 Gi-Rim Park
Gi-Rim Park Seon-Hwa Bae
Seon-Hwa Bae Beom-Kyu Kang
Beom-Kyu Kang Jeong-Hyun Seo
Jeong-Hyun Seo Jae-Hyeon Oh
Jae-Hyeon Oh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 26 March 2025
Sec. Genomics of Plants and the Phytoecosystem
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1564160
Introduction: Drought stress significantly reduces soybean yield, underscoring the need to develop drought-resistant varieties and identify the underlying genetic mechanisms. However, the specific genes and pathways contributing to drought tolerance remain poorly understood. This study aimed to identify candidate genes associated with drought tolerance in soybean using a recombinant inbred line (RIL) population derived from PI416937 and Cheongsang.
Methods: A quantitative trait loci (QTL) mapping study using a 180K high-quality SNP array and composite interval mapping on 140 recombinant inbred lines, coupled with RNA sequencing of treated and control groups, was conducted to identify candidate genes for drought tolerance in soybean.
Results and Discussion: Through QTL mapping and differential gene expression profiling, five candidate genes were identified, with two (Glyma.06G076100 and Glyma.10G029600) highlighted as putative candidates based on functional annotations. These genes appear to play critical roles in stress tolerance, including ion homeostasis and the regulation of plasma membrane ATPase, as well as the synthesis of heat shock proteins (HSPs) that mitigate dehydration and thermal stress. These findings advance our understanding of the genetic basis of drought tolerance in soybean and provide valuable targets for breeding programs aimed at developing resilient cultivars.
Drought stress is a major abiotic factor that significantly reduces soybean yield, with losses exceeding 50% in severe cases (Wang et al., 2003). Combined with flooding, drought contributes to 40%–60% of global yield loss in soybeans and other major crops, including corn, rice, and wheat (Valliyodan and Nguyen, 2006; Krasensky and Jonak, 2012; Ahmed et al., 2013). With the increasing frequency and intensity of droughts due to climate change, developing drought-tolerant soybean cultivars is critical to ensuring food security and agricultural sustainability.
Drought tolerance is a complex trait regulated by networks of genes that control key physiological processes such as water uptake, osmotic adjustment, stomatal regulation, and stress signaling (Harb et al., 2010; Agurla et al., 2018; Abhilasha and Roy Choudhury, 2021; Haghpanah et al., 2024). These processes are influenced by genetic and environmental factors, complicating the identification of stable and reliable markers for drought tolerance (Dhungana et al., 2021; Haghpanah et al., 2024; Saeed et al., 2025). Soybean cultivars exhibit considerable genetic diversity in their ability to tolerate water stress, making breeding programs increasingly focused on specific traits associated with drought tolerance. Key physiological traits linked to drought tolerance include root system architecture, osmotic adjustment, stomatal regulation, and leaf area reduction. For instance, the drought-tolerant accession PI416937 is notable for its fibrous root system, which enables deeper soil water access and enhances drought resilience compared to other cultivars (Fallen et al., 2023; Abdel-Haleem et al., 2011; Gilbert et al., 2011). Extensive research on this accession has identified several QTLs associated with root structure, osmotic regulation, and slow wilting (Abdel-Haleem et al., 2011; Carpentieri-Pipolo et al., 2012; Ye et al., 2020; Kwon et al., 2024).
Drought tolerance is a polygenic trait influenced by multiple genes and environmental factors, making it challenging to evaluate based on a single trait. To address this complexity, in both our previous and current studies, we calculate weighted drought coefficient (WDC) values through statistical analysis of vegetative and reproductive traits. These traits include plant height (PH), number of nodes on the main stem (NN), number of branches (BN), number of pods (PN), biomass (BM), and leaf area (LA), all of which are highly sensitive to drought stress. This approach aims to provide a holistic understanding of the genetic basis of drought tolerance in soybean.
Advances in molecular breeding and next-generation sequencing technologies have facilitated the identification of quantitative trait loci (QTLs) linked to agronomically important traits (Rasheed et al., 2022). Identified QTLs, such as qSWPP19 and qSWPP17 for seed weight (Ren et al., 2020), qPH7-4 and qPH7-6 for plant height (Zhang et al., 2022), qCT3 for canopy temperature regulation (Bazzer and Purcell, 2020), and qSW for slow wilting (Ye et al., 2020; Kwon et al., 2024), are pivotal to drought tolerance. These discoveries, enabled by recombinant inbred line (RIL) analysis and genome-wide association studies (GWAS), provide valuable tools for marker-assisted selection (MAS) and QTL pyramiding to improve drought resilience in soybean cultivars (Rasheed et al., 2022). However, the genetic basis of drought tolerance remains highly complex, involving numerous interacting genes that complicate the effective integration of QTLs into breeding programs.
Recent advancements in genomic technologies, including high-density SNP genotyping, have enhanced the precision of QTL and gene identification associated with drought tolerance, accelerating the development of resilient cultivars. Transcriptomic approaches, such as RNA sequencing (RNA-seq), have advanced our understanding by enabling detailed analysis of gene expression changes under drought stress (Liu et al., 2015; Fracasso et al., 2016; Zhao et al., 2020; Dhungana et al., 2021; Aleem et al., 2021; Chu et al., 2021; Yang et al., 2023). These technologies provide insights into the regulatory networks governing drought tolerance. They demonstrate how genetic variation in drought-responsive genes—involved in water use efficiency, stomatal regulation, and cellular protection—contributes to increased resilience. High-throughput genome and transcriptome sequencing has also facilitated the identification of transcriptional regulators and stress-responsive genes in soybean. Prominent transcription factor families, including Dehydration-Responsive Element Binding (DREB), NAC, WRKY, MYB, and AREB (ABA-Responsive Element Binding), play crucial roles in activating stress-responsive pathways and mediating plant responses to environmental stress (Chen et al., 2007; Fuganti-Pagliarini et al., 2017; Nguyen et al., 2019; Wei et al., 2019; Zhou et al., 2020). These transcription factors regulate processes such as osmoregulation and antioxidant defense. However, the stability of candidate genes across different environmental conditions remains variable, highlighting the complexity of drought tolerance as a quantitative trait. Further research is needed to discover and validate genomic regions associated with drought tolerance, which will provide deeper insights into its mechanisms and practical applications for crop improvement.
Despite progress in identifying QTLs and candidate genes for drought tolerance in soybean, the genetic basis of this trait remains poorly understood due to its complexity and environmental variability. To address these gaps, this study investigates the genetic architecture of drought tolerance using a recombinant inbred line (RIL) population derived from a cross between PI416937, a drought-tolerant accession, and Cheonsang, a drought-sensitive cultivar. By integrating QTL mapping, RNA-seq analysis, and ortholog analysis based on Arabidopsis genes, we aim to identify key loci and genes that contribute to drought resilience. Our findings are expected to provide novel insights into the genetic mechanisms underlying drought tolerance, offering valuable targets for soybean breeding programs.
An RIL population consisting of 140 lines, advanced to the F8 generation through selfing, was developed from a cross between the drought-tolerant accession PI416937 and the drought-sensitive cultivar Cheonsang. This population was used to analyze genomic regions associated with drought tolerance. The parents and RIL population were cultivated in a greenhouse at the National Institute of Crop Science, Rural Development Administration, Miryang (N35°29′32″ E128°44′35″) from 2017 to 2019. Seeds were sown with a spacing of 25 cm between plants and 30 cm between rows. Irrigation was provided via a drip hose system. To simulate drought conditions, irrigation was discontinued at the V4-R4 stage in the drought treatment plots, whereas it was maintained in the control plots. The experiments were organized into two blocks, each containing two replicates. Soil moisture content in control and drought-stressed plots was measured using a TDR 300 soil moisture meter (Sperctrum Technologies, Plainfield, IL, United States).
Phenotyping was conducted for parental lines and 140 RILs across eight traits: PH, number of nodes (ND), number of branches (BR), number of pods (PD), biomass (BM), LA, 100-seed-weight (SW), and seed yield (YD). LA was measured at the R3-R4 stage on the 3rd node below the fresh trifoliate leaves at the apex. PH, nodes (ND), branches (BR), and pods (PD) were measured at the R6 stage, while biomass (BM), 100-SW, and seed yield (YD) were measured at harvest (R8 stage). The drought coefficient (DC) for each trait was calculated as the ratio of the trait value under drought conditions to that under control conditions, using the equation:
The WDC was statistically calculated based on the eight traits using the formula described by Dhungana et al. (2021). Phenotypic data collected from 2017 to 2019 for the parental lines and 140 RILs were combined. Traits were grouped into combinations of 2, 3, 5, 6, and 8 for WDC calculation, resulting in 22 drought coefficients that reflected the years of investigation and phenotype combinations. These coefficients were utilized for QTL analysis.
Genomic DNA was extracted from fresh trifoliate leaves using an Exgene Plant SV Miniprep kit (GeneAll, Seoul, Korea), following the manufacturer’s instructions. The parents and RILs were genotyped using the 180K Axiom® Soya SNP array (Lee et al., 2015) to construct a high-density linkage map.
Polymorphic markers between the parents were selected from the 180K SNP dataset and analyzed for redundancy. Redundant markers with identical segregation patterns and clustered at the same genetic positions were removed using the Bin function in ICIMapping version 4.1 (Meng et al., 2015). During this process, markers with significant segregation distortion (p < 0.05) and a missing rate exceeding 5% were excluded. The linkage map was constructed using the Kosambi mapping function, with adjusted parameters including a grouping LOD threshold of 3.0, marker ordering by nnTwoOpt, and rippling based on adjacent recombination fractions.
QTLs for drought tolerance were identified using composite interval mapping (CIM) in QTL Cartographer version 2.5 (available at https://statgen.ncsu.edu/qtlcart). The CIM analysis was performed with Model 6, incorporating forward and backward regression, a scanning interval of 1.0 cM between markers, and a window size of 10 cM. The LOD threshold for each trait was determined using 1,000 permutation tests at p < 0.05. QTLs were named according to the method described by Dhungana et al. (2020).
Candidate genes within QTL regions were queried using the National Center for Biotechnology Information database, Kyoto Encyclopedia of Genes and Genomes Pathway Database (www.genome.jp/kegg/pathway.html), and Phytozome (www.phytozome.net) to analyze sequence similarity based on orthologs. Comparative analyses were conducted using BLASTX with an e-value threshold of 1e-10, a minimum sequence identity of 40%, and a query coverage of at least 40%. Soybean genes aligning with Arabidopsis thaliana genes were identified and prioritized for further analysis.
Two parental lines of the RIL population, representing drought tolerance and susceptibility, were selected along with two RILs showing high tolerance and high susceptibility to drought. To analyze differences in gene expression levels, both treated and control groups were used. RNA was extracted from three plants of each line, and six samples per line (three replicates for each group) were analyzed, resulting in a total of 36 samples (Table 1). Total RNA was isolated from frozen leaf samples using a Qiagen RNA isolation kit, and mRNA was converted into library templates using the TruSeq RNA Sample Prep Kit v2 (Illumina, Cat No. 15026495 Rev. F). Library preparation included mRNA purification, strand synthesis, end repair, adapter ligation, and PCR enrichment. Enriched libraries were quantified and sequenced using the Illumina HiSeq 4000 platform.

Table 1. Parental lines and recombinant inbred lines used for transcriptome analysis.
The raw sequence in FASTQ format, generated by the Illumina platform, was processed using open-source bioinformatics tools. Quality assessment was performed to calculate base quality scores, and sequences with a quality score (Q) below 15 were trimmed. Adapter sequences were removed using Cutadapt (http://code.google.com/p/cutadapt) (Supplementary Table S1). After trimming, sequences were mapped to the Glycine max Wm82.a2.v1 reference genome using HISAT. Transcription expression levels were quantified by calculating read counts using StringTie (Pertea et al., 2016). Normalization was performed using the DESeq R package (Anders and Huber, 2010) to generate expression values and estimates of variability.
The drought-tolerant parent PI416397 consistently exhibited higher WDC values than the drought-susceptible parent Cheonsang across all trait combinations. The mean WDC values for PI416937, calculated using two, three, and six traits, were 0.75, 0.80, and 0.79, respectively, compared with 0.42, 0.52, and 0.57 for Cheonsang. The WDC distribution of the RIL population over a continuous 3-year period exhibited a normal distribution, suggesting transgressive segregation patterns (Table 2).
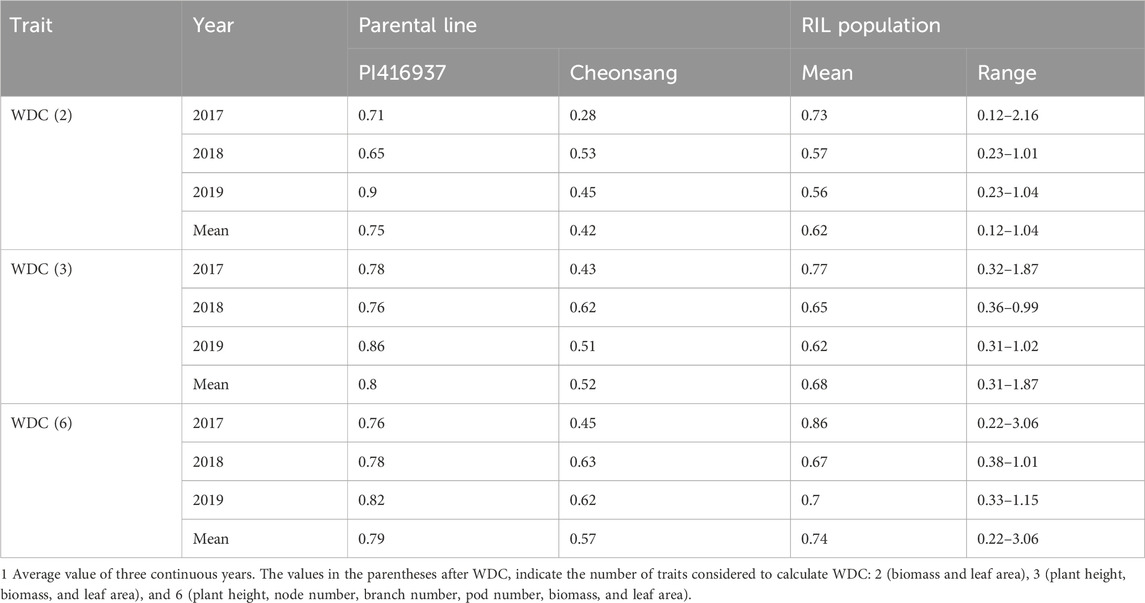
Table 2. Weighted drought coefficient (WDC) of the parental lines and recombinant inbred lines (RILs) for 3 years (2017–2019).
A total of 2,595 SNPs were used to construct the linkage maps of 20 soybean chromosomes. The total map spanned 3,610.36 cM, with an average marker spacing of 1.39 cM. Chromosome 3 had the longest linkage map at 219.35 cM, whereas Chromosome 5 had the shortest at 110.68 cM (Supplementary Table S2). Nine QTLs (LOD ≥ 3) associated with drought tolerance were identified across nine combinations of WDCs, distributed on Chromosomes 1, 6, 7, 10, and 19. The LOD values of these QTLs ranged from 3.00 to 4.54, and the phenotypic variation explained (PVE) ranged from 6.78% to 14.69% (Table 3). Of the nine QTLs, one was located on chromosomes 6 and 19, two were found on chromosomes 1 and 10, and three were identified on chromosome 7 (Figure 1). Notably, qWDC7-2, qWDC7-3, and qWDC10-1 were commonly detected in two trait combinations (biomass and leaf area), whereas qWDC19-1 was identified in three combinations (PH, biomass, and leaf area), suggesting stability across different trait analyses.
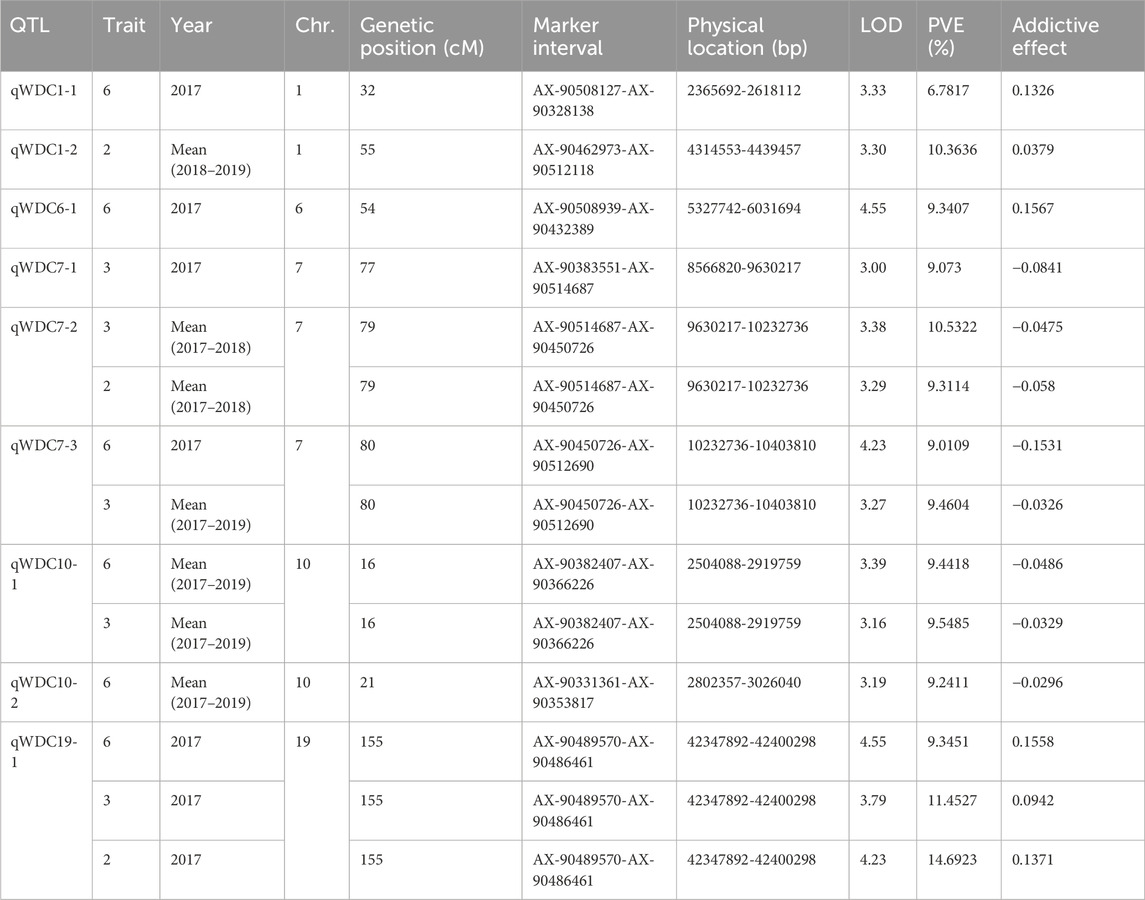
Table 3. QTLs for drought tolerance identified in a recombination inbred line population derived from a drought-tolerant “PI416937” and susceptible “Cheonsang” cultivar.

Figure 1. High-density genetic maps for 5 soybean chromosomes (i.e., 1, 6, 7, 10, and 19) developed using the PI416937/Cheonsang RIL population. Shown are QTL locations for all WDC plants.
A total of 223 genes were identified within the QTL regions, distributed as follows: 25 genes on chromosome 1, 69 genes on chromosome 6, 85 genes on chromosome 7, 39 genes on chromosome 10, and 5 genes on chromosome 19. Of them, 35 genes aligned with drought-related A. thaliana genes, including heat shock transcription factors and members of the glutathione S-transferase family. Additionally, 176 genes were annotated as having unknown functions, and 12 were identified with other known functions. The overall analysis workflow used to identify these genes in summarized in Figure 2.
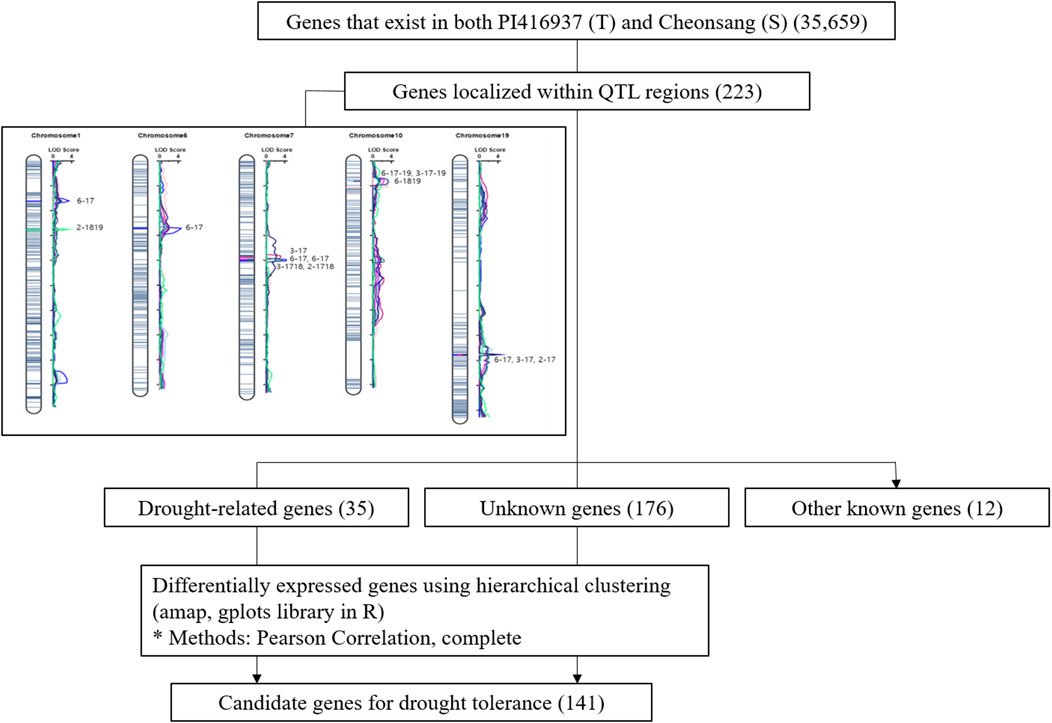
Figure 2. Flow chart of the process used to identify candidate genes related drought tolerance.
For transcriptomic analysis, raw data generated by Illumina sequencing were initially filtered based on the average Phred score value of the sequences, measured in units of 4 bp. Sequences with an average Phred score of 20 or less were removed, resulting in the exclusion of approximately 20% of the raw data. The resulting dataset included paired sequences with a minimum fragment length of 36 bp. Sequence alignment was performed using Glycine max Wm82.a2.v1 as the reference genome, achieving an average mapping rate of 87.8% (Supplementary Table S3).
Transcript expression levels were quantified as read counts using StringTie2. For DEG analysis in DES eq 2, 3 biological replicates within the control and treatment groups were averaged, resulting in 12 samples analyzed in total. Of the 75,384 transcripts identified with read counts greater than or equal to 1, transcripts were considered differentially expressed if their absolute log 2 fold change was ≥ 2 and the adjusted p-value (p-adj) was <0.05. These transcripts were included in downstream analyses. To investigate gene expression changes induced by drought treatment, comparisons were made between drought-treated tolerant lines (dCS, dR56, and dR84) and the PI control lines. A total of 1,826 transcripts showed increased expression in response to drought treatment; however, 38 transcripts were excluded because their expression levels also increased in drought-treated susceptible lines.
To identify phenotype-associated genes located within the QTL regions, clustering was performed on RNA expression data for 211 genes, comprising 35 drought-related genes identified through ortholog analysis and 176 genes with unknown functions. Prior to clustering, raw read counts were normalized using the variance-stabilizing transformation method in DESeq2 to ensure comparability across samples. Hierarchical clustering was conducted based on Person correlation distance and complete linkage to group genes with similar expression patterns. The clustering results were visualized using a heatmap (Figure 3), providing insights into gene expression differences between drought-sensitive and drought-tolerant samples. A total of 141 genes were found to belong to cluster C2-C5, exhibiting distinct expression patterns between drought-sensitive and drought-tolerant samples. These genes were subsequently selected as candidate genes for drought tolerance (Figure 3).
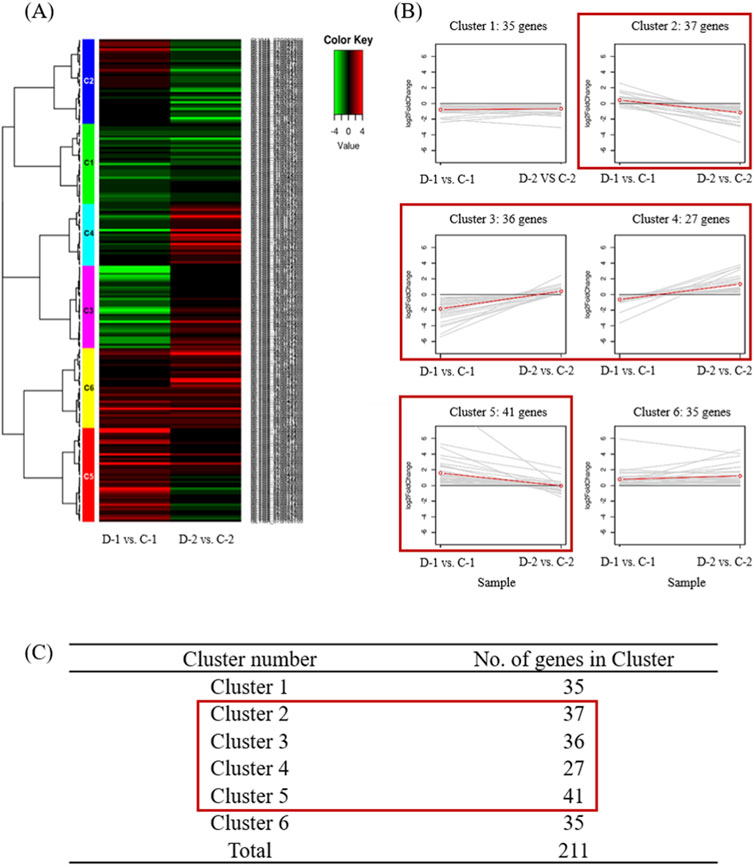
Figure 3. Clustering analysis of gene expression patterns in sensitivity and drought-tolerant cultivars using 211 genes. (A) Differentially expressed gene (DEG) analysis showed six clusters that were differentially expressed in D-1 vs C-1 and D-2 vs C-2 samples. Gene expression profiles of D-1 vs C-1 and D-2 vs C-2 combinations. (B) Line plots of clusters. Expression patterns of genes in the six clusters, namely, Cluster 1–Cluster 6, corresponding to the hierarchical heatmap. Red boxes indicate clusters that showed significant differential expression. (C) Number of genes in the clustering set; 141 candidate genes were found in Clusters 2 to 5.
Through QTL analysis, a total of 223 genes were identified within the QTL regions. Of them, 141 genes exhibited differential expression between drought-tolerant and drought-sensitive lines, as confirmed by clustering analysis. To refine the list of candidate genes, we prioritized those with increased expression in tolerant lines and decreased expression in susceptible lines under drought conditions. Among the 141 genes, 47 showed differential expression patterns between the lines (Supplementary Table S4). Based on normalized counts and functional annotation, five putative candidate genes were ultimately selected (Table 4). These genes are located on chromosomes 1, 6, 7, and 10. Two genes, Glyma.10G029400 and Glyma.10G029600, are situated within the qWDC10-1 region, a stable QTL detected across multiple trait combinations. Additionally, Glyma.06G076100 and Glyma.10G029600 were identified with plasma membrane ATPase 4 and heat stress transcription factor Hsf-21, respectively.
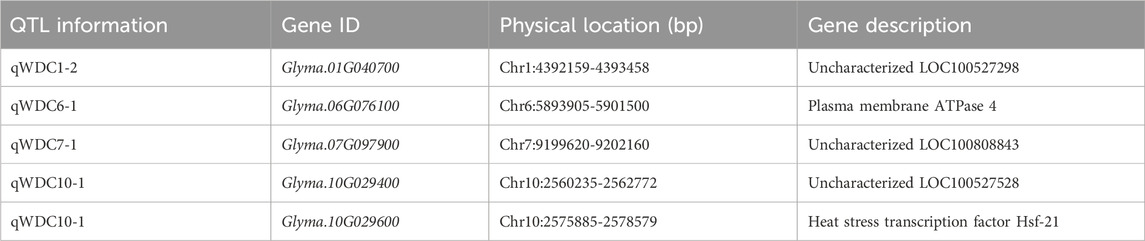
Table 4. List of putative candidate genes based on QTL analysis and RNA-seq.
Drought tolerance is a complex trait influenced by both genetic and environmental factors, making it challenging to identify key genes involved in this process. Recent advancements in genomic technologies, such as high-density SNP genotyping and RNA-seq, have significantly improved our ability to identify QTLs and gene expression changes associated with drought tolerance in crops like soybean (Waheed et al., 2021; Ouyang et al., 2022; Manjunath et al., 2024). These advancements provide insights into the genetic basis of drought tolerance and are crucial for developing resilient crop varieties. In this study, we utilized the 180K SNP chip, which offers high resolution and accuracy, thereby improving data reliability. Moreover, a WDC, which integrates multiple drought-related traits with assigned weights, was used for phenotypic evaluation to capture a more comprehensive response to drought stress.
In previous studies, over 30 QTLs related to drought tolerance in soybean have been identified on chromosomes 1, 2, 4, 7, 10, and 12. These QTLs have been associated with traits such as slow canopy wilting, root thickness, and drought susceptibility index (Du et al., 2009; Brensha et al., 2012; Ye et al., 2020; Arya et al., 2021). For instance, slow canopy wilting has been extensively studied using drought-tolerant lines such as PI416937, PI567690, and PI567331. Several QTLs have been mapped for this trait, including stable loci on chromosomes 6 and 10 (qSW_Gm06 and qSW_Gm10), which account for 20%–30% of the phenotypic variation across diverse environmental conditions (Ye et al., 2020; Rasheed et al., 2022).
In this study, 9 QTLs associated with drought tolerance were identified on chromosomes 1, 6, 7, 10, and 19. Notably, the QTLs identified on chromosomes 6 and 10, as reported in previous findings (Ye et al., 2020; Kwon et al., 2024), suggest their consistent role in conferring drought tolerance. A total of 223 genes were initially mapped within these QTL regions, of which 141 displayed differential expression between drought-tolerant and susceptible plant lines. Among these 141 genes showing differential expression, 47 exhibited significant expression changes under drought stress, and five were selected as putative candidates based on their expression patterns and functional annotations. These candidate genes, located on chromosomes 1, 6, 7, and 10, reside in stable QTL regions consistently linked to drought tolerance across various environmental conditions.
According to Dhungana et al. (2021), QTL analysis using the same segregating population and phenotypic evaluation methods identified genomic regions on five chromosomes: 2, 7, 10, 15, and 20. Chromosomes 7 and 10, in particular, showed consistent results with those of previous studies. Differences in identified QTL regions may result from variations in SNP filtering criteria. Nonetheless, the regions on chromosomes 7 and 10, common to both analyses, likely play a significant role in drought tolerance. In this study, a distinct strategy was adopted to select candidate genes strongly associated with drought tolerance traits. Instead of following the approach of previous studies, we focused on genes located within QTL regions and explored their expression differences in tolerant and susceptible lines before and after drought treatment.
A comparison of gene expression levels revealed that the drought-tolerant accession exhibited a significant upregulation of stress-responsive genes, with an average fold change exceeding 3.8-fold. In contrast, the drought-sensitive accession showed a decrease in gene expression, with an average reduction of 0.4-fold, highlighting its limited transcriptional response to drought stress. Ortholog analysis identified two candidate genes, Glyma.06G076100 and Glyma.10G029600, which are closely associated with drought-related genes in A. thaliana. Glyma.06G076100, annotated as a Plasma Membrane ATPase 4 (PMA4), plays a crucial role in maintaining ion homeostasis and osmotic balance under stress conditions by regulating ATPase activity in the plasma membrane (Baxter et al., 2003). This gene has been shown to enhance plant stress tolerance by facilitating the management of ionic gradients and maintaining cellular functions during environmental stress (Gévaudant et al., 2007; Li et al., 2022). Similar findings in other crops, such as rice, have confirmed the role of PMA4 and its orthologs in drought tolerance (Duby and Boutry, 2009; Zhang et al., 2013), supporting its potential for improving drought resilience in soybean. Furthermore, although located in a genomic region distinct from this study, Glyma.10g019000—a key gene associated with ATPase activity within the qSW_Gm10 region—has been implicated in drought tolerance through the regulation of stomatal opening (Zhao et al., 2019; Kwon et al., 2024).
Glyma.10g029600 may contribute to drought tolerance through the synthesis of stress-related proteins, particularly heat shock proteins (HSPs). These proteins are crucial for maintaining proper protein folding and cellular integrity during stress (Ahuja et al., 2010; Tiwari et al., 2015; Bakthisaran et al., 2015; Ul Haq et al., 2019). HSPs protect plant cells from dehydration and thermal stress by preventing protein aggregation and ensuring proper protein functionality (Ul Haq et al., 2019; Ding et al., 2023). The upregulation of genes encoding HSPs under drought conditions suggests that Glyma.10g29600 may play a role in these protective mechanisms, possibly by regulating osmotic balance and stabilizing cellular structures. In soybean, heat stress transcription factor (Hsf) regulates plant responses to multiple abiotic stresses, including heat, drought, and salinity (Li et al., 2014; Guo et al., 2016; Wang et al., 2021). Hsf activates stress-responsive genes, underscoring its importance in mitigating the adverse effects of environmental stresses (Joshi et al., 2016; Guo et al., 2016; Song et al., 2017; Hussain et al., 2021).
The identification of Glyma.06G076100 and Glyma.10G029600 as key candidate genes highlights their potential utility in breeding programs. For example, PMA4 has been implicated in maintaining ion homeostasis, a critical component of drought resilience, while HSPs play a pivotal role in stabilizing cellular structures under stress. Incorporating these genes into MAS frameworks could accelerate the development of drought-tolerant soybean cultivars. Furthermore, exploring the expression patterns of novel genes identified in this study may uncover previously uncharacterized pathways involved in drought response. These findings underscore the importance of integrating QTL mapping, transcriptomic analysis, and functional genomics to address the growing challenges of climate change and ensure agricultural sustainability.
In addition to these well-characterized genes, other genes within the identified QTL regions exhibited significant differential expression between drought-tolerant and drought-sensitive accessions. These genes may represent novel candidates for drought tolerance that remain unexplored. While their precise roles in stress response mechanisms remain unclear, their differential expression patterns suggest involvement in physiological processes contributing to drought tolerance. Further functional validation of these genes is essential to elucidate their specific roles and evaluate their potential as targets for breeding drought-tolerant soybean cultivars.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
G-RP: Visualization, Writing–original draft, Writing–review and editing. S-HB: Data curation, Writing–original draft, Writing–review and editing. B-KK: Investigation, Methodology, Project administration, Writing–review and editing. J-HS: Funding acquisition, Supervision, Visualization, Writing–original draft, Writing–review and editing. J-HO: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the project “QTL analysis and development of elite lines for drought tolerance in soybean” (No. PJ011868022020) of the Frontiers in Genetics 09 frontiersin.org Park et al. 10.3389/fgene.2025.1564160 National Institute of Crop Science, Rural Development Administration of the Republic of Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1564160/full#supplementary-material
Abdel-Haleem, H., Lee, G.-J., and Boerma, R. H. (2011). Identification of QTL for increased fibrous roots in soybean. Züchter Genet. Breed. Res. 122, 935–946. doi:10.1007/s00122-010-1500-9
Abhilasha, A., and Roy Choudhury, S. (2021). Molecular and physiological perspectives of abscisic acid mediated drought adjustment strategies. Plants 10, 2769. doi:10.3390/plants10122769
Agurla, S., Gahir, S., Munemasa, S., Murata, Y., and Raghavendra, A. S. (2018). “Mechanism of stomatal closure in plants exposed to drought and cold stress,” in Advances in experimental medicine and biology (Singapore: Springer Singapore), 215–232.
Ahmed, F., Rafii, M. Y., Ismail, M. R., Juraimi, A. S., Rahim, H. A., Asfaliza, R., et al. (2013). Waterlogging tolerance of crops: breeding, mechanism of tolerance, molecular approaches, and future prospects. Biomed. Res. Int. 2013, 1–10. doi:10.1155/2013/963525
Ahuja, I., de Vos, R. C. H., Bones, A. M., and Hall, R. D. (2010). Plant molecular stress responses face climate change. Trends Plant Sci. 15, 664–674. doi:10.1016/j.tplants.2010.08.002
Aleem, M., Raza, M. M., Haider, M. S., Atif, R. M., Ali, Z., Bhat, J. A., et al. (2021). Comprehensive RNA-seq analysis revealed molecular pathways and genes associated with drought tolerance in wild soybean (Glycine soja Sieb. and Zucc.). Physiol. Plant. 172, 707–732. doi:10.1111/ppl.13219
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi:10.1186/gb-2010-11-10-r106
Arya, H., Singh, M. B., and Bhalla, P. L. (2021). Towards developing drought-smart soybeans. Front. Plant Sci. 12, 750664. doi:10.3389/fpls.2021.750664
Bakthisaran, R., Tangirala, R., and Rao, C. M. (2015). Small heat shock proteins: role in cellular functions and pathology. Biochim. Biophys. Acta Proteins Proteom 1854, 291–319. doi:10.1016/j.bbapap.2014.12.019
Baxter, I., Tchieu, J., Sussman, M. R., Boutry, M., Palmgren, M. G., Gribskov, M., et al. (2003). Genomic comparison of P-type ATPase ion pumps in Arabidopsis and rice. Plant Physiol. 132, 618–628. doi:10.1104/pp.103.021923
Bazzer, S. K., and Purcell, L. C. (2020). Identification of quantitative trait loci associated with canopy temperature in soybean. Sci. Rep. 10, 17604. doi:10.1038/s41598-020-74614-8
Brensha, W., Kantartzi, S. K., Meksem, K., Grier, I. V. R. L., Barakat, A., Lightfoot, D. A., et al. (2012). Genetic analysis of root and shoot traits in the ‘Essex’by ‘Forrest’recombinant inbred line (RIL) population of soybean [Glycine max (L.) Merr.]. Plant Genet. Genomics Biotechnol. 1, 1–9. doi:10.5147/pggb.v1i1.146
Carpentieri-Pipolo, V., Pipolo, A. E., Abdel-Haleem, H., Boerma, H. R., and Sinclair, T. R. (2012). Identification of QTLs associated with limited leaf hydraulic conductance in soybean. Euphytica 186, 679–686. doi:10.1007/s10681-011-0535-6
Chen, M., Wang, Q.-Y., Cheng, X.-G., Xu, Z.-S., Li, L.-C., Ye, X.-G., et al. (2007). GmDREB2, a soybean DRE-binding transcription factor, conferred drought and high-salt tolerance in transgenic plants. Biochem. Biophys. Res. Commun. 353, 299–305. doi:10.1016/j.bbrc.2006.12.027
Chu, C., Wang, S., Paetzold, L., Wang, Z., Hui, K., Rudd, J. C., et al. (2021). RNA-seq analysis reveals different drought tolerance mechanisms in two broadly adapted wheat cultivars ‘TAM 111’ and ‘TAM 112. Sci. Rep. 11, 4301. doi:10.1038/s41598-021-83372-0
Dhungana, S. K., Kim, H.-S., Kang, B.-K., Seo, J.-H., Kim, H.-T., Shin, S.-O., et al. (2020). Quantitative trait loci mapping for flooding tolerance at an early growth stage of soybean recombinant inbred line population. Plant Breed. 139, 626–638. doi:10.1111/pbr.12790
Dhungana, S. K., Park, J.-H., Oh, J.-H., Kang, B.-K., Seo, J.-H., Sung, J.-S., et al. (2021). Quantitative trait locus mapping for drought tolerance in soybean recombinant inbred line population. Plants 10, 1816. doi:10.3390/plants10091816
Ding, X., Lv, M., Liu, Y., Guo, Q., Gai, J., and Yang, S. (2023). A small heat shock protein GmHSP18.5a improves the male fertility restorability of cytoplasmic male sterility-based restorer line under high temperature stress in soybean. Plant Sci. 337, 111867. doi:10.1016/j.plantsci.2023.111867
Du, W., Wang, M., Fu, S., and Yu, D. (2009). Mapping QTLs for seed yield and drought susceptibility index in soybean (Glycine max L.) across different environments. J. Genet. Genomics 36, 721–731. doi:10.1016/s1673-8527(08)60165-4
Duby, G., and Boutry, M. (2009). The plant plasma membrane proton pump ATPase: a highly regulated P-type ATPase with multiple physiological roles. Pflugers Arch. 457, 645–655. doi:10.1007/s00424-008-0457-x
Fallen, B. D., Mian, M. A. R., Robertson, M. H., Powell, E., and Carter, T. E. (2023). Registration of USDA-N7006 soybean germplasm with increased tolerance to drought stress and 37.5% pedigree from Asian accessions PI 416937 and PI 407859-2. J. Plant Regist. 17, 573–579. doi:10.1002/plr2.20323
Fracasso, A., Trindade, L. M., and Amaducci, S. (2016). Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol. 16, 115. doi:10.1186/s12870-016-0800-x
Fuganti-Pagliarini, R., Ferreira, L. C., Rodrigues, F. A., Molinari, H. B. C., Marin, S. R. R., Molinari, M. D. C., et al. (2017). Characterization of soybean genetically modified for drought tolerance in field conditions. Front. Plant Sci. 8, 448. doi:10.3389/fpls.2017.00448
Gévaudant, F., Duby, G., von Stedingk, E., Zhao, R., Morsomme, P., and Boutry, M. (2007). Expression of a constitutively activated plasma membrane H+-ATPase alters plant development and increases salt tolerance. Plant Physiol. 144, 1763–1776. doi:10.1104/pp.107.103762
Gilbert, M. E., Holbrook, N. M., Zwieniecki, M. A., Sadok, W., and Sinclair, T. R. (2011). Field confirmation of genetic variation in soybean transpiration response to vapor pressure deficit and photosynthetic compensation. Field Crops Res. 124, 85–92. doi:10.1016/j.fcr.2011.06.011
Guo, M., Liu, J.-H., Ma, X., Luo, D.-X., Gong, Z.-H., and Lu, M.-H. (2016). The plant heat stress transcription factors (HSFs): structure, regulation, and function in response to abiotic stresses. Front. Plant Sci. 7, 114. doi:10.3389/fpls.2016.00114
Haghpanah, M., Hashemipetroudi, S., Arzani, A., and Araniti, F. (2024). Drought tolerance in plants: physiological and molecular responses. Plants 13, 2962. doi:10.3390/plants13212962
Harb, A., Krishnan, A., Ambavaram, M. M. R., and Pereira, A. (2010). Molecular and physiological analysis of drought stress in Arabidopsis reveals early responses leading to acclimation in plant growth. Plant Physiol. 154, 1254–1271. doi:10.1104/pp.110.161752
Hussain, Q., Asim, M., Zhang, R., Khan, R., Farooq, S., and Wu, J. (2021). Transcription factors interact with ABA through gene expression and signaling pathways to mitigate drought and salinity stress. Biomolecules 11, 1159. doi:10.3390/biom11081159
Joshi, R., Wani, S. H., Singh, B., Bohra, A., Dar, Z. A., Lone, A. A., et al. (2016). Transcription factors and plants response to drought stress: current understanding and future directions. Front. Plant Sci. 7, 1029. doi:10.3389/fpls.2016.01029
Krasensky, J., and Jonak, C. (2012). Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 63, 1593–1608. doi:10.1093/jxb/err460
Kwon, H., Kim, M. Y., Yang, X., and Lee, S.-H. (2024). Unveiling synergistic QTLs associated with slow wilting in soybean (Glycine max [L.] Merr.). Züchter Genet. Breed. Res. 137, 85. doi:10.1007/s00122-024-04585-1
Lee, Y. G., Jeong, N., Kim, J. H., Lee, K., Kim, K. H., Pirani, A., et al. (2015). Development, validation and genetic analysis of a large soybean SNP genotyping array. Plant J. 81, 625–636. doi:10.1111/tpj.12755
Li, J., Guo, Y., and Yang, Y. (2022). The molecular mechanism of plasma membrane H+-ATPases in plant responses to abiotic stress. J. Genet. Genomics 49, 715–725. doi:10.1016/j.jgg.2022.05.007
Li, P.-S., Yu, T.-F., He, G.-H., Chen, M., Zhou, Y.-B., Chai, S.-C., et al. (2014). Genome-wide analysis of the Hsf family in soybean and functional identification of GmHsf-34 involvement in drought and heat stresses. BMC Genomics 15, 1009. doi:10.1186/1471-2164-15-1009
Liu, Y., Zhou, M., Gao, Z., Ren, W., Yang, F., He, H., et al. (2015). RNA-seq analysis reveals MAPKKK family members related to drought tolerance in maize. PLoS One 10, e0143128. doi:10.1371/journal.pone.0143128
Manjunath, K. K., Krishna, H., Devate, N. B., Sunilkumar, V. P., Sahana, P. P., Chauhan, D., et al. (2024). QTL mapping: insights into genomic regions governing component traits of yield under combined heat and drought stress in wheat. Front. Genet. 14, 1282240. doi:10.3389/fgene.2023.1282240
Meng, L., Li, H., Zhang, L., and Wang, J. (2015). QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 3, 269–283. doi:10.1016/j.cj.2015.01.001
Nguyen, N. C., Hoang, X. L. T., Nguyen, Q. T., Binh, N. X., Watanabe, Y., Thao, N. P., et al. (2019). Ectopic expression of Glycine max GmNAC109 enhances drought tolerance and ABA sensitivity in Arabidopsis. Biomolecules 9, 714. doi:10.3390/biom9110714
Ouyang, W., Chen, L., Ma, J., Liu, X., Chen, H., Yang, H., et al. (2022). Identification of quantitative trait locus and candidate genes for drought tolerance in a soybean recombinant inbred line population. Int. J. Mol. Sci. 23, 10828. doi:10.3390/ijms231810828
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi:10.1038/nprot.2016.095
Rasheed, A., Mahmood, A., Maqbool, R., Albaqami, M., Sher, A., Sattar, A., et al. (2022). Key insights to develop drought-resilient soybean: a review. J. King Saud. Univ. Sci. 34, 102089. doi:10.1016/j.jksus.2022.102089
Ren, H., Han, J., Wang, X., Zhang, B., Yu, L., Gao, H., et al. (2020). QTL mapping of drought tolerance traits in soybean with SLAF sequencing. Crop J. 8, 977–989. doi:10.1016/j.cj.2020.04.004
Saeed, A., Ahmed, H. G. M.-D., Zeng, Y., Fatima, N., Hussain, G. S., Akram, M. I., et al. (2025). Genetic evaluation and breeding strategies under water deficit environment to develop the drought tolerant wheat germplasm. Pol. J. Environ. Stud. 34, 1709–1720. doi:10.15244/pjoes/188056
Song, K., Yim, W. C., and Lee, B.-M. (2017). Expression of heat shock proteins by heat stress in soybean. Plant Breed. Biotechnol. 5, 344–353. doi:10.9787/pbb.2017.5.4.344
Tiwari, S., Thakur, R., and Shankar, J. (2015). Role of heat-shock proteins in cellular function and in the biology of fungi. Biotechnol. Res. Int. 2015, 132635. doi:10.1155/2015/132635
Ul Haq, S., Khan, A., Ali, M., Khattak, A. M., Gai, W.-X., Zhang, H.-X., et al. (2019). Heat shock proteins: dynamic biomolecules to counter plant biotic and abiotic stresses. Int. J. Mol. Sci. 20, 5321. doi:10.3390/ijms20215321
Valliyodan, B., and Nguyen, H. T. (2006). Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr. Opin. Plant Biol. 9, 189–195. doi:10.1016/j.pbi.2006.01.019
Waheed, R., Ignacio, J. C., Arbelaez, J. D., Juanillas, V. M., Asif, M., Henry, A., et al. (2021). Drought response QTLs in a Super Basmati × Azucena population by high-density GBS-based SNP linkage mapping. Plant Breed. 140, 758–774. doi:10.1111/pbr.12961
Wang, J., Chen, L., Long, Y., Si, W., Cheng, B., and Jiang, H. (2021). A novel heat shock transcription factor (ZmHsf08) negatively regulates salt and drought stress responses in maize. Int. J. Mol. Sci. 22, 11922. doi:10.3390/ijms222111922
Wang, W., Vinocur, B., and Altman, A. (2003). Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218, 1–14. doi:10.1007/s00425-003-1105-5
Wei, W., Liang, D.-W., Bian, X.-H., Shen, M., Xiao, J.-H., Zhang, W.-K., et al. (2019). GmWRKY54 improves drought tolerance through activating genes in abscisic acid and Ca2+ signaling pathways in transgenic soybean. Plant J. 100, 384–398. doi:10.1111/tpj.14449
Yang, X., Kwon, H., Kim, M. Y., and Lee, S.-H. (2023). RNA-seq profiling in leaf tissues of two soybean (Glycine max [L.] Merr.) cultivars that show contrasting responses to drought stress during early developmental stages. Mol. Breed. 43, 42. doi:10.1007/s11032-023-01385-1
Ye, H., Song, L., Schapaugh, W. T., Ali, M. L., Sinclair, T. R., Riar, M. K., et al. (2020). The importance of slow canopy wilting in drought tolerance in soybean. J. Exp. Bot. 71, 642–652. doi:10.1093/jxb/erz150
Zhang, H., Niu, X., Liu, J., Xiao, F., Cao, S., and Liu, Y. (2013). RNAi-directed downregulation of vacuolar H+-ATPase subunit A results in enhanced stomatal aperture and density in rice. PLoS One 8, e69046. doi:10.1371/journal.pone.0069046
Zhang, Y., Liu, Z., Wang, X., Li, Y., Li, Y., Gou, Z., et al. (2022). Identification of genes for drought resistance and prediction of gene candidates in soybean seedlings based on linkage and association mapping. Crop J. 10, 830–839. doi:10.1016/j.cj.2021.07.010
Zhao, W., Jung, S., and Schubert, S. (2019). Transcription profile analysis identifies marker genes to distinguish salt shock and salt stress after stepwise acclimation in Arabidopsis thaliana and Zea mays. Plant Physiol. Biochem. 143, 232–245. doi:10.1016/j.plaphy.2019.09.001
Zhao, X., Bai, S., Li, L., Han, X., Li, J., Zhu, Y., et al. (2020). Comparative transcriptome analysis of two Aegilops tauschii with contrasting drought tolerance by RNA-seq. Int. J. Mol. Sci. 21, 3595. doi:10.3390/ijms21103595
Keywords: soybean, drought tolerance, QTL, RNA sequencing, candidate genes
Citation: Park G-R, Bae S-H, Kang B-K, Seo J-H and Oh J-H (2025) Identification of candidate genes for drought tolerance in soybean through QTL mapping and gene expression analysis. Front. Genet. 16:1564160. doi: 10.3389/fgene.2025.1564160
Received: 21 January 2025; Accepted: 17 March 2025;
Published: 26 March 2025.
Edited by:
Purushothaman Natarajan, University of Maryland Eastern Shore, United StatesReviewed by:
Liza Van der Laan, Iowa State University, United StatesCopyright © 2025 Park, Bae, Kang, Seo and Oh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeong-Hyun Seo, bmV4dDA1MDFAa29yZWEua3I=; Jae-Hyeon Oh, amhvaDgyODhAa29yZWEua3I=
† These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.