 Wenwen Zhang
Wenwen Zhang Shengjun Zhao1†
Shengjun Zhao1† Hongyi Liu
Hongyi Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 17 March 2025
Sec. Evolutionary and Population Genetics
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1566634
The phylogeny of Diplopoda, a group of ancient arthropod and an important component of modern terrestrial ecosystems, remains unclear. Here, the complete mitogenome of Spirobolus walkeri was determined. The newly sequenced complete mitogenome was circular DNA molecules with sizes of 14,879 bp. The mitogenome was composed of 37 genes and one control region. Negative AT-skews and positive GC-skews were found in whole mitogenome. The gene COX1 used CGA as the start codon, while the other PCGs utilized ATN (A, T, G) as the start codons; however, the sequence of the stop codon was variable. The Ser2 exhibited the highest usage bias. All tRNAs have typical cloverleaf structures, except trnS-AGC and trnM. Phylogenetic analysis showed that S. walkeri and Spirobolus bungii shared a close relationship and that they were also closely related with Narceus annularus. This study helps resolve taxonomic ambiguities among morphologically similar species and provides data to support the establishment of evolutionary benchmarks for millipedes, including gene rearrangements and variations in tRNA structure.
Hitherto, the Diplopoda contained more than 18,000 species worldwide (Golovatch and Liu, 2019). They play the role of decomposers in ecosystems and are an important component of modern terrestrial ecosystems. Because of the large variety of species and the morphological similarity of some different species, the phylogenetic relationships among the millipede orders are uncertain despite their self-evident importance (Woo et al., 2007).
Mitochondria are fundamental organelles found in eukaryotic cells, exhibiting autonomous and preserved genetic characteristics. The mitochondrial genome (mitogenome) of most animals is circular in structure and varies in length from 13 to 17 kbp. This biomolecule typically consists of 37 coding segments (Xu et al., 2025). Specifically, it includes 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and one control region (Xu et al., 2024a). In recent years, mitochondrial genes as well as mitogenomes have been extensively utilized in research regarding molecular systematics, population genetics, molecular evolution, and the application of molecular barcodes in environmental investigations (Xu et al., 2024b; Xu et al., 2024c). Contrasted with mitochondrial genes, which are prone to loss, mitogenomes exhibit multiple characteristic variations that enhance the precision of inferring phylogenetic relationships. Among the features of mitogenomes, gene rearrangements are notably complex and diverse, offering significant value for phylogenetic analysis. Molecular phylogenetic methods based on mitochondrial genomes have only recently been employed to reconstruct the phylogenetic relationships of arthropods (Xu et al., 2025). Furthermore, Myriapoda species serve as excellent model organisms for exploring the relationship between gene rearrangement and phylogenetic analysis. This is due to their high frequency of mitogenome rearrangement (Woo et al., 2007). Despite their potential, studies on the mitogenomes of Diplopod species have been relatively limited. Fewer than 50 millipede species have had their mitogenomes sequenced. To conduct a comprehensive and systematic phylogenetic analysis of the class Diplopoda, a substantial number of complete mitogenomes from its subordinate species is required (Golovatch and Liu, 2019). The scarcity of mitogenome data has resulted in inadequate research into the evolutionary significance of gene rearrangement in Diplopoda and their phylogenetic relationships.
The genus Spirobolus is a relatively common millipede found in eastern China. Like other millipedes, they play a crucial ecological role in the formation of humus by promoting the decomposition of organic litter and maintaining soil fertility. However, few studies have focused on their evolutionary relationships, and classification methods based solely on morphology may lead to potential classification issues. Our understanding of the higher-level taxonomic relationships within the genus Spirobolus and their genetic composition remains limited.
Spirobolus walkeri (Spirobolida: Spirobolidae) was found in the eastern part of China and firstly reported and described by Pocock (PocockBassett-Smith et al., 1895). In this study, we determined the complete mitogenomes of S. walkeri and analyzed the mitogenomic structure with respect to reveal its phylogenetic characteristics and evolutionary relationship in Diplopoda. The purpose of this study is to provide new data for the characterization of mitotic genomes in the millipede family, to further refine the phylogenetic relationships between these groups.
Specimen of S. walkeri was collected from the Mount Wuyi in Fujian Province, China (27°34′15″N, 117°50′11″E). The sampling site was protected, and we had the permission for the sampling approved by Biodiversity Comprehensive Observation Station for Wuyi Mountains. The sample is 6 mm wide and 84 mm long, with 49 segments. The sixth and seventh segments are swollen. The first segment is covered in small indentations and striae above, laterally narrowed to 45°, with a rounded apex. The sample has a characteristic red half-ring. The specimen was identified based on the morphological characteristics described by Pocock (PocockBassett-Smith et al., 1895). Fresh tissues were dissected and stored at −20°C. The specimen was preserved in a cryogenic freezer at −80°C at the Zoology Laboratory of Nanjing Forestry University with the voucher ID H005.
Genomic DNA was extracted using a FastPure Cell/Tissue DNA Isolation Mini Kit (Vazyme, Nanjing, China). The complete genomic of S. walkeri was sequenced using the NovaSeq 6,000 sequencer on the Illumina platform (Personal, Shanghai, China) with an insert size of 300 bp (about 2 Gb of raw data). To get clean data, low-quality sequences were removed. Then, 30,878,964 reads were assembled to obtain a complete mitogenome using Geneious Prime (Version. 2021.1). The mitogenome of Spirobolus bungii (Accession: MT767838) was used as templates for the assembly. Sequence contigs were assembled and trimmed using the medium sensitivity/fast option in the Geneious Prime (Version. 2021.1). Consensus sequences were constructed in Geneious Prime using a 99% base call threshold. The complete mitogenome of S. walkeri was obtained (Accession: OR078377).
MITOS WebServer (Bernt et al., 2013) was used for sequence annotation, and all parts of the sequence were confirmed using BLAST. The RNA secondary structure was described by ViennaRNA Web Services (Kerpedjiev et al., 2015). Circular gene maps were generated using the CGView Server (Stothard et al., 2019). MEGA11 (Tamura et al., 2019) was used to determine the base composition, the relative synonymous codon usage (RSCU) of each codon, and the nonsynonymous (Ka) and synonymous substitutions (Ks) (Tamura et al., 2019). Nucleotide compositional skewness was calculated according to the formula proposed by Perna and Kocher (Ojala et al., 1981).
The phylogenetic relationships were reconstructed using 27 Diplopoda mitogenomes with a Chilopoda mitogenome as the outgroup Bayesian inference (BI) and maximum likelihood (ML) methods were used for phylogenetic analyses with the nucleotide sequences of the 13 PCGs (Supplementary Table S3). TreeSuite was used for checking saturation. MAFFT v.7.313 (Rozewicki et al., 2019) was used to align all PCGs, and the best substitution model was identified by ModelFinder (Kalyaanamoorthy et al., 2017). The best fit models for BI and ML trees were shown in Supplementary Table S1. Phylogenetic analysis was performed using PhyloSuite v.1.2.2 (Zhang et al., 2020). The BI method was used to construct a tree using the software MrBayes (2 parallel runs, 1,000,000 generations), in which the initial 25% of sampled data were discarded as burn-in (Ronquist et al., 2012). The PSRF of the tree is close to 1 and therefore converges (Supplementary Table S2). The ML tree was constructed using IQ-TREE with 100,000 ultrafast bootstraps (Nguyen et al., 2015). The phylogenetic trees were visualized and edited using the Interactive Tree of Life Web Services (Zhou et al., 2023).
The complete mitogenome of S. walkeri (Accession: OR078377) was a double-stranded circular DNA molecule with 14,879 bp in size, which contained 37 typical mitochondrial genes (13 PCGs, 22 tRNAs, and 2 rRNAs) and one control region. Two rRNAs, 4 PCGs (ND1, ND4L, ND4 and ND5), and 9 tRNAs (trnV, trnL-CUA, trnL-UUA, trnP, trnH, trnF, trnY, trnQ, and trnC) were found to be encoded on the major strand (J-chain), whereas the remaining genes were located on the minor strand (N-chain) (Figure 1). The mitogenome had 7 intervals and 11 overlaps. All intervals length ranged from 1 to 40 bp, and the longest interval occurred between trnQ and trnT. All overlaps length ranges from 1 to 8 bp, and the longest overlap occurs between Cytb and ND6, and between trnC and trnW (Supplementary Table S4).
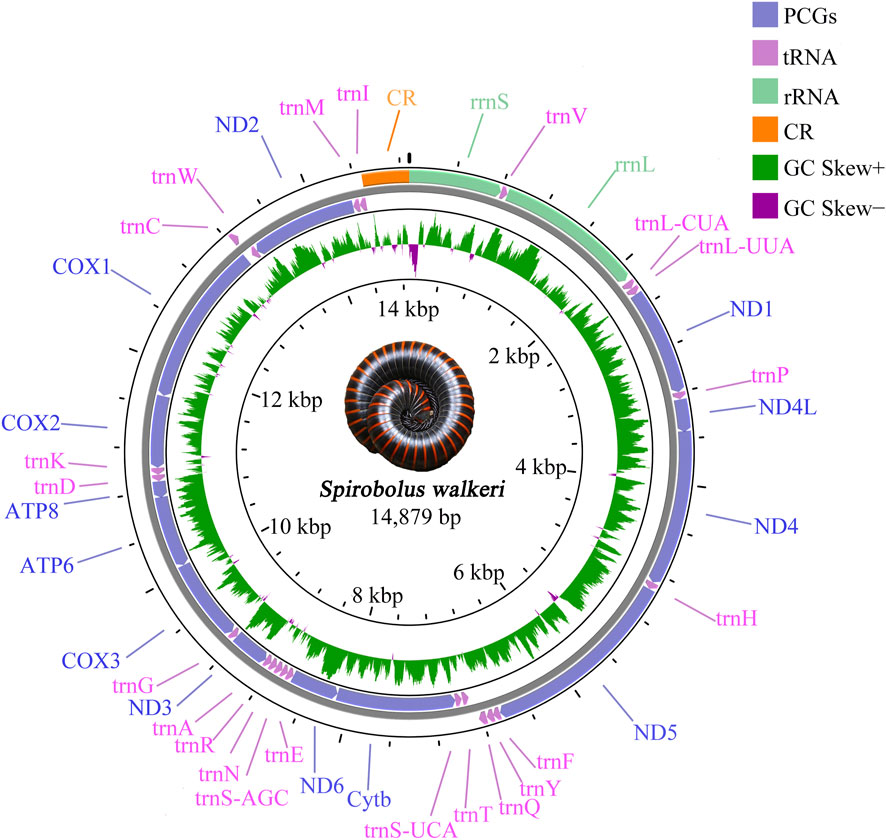
Figure 1. Gene map of the sequenced mitogenome of Spirobolus walkeri. Genes encoded by the J-chain are shown outside the circle, and those encoded by the N-chain are shown inside the circle. Different gene types are shown as filled boxes in different colours.
The base composition of complete mitochondrial genome was 27.30% for A, 33.50% for T, 27.80% for G, and 11.50% for C (Supplementary Table S5). The AT-skew of S. walkeri was negative, while the GC-skew was positive. The AT-skew of PCGs and rRNAs showed similar features to the complete mitogenome except tRNAs. The CG-skew of complete mitogenome was negative and so is rRNAs and tRNAs, except for PCGs (Supplementary Table S6).
The total length of the 13 PCGs of S. walkeri was 10,988 bp. The AT-skew of the 13 PCGs was −0.128 and the GC-skew was −0.063. The base composition of the PCGs was A = 25.5%, T = 33.1%, G = 19.4%, and C = 22.0% (Supplementary Table S5). Six PCGs (ND4L, ND4, CYTB, COX3, ATP6 and COX2) used ATG as the initiation codon; Three PCGs (ND1, ND3 and ND2) used ATA as the initiation codon; and three PCGs (ND5, ND6 and ATP8) used ATT as the initiation codon. A nonstandard initiation codon CGA was observed in COX1 in the mitogenome of S. walkeri. Five PCGs (ND4L, ND6, ATP8, COX2 and COX1) used TAN (TAA and TAG) as termination codon, and the other PCGs used T as the termination codon (Supplementary Table S4).
The RSCU values of Spirobolida species are summarized in Figure 2. According to the analysis of RSCU and codon distribution, Leu, Val, and Gly were the three most frequently utilized amino acids, and Cys had the lowest concentration. Nine of the twenty-two amino acids had four codons, while the others had two.
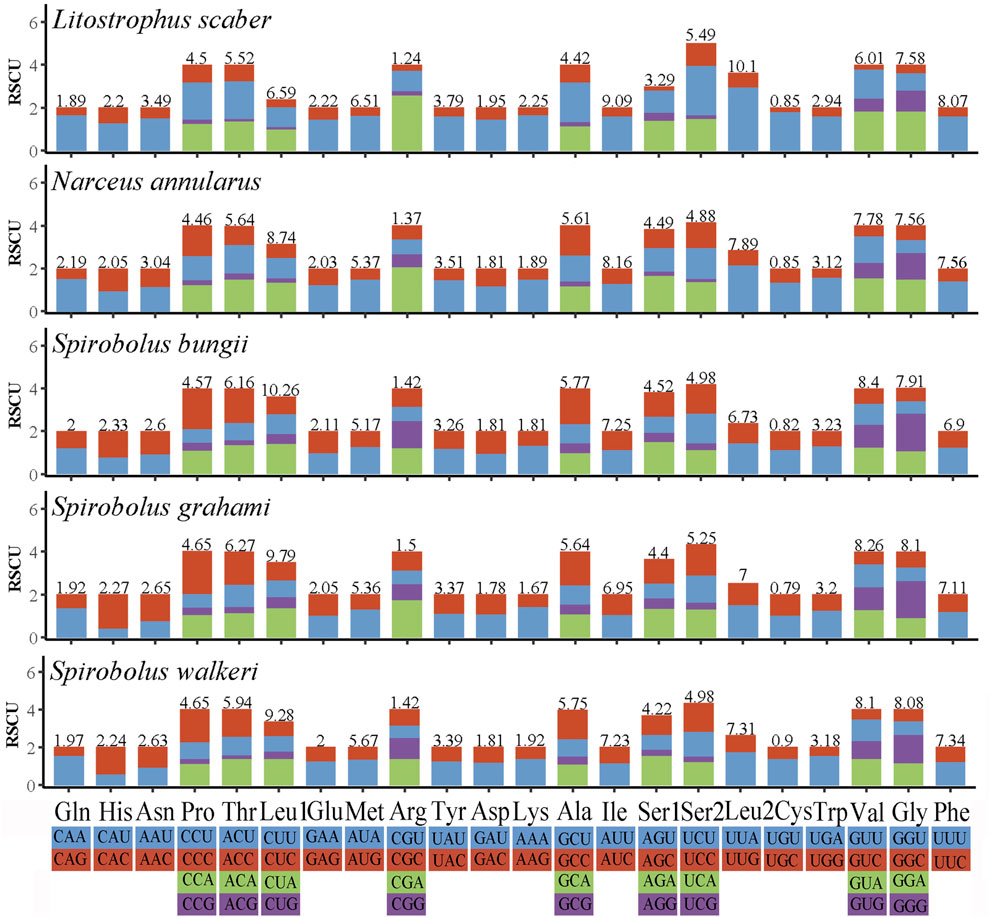
Figure 2. Relative synonymous codon usage in five Spirobolida mitogenomes. Different colors correspond to different third base.
To examine the evolutionary rate of the PCGs, we evaluated the Ka/Ks values of Spirobolida species (Figure 3). The COX3 gene (1.40), ND4 gene (1.56), and ND5 gene (1.55) exhibited an average Ka/Ks ratio greater than 1, while the COX1 gene had the lowest average Ka/Ks value.
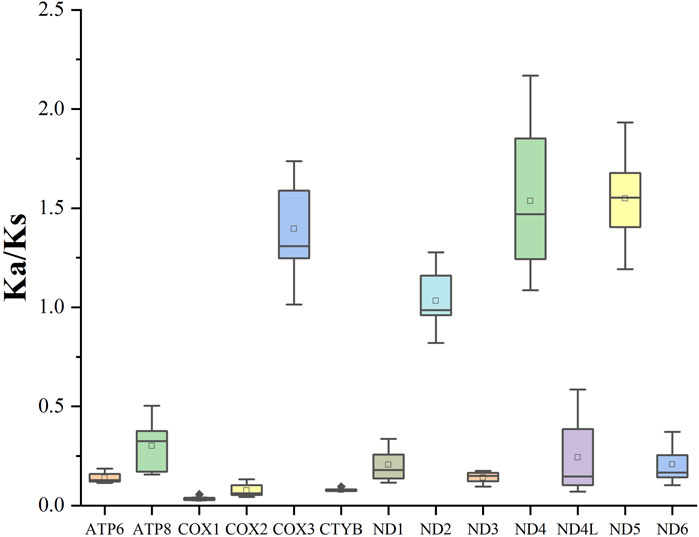
Figure 3. Ka/Ks values for the 13 PCGs of the order Spirobolida.
The total size of the 22 rRNAs was 1,378 bp, with an A + T content of 66.14%, an AT-skew of 0.052, and a GC-skew of 0.075. The sizes of the 22 tRNAs ranging from 59 bp (trnV and trnS-AGC) to 68 bp (trnQ). All tRNAs were of the standard cloverleaf structure, except trnM and trnS-AGC. Only trnM showed loss of the pseudo uracil (TΨC) arm, which was lack of a simple loop (Supplementary Figure S1). Moreover, 14 U-G wobble pairs were found in tRNAs.
The length of rrnL was 1,270 bp, whereas rrnS was 802 bp. The total size of the two rRNAs was 2,072 bp, with an A + T content of 66.1%, an AT-skew of −0.063, and a GC-skew of 0.361 (Supplementary Table S6).
Based on ML and BI analyses of nucleotide data of the 13 PCGs, we reconstructed the phylogenetic relationships of 27 species of Diplopoda, with Scolopendra subspinipes as an outgroup. As shown in Figure 4, the trees generated by BI and ML had identical topology and nodal support. S. walkeri clustered together with two other species in the genus, S. bungii, and S. grahami. The genus Spirobolus may have a sister-group relationship with the genus Narceus.
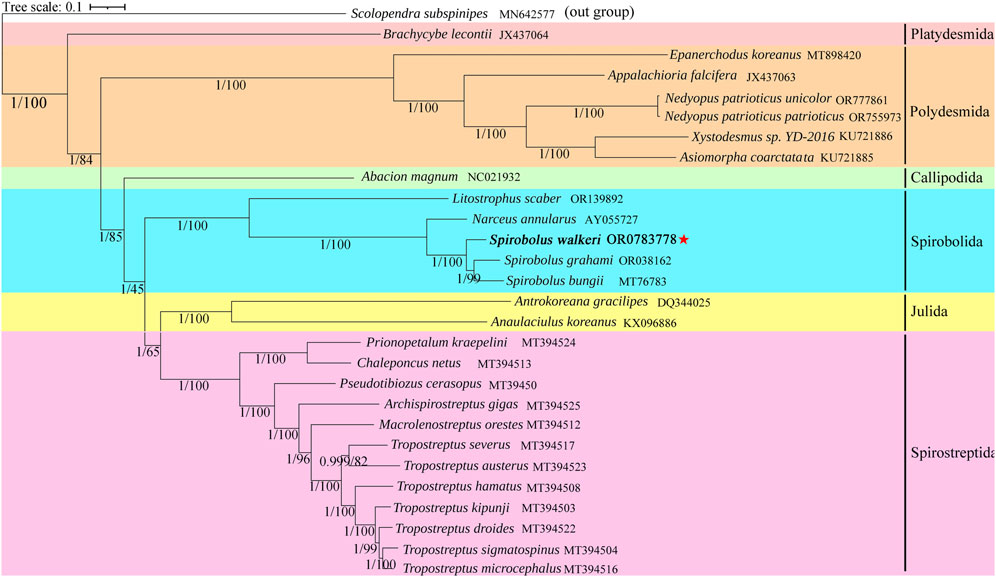
Figure 4. Phylogenetic tree based on 13 PCGs of 27 Diplopoda species and an outgroup. Numbers at nodes represent the posterior probability and bootstrap values for BI and ML analysis, respectively. Reported species is highlighted using bold font and red five-pointed stars.
The mitogenome structure of S. walkeri is similar to that of other millipede mitogenomes (19). The A + T base content (61.8%) is greater than that of G + C bases (39.3%), revealing an AT preference, as observed in other Millipedes (Xu et al., 2022; Zhang WW. et al., 2024), such as Litostrophus scaber (Zhang et al., 2024b) and Nedyopus patrioticus (Zhang et al., 2024c). Additionally, the A + T% of PCGs, rRNAs, and tRNAs in Spirobolida species were also higher than the G + C%. Skewness analysis based on base composition was used to estimate the relative numbers of A to T and G to C. The results of the skewness analysis indicated that the AT-skews of Spirobolida species were negative, along with the tRNA genes, while the GC-skews were positive in relation to PCGs. This pattern is consistent with findings in other millipede species (Xu et al., 2022; Zhang WW. et al., 2024). The asymmetries in nucleotide composition are typically regarded as indicators of gene direction and replication orientation during the processes of replication and transcription (Perna and Kocher, 1995).
In the mitogenome of S. walkeri, PCGs are present on both the H- and L-strands. Like in S. bungii (Zhang WW. et al., 2024), the majority of PCGs in the S. walkeri mitogenome are located on the H-strand, rendering them prone to hydrolysis and oxidation. Uncommon initiation codons in S. walkeri have also been observed on other millipedes. For example, in N. patrioticus, the ND1 gene utilizes GTA as its initiation codon (Zhang et al., 2024c). The use of T as a specific termination codon is not exclusive to one species; it is also present in other arthropods. This base can be transcribed into either TAA or TAG, serving its function during the transcription process (Ojala et al., 1981).
The RSCU values and codon distribution of S. walkeri were similar to species in the same order (Zhang et al., 2024b). The usage of codons ending in A or U was significantly higher than the usage of codons ending in C or G. This reflects a strong bias towards A and T nucleotides in the third position of the codons. Similarly, the preference for A and T nucleotides was reflected in the frequencies of codons. Based on the assumption of neutral protein-level evolution, the values of Ka and Ks should be equivalent, resulting in a Ka/Ks ratio of 1. When the Ka/Ks ratio is less than 1, it indicates the presence of purifying or stabilizing selection, suggesting a tendency to resist change. Conversely, a ratio greater than 1 signifies positive or Darwinian selection, which encourages evolutionary change (Hurst, 2002). Higher Ka/Ks values indicate that the genes COX3, ND4, and ND5 are experiencing positive selection, potentially due to environmental changes or interspecific competition. Ka/Ks values close to 1 indicate that the gene ND2 is undergoing neutral evolution, which suggests that the ND2 protein function in order Spirobolida has been highly optimized. The lowest Ka/Ks value of the COX1 gene indicates that it has the slowest evolutionary rate, making it a useful marker for species identification (Zhang et al., 2024c).
The tRNA result is consistent with previous studies on the class Myriapoda (Ding et al., 2023; Nunes et al., 2020). All tRNAs have typical cloverleaf structures, except trnS-AGC and trnM. Moreover, trnS-AGC was absence of a dihydrouridine (DHU) arm, which seems to be a common phenomenon in arthropods (Bai et al., 2022; Ge et al., 2022; Jiang et al., 2022; Wu et al., 2020).
Related genes in mitogenomes are currently widely used in phylogenetic research and species classification. Mwabvu et al. (Tarombera et al., 2013) constructed a phylogenetic tree based on rrnL genes to study the evolutionary status of Bicoxidens flavicollis. With the widespread use of molecular biology methods, domestic and foreign researchers are increasingly studying the phylogenetics of millipedes. Nielsen et al. (2022) observed a clear genetic structure in Tropostreptus, with distinct lineages (both inter- and intraspecific) being defined by the mountain blocks. Moreover, phylogenetic trees provide support for the classification of the genus Tropostreptus. Previous research on millipede mitochondria has indicated that the genus Tropostreptus is phylogenetically more closely related to Archispirostreptus gigas and Macrolenostreptus orestes (Nielsen et al., 2022). Current taxonomies classify Julida, Spirostreptida, and Spirobolida as part of the superorder Juliformia; however, the precise relationships among these three groups remain controversial (Zhang et al., 2024b). A study based on mitogenome confirmed that Julida and Spirostreptida share a sister-group relationship (Zuo et al., 2022). However, morphological studies suggest that Spirobolida and Julida have a sister-group relationship (Blanke and Wesener, 2014). Our results align with previous phylogenetic studies based on mitogenome. Although mitogenome exhibited several distinctive variations that improved the accuracy of inferring phylogenetic relationships, our results were relatively weak, possibly due to limited use of mitogenome data. More mitogenomes would significantly enhance phylogenetic studies of Diplopoda species.
In general, the study of S. walkeri’s mitogenome elucidate its evolutionary dynamics through gene arrangement, selection pressures, and nucleotide diversity, thereby revealing its divergence within the Spirobolus genus and its environmental adaptations. The complete mitogenome of S. walkeri will benefit resolving taxonomic ambiguities among morphologically similar species and provides data to support the establishment of evolutionary benchmarks for millipedes, including gene rearrangements and variations in tRNA structure. This research addresses gaps in genetic resources and provides a significant molecular foundation for soil invertebrate conservation, biogeography, and studies on global change.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OR078377.
The animal study was approved by the Experimental Animal Welfare and Ethics Committee, Nanjing Forestry University. The study was conducted in accordance with the local legislation and institutional requirements.
WZ: Formal Analysis, Funding acquisition, Resources, Writing–original draft. SZ: Formal Analysis, Resources, Writing–original draft. LL: Methodology, Writing–original draft. YL: Methodology, Writing–original draft. HL: Formal Analysis, Writing–original draft. PC: Conceptualization, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Open Foundation of State Environmental Protection Scientific Observation and Research Station for Ecological Environment of Wuyi Mountains.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1566634/full#supplementary-material
Bai, Y., Yang, K., Ye, L., and Gao, X. Y. (2022). Complete mitogenome and phylogenetic analyses of Galerita orientalis schmidt-goebel, 1846 (insecta: Coleoptera: carabidae: galeritini). Genes 13, 2199. doi:10.3390/genes13122199
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 69, 313–319. doi:10.1016/j.ympev.2012.08.023
Blanke, A., and Wesener, T. (2014). Revival of forgotten characters and modern imaging techniques help to produce a robust phylogeny of the Diplopoda (Arthropoda, Myriapoda). Arthropod Struct. and Dev. 43, 63–75. doi:10.1016/j.asd.2013.10.003
Ding, J. Y., Lan, H., Xu, W., Chen, Y. N., Wu, H., Jiang, H. M., et al. (2023). Two complete mitochondrial genomes in Scolopendra and a comparative analysis of tRNA rearrangements in centipedes. Mol. Biol. Rep. 49, 6173–6180. doi:10.1007/s11033-022-07409-x
Ge, X., Wang, L., and Wu, Y. (2022). The complete mitochondrial genome of Matsumuramata muiri (Hemiptera: Delphacidae). Mitochondrial DNA Part B 7, 294–296. doi:10.1080/23802359.2021.1955026
Golovatch, S. I., and Liu, W. X. (2019). Diversity, distribution patterns, and fauno-genesis of the millipedes (Diplopoda) of mainland China. Zookeys 930, 153–198. doi:10.3897/zookeys.930.47513
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486–487. doi:10.1016/s0168-9525(02)02722-1
Jiang, J., Wu, T., Deng, J., and Peng, L. A. (2022). A compositional heterogeneity analysis of mitochondrial phylogenomics in chalcidoidea involving two newly sequenced mitogenomes of eupelminae (hymenoptera: chalcidoidea). Genes 13, 2340. doi:10.3390/genes13122340
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi:10.1038/nmeth.4285
Kerpedjiev, P., Hammer, S., and Hofacker, I. L. (2015). Forna (force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics 31, 3377–3379. doi:10.1093/bioinformatics/btv372
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/molbev/msu300
Nielsen, M., Margaryan, A., Nielsen, T. L., Enghoff, H., and Allentoft, M. E. (2022). Complete mitochondrial genomes from museum specimens clarify millipede evolution in the Eastern Arc Mountains. Zoological J. Linn. Soc. 196, 924–939. doi:10.1093/zoolinnean/zlac058
Nunes, G. L., Oliveira, R. R. M., Pires, E. S., Pietrobon, T., Prous, X., Oliveira, G., et al. (2020). Complete mitochondrial genome of Glomeridesmus spelaeus (Diplopoda, Glomeridesmida), a troglobitic species from iron-ore caves in Eastern Amazon. Mitochondrial DNA Part B 5, 3272–3273. doi:10.1080/23802359.2020.1812450
Ojala, D., Montoya, J., and Attardi, G. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. doi:10.1038/290470a0
Perna, N. T., and Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. doi:10.1007/BF00186547
PocockBassett-Smith, R. I. P. W., Surgeon, R. N., Walker, J. J., Esq, R. N., and Commander, W. U. (1895). Moore commanding. Ann. Mag. Nat. Hist. 15, 346–368. doi:10.1080/00222939508677895
Ronquist, F., Teslenko, M., Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi:10.1093/sysbio/sys029
Rozewicki, J., Li, S. L., Amada, K. M., Standley, D. M., and Katoh, K. (2019). MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5-W10–W10. doi:10.1093/nar/gkz342
Stothard, P., Grant, J. R., and Van, D. G. (2019). Visualizing and comparing circular genomes using the CGView family of tools. Briefings Bioinforma. 20, 1576–1582. doi:10.1093/bib/bbx081
Tamura, K., Stecher, G., and Kumar, S. (2019). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi:10.1093/molbev/msab120
Tarombera, M., Jenny, L., Rob, S., Michelle, H., and David, B. (2013). Millipede taxonomy based on gonopod morphology too inclusive? Observations on genetic variation and cryptic speciation in Bicoxidens flavicollis (Diplopoda: Spirostreptida: Spirostreptidae). Afr. Invertebr. 54, 349–356. doi:10.5733/afin.054.0203
Woo, H. J., Lee, Y. S., Park, S. J., Lim, J. T., Jang, K. H., Choi, E. H., et al. (2007). Complete mitochondrial genome of a troglobite millipede Antrokoreana gracilipes (Diplopoda, Juliformia, Julida), and juliformian phylogeny. Mol. CELLS 23, 182–191. doi:10.1016/s1016-8478(23)07372-7
Wu, Y. P., Lu, J. J., Yang, J., Wang, J. P., Cao, T. W., and Fan, R. J. (2020). Complete mitochondrial genome of Mycalesis intermedia (Lepidoptera: nymphalidae). Mitochondrial DNA Part B 5, 703–704. doi:10.1080/23802359.2020.1714491
Xu, H., Fang, Y., Cao, G., Shen, C., Liu, H. Y., and Ruan, H. H. (2022). The complete mitochondrial genome of Spirobolus bungii (Diplopoda, Spirobolidae): the first sequence for the genus Spirobolus. Genes 13, 1587. doi:10.3390/genes13091587
Xu, T. J., Xu, W., Zhang, G. J., Liu, Z. Y., and Liu, H. Y. (2025). Characterization of the complete mitochondrial genomes of four tarantulas (Arachnida: theraphosidae) with phylogenetic analysis. diversity 15, 148954. doi:10.1016/j.gene.2024.148954
Xu, W., Bai, Y. W., Xu, T. J., Chen, Y. K., Wang, J. C., Li, T. T., et al. (2024b). Revealing potential mechanisms of native and non-native snail coexistence through fecal microbiomes and dietary compositions. Sci. Total Environ. 20, 177774. doi:10.1016/j.scitotenv.2024.177774
Xu, W., Tai, J. Z., He, K., Xu, T. J., Zhang, G. J., Xu, B. Y., et al. (2024c). Complete mitochondrial genomes of nannostomus pencilfish: genome characterization and phylogenetic analysis. animals 14, 1598. doi:10.3390/ani14111598
Xu, W., Zhang, G. J., Xu, T. J., He, K., Wang, J. C., Liu, Z. Y., et al. (2025). Comparative analysis of mitochondrial genomes from buthidae: gene rearrangement and phylogenetic implications. Arthropod Syst. and Phylogeny 82, 3–13. doi:10.3897/asp.83.e140421
Zhang, D., Gao, F., Jakovlić, I., Zhou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi:10.1111/1755-0998.13096
Zhang, G. J., Gao, M., Chen, Y. K., Wang, Y. N., Gan, T. Y., Zhu, F. Y., et al. (2024b). The first complete mitochondrial genome of the genus Litostrophus: insights into the rearrangement and evolution of mitochondrial genomes in Diplopoda. Genes 15, 254. doi:10.3390/genes15020254
Zhang, G. J., Xu, T. J., Chen, Y. K., Xu, W., Wang, Y. N., Li, Y. Y., et al. (2024c). Complete mitochondrial genomes of Nedyopus patrioticus: new insights into the color polymorphism of millipedes. Curr. Issues Mol. Biol. 4, 2514–2527. doi:10.3390/cimb46030159
Zhang, W. W., Gan, T. Y., Xu, T. J., Wang, P., Tai, J. Z., and Ma, F. Z. (2024a). Characterization of the complete mitochondrial genome of Spirobolus grahami (Diplopoda: Spirobolidae) with phylogenetic analysis. Sci. Rep. 14, 7541. doi:10.1038/s41598-024-57421-3
Zhou, T., Xu, K. D., Zhao, F., Liu, W. Y., Li, L. Z., Hua, Z. Y., et al. (2023). itol.toolkit accelerates working with iTOL (Interactive Tree of Life) by an automated generation of annotation files. Bioinformatics 39, btad339. doi:10.1093/bioinformatics/btad339
Keywords: diplopoda, mitogenome, phylogeny, Spirobolus walkeri, genomic features
Citation: Zhang W, Zhao S, Li L, Li Y, Liu H and Cui P (2025) Structural characteristics of mitochondrial genome of Spirobo-lus walkeri (Spirobolida: Spirobolidae), and phylogenetic analysis of Diplopoda. Front. Genet. 16:1566634. doi: 10.3389/fgene.2025.1566634
Received: 25 January 2025; Accepted: 03 March 2025;
Published: 17 March 2025.
Edited by:
Karine Frehner Kavalco, Universidade Federal de Viçosa, BrazilReviewed by:
Sofia Priyadarsani Das, National Taiwan Ocean University, TaiwanCopyright © 2025 Zhang, Zhao, Li, Li, Liu and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Cui, Y3VpcGVuZzExMjZAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.