the risk of both on- and off-target genome edits in an unintended cell type such as the germline or other tissues.
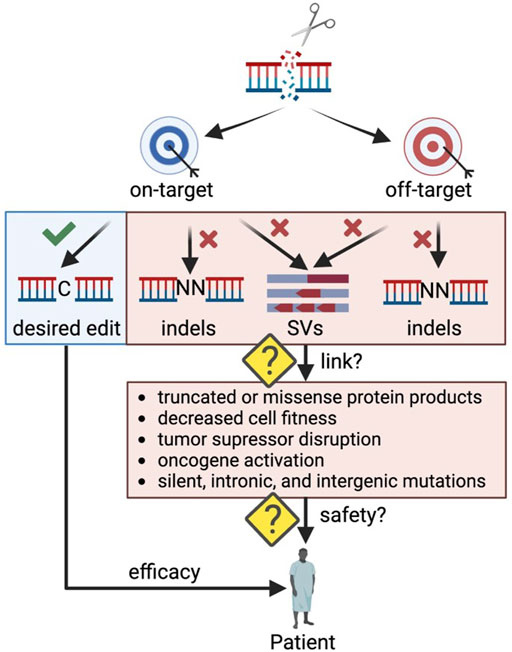
FIGURE 1. Outstanding questions to ensure safety in therapeutic genome editing applications. Indels: insertions and deletions; SVs: structural variants. Created with BioRender.com.
This field is rapidly evolving and new technologies to quantify unintended modifications are continuously being developed and evaluated. Furthermore, there is no broad consensus on the most appropriate measures needed to be taken to comprehensively assess the frequency and risks of CRISPR off-target activity, both for publication in high-impact journals or pre-clinically for the FDA. This review will therefore summarize the state of the field in terms of the current methods to evaluate on- and off-target gene edits, recent advances in method development for both ex vivo and in vivo editing workflows, and strategies for mitigation and reduction of off-target and off-tissue edits altogether.
One main advantage of CRISPR-Cas editing over viral genome addition is that it is specifically targeted to a gene locus rather than dependent on random integration. However, as the genome encompasses billions of base pairs, it is possible that the CRISPR-targeted sequence has a near-match elsewhere. Indeed, it has been shown that Cas9 and other nucleases will often cut highly homologous sequences depending on the location of the mismatch and the genomic context (Mali et al., 2013a; Fu et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013). Finding these off-target sites is critical so that the risk of unintended genomic events can be assessed and minimized (Figure 2).
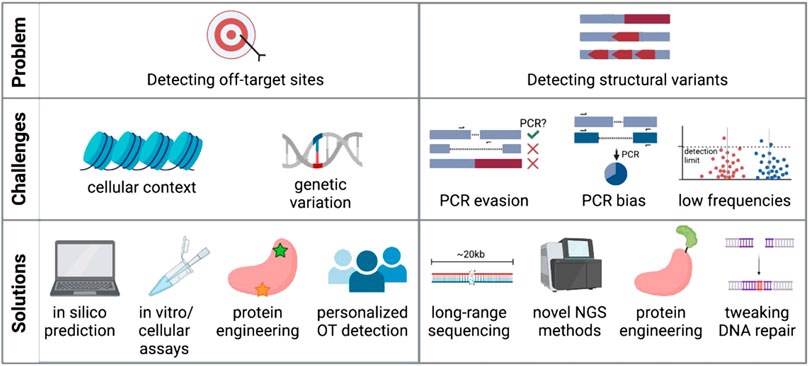
FIGURE 2. Challenges and potential solutions of current off-target detection methods. Created with BioRender.com.
Many computational tools are available to identify highly homologous genomic sequences and thus predict potential off-target sites for CRISPR-Cas9 activity (Bae et al., 2014; Cradick et al., 2014; Heigwer et al., 2014; Montague et al., 2014; Xiao et al., 2014; Zhu et al., 2014; Singh et al., 2015; Stemmer et al., 2015; Concordet and Haeussler, 2018; Aprilyanto et al., 2021). The application of machine learning to large experimental datasets has further improved the predictive power of these bioinformatic tools (Allen et al., 2018; Shen et al., 2018; Xiang et al., 2021), although these algorithms are biased towards the input gRNAs and reference genomes used to build their predictions. After identifying possible regions of off-target activity, screening must be done in genome-edited cells to confirm whether these sites show signs of CRISPR activity. This is most often done using targeted sequencing of candidate sites with standard sequencing panels achieving detection of variants at or below 5% frequency (Starks et al., 2021), while the detection limit for indels might be lower (0.2-1%) depending on sequencing depth (Chaudhari et al., 2020).
Alternatively, a number of experimental methods have been developed to find off-target sites that may not have been bioinformatically predicted based on homology to the gRNA. These methods vary widely in their approach and even starting material, using cell-free genomic DNA in vitro (Kim et al., 2015; Cameron et al., 2017; Tsai et al., 2017; Kim and Kim, 2018; Lazzarotto et al., 2020), in intact live cells ex vivo (Crosetto et al., 2013; Tsai et al., 2015; Yan et al., 2017; Wienert et al., 2019; 2020; Zhu et al., 2019; Dobbs et al., 2022), and in vivo animal models (Akcakaya et al., 2018; Wienert et al., 2019; Liang et al., 2022). Methods that use cell- and nucleosome-free DNA generally report the highest number of off-targets, many of which cannot be verified in a cellular context (Cromer et al., 2022a). Furthermore, methods such as GUIDE-Seq have been shown to identify more off-target sites in immortalized cell lines than when assaying primary cells (Shapiro et al., 2020). This highlights the importance of chromatin context and DNA repair factors in determining therapeutically relevant off-target activity. And even when using intact cells as input into these assays, conclusions drawn from immortalized cell lines with accumulated variants, distorted karyotypes, and dysfunctional DNA repair pathways (Mittelman and Wilson, 2013; Passerini et al., 2016) may confound the clinical relevance of identified off-target edits.
In spite of these myriad strategies for detection of off-target indels, recent work has shown that ex vivo editing in HSPCs elicits very few bona fide off-target sites (<1 true off-target site per gRNA) when using clinically relevant workflows (Cromer et al., 2022b). Of bona fide sites, all were highly homologous to the target sequence and predicted by the majority of in silico methods included in the study. However, several sources have shown that genetic variation amongst people can impact off-target activity (Lessard et al., 2017; Lazzarotto et al., 2020; Cromer et al., 2022a). Therefore, implementing a personalized patient-specific workflow in gene therapy products may be needed to circumvent the issue. Common SNPs can be taken into account for in silico prediction, in vitro methods could be personalized by using genomic DNA extracted from a patient sample (Tsai et al., 2017; Lazzarotto et al., 2020), and some cellular methods are amenable to use on primary cells from patients (Wienert et al., 2019). However, this type of personalized off-target analysis is limited by cost, logistical feasibility, and the availability of patient material.
Taken together, researchers have a broad range of tools available that allow them to identify potential off-target sites in silico, in cell-free DNA in vitro, in live cells ex vivo, and in animal models. By applying these tools sensibly in the experimental design of therapeutic genome editing strategies, off-target gene editing can be identified, and measures can be taken to minimize these unintended events.
While the off-target detection methods described above are most useful for identifying localized effects of DNA double-strand breaks (DSBs), larger scale off-target effects have been observed. These include gross chromosomal rearrangements such as translocations (Bothmer et al., 2020; Stadtmauer et al., 2020; Samuelson et al., 2021), chromothripsis (Leibowitz et al., 2021), and aneuploidy (Amendola et al., 2022; Nahmad et al., 2022). Translocation events most often occur as a consequence of: 1) on-target cleavage and recombination with a homologous region of the genome (Turchiano et al., 2021); 2) simultaneous cleavage at an on-target and off-target sequence (Lattanzi et al., 2021); or 3) following multiple on-target cleavage events in multiplexed editing workflows (Qasim et al., 2017; Bothmer et al., 2020; Stadtmauer et al., 2020; Samuelson et al., 2021; Diorio et al., 2022). In addition, large-scale deletions either surrounding the cut site or of the distal end of the chromosome can occur (Cullot et al., 2019), as well as copy-neutral loss-of-heterozygosity (Boutin et al., 2021).
Although targeted amplicon sequencing is commonly used to report on small indels at the cut site, most standard sequencing methods only allow sequencing of relatively short amplicons (hundreds of base pairs). Detecting and quantifying large-scale, multi-kilobase events with PCR-based sequencing methods thus remains challenging (Figure 2B). This is due to several reasons: 1) any deletion that eliminates primer binding sites would not be efficiently amplified with standard sequencing methods and would be missed; 2) if the primer binding sites are preserved larger deletions could skew the PCR reaction towards shorter amplicons and overestimate deletion events; and 3) other undesired on-target events may include inversions, gene duplications, and large insertions (Skryabin et al., 2020) that may also evade detection by PCR-based methods. New No-Amp long-range sequencing protocols avoid PCR and instead use CRISPR-Cas9 to enrich for the sequence of interest up to tens of kilobases. This PCR-free strategy circumvents size bias and can identify large deletions and other structural variants at the target site. However, limited read depth can make it difficult to detect and quantify low-frequency events and low base-calling accuracy of some methods may not achieve single base pair resolution (Lang et al., 2020). However, if these sequencing methods are able to improve and become cheaper, they may become the standard for evaluating structural variants after genome editing.
In addition to large deletions, other genomic abnormalities remain technically challenging to capture, especially when occurring at low frequency. To identify translocations of the on-target site with other genomic regions, several assays have been developed (Zheng et al., 2014; Hu et al., 2016; Qasim et al., 2017; Giannoukos et al., 2018; Yin et al., 2019; Turchiano et al., 2021) which use a sequence at the on-target site as “bait” and next-generation sequencing and bioinformatics to identify the “prey.” This allows identification of genomic sequences that have been fused to the on-target site. Another bioinformatic approach analyzes multiplexed-PCR data for on- and off-target sites using a pipeline specific for translocation detection (Amit et al., 2021) which could allow these events to be quantified from pre-existing data.
The delivery modality of the genome editing tools can also introduce unintended effects. For instance, if the nuclease or DNA repair template is delivered by adeno-associated virus (AAV), there is the possibility that non-homologous integration of inverted terminal repeats (ITRs) could occur (Hanlon et al., 2019; Nelson et al., 2019), however this may have minimal effect on adjacent genes if the template is promoter-less. The introduction of DSBs by a nuclease increases the amount of non-homologous integration of AAV vector sequences (Miller et al., 2003; Miller et al., 2004), however the overall frequency seems to be determined by the genomic context and can range between 0.06% and 12.5% of total events (Hanlon et al., 2019). These rare events can be captured with long-range sequencing methods or by a recently developed next-generation sequencing method, named ITR-Seq (Breton et al., 2020), which can identify and quantify ITR integrations on a genome-wide basis independent of the on-target site.
In summary, the field has made great progress in developing methods that can identify structural variants including deletions, inversions, duplications, insertions, and translocations. However, absolute quantification of these events remains challenging due to their low frequency of occurrence. While promising, novel long-range sequencing strategies are still lacking read depth and quality compared to traditional sequencing methods. As structural variants are diverse it is currently not possible for a single assay to capture all possible events, but future advances in sequencing technology could allow for this to become a reality.
Most CRISPR-based therapies currently in the clinic rely on isolation of patient-derived stem cells, ex vivo modification, and re-transplantation. This approach thereby addresses the limited availability of matched donors and risk of immune rejection or graft-versus-host-disease associated with allogeneic transplantation. However, these strategies are only compatible with cell types that may be safely isolated, modified ex vivo, and transplanted back into the patient, such as HSPCs. Therefore, the next frontier will be to deliver genome-editing components to modify cells directly where they reside in the body.
Toward this end, many delivery modalities have been developed and optimized to transduce clinically relevant cell types in vivo (Long et al., 2016; Goldstein et al., 2019; Mangeot et al., 2019; Gillmore et al., 2021). These platforms are now being used to package and deliver CRISPR-based editing tools in vivo, which has shown initial success in the first human clinical trials. One of the most prominent of these trials was conducted by Intellia where a liver-tropic lipid nanoparticle (LNP) was used to deliver Cas9 mRNA along with a gRNA specific to the TTR gene in order to treat transthyretin amyloidosis (Gillmore et al., 2021). This strategy effectively lowered serum TTR levels up to 87% from baseline in human patients, serving as a landmark study for efficacy of Cas9 to achieve a clinical endpoint. While in vivo Cas9 delivery was found to be quite effective in this instance, there was limited data collected to confirm safety aside from the absence of severe adverse events in these patients.
When delivering editing tools in vivo, there is only a single desired outcome—on-target editing at the intended CRISPR cleavage site in the intended target tissue (Figure 3). However, a number of unintended consequences can occur following delivery of editing tools to patients in vivo, such as: 1) off-target genomic activity in the intended target tissue (off-target, on-tissue); 2) on-target genomic activity in unintended tissue types (on-target, off-tissue); and 3) off-target genomic activity in unintended tissue types (off-target, off-tissue). Off-tissue events in the gonads are of particular clinical and ethical concern since these could result in changes to the germline which may be transmitted to a patient’s offspring (Turocy et al., 2021). Despite these fears and the use of methods to detect off-target Cas9 activity in vivo in animal models (Akcakaya et al., 2018; Wienert et al., 2020; Liang et al., 2022), no study to date has investigated the frequency of unintended events following delivery of editing tools to human patients in a clinical context in vivo. In the seminal TTR Cas9-LNP paper, the only investigation into off-target activity was done by performing GUIDE-Seq ex vivo in hepatocytes (Gillmore et al., 2021). While this is helpful in locating sites of potential off-target activity in the patient’s genome, these results were not validated in vivo. In the simplest form, clinically routine liver biopsies could have been performed pre- and post-delivery (perhaps at sites both near and far from the hepatic artery where the LNP would have entered the liver) to quantify the frequency of on- and off-target activity at the on-tissue site. However, this approach would yield little insight into CRISPR activity outside the liver, even though this LNP was reported to edit the spleen, adrenal glands, and ovaries at detectable frequencies. While the liver may be easily biopsied, this is not a routine procedure for many other tissues, particularly the ovaries. This therefore presents a major barrier to ensuring the safety of in vivo CRISPR delivery.
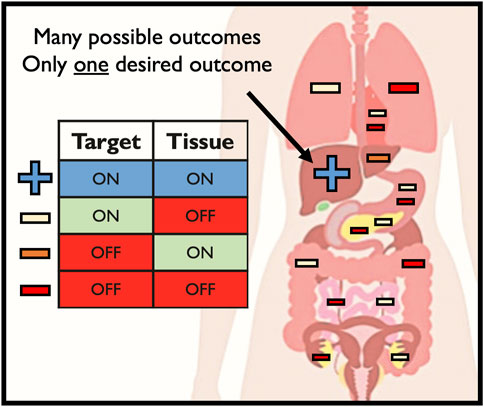
FIGURE 3. Possible outcomes of in vivo genome editing.
A potential source of genetic material that could be obtained in a minimally invasive fashion to determine the frequency of intended and unintended editing events following in vivo CRISPR delivery is cell-free genomic DNA (cfDNA). CfDNA is primarily derived from dying cells that release their genomic material into the bloodstream. Because of this, assaying cfDNA may be a powerful approach for detecting potentially pathogenic effects of CRISPR delivery, either in terms of genotoxic or oncogenic editing events. While cfDNA has primarily been used in the diagnostic space to detect occurrence/relapse of cancer (Bronkhorst et al., 2019), this technology is sensitive enough to sample maternal blood to discover de novo mutations in the fetus during pregnancy (Kitzman et al., 2012). In fact, a proof-of-concept study used cfDNA to map insertion sites following in vivo delivery of lentiviral vectors (Cesana et al., 2021). Therefore, a comparable strategy could be employed to quantify the frequency of on- and off-target cleavage activity following in vivo CRISPR delivery. However, unlike the workflow developed for mapping lentiviral insertions which relies on sequencing outward from the vector backbone, mapping sites of CRISPR activity may be aided by defining high-likelihood regions of activity. This could be done using in silico or empirical off-target detection methods defined above, and candidate regions could then be probed for indels by targeted deep sequencing of cfDNA. An alternative approach that would not require defining CRISPR off-target sites a priori could be to use translocations as a surrogate for on- and off-target activity by adapting technologies such as LAM-HTGTS (Hu et al., 2016), CAST-Seq (Turchiano et al., 2021), or PEM-Seq (Yin et al., 2019) to use patient cells or cfDNA as input. Furthermore, if using cfDNA isolated from peripheral blood is successful, a similar approach could be used to detect occurrence of CRISPR activity in cerebrospinal fluid (CSF) as well to quantify the ability of genome editing tools to cross the blood-brain barrier and edit cells residing in the brain, which may not be safely biopsied.
While cfDNA presents an opportunity to quantify on-target and off-target editing, it may give little insight into the tissue of origin. To shed light on this without invasive biopsies, the use of cell-free RNA (cfRNA) is a possibility. Analogous to the use of cfDNA, workflows to isolate cfRNA from the peripheral blood have been developed that allow insight into the tissue of origin due to the predominant expression patterns of cells releasing DNA and RNA into the bloodstream (Cheung et al., 2019). While the degree to which tissue-of-origin could be gleaned from this approach has yet to be fully explored, the investigation of on- and off-target CRISPR activity at expected cleavage sites in cfRNA could determine whether intended or unintended genome editing results in changes to the transcriptome. Since sites of CRISPR off-target activity typically reside in intergenic regions of the genome with no known functional significance (Cromer et al., 2022b), it is possible that CRISPR activity will be detected in cfDNA, but not in transcribed cfRNA. This could be an important measure to assay efficacy and safety of in vivo editing immediately following therapeutic delivery as well as over time. The combined cfDNA/RNA approach could also be an effective way to detect pathogenic clonal expansion of edited cells following treatment. In this specific use case, it may not be necessary to have identified the initiating driver genomic event, but even oligoclonality of passenger events—such as a particular indel at the on- or common off-target sites—could allow us to infer that clonal expansion is occurring. Importantly, the strategies proposed are most likely to capture and monitor the frequency of small site-specific indels, and more sophisticated methods (like those described earlier) may be needed to identify large genomic aberrations in cfDNA/RNA.
Even when we can successfully identify off-target CRISPR effects, determining if an unintended editing event is of concern to the patient’s health remains challenging (Figure 1). Broadly speaking, off-target genomic events are most likely to either elicit no effect or result in a loss or gain of fitness. While loss of fitness will likely result in drop out of the cell carrying the undesired event, gains of fitness are of greater concern due to the possibility of oncogenicity. Although site-specific off-target effects are infrequent (Cromer et al., 2022b), in the event that they do occur, the likelihood of directly disrupting another gene is small (only 1% of the human genome is coding DNA and only 7.2% of predicted off-target sites for exon-targeting gRNAs fall in exonic regions). And while modifications to non-coding DNA sequences may alter gene expression patterns or modify elements with as-yet-unknown important functions (ENCODE Project Consortium, 2012) interpreting non-coding genomic disruptions is difficult. As our understanding of the function of non-coding regions of DNA improves, we may better predict the impact of off-targets modifications in the future. Until then, we must rely on methods that can measure oncogenicity and toxicity from off-target modifications events in vitro or in vivo.
The most conventional approach to assess tumorigenicity of cell products is implanting cells at an ectopic site in immunodeficient mice followed by monitoring for tumor growth and other adverse events (Human Gene Therapy Products Incorporating Human Genome Editing | FDA). One major caveat of this method is that it has limited sensitivity, depends on the animal model used, and may miss low frequency events (Sato et al., 2019). Alternative in vivo approaches have developed technology to track clonality of cell-based products following ex vivo HSPC editing and transplantation through barcodes included in the HDR template (Ferrari et al., 2021; Sharma et al., 2021) or by tracking indel diversity (Magis et al., 2022). These technologies can identify clones that have expanded abnormally and hint towards genomic events that led to the oncogenic transformation. Currently these approaches have been limited to research applications but could potentially also be incorporated in therapeutic workflows in the future. However, in vivo studies are time- and cost-intensive and can slow down the drug development process immensely. Thus, in vitro studies that measure oncogenicity or genomic instability would be preferred, though these may not properly recapitulate in vivo activity. While performing whole genome sequencing on every cell product for every patient would ensure an unbiased approach of variant discovery across the whole genome, the currently limited coverage per base pair would miss low-frequency events. Using an intermediate approach of exome sequencing the most commonly mutated oncogenes and tumor suppressors increases read depth significantly and could provide a feasible alternative to assess the safety of ex vivo gene therapy drug products (Cromer et al., 2022a).
While the above work focuses on unintended off-target effects, even unintended on-target effects can lead to adverse effects. For instance, when a therapeutic editing approach targets a coding sequence—like knocking out a pathogenic gene to correct a disease phenotype (Gillmore et al., 2021)—an array of indels will form at the cut site. A recent report has shown that Cas9-induced indels can result in the formation of disrupted, non-natural mRNAs, which can be translated into aberrant protein products (Tuladhar et al., 2019). This study found that indels can induce internal ribosome entry sites to produce alternative mRNAs or induce exon skipping by disrupting exon-splicing enhancers. The same study also provided a bioinformatic tool to help design gRNAs to avoid such events (Tuladhar et al., 2019). Since truncated protein products could exert a dominant negative function (Savas et al., 2006), potential undesired translated proteins should be studied carefully. Properly characterizing the genome-edited cell population by combining on-target amplicon sequencing with mRNA sequencing and proteomics may allow us to identify and develop strategies to reduce the occurrence of such events.
Taken together, linking genomic events to oncogenicity is difficult and currently available in vitro and in vivo assays often lack sensitivity. Progress has been made to develop barcoding technologies that can track transformed cells and next-generation sequencing methods such as exome and RNA sequencing can also help identify oncogenic events.
As we learn more about the types of editing events that can occur at on- and off-target sites, many researchers are developing methods to reduce off-target effects altogether. These efforts range from protein engineering to make nucleases more specific to the discovery of novel, more specific delivery vehicles of genome editing reagents in vivo (Figure 4).
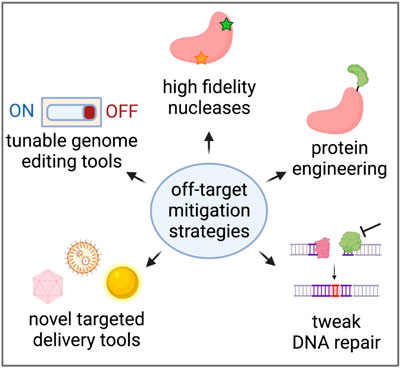
FIGURE 4. Strategies to reduce unintended genome editing events. Created with BioRender.com.
Careful nuclease selection and gRNA selection is often the first step when designing a de novo genome editing strategy (Lee et al., 2016). While a number of CRISPR nucleases have been discovered (Swarts and Jinek, 2018; Li and Peng, 2019), the majority of clinical efforts to date have used one of the original enzymes characterized, Streptococcus pyogenes Cas9 (SpCas9) (Jinek et al., 2012). This nuclease is one of the most common due to its relatively unrestrictive protospacer adjacent motif (PAM) and its high frequency of cleavage activity at a wide variety of loci in a range of cell types across a number of organisms, from humans to Arabidopsis (Mali et al., 2013b; Miki et al., 2018). While this nuclease typically has few genomic sites of off-target activity, some of these can be cut at high frequencies (>30% of alleles harboring indels), depending on the specificity of the particular gRNA (Cromer et al., 2022a). To address this, more specific versions of Cas9 have been engineered which reduce off-target activity by > 20-fold (Chen et al., 2017; Vakulskas et al., 2018; Bravo et al., 2022; Kulcsar et al., 2022). In doing so, incorporation of these higher-fidelity Cas9 variants has been shown to reduce the risk of large-scale genomic rearrangements (Turchiano et al., 2021). In addition to engineering more specific nucleases, a study that fused Cas9 to the exonuclease TREX2 in order to prevent perfect DNA repair reported significantly fewer large deletions and nearly eliminated chromosomal translocations during multiplex editing in T cells (Yin et al., 2022).
The format in which Cas9 is delivered—most often DNA, mRNA, or ribonucleoprotein (RNP)—will alter Cas9 expression and duration of exposure. This in turn has been shown to impact off-target activity, with short half-life RNP and mRNA resulting in lower off-target activity than longer-lived DNA formulations (Cameron et al., 2017; Lu et al., 2019; Zhang et al., 2021). In addition, tunable/inducible control strategies have been incorporated to regulate CRISPR expression using bioavailable small molecules, (Truong et al., 2015; Zhao et al., 2018), light (Nihongaki et al., 2015), and even magnets (Hsu and Hu, 2019). Similarly, other groups have developed self-inactivating Cas9 and AAV delivery vectors that may prevent prolonged exposure to genome editing tools and therefore reduce the likelihood of unintended activity or genomic events (Li et al., 2019; Ibraheim et al., 2021). However, depending on the tools these strategies are applied to, there remains the potential for off-target activity or large-scale genomic rearrangements following creation of DSBs. Furthermore, in their current forms, most approaches are only compatible with ex vivo editing workflows where high efficiency delivery of large payloads is possible.
All nuclease-based genome editing applications rely on DSB resolution, therefore modifying the cell’s natural DNA damage repair pathways has emerged as a strategy for increasing the ratio of intended to unintended genomic events (Yeh et al., 2019; Xue and Greene, 2021). For example, transiently inhibiting non-homologous end joining (NHEJ)-mediators such as 53BP1 or DNA-dependent protein kinase catalytic subunit can decrease indels and increase precise genome editing outcomes through homology-directed repair (Robert et al., 2015; Canny et al., 2018; Riesenberg and Maricic, 2018; Riesenberg et al., 2019). While some work has been done to determine which DNA repair enzymes are responsible for the formation of small indels (Hussmann et al., 2021), less is known about the factors that promote large deletions. Recently, a study that used a clonal library of embryonic stem cells deficient for DNA repair genes found that inhibition of NHEJ-mediating enzymes increased frequencies of large deletions, while inhibition of microhomology-mediated end joining-mediating proteins decreased them (Kosicki et al., 2022). Of course, it is crucial to ensure that temporarily inhibiting DNA repair enzymes does not affect other regions of the cell’s genome. Another study has shown that the presence of an HDR template such as a single-stranded oligodeoxynucleotide or AAV donor can reduce the frequency of large deletions by 50%–80% (Wen et al., 2021), emphasizing the importance of testing unintended editing outcomes in the context of both the nuclease and the DNA donor.
While Cas9 nuclease technology continues to improve, recent editing tools replace this nuclease with a nickase to introduce single base pair changes or small site-specific modifications, most commonly in the form of single or dual nickase editing (Ran et al., 2013), base editing (Komor et al., 2016), or prime editing (Anzalone et al., 2019). Although these tools avoid formation of DSBs and likely reduce the frequency of large-scale genomic rearrangements, there is still the possibility of unintended off-target activity. In the case of base and prime editors, this arises from the tethering of Cas9 with deaminases and reverse transcriptases, respectively. In fact, some studies have reported that base editors can initiate off-target activity at sites with little homology to the gRNA (Jin et al., 2019; Zuo et al., 2019; Lei et al., 2021) and in a significant proportion of cellular mRNA (Grünewald et al., 2019a). In addition, base editor-induced modifications are often single nucleotide variants which are more difficult to detect by next-generation sequencing than localized indels introduced by traditional CRISPR nucleases. Ongoing efforts continue to engineer improved versions of these base editors to reduce off-target activity (Rees et al., 2017; Grünewald et al., 2019b; Li et al., 2022).
The above advances primarily concern the editing tools themselves, which is most likely to boost on-target effects and reduce unintended off-target consequences. However, these improvements will likely have limited impact on the ratio of on-tissue to off-tissue activity following in vivo delivery of editing tools. Toward this end, many groups are working to improve specificity of the delivery modalities themselves. This includes screening for vectors or nanoparticles that have specific tissue tropisms, such as those optimized to cross the blood-brain-barrier, to transduce vascular tissue, and more (White et al., 2004; Choudhury et al., 2016; Sago et al., 2018; Boehnke et al., 2022). There also are efforts to conjugate cell type-specific antibodies to delivery vectors to improve targeted in vivo delivery (Tombácz et al., 2021). While preliminary, this approach may be an effective means to improve on-tissue editing when injecting delivery vectors systemically.
In the early stages of development, but with great translational potential, are strategies to encode logic into cells (i.e., to introduce DNA code capable of responding to a given cellular state). As with CRISPR, many of these efforts use RNA-based homology to facilitate downstream expression of transgenes in the presence of a user-defined RNA sequence (Green et al., 2014, 2017; Kaseniit et al., 2022). Several proof-of-concept studies demonstrated that this technology could be used to encode complex logic into cells, such as multi-input OR, AND, and NOT gates. While much of this work was done in E. coli or human cell lines, if an analogous system was ported to clinically relevant primary cells it could allow genome editing tools to only be expressed in cells with a particular gene expression profile—effectively reducing or eliminating off-tissue activity.
All the above efforts have been aimed at reducing unintended off-target and off-tissue activity. However, because millions or billions of cells are transplanted with ex vivo therapies, and billions or trillions of cells may be transduced with in vivo delivery vectors, any degree of unintended activity has the potential to be deleterious. Jennifer Doudna stated that one day she hopes to see CRISPR become a “standard of care” (Jennifer Doudna and William Kearney). If this is ever to become a reality, how do we make these therapies safe enough to be delivered routinely to patients?
While any manipulation to the genome opens the possibility for unwanted genetic events, we believe advances in off-target/off-tissue detection methods and improvements in genome editing tools and delivery modalities will ultimately allow personalized medicine to become a reality. As the development of advanced tools allows us to introduce increasingly complex features (and even logic) into cells, we will likely have to establish increasingly complex safety mechanisms as well. These may include automatic or inducible safety switches that provide a necessary safeguard in the instance of adverse clinical events (Di Stasi et al., 2011; Liang et al., 2018; Martin et al., 2020). And while initial data from CRISPR-based therapies in the clinic (both ex vivo and in vivo) has shown incredible promise, as greater numbers of patients receive genome editing treatments, we must ensure that editing safety keeps pace with efficacy. If CRISPR is ever to become the standard-of-care, then all of us—basic biologists, synthetic biologists, bioinformaticists, and clinicians—will have to combine efforts to ensure that genome editing therapies are as safe as possible.
BW and MKC generated figures and wrote the manuscript.
MKC is funded by an American Society of Gene and Cell Therapy Career Development Award as well as National Heart, Lung, and Blood Institute R01 (HL135607).
BW is current employee and equity holder in Graphite Bio, Inc., a for-profit company.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akcakaya, P., Bobbin, M. L., Guo, J. A., Malagon-Lopez, J., Clement, K., Garcia, S. P., et al. (2018). In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561, 416–419. doi:10.1038/s41586-018-0500-9
Allen, F., Crepaldi, L., Alsinet, C., Strong, A. J., Kleshchevnikov, V., De Angeli, P., et al. (2018). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 37, 64–72. doi:10.1038/nbt.4317
Amendola, M., Brusson, M., and Miccio, A. (2022). CRISPRthripsis: The risk of CRISPR/Cas9-induced chromothripsis in gene therapy. Stem Cells Transl. Med. 11, 1003–1009. doi:10.1093/stcltm/szac064
Amit, I., Iancu, O., Levy-Jurgenson, A., Kurgan, G., McNeill, M. S., Rettig, G. R., et al. (2021). CRISPECTOR provides accurate estimation of genome editing translocation and off-target activity from comparative NGS data. Nat. Commun. 12, 3042. doi:10.1038/s41467-021-22417-4
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. doi:10.1038/s41586-019-1711-4
Aprilyanto, V., Aditama, R., Tanjung, Z. A., Utomo, C., and Liwang, T. (2021). Crop: A CRISPR/Cas9 guide selection program based on mapping guide variants. Sci. Rep. 11, 1504. doi:10.1038/s41598-021-81297-2
Bae, S., Park, J., and Kim, J.-S. (2014). Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. doi:10.1093/bioinformatics/btu048
Blattner, G., Cavazza, A., Thrasher, A. J., and Turchiano, G. (2020). Gene editing and genotoxicity: Targeting the off-targets. Front. Genome Ed. 2, 613252. doi:10.3389/fgeed.2020.613252
Boehnke, N., Straehla, J. P., Safford, H. C., Kocak, M., Rees, M. G., Ronan, M., et al. (2022). Massively parallel pooled screening reveals genomic determinants of nanoparticle delivery. Science 377, eabm5551. doi:10.1126/science.abm5551
Bothmer, A., Gareau, K. W., Abdulkerim, H. S., Buquicchio, F., Cohen, L., Viswanathan, R., et al. (2020). Detection and modulation of DNA translocations during multi-gene genome editing in T cells. CRISPR J. 3, 177–187. doi:10.1089/crispr.2019.0074
Boutin, J., Rosier, J., Cappellen, D., Prat, F., Toutain, J., Pennamen, P., et al. (2021). CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 12, 4922. doi:10.1038/s41467-021-25190-6
Bravo, J. P. K., Liu, M.-S., Hibshman, G. N., Dangerfield, T. L., Jung, K., McCool, R. S., et al. (2022). Structural basis for mismatch surveillance by CRISPR-Cas9. Nature 603, 343–347. doi:10.1038/s41586-022-04470-1
Breton, C., Clark, P. M., Wang, L., Greig, J. A., and Wilson, J. M. (2020). ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genomics 21, 239. doi:10.1186/s12864-020-6655-4
Bronkhorst, A. J., Ungerer, V., and Holdenrieder, S. (2019). The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 17, 100087. doi:10.1016/j.bdq.2019.100087
Cameron, P., Fuller, C. K., Donohoue, P. D., Jones, B. N., Thompson, M. S., Carter, M. M., et al. (2017). Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods 14, 600–606. doi:10.1038/nmeth.4284
Canny, M. D., Moatti, N., Wan, L. C. K., Fradet-Turcotte, A., Krasner, D., Mateos-Gomez, P. A., et al. (2018). Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 36, 95–102. doi:10.1038/nbt.4021
Cesana, D., Calabria, A., Rudilosso, L., Gallina, P., Benedicenti, F., Spinozzi, G., et al. (2021). Retrieval of vector integration sites from cell-free DNA. Nat. Med. 27, 1458–1470. doi:10.1038/s41591-021-01389-4
Chaudhari, H. G., Penterman, J., Whitton, H. J., Spencer, S. J., Flanagan, N., Lei Zhang, M. C., et al. (2020). Evaluation of homology-independent CRISPR-Cas9 off-target assessment methods. The CRISPR Journal 3, 440–453. doi:10.1089/crispr.2020.0053
Chen, J. S., Dagdas, Y. S., Kleinstiver, B. P., Welch, M. M., Sousa, A. A., Harrington, L. B., et al. (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410. doi:10.1038/nature24268
Cheung, K. W. E., Choi, S.-Y. R., Lee, L. T. C., Lee, N. L. E., Tsang, H. F., Cheng, Y. T., et al. (2019). The potential of circulating cell free RNA as a biomarker in cancer. Expert Rev. Mol. diagn. 19, 579–590. doi:10.1080/14737159.2019.1633307
Choudhury, S. R., Fitzpatrick, Z., Harris, A. F., Maitland, S. A., Ferreira, J. S., Zhang, Y., et al. (2016). In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol. Ther. 24, 1247–1257. doi:10.1038/mt.2016.84
Concordet, J.-P., and Haeussler, M. (2018). Crispor: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245. doi:10.1093/nar/gky354
Cradick, T. J., Qiu, P., Lee, C. M., Fine, E. J., and Bao, G. (2014). Cosmid: A web-based tool for identifying and validating CRISPR/cas off-target sites. Mol. Ther. Nucleic Acids 3, e214. doi:10.1038/mtna.2014.64
Cromer, M. K., Barsan, V. V., Jaeger, E., Wang, M., Hampton, J. P., Chen, F., et al. (2022a). Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat. Commun. 13, 4724. doi:10.1038/s41467-022-32233-z
Cromer, M. K., Majeti, K. R., Rettig, G. R., Murugan, K., Kurgan, G. L., Hampton, J. P., et al. (2022b). Comparative analysis of CRISPR off-target activity discovery tools following ex vivo editing of CD34+ hematopoietic stem and progenitor cells. BioRxiv. doi:10.1101/2022.09.09.507306
Crosetto, N., Mitra, A., Silva, M. J., Bienko, M., Dojer, N., Wang, Q., et al. (2013). Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 10, 361–365. doi:10.1038/nmeth.2408
Cullot, G., Boutin, J., Toutain, J., Prat, F., Pennamen, P., Rooryck, C., et al. (2019). CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 10, 1136. doi:10.1038/s41467-019-09006-2
Di Stasi, A., Tey, S.-K., Dotti, G., Fujita, Y., Kennedy-Nasser, A., Martinez, C., et al. (2011). Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 365, 1673–1683. doi:10.1056/NEJMoa1106152
Diorio, C., Murray, R., Naniong, M., Barrera, L., Camblin, A., Chukinas, J., et al. (2022). Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood 140, 619–629. doi:10.1182/blood.2022015825
Dobbs, F. M., van Eijk, P., Fellows, M. D., Loiacono, L., Nitsch, R., and Reed, S. H. (2022). Precision digital mapping of endogenous and induced genomic DNA breaks by INDUCE-seq. Nat. Commun. 13, 3989. doi:10.1038/s41467-022-31702-9
Doudna, Jennifer, and Kearney, William.2020 An interview with nobel prize-winner jennifer Doudna. Issues Sci. Technol.XXXVI Available at: https://issues.org/jennifer-doudna-interview/ (Accessed August 17, 2022).
ENCODE Project Consortium, (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi:10.1038/nature11247
FDA, Human gene therapy products incorporating human genome editing | FDA.2022 Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-products-incorporating-human-genome-editing (Accessed August 31, 2022).
Ferrari, S., Beretta, S., Jacob, A., Cittaro, D., Albano, L., Merelli, I., et al. (2021). BAR-Seq clonal tracking of gene-edited cells. Nat. Protoc. 16, 2991–3025. doi:10.1038/s41596-021-00529-x
Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y.-S., Domm, J., Eustace, B. K., et al. (2021). CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260. doi:10.1056/NEJMoa2031054
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. doi:10.1038/nbt.2623
Giannoukos, G., Ciulla, D. M., Marco, E., Abdulkerim, H. S., Barrera, L. A., Bothmer, A., et al. (2018). UDiTaSTM, a genome editing detection method for indels and genome rearrangements. BMC Genomics 19, 212. doi:10.1186/s12864-018-4561-9
Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M. L., et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502. doi:10.1056/NEJMoa2107454
Goldstein, J. M., Tabebordbar, M., Zhu, K., Wang, L. D., Messemer, K. A., Peacker, B., et al. (2019). In situ modification of tissue stem and progenitor cell genomes. Cell Rep. 27, 1254–1264. e7. doi:10.1016/j.celrep.2019.03.105
Green, A. A., Kim, J., Ma, D., Silver, P. A., Collins, J. J., and Yin, P. (2017). Complex cellular logic computation using ribocomputing devices. Nature 548, 117–121. doi:10.1038/nature23271
Green, A. A., Silver, P. A., Collins, J. J., and Yin, P. (2014). Toehold switches: De-novo-designed regulators of gene expression. Cell 159, 925–939. doi:10.1016/j.cell.2014.10.002
Grünewald, J., Zhou, R., Garcia, S. P., Iyer, S., Lareau, C. A., Aryee, M. J., et al. (2019a). Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437. doi:10.1038/s41586-019-1161-z
Grünewald, J., Zhou, R., Iyer, S., Lareau, C. A., Garcia, S. P., Aryee, M. J., et al. (2019b). CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048. doi:10.1038/s41587-019-0236-6
Hacein-Bey-Abina, S., Garrigue, A., Wang, G. P., Soulier, J., Lim, A., Morillon, E., et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 118, 3132–3142. doi:10.1172/JCI35700
Hanlon, K. S., Kleinstiver, B. P., Garcia, S. P., Zaborowski, M. P., Volak, A., Spirig, S. E., et al. (2019). High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 10, 4439. doi:10.1038/s41467-019-12449-2
Heigwer, F., Kerr, G., and Boutros, M. (2014). E-CRISP: Fast CRISPR target site identification. Nat. Methods 11, 122–123. doi:10.1038/nmeth.2812
Hsu, M.-N., and Hu, Y.-C. (2019). Local magnetic activation of CRISPR. Nat. Biomed. Eng. 3, 83–84. doi:10.1038/s41551-019-0354-y
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi:10.1038/nbt.2647
Hu, J., Meyers, R. M., Dong, J., Panchakshari, R. A., Alt, F. W., and Frock, R. L. (2016). Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 11, 853–871. doi:10.1038/nprot.2016.043
Hussmann, J. A., Ling, J., Ravisankar, P., Yan, J., Cirincione, A., Xu, A., et al. (2021). Mapping the genetic landscape of DNA double-strand break repair. Cell 184, 5653–5669.e25. e25. doi:10.1016/j.cell.2021.10.002
Ibraheim, R., Tai, P. W. L., Mir, A., Javeed, N., Wang, J., Rodríguez, T. C., et al. (2021). Self-inactivating, all-in-one AAV vectors for precision Cas9 genome editing via homology-directed repair in vivo. Nat. Commun. 12, 6267. doi:10.1038/s41467-021-26518-y
Jin, S., Zong, Y., Gao, Q., Zhu, Z., Wang, Y., Qin, P., et al. (2019). Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295. doi:10.1126/science.aaw7166
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi:10.1126/science.1225829
Kaseniit, K. E., Katz, N., Kolber, N. S., Call, C. C., Wengier, D. L., Cody, W. B., et al. (2022). Modular and programmable RNA sensing using ADAR editing in living cells. BioRxiv. doi:10.1101/2022.01.28.478207
Kim, D., Bae, S., Park, J., Kim, E., Kim, S., Yu, H. R., et al. (2015). Digenome-seq: Genome-wide profiling of CRISPR-cas9 off-target effects in human cells. Nat. Methods 12 (237–43), 237–243. 1 p following 243. doi:10.1038/nmeth.3284
Kim, D., and Kim, J.-S. (2018). DIG-Seq: A genome-wide CRISPR off-target profiling method using chromatin DNA. Genome Res. 28, 1894–1900. doi:10.1101/gr.236620.118
Kitzman, J. O., Snyder, M. W., Ventura, M., Lewis, A. P., Qiu, R., Simmons, L. E., et al. (2012). Noninvasive whole-genome sequencing of a human fetus. Sci. Transl. Med. 4, 137ra76. doi:10.1126/scitranslmed.3004323
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi:10.1038/nature17946
Kosicki, M., Allen, F., Steward, F., Tomberg, K., Pan, Y., and Bradley, A. (2022). Cas9-induced large deletions and small indels are controlled in a convergent fashion. Nat. Commun. 13, 3422. doi:10.1038/s41467-022-30480-8
Kulcsar, P. I., Talas, A., Welker, E., and Krausz, S. L. (2022). SuperFi-Cas9 exhibits extremely high fidelity but reduced activity in mammalian cells. BioRxiv. doi:10.1101/2022.05.27.493683
Lang, D., Zhang, S., Ren, P., Liang, F., Sun, Z., Meng, G., et al. (2020). Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of pacific biosciences sequel II system and ultralong reads of oxford nanopore. Gigascience 9, giaa123–7. doi:10.1093/gigascience/giaa123
Lattanzi, A., Camarena, J., Lahiri, P., Segal, H., Srifa, W., Vakulskas, C. A., et al. (2021). Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 13. doi:10.1126/scitranslmed.abf2444
Lazzarotto, C. R., Malinin, N. L., Li, Y., Zhang, R., Yang, Y., Lee, G., et al. (2020). CHANGE-seq reveals genetic and epigenetic effects on CRISPR-Cas9 genome-wide activity. Nat. Biotechnol. 38, 1317–1327. doi:10.1038/s41587-020-0555-7
Lee, C. M., Cradick, T. J., Fine, E. J., and Bao, G. (2016). Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol. Ther. 24, 475–487. doi:10.1038/mt.2016.1
Lei, Z., Meng, H., Lv, Z., Liu, M., Zhao, H., Wu, H., et al. (2021). Detect-seq reveals out-of-protospacer editing and target-strand editing by cytosine base editors. Nat. Methods 18, 643–651. doi:10.1038/s41592-021-01172-w
Leibowitz, M. L., Papathanasiou, S., Doerfler, P. A., Blaine, L. J., Sun, L., Yao, Y., et al. (2021). Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 53, 895–905. doi:10.1038/s41588-021-00838-7
Lessard, S., Francioli, L., Alfoldi, J., Tardif, J.-C., Ellinor, P. T., MacArthur, D. G., et al. (2017). Human genetic variation alters CRISPR-Cas9 on- and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. U. S. A. 114, E11257–E11266. doi:10.1073/pnas.1714640114
Li, A., Lee, C. M., Hurley, A. E., Jarrett, K. E., De Giorgi, M., Lu, W., et al. (2019). A self-deleting AAV-CRISPR system for in vivo genome editing. Mol. Ther. Methods Clin. Dev. 12, 111–122. doi:10.1016/j.omtm.2018.11.009
Li, A., Mitsunobu, H., Yoshioka, S., Suzuki, T., Kondo, A., and Nishida, K. (2022). Cytosine base editing systems with minimized off-target effect and molecular size. Nat. Commun. 13, 4531. doi:10.1038/s41467-022-32157-8
Li, Y., and Peng, N. (2019). Endogenous CRISPR-cas system-based genome editing and antimicrobials: Review and prospects. Front. Microbiol. 10, 2471. doi:10.3389/fmicb.2019.02471
Liang, Q., Monetti, C., Shutova, M. V., Neely, E. J., Hacibekiroglu, S., Yang, H., et al. (2018). Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature 563, 701–704. doi:10.1038/s41586-018-0733-7
Liang, S.-Q., Liu, P., Smith, J. L., Mintzer, E., Maitland, S., Dong, X., et al. (2022). Genome-wide detection of CRISPR editing in vivo using GUIDE-tag. Nat. Commun. 13, 437. doi:10.1038/s41467-022-28135-9
Lindeboom, R. G. H., Vermeulen, M., Lehner, B., and Supek, F. (2019). The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651. doi:10.1038/s41588-019-0517-5
Long, C., Amoasii, L., Mireault, A. A., McAnally, J. R., Li, H., Sanchez-Ortiz, E., et al. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403. doi:10.1126/science.aad5725
Lu, B., Javidi-Parsijani, P., Makani, V., Mehraein-Ghomi, F., Sarhan, W. M., Sun, D., et al. (2019). Delivering SaCas9 mRNA by lentivirus-like bionanoparticles for transient expression and efficient genome editing. Nucleic Acids Res. 47, e44. doi:10.1093/nar/gkz093
Lu, Y., Xue, J., Deng, T., Zhou, X., Yu, K., Deng, L., et al. (2020). Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 26, 732–740. doi:10.1038/s41591-020-0840-5
Magis, W., DeWitt, M. A., Wyman, S. K., Vu, J. T., Heo, S.-J., Shao, S. J., et al. (2022). High-level correction of the sickle mutation is amplified in vivo during erythroid differentiation. iScience 25, 104374. doi:10.1016/j.isci.2022.104374
Mali, P., Aach, J., Stranges, P. B., Esvelt, K. M., Moosburner, M., Kosuri, S., et al. (2013a). CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838. doi:10.1038/nbt.2675
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013b). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi:10.1126/science.1232033
Mangeot, P. E., Risson, V., Fusil, F., Marnef, A., Laurent, E., Blin, J., et al. (2019). Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 10, 45. doi:10.1038/s41467-018-07845-z
Martin, R. M., Fowler, J. L., Cromer, M. K., Lesch, B. J., Ponce, E., Uchida, N., et al. (2020). Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 11, 2713. doi:10.1038/s41467-020-16455-7
Miki, D., Zhang, W., Zeng, W., Feng, Z., and Zhu, J.-K. (2018). CRISPR/Cas9-mediated gene targeting in Arabidopsis using sequential transformation. Nat. Commun. 9, 1967. doi:10.1038/s41467-018-04416-0
Miller, D. G., Petek, L. M., and Russell, D. W. (2004). Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 36, 767–773. doi:10.1038/ng1380
Mittelman, D., and Wilson, J. H. (2013). The fractured genome of HeLa cells. Genome Biol. 14, 111. doi:10.1186/gb-2013-14-4-111
Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., and Valen, E. (2014). CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407. doi:10.1093/nar/gku410
Nahmad, A. D., Reuveni, E., Goldschmidt, E., Tenne, T., Liberman, M., Horovitz-Fried, M., et al. (2022). Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. doi:10.1038/s41587-022-01377-0
Nelson, C. E., Wu, Y., Gemberling, M. P., Oliver, M. L., Waller, M. A., Bohning, J. D., et al. (2019). Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 25, 427–432. doi:10.1038/s41591-019-0344-3
Nihongaki, Y., Kawano, F., Nakajima, T., and Sato, M. (2015). Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760. doi:10.1038/nbt.3245
Passerini, V., Ozeri-Galai, E., de Pagter, M. S., Donnelly, N., Schmalbrock, S., Kloosterman, W. P., et al. (2016). The presence of extra chromosomes leads to genomic instability. Nat. Commun. 7, 10754. doi:10.1038/ncomms10754
Pattanayak, V., Lin, S., Guilinger, J. P., Ma, E., Doudna, J. A., and Liu, D. R. (2013). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31, 839–843. doi:10.1038/nbt.2673
Qasim, W., Zhan, H., Samarasinghe, S., Adams, S., Amrolia, P., Stafford, S., et al. (2017). Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 9, eaaj2013. doi:10.1126/scitranslmed.aaj2013
Ran, F. A., Hsu, P. D., Lin, C.-Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. doi:10.1016/j.cell.2013.08.021
Rees, H. A., Komor, A. C., Yeh, W.-H., Caetano-Lopes, J., Warman, M., Edge, A. S. B., et al. (2017). Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 8, 15790. doi:10.1038/ncomms15790
Riesenberg, S., Chintalapati, M., Macak, D., Kanis, P., Maricic, T., and Pääbo, S. (2019). Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 47, e116. doi:10.1093/nar/gkz669
Riesenberg, S., and Maricic, T. (2018). Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 9, 2164. doi:10.1038/s41467-018-04609-7
Robert, F., Barbeau, M., Éthier, S., Dostie, J., and Pelletier, J. (2015). Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 7, 93. doi:10.1186/s13073-015-0215-6
Sago, C. D., Lokugamage, M. P., Paunovska, K., Vanover, D. A., Monaco, C. M., Shah, N. N., et al. (2018). High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. U. S. A. 115, E9944–E9952. doi:10.1073/pnas.1811276115
Samuelson, C., Radtke, S., Zhu, H., Llewellyn, M., Fields, E., Cook, S., et al. (2021). Multiplex CRISPR/Cas9 genome editing in hematopoietic stem cells for fetal hemoglobin reinduction generates chromosomal translocations. Mol. Ther. Methods Clin. Dev. 23, 507–523. doi:10.1016/j.omtm.2021.10.008
Sato, Y., Bando, H., Di Piazza, M., Gowing, G., Herberts, C., Jackman, S., et al. (2019). Tumorigenicity assessment of cell therapy products: The need for global consensus and points to consider. Cytotherapy 21, 1095–1111. doi:10.1016/j.jcyt.2019.10.001
Savas, S., Tuzmen, S., and Ozcelik, H. (2006). Human SNPs resulting in premature stop codons and protein truncation. Hum. Genomics 2, 274–286. doi:10.1186/1479-7364-2-5-274
Shapiro, J., Iancu, O., Jacobi, A. M., McNeill, M. S., Turk, R., Rettig, G. R., et al. (2020). Increasing CRISPR efficiency and measuring its specificity in hspcs using a clinically relevant system. Mol. Ther. Methods Clin. Dev. 17, 1097–1107. doi:10.1016/j.omtm.2020.04.027
Sharma, R., Dever, D. P., Lee, C. M., Azizi, A., Pan, Y., Camarena, J., et al. (2021). The TRACE-Seq method tracks recombination alleles and identifies clonal reconstitution dynamics of gene targeted human hematopoietic stem cells. Nat. Commun. 12, 472. doi:10.1038/s41467-020-20792-y
Shen, M. W., Arbab, M., Hsu, J. Y., Worstell, D., Culbertson, S. J., Krabbe, O., et al. (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. doi:10.1038/s41586-018-0686-x
Singh, R., Kuscu, C., Quinlan, A., Qi, Y., and Adli, M. (2015). Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 43, e118. doi:10.1093/nar/gkv575
Skryabin, B. V., Kummerfeld, D.-M., Gubar, L., Seeger, B., Kaiser, H., Stegemann, A., et al. (2020). Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9-mediated genome editing events. Sci. Adv. 6, eaax2941. doi:10.1126/sciadv.aax2941
Stadtmauer, E. A., Fraietta, J. A., Davis, M. M., Cohen, A. D., Weber, K. L., Lancaster, E., et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365. doi:10.1126/science.aba7365
Starks, E. R., Swanson, L., Docking, T. R., Bosdet, I., Munro, S., Moore, R. A., and Karsan, A. (2021). Assessing limit of detection in clinical sequencing. J. Mol. Diagn. 23, 455–466. doi:10.1016/j.jmoldx.2020.12.010
Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., and Mateo, J. L. (2015). Cctop: An intuitive, flexible and reliable crispr/cas9 target prediction tool. PLoS ONE 10, e0124633. doi:10.1371/journal.pone.0124633
Swarts, D. C., and Jinek, M. (2018). Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA 9, e1481. doi:10.1002/wrna.1481
Tombácz, I., Laczkó, D., Shahnawaz, H., Muramatsu, H., Natesan, A., Yadegari, A., et al. (2021). Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol. Ther. 29, 3293–3304. doi:10.1016/j.ymthe.2021.06.004
Truong, D.-J. J., Kühner, K., Kühn, R., Werfel, S., Engelhardt, S., Wurst, W., et al. (2015). Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 43, 6450–6458. doi:10.1093/nar/gkv601
Tsai, S. Q., Nguyen, N. T., Malagon-Lopez, J., Topkar, V. V., Aryee, M. J., and Joung, J. K. (2017). CIRCLE-Seq: A highly sensitive in vitro screen for genome-wide CRISPR-cas9 nuclease off-targets. Nat. Methods 14, 607–614. doi:10.1038/nmeth.4278
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. doi:10.1038/nbt.3117
Tuladhar, R., Yeu, Y., Tyler Piazza, J., Tan, Z., Rene Clemenceau, J., Wu, X., et al. (2019). CRISPR-Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat. Commun. 10, 4056. doi:10.1038/s41467-019-12028-5
Turchiano, G., Andrieux, G., Klermund, J., Blattner, G., Pennucci, V., El Gaz, M., et al. (2021). Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell 28, 1136–1147.e5. e5. doi:10.1016/j.stem.2021.02.002
Turocy, J., Adashi, E. Y., and Egli, D. (2021). Heritable human genome editing: Research progress, ethical considerations, and hurdles to clinical practice. Cell 184, 1561–1574. doi:10.1016/j.cell.2021.02.036
Vakulskas, C. A., Dever, D. P., Rettig, G. R., Turk, R., Jacobi, A. M., Collingwood, M. A., et al. (2018). A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224. doi:10.1038/s41591-018-0137-0
Wen, W., Quan, Z.-J., Li, S.-A., Yang, Z.-X., Fu, Y.-W., Zhang, F., et al. (2021). Effective control of large deletions after double-strand breaks by homology-directed repair and dsODN insertion. Genome Biol. 22, 236. doi:10.1186/s13059-021-02462-4
White, S. J., Nicklin, S. A., Büning, H., Brosnan, M. J., Leike, K., Papadakis, E. D., et al. (2004). Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation 109, 513–519. doi:10.1161/01.CIR.0000109697.68832.5D
Wienert, B., Wyman, S. K., Richardson, C. D., Yeh, C. D., Akcakaya, P., Porritt, M. J., et al. (2019). Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364, 286–289. doi:10.1126/science.aav9023
Wienert, B., Wyman, S. K., Yeh, C. D., Conklin, B. R., and Corn, J. E. (2020). CRISPR off-target detection with DISCOVER-seq. Nat. Protoc. 15, 1775–1799. doi:10.1038/s41596-020-0309-5
Xiang, X., Corsi, G. I., Anthon, C., Qu, K., Pan, X., Liang, X., et al. (2021). Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 12, 3238. doi:10.1038/s41467-021-23576-0
Xiao, A., Cheng, Z., Kong, L., Zhu, Z., Lin, S., Gao, G., et al. (2014). CasOT: A genome-wide cas9/gRNA off-target searching tool. Bioinformatics 30, 1180–1182. doi:10.1093/bioinformatics/btt764
Xue, C., and Greene, E. C. (2021). DNA repair pathway choices in CRISPR-cas9-mediated genome editing. Trends Genet. 37, 639–656. doi:10.1016/j.tig.2021.02.008
Yan, W. X., Mirzazadeh, R., Garnerone, S., Scott, D., Schneider, M. W., Kallas, T., et al. (2017). BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 8, 15058. doi:10.1038/ncomms15058
Yeh, C. D., Richardson, C. D., and Corn, J. E. (2019). Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 21, 1468–1478. doi:10.1038/s41556-019-0425-z
Yin, J., Liu, M., Liu, Y., Wu, J., Gan, T., Zhang, W., et al. (2019). Optimizing genome editing strategy by primer-extension-mediated sequencing. Cell Discov. 5, 18. doi:10.1038/s41421-019-0088-8
Yin, J., Lu, R., Xin, C., Wang, Y., Ling, X., Li, D., et al. (2022). Cas9 exo-endonuclease eliminates chromosomal translocations during genome editing. Nat. Commun. 13, 1204. doi:10.1038/s41467-022-28900-w
Zhang, S., Shen, J., Li, D., and Cheng, Y. (2021). Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 11, 614–648. doi:10.7150/thno.47007
Zhao, C., Zhao, Y., Zhang, J., Lu, J., Chen, L., Zhang, Y., et al. (2018). HIT-Cas9: A CRISPR/cas9 genome-editing device under tight and effective drug control. Mol. Ther. Nucleic Acids 13, 208–219. doi:10.1016/j.omtn.2018.08.022
Zheng, Z., Liebers, M., Zhelyazkova, B., Cao, Y., Panditi, D., Lynch, K. D., et al. (2014). Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 20, 1479–1484. doi:10.1038/nm.3729
Zhu, L. J., Holmes, B. R., Aronin, N., and Brodsky, M. H. (2014). CRISPRseek: A bioconductor package to identify target-specific guide RNAs for CRISPR-cas9 genome-editing systems. PLoS ONE 9, e108424. doi:10.1371/journal.pone.0108424
Zhu, Y., Biernacka, A., Pardo, B., Dojer, N., Forey, R., Skrzypczak, M., et al. (2019). qDSB-Seq is a general method for genome-wide quantification of DNA double-strand breaks using sequencing. Nat. Commun. 10, 2313. doi:10.1038/s41467-019-10332-8
Keywords: Gene therapy, off-target activity, in vivo delivery, genome editing, CRISPR/Cas9, next-generating sequencing
Citation: Wienert B and Cromer MK (2022) CRISPR nuclease off-target activity and mitigation strategies. Front. Genome Ed. 4:1050507. doi: 10.3389/fgeed.2022.1050507
Received: 21 September 2022; Accepted: 26 October 2022;
Published: 10 November 2022.
Edited by:
Ayal Hendel, Bar-Ilan University, IsraelReviewed by:
Toni Cathomen, Medical Center—University of Freiburg, GermanyCopyright © 2022 Wienert and Cromer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Kyle Cromer, a3lsZS5jcm9tZXJAdWNzZi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.