 Yuying Zhu1†
Yuying Zhu1† Ke Wu
Ke Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 05 March 2025
Sec. Molecular Cytogenetics
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1544565
Uniparental disomy (UPD) occurs when both homologous chromosomes are inherited from a single parent. To date, the UPD of all autosomes and the X chromosome has been recorded. A few cases of UPD of chromosome 21 have been documented. At 15 weeks of gestation, a 25-year-old pregnant woman’s non-invasive prenatal screening revealed a high risk of trisomy 21. Although no anomalies were detected in the fetal ultrasonography, amniocentesis was performed, and the fetal karyotype analysis was found normal. A single-nucleotide polymorphism (SNP) array revealed that the fetus had the copy-neutral region of homozygosity (ROH) in the long arm of chromosome 21. Subsequently, single whole-exome sequencing was performed due to the risk of recessive gene variants in ROH, and no homozygous like pathogenic or pathogenic variants were found on the long arm of chromosome 21. After genetic counseling, the parents decided to continue this pregnancy. At 37 weeks of gestation, a live male infant was delivered by Cesarean section. Copy number variation sequencing showed that the placental tissue was mosaic for trisomy 21. At the final follow-up evaluation, the 6-month-old boy had a normal phenotype.
In uniparental disomy (UPD), both homologous chromosomes are inherited from a single parent. Based on whether both homologous chromosomes from one parent are identical, there are two subtypes of UPD: uniparental heterodisomy (UPhD) and uniparental isodisomy (UPiD). Mechanisms leading to UPD include trisomic/monosomic rescue, gamete complementation, and postfertilization errors (Liehr, 2022). Genomic imprinting depends on the parental origin of the imprinted genes, thereby resulting in the non-equivalent expression of maternal and paternal genomes (Eggermann, 2024). UPD could lead to imprinting disorders. To date, the UPD of all autosomes and the X chromosome has already been recorded. Studies have reported UPiD-caused autosomal recessive diseases detected by whole-exome sequencing. Few cases of UPD of chromosome 21 have been documented. Herein, we report a phenotypically normal infant with UPiD (21), explore previously published cases, and aim to provide useful lessons for clinical diagnosis in the future.
A 25-year-old pregnant woman (gravida 0, para 0) was referred to the Center of Prenatal Diagnosis at Quzhou Maternal and Children Hospital for genetic counseling. At 15 weeks of gestation, the pregnant woman’s non-invasive prenatal screening (NIPS) showed a high risk of trisomy 21 (Z-score, 6). The patient signed an informed consent for her genetic analysis and amniocentesis. The fetal ultrasonography indicated no anomalies before the amniocentesis. Subsequently, the amniocentesis was performed at 18 weeks of gestation, and the fetal sample was detected by single-nucleotide polymorphism (SNP) array analysis, and G-banding karyotype analysis with the 400-band level.
To examine the reason behind the false positive of NIPS, copy number variation sequencing (CNV-seq) was performed with low read-depth (3×) on placental tissues, umbilical cord, and cord blood for detecting the ploidy (number of sets of chromosomes in a cell or organism). Soybean-sized placental tissues symmetrically positioned at specific depths were obtained from the fetal and maternal sides of the placenta, respectively. Six samples were collected: two from the maternal side of the center of the placenta, two from the fetal side of the edge of the placenta, one umbilical cord, and one cord blood sample.
The G-banding karyotype analysis of 30 metaphases revealed a normal fetal amniotic fluid.
The chromosomal microarray analysis (CMA) was done using an SNP array (Affymetrix CytoScan 750K Array, Santa Clara, California). It revealed that the fetus had the copy-neutral region of homozygosity (ROH) in the long arm of chromosome 21 (Figure 1A).
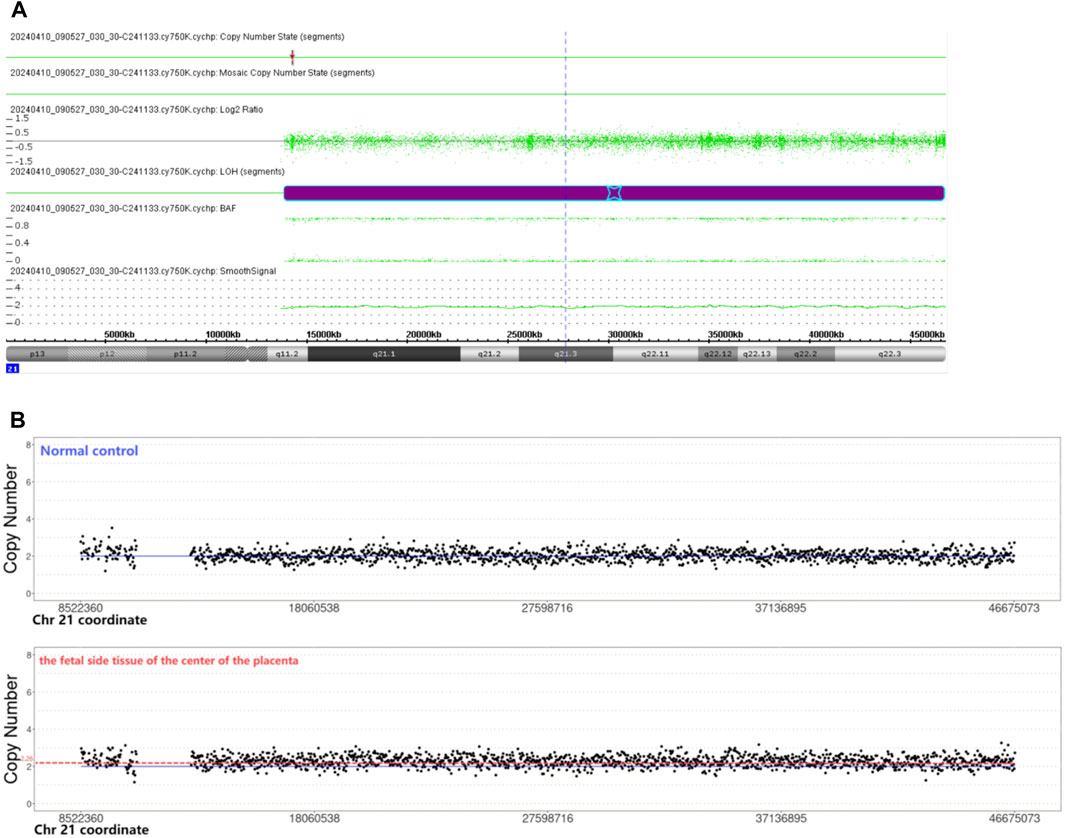
Figure 1. (A) Chromosomal microarray analysis indicated the copy-neutral region of homozygosity (ROH) in the long arm of chromosome 21. Smooth signal representing a normal copy number (green line) along the arm of chromosome 21. The B Allele Frequency (BAF) representing AA, and BB alleles (two green lines). (B) CNV-seq revealed the copy number of the fetal side tissue on the center of the placenta was 2.26. The copy number of the normal control was 2.
Due to the risk of recessive gene variants in ROH, single WES was recommended, and WES found no homozygous likely pathogenic or pathogenic variants on the long arm of chromosome 21.
The pregnant woman was informed of these genetic results. There were no abnormal findings on the ultrasound throughout the entire pregnancy. After genetic counseling, this family decided to continue the pregnancy of the women. At 37 weeks of gestation, a live male infant was delivered by Cesarean section, with a length of 50 cm and a weight of 3,250 g. The 1-min and 5-min Apgar score were all 10. At the final follow-up evaluation, the 6-month-old male newborn demonstrated a normal phenotype.
CNV-seq revealed mosaic trisomy 21 in only the fetal side tissue on the center of the placenta, the percentage of trisomy 21 mosaicism was about 26% (Figure 1B). The other five samples were all euploid.
Two copies of a single chromosome or chromosome segment are inherited from one parent, and no copy is inherited from the other parent, which is called UPiD (21). It was concluded that the positive result of NIPS was caused by the fetal side of the placenta of mosaic trisomy 21. It was presumed that ROH in the long arm of chromosome 21 is caused by a postzygotic trisomy 21 self-rescue event, the two remaining chromosomal 21 copies originated from the same parent, thereby resulting in UPiD (21). One in four placental samples was mosaic for trisomy 21; it suggests that postzygotic trisomy 21 trophectoderm does not rescue completely like inner cell mass, and collecting more than one placental sample is important to explore the mechanism of UPD.
There are two imprinted genes (MIR125B2, DSCAM) and one predicted imprinted gene (SIM2) found on chromosomal 21 according to the Geneimprint database (http://www.geneimprint.com/). The MIRN125B2 gene maps to chromosome 21q21.1. The paternal expression of MIR125B2 is ubiquitous in human tissues (Sonkoly et al., 2007). Chou et al. (2023) demonstrated that MIR125B2 was only imprinted in the human brain, and is associated with cognitive impairment and brain hypotrophy. Patients with Down syndrome (DS) displayed an increased level of miR-125b-2 (Farroni et al., 2018). The DSCAM gene which maps to chromosome 21q22.2-q22.3 is a paternally expressed imprinted gene in the human placenta, which would not be affected by the presence of the supernumerary chromosome 21 (Allach El Khattabi et al., 2019). DSCAM may be a candidate gene responsible for intellectual disability (Yamakawa et al., 1998), and cardiac and visceral malformations (Jannot et al., 2013).
To date, a few published cases of UPD(21) have been reported. We excluded UPD(21) cases with mosaic trisomy 21 (Bruyere et al., 2000; Chen et al., 2020; Chen et al., 2022; Chen et al., 2023), ring chromosome 21 (Bartsch et al., 1994) or a de novo mutation on the Y chromosome (Mansuet-Lupo et al., 2009). UPD(21) cases without available detailed clinical information were also excluded from the study (Nakka et al., 2019; Cavalheiro et al., 2020; Semikhodskii et al., 2023). So, only seven previously published cases of “pure” UPD(21) without mosaicism or other variations were assessed (Table 1).
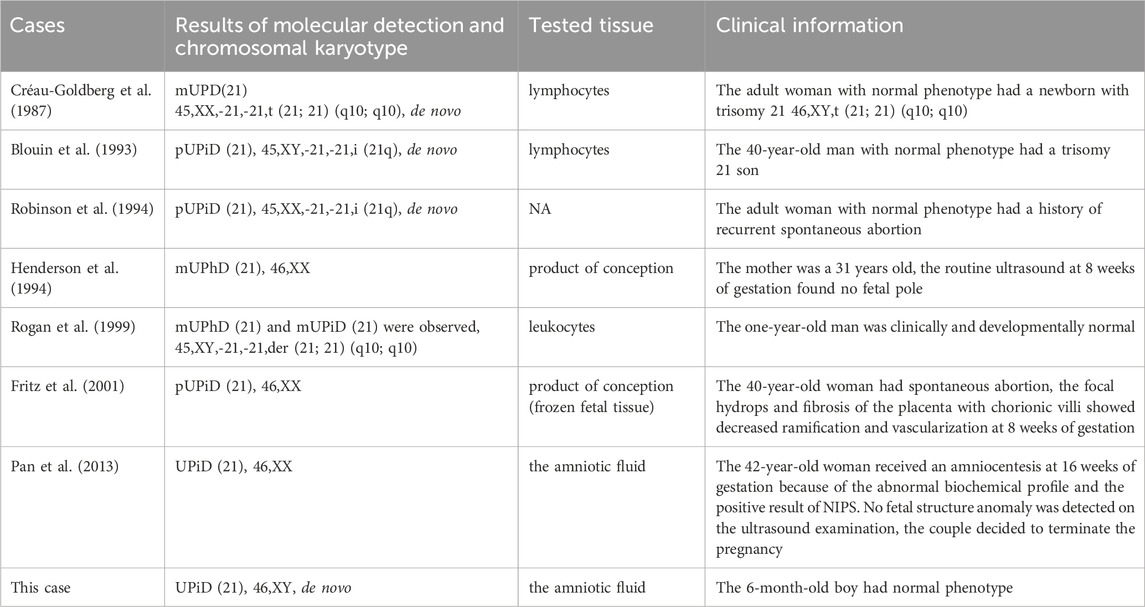
Table 1. Clinical information on previously published cases of UPD(21) without mosaicism or other variations.
Two cases were products of conception with normal karyotypes. Four cases were postnatal cases with normal phenotypes and abnormal karyotypes. Though one case had a prenatal fetus with a normal karyotype, the couple decided to terminate the pregnancy.
We reported one case with UPiD (21) that is attributed to the mechanism of trisomic rescue, and reviewed previously published cases of UPD(21). Some findings from these cases are documented as under:
1) These two imprinted genes on chromosomal 21 might not be associated with abnormal phenotype or human disease, so the presence of UPD(21) in prenatal diagnosis would be considered a favorable outcome, thereby potentially influencing the decision regarding termination of pregnancy.
2) UPiD-caused autosomal recessive diseases detected by WES have been reported previously (Zhou et al., 2024; Lopez-Garrido et al., 2022). Although UPiD (21)-caused autosomal recessive diseases have not been reported, the utilization of WES is recommended for detecting homozygous likely pathogenic or pathogenic variants on chromosome 21.
3) If the NIPS suggests a high risk of trisomy 21, the presence of confined placental mosaicism (CPM) should be considered. However, CPM involving trisomy 21 has not shown an unfavorable effect on pregnancy outcomes (Thomsen et al., 2024; Grati et al., 2020).
4) The possibility of considering the chromosome-balanced translocation should be taken into account. UPD (21) can coexist with chromosome-balanced translocations, typically der (21; 21) (q10; q10). It is likely that these carriers may encounter recurrent spontaneous abortion and have a high risk of pregnancy with trisomy 21. Therefore, chromosomal karyotype analysis is also recommended.
Overall, we also describe a phenotypically normal 6-month-old boy with UPiD (21). We also review previously published cases and sum up some useful lessons for clinical diagnosis and prenatal diagnosis.
Datasets are available on request: the raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. Requests to access these datasets should be directed to [Qiumin Zhu, OTQ2MzQ2NDM5QHFxLmNvbQ==].
The studies involving humans were approved by the Ethics Committee of Quzhou Maternal and Child Healthcare Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Prior written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YZ: writing–original draft and validation. KW: writing–original draft and writing–review and editing. CJ: conceptualization, data curation, methodology, and writing–original draft. QZ: formal analysis, funding acquisition, project administration, and writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants from projects of Science and Technology of Quzhou (2023ZD084).
We are grateful to the patients and their families for their participation in this study, as well as for the help of all the physicians extended in the course of the medical treatment. We wish to thank the staff of Shanghai We-Health Biomedical Technology Co. Ltd. for assisting with sequencing data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Allach El Khattabi, L., Backer, S., Pinard, A., Dieudonné, M. N., Tsatsaris, V., Vaiman, D., et al. (2019). A genome-wide search for new imprinted genes in the human placenta identifies DSCAM as the first imprinted gene on chromosome 21. Eur. J. Hum. Genet. 27 (1), 49–60. doi:10.1038/s41431-018-0267-3
Bartsch, O., Petersen, M. B., Stuhlmann, I., Mau, G., Frantzen, M., Schwinger, E., et al. (1994). Compensatory uniparental disomy of chromosome 21 in two cases. J. Med. Genet. 31 (7), 534–540. doi:10.1136/jmg.31.7.534
Blouin, J. L., Avramopoulos, D., Pangalos, C., and Antonarakis, S. E. (1993). Normal phenotype with paternal uniparental isodisomy for chromosome 21. Am. J. Hum. Genet. 53 (5), 1074–1078.
Bruyère, H., Rupps, R., Kuchinka, B. D., Friedman, J. M., and Robinson, W. P. (2000). Recurrent trisomy 21 in a couple with a child presenting trisomy 21 mosaicism and maternal uniparental disomy for chromosome 21 in the euploid cell line. Am. J. Med. Genet. 94 (1), 35–41. doi:10.1002/1096-8628(20000904)94:1<35::aid-ajmg8>3.0.co;2-9
Cavalheiro, C. P., Avila, E., Gastaldo, A. Z., Graebin, P., Motta, C. H. A., Rodenbusch, R., et al. (2020). Uniparental disomy of chromosome 21: a statistical approach and application in paternity tests. Forensic Sci. Int. Genet. 49, 102368. doi:10.1016/j.fsigen.2020.102368
Chen, C. P., Hsu, T. Y., Chern, S. R., Wu, P. S., Chen, S. W., Wang, L. K., et al. (2023). Mosaic trisomy 21 at amniocentesis in a twin pregnancy associated with a favorable fetal outcome, maternal uniparental disomy 21 and postnatal decrease of the trisomy 21 cell line. Taiwan J. Obstet. Gynecol. 62 (1), 137–141. doi:10.1016/j.tjog.2022.01.012
Chen, C. P., Ko, T. M., Chen, Y. Y., Chern, S. R., Wu, P. S., Chen, S. W., et al. (2020). Prenatal diagnosis of low-level mosaicism for trisomy 21 by amniocentesis in a pregnancy associated with maternal uniparental disomy of chromosome 21 in the fetus and a favorable outcome. Taiwan J. Obstet. Gynecol. 59 (5), 754–757. doi:10.1016/j.tjog.2020.07.023
Chen, C. P., Liou, J. D., Chern, S. R., Wu, P. S., Chen, S. W., Wu, F. T., et al. (2022). Prenatal diagnosis of maternal uniparental disomy 21 in association with low-level mosaic trisomy 21 at amniocentesis in a pregnancy associated with intrauterine growth restriction and a favorable outcome. Taiwan J. Obstet. Gynecol. 61 (1), 146–149. doi:10.1016/j.tjog.2021.11.025
Chou, M. Y., Cao, X., Hou, K. C., Tsai, M. H., Lee, C. Y., Kuo, M. F., et al. (2023). Mir125b-2 imprinted in human but not mouse brain regulates hippocampal function and circuit in mice. Commun. Biol. 6 (1), 267. doi:10.1038/s42003-023-04655-y
Créau-Goldberg, N., Gegonne, A., Delabar, J., Cochet, C., Cabanis, M. O., Stehelin, D., et al. (1987). Maternal origin of a de novo balanced t(21q21q) identified by ets-2 polymorphism. Hum. Genet. 76 (4), 396–398. doi:10.1007/BF00272452
Eggermann, T. (2024). Human reproduction and disturbed genomic imprinting. Genes (Basel) 15 (2), 163. doi:10.3390/genes15020163
Farroni, C., Marasco, E., Marcellini, V., Giorda, E., Valentini, D., Petrini, S., et al. (2018). Dysregulated miR-155 and miR-125b are related to impaired B-cell responses in Down syndrome. Front. Immunol. 9, 2683. doi:10.3389/fimmu.2018.02683
Fritz, B., Aslan, M., Kalscheuer, V., Ramsing, M., Saar, K., Fuchs, B., et al. (2001). Low incidence of UPD in spontaneous abortions beyond the 5th gestational week. Eur. J. Hum. Genet. 9 (12), 910–916. doi:10.1038/sj.ejhg.5200741
Grati, F. R., Ferreira, J., Benn, P., Izzi, C., Verdi, F., Vercellotti, E., et al. (2020). Outcomes in pregnancies with a confined placental mosaicism and implications for prenatal screening using cell-free DNA. Genet. Med. 22 (2), 309–316. doi:10.1038/s41436-019-0630-y
Henderson, D. J., Sherman, L. S., Loughna, S. C., Bennett, P. R., and Moore, G. E. (1994). Early embryonic failure associated with uniparental disomy for human chromosome 21. Hum. Mol. Genet. 3 (8), 1373–1376. doi:10.1093/hmg/3.8.1373
Jannot, A. S., Pelet, A., Henrion-Caude, A., Chaoui, A., Masse-Morel, M., Arnold, S., et al. (2013). Chromosome 21 scan in Down syndrome reveals DSCAM as a predisposing locus in Hirschsprung disease. PLoS One 8 (5), e62519. doi:10.1371/journal.pone.0062519
Liehr, T. (2022). Uniparental disomy is a chromosomic disorder in the first place. Mol. Cytogenet 15 (1), 5. doi:10.1186/s13039-022-00585-2
López-Garrido, M. P., Carrascosa-Romero, M. C., Montero-Hernández, M., Serrano-Martínez, C. M., and Sánchez-Sánchez, F. (2022). Case Report: precision genetic diagnosis in a case of Dyggve-Melchior-Clausen syndrome reveals paternal isodisomy and heterodisomy of chromosome 18 with imprinting clinical implications. Front. Genet. 13, 1005573. doi:10.3389/fgene.2022.1005573
Mansuet-Lupo, A., Henke, J., Henke, L., Blank, C., Ernsting, A., Kozlowski, P., et al. (2009). A paternity case with three genetic incompatibilities between father and child due to maternal uniparental disomy 21 and a mutation at the Y chromosome. Forensic Sci. Int. Genet. 3 (2), 141–143. doi:10.1016/j.fsigen.2008.09.010
Nakka, P., Pattillo Smith, S., O'Donnell-Luria, A. H., McManus, K. F., Mountain, J. L., et al. (2019). Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105 (5), 921–932. doi:10.1016/j.ajhg.2019.09.016
Pan, M., Li, F. T., Li, Y., Jiang, F. M., Li, D. Z., Lau, T. K., et al. (2013). Discordant results between fetal karyotyping and non-invasive prenatal testing by maternal plasma sequencing in a case of uniparental disomy 21 due to trisomic rescue. Prenat. Diagn 33 (6), 598–601. doi:10.1002/pd.4069
Robinson, W. P., Bernasconi, F., Basaran, S., Yüksel-Apak, M., Neri, G., Serville, F., et al. (1994). A somatic origin of homologous Robertsonian translocations and isochromosomes. Am. J. Hum. Genet. 54 (2), 290–302.
Rogan, P. K., Sabol, D. W., and Punnett, H. H. (1999). Maternal uniparental disomy of chromosome 21 in a normal child. Am. J. Med. Genet. 83 (1), 69–71. doi:10.1002/(sici)1096-8628(19990305)83:1<69::aid-ajmg14>3.0.co;2-q
Semikhodskii, A., Makarova, T., and Sutyagina, D. (2023). Maternal uniparental disomy of chromosome 21 as a cause of pseudo-exclusion from paternity. Mol. Genet. Genomics 298 (6), 1389–1394. doi:10.1007/s00438-023-02064-8
Sonkoly, E., Wei, T., Janson, P. C., Sääf, A., Lundeberg, L., Tengvall-Linder, M., et al. (2007). MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PLoS One 2 (7), e610. doi:10.1371/journal.pone.0000610
Thomsen, S. H., Lund, I. C. B., Bache, I., Becher, N., and Vogel, I. (2024). Placental mosaicism for autosomal trisomies: comprehensive follow-up of 528 Danish cases (1983-2021). Am. J. Obstet. Gynecol. MFM, 101497. doi:10.1016/j.ajogmf.2024.101497
Yamakawa, K., Huot, Y. K., Haendelt, M. A., Hubert, R., Chen, X. N., Lyons, G. E., et al. (1998). DSCAM: a novel member of the immunoglobulin superfamily maps in a Down syndrome region and is involved in the development of the nervous system. Hum. Mol. Genet. 7 (2), 227–237. doi:10.1093/hmg/7.2.227
Keywords: uniparental isodisomy, region of homozygosity, whole-exome sequencing, genetic counseling, mosaicism
Citation: Zhu Y, Wu K, Jiang C and Zhu Q (2025) Lessons from a phenotypically normal infant with uniparental isodisomy of chromosome 21: a Case Report and review. Front. Genet. 16:1544565. doi: 10.3389/fgene.2025.1544565
Received: 13 December 2024; Accepted: 12 February 2025;
Published: 05 March 2025.
Edited by:
Kornsorn Srikulnath, Kasetsart University, ThailandReviewed by:
Filipe Brum Machado, Minas Gerais State University, BrazilCopyright © 2025 Zhu, Wu, Jiang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiumin Zhu, OTQ2MzQ2NDM5QHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.