 Meiqi Zhang
Meiqi Zhang Xiaoou Zhai2,3†
Xiaoou Zhai2,3† Lianqing He
Lianqing He Zhen Wang
Zhen Wang Panpan Wang
Panpan Wang Weichao Ren
Weichao Ren Wei Ma
Wei Ma
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 26 February 2025
Sec. Statistical Genetics and Methodology
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1544062
This article is part of the Research Topic Expanding Insights Into Structure, Function, and Disorder of Genome by the Power of Artificial Intelligence in Bioinformatics View all articles
Introduction: Syringa plants are highly valued for their ornamental qualities. However, traditional morphological identification methods are inefficient for discriminating Syringa species. DNA barcoding has emerged as a powerful alternative for species identification, but research on Syringa DNA barcodes is still limited.
Methods: This study employed a multi-locus strategy, combining the nuclear ITS2 region with chloroplast genome regions psbA-trnH, trnL-trnF, and trnL to evaluate the effectiveness of Syringa DNA barcodes. The assessment involved genetic distance analysis, BLAST searches in NCBI, sequence character analysis, and phylogenetic tree construction, examining both individual and combined sequences.
Results: The genetic distance analysis showed that the sequence combination of ITS2 + psbA-trnH + trnL-trnF exhibited a variation pattern where most interspecific genetic distances were greater than intraspecific genetic distances. The Wilcoxon signed-rank test results indicated that, except for psbA-trnH, the interspecific differences of the ITS2 + psbA-trnH + trnL-trnF sequence were greater than those of all single and combined sequences. BLAST analysis revealed that the identification rate for nine Syringa species using ITS2 + psbA-trnH + trnL-trnF could reach 98.97%. The trait-based method also demonstrated that ITS2 + psbA-trnH + trnL-trnF could effectively identify the nine Syringa species. Furthermore, the neighbor-joining (NJ) tree based on ITS2 + psbA-trnH + trnL-trnF clustered each of the nine Syringa species into distinct clades.
Discussion: The study ultimately selected the barcode ITS2 + psbA-trnH + trnL-trnF, with an identification rate of 93.6%, as the optimal barcode for identifying nine species of Syringa trees. This combination proved to be highly effective in discriminating Syringa species, highlighting the potential of DNA barcoding as a reliable tool for species identification in Syringa. Future research could focus on expanding the sample size and exploring additional genetic markers to further enhance the accuracy and applicability of DNA barcoding in Syringa species identification.
Syringa, a genus of deciduous shrubs or small trees, belongs to the Oleaceae family. With approximately 27 species globally, Syringa is predominantly found in East Asia, Central Asia, and Europe. They are renowned for their diverse flower colors and distinctive fragrance, which have made them a common choice for landscaping worldwide (Ming et al., 2007). Beyond their aesthetic appeal, Syringa species are also valued for the diverse chemical constituents found in their flowers, stems, leaves, roots, and fruits, which are used as premium raw materials in the medical and cosmetic industries (Wang et al., 2018). At the turn of the 21st century, the dried leaves of several Syringa species were officially recognized and incorporated into the standards of traditional Chinese medicinal materials. This recognition was based on the significant hypoglycemic, anti-inflammatory, antiviral, antioxidant, antitumor, and protective properties of Syringa leaves, particularly beneficial to the liver, heart, and nervous system (Zhang et al., 2006; Zheng and Guo, 2013; Zhao et al., 2016; Zhu et al., 2021). Since their inclusion in various pharmacopeias, research into the chemical composition and pharmacological effects of Syringa leaves has expanded, with comprehensive reviews detailing the chemical constituents and pharmacological activities of these species (Su et al., 2015), with active components such as phenylpropanoids and iridoids being employed in the treatment of gynecological inflammation, vomiting, diarrhea, cough, and bronchitis (Liu et al., 2020; Yang et al., 2021). The chemical composition varies significantly among different Syringa species, complicating the identification process during the procurement of raw materials for traditional Chinese patent medicines. This can lead to the adulteration of inferior or counterfeit products in the market, underscoring the need for efficient and accurate identification methods to support quality control and market regulation (Wang et al., 2003). Taxonomic studies on Syringa species have been complicated by long-term cultivation, outcrossing, and natural hybridization, resulting in unclear species boundaries within the genus. Correct and effective differentiation of Syringa species is therefore of paramount importance (Chen, 2006). Morphological identification, which requires specialized taxonomic knowledge and detailed descriptions of species morphology at various developmental stages, has several limitations. It may not fully capture potential genetic variations, especially in closely related species with intermediate and similar phenotypes, as well as recently diverged or hybrid-derived species (Chen et al., 2009; Hu et al., 2009; Lattier and Contreras, 2017). To address the challenges in the identification of Syringa species, in addition to traditional morphological identification, some studies have opted to use molecular methods for species determination. Research has identified nine new polymorphic microsatellite sequence markers for distinguishing common Syringa varieties (Juntheikki-Palovaara et al., 2013). Currently, there is a scarcity of research employing molecular methods for the identification of Syringa species. These limitations highlight the need for more reliable and effective methods for Syringa species identification, given their medicinal value and the varying conclusions drawn from earlier taxonomic studies.
DNA barcoding technology utilizes one or several standardized short DNA regions to identify taxonomic groups, providing a precise and rapid method for species identification (Fu et al., 2010; Cheng et al., 2011). This technology has been widely applied in the classification and evolutionary studies of various forest trees and medicinal plants, demonstrating high accuracy (Liu et al., 2019; Frigerio et al., 2021; Liu et al., 2021). Despite its widespread use, there is currently no consensus on the ideal barcode for Syringa species. One of the main challenges faced by barcodes is the identification of sister species. In forest trees, chloroplast genomic coding sequences such as rbcL, matK genes, intergenic spacers psbA-trnH, trnL-trnF, and introns trnL are commonly used for phylogenetic and kinship analyses (Bafeel et al., 2012; Liu et al., 2012; Yang et al., 2012; Yang et al., 2018; Cai et al., 2022; Wei et al., 2024). MatK and rbcL are two standard plant DNA barcode loci recommended by the Consortium for the Barcode of Life (CBOL). Numerous experiments have been conducted using these markers across various taxonomic units and species. However, the identification results have not been satisfactory. Some researchers have indicated that the universality and discriminatory power of matK primers are not ideal (Chase et al., 2007). In another research, it is emphasized that matK and rbcL are predominantly employed for taxonomic ranks above the genus level (Bi et al., 2020). In the analysis and identification of Ligustrum lucidum within the Oleaceae family, the DNA barcoding fragments matK and rbcL were utilized for species identification. The results indicated that both sequences exhibited low efficiency in species discrimination (Wang et al., 2023). To establish a DNA barcode suitable for the identification of Syringa plants, the ITS sequence from the nuclear genome was initially selected. However, it was found that the ITS sequence exhibited specific amplification bands during the amplification process and showed double peaks during sequencing, making it unsuitable for identification studies as a barcode for the genus Syringa. Additionally, the nuclear ITS2 region is recognized as an effective barcode for species identification (Li et al., 2013; Zhu and Gao, 2014; Chen et al., 2016; Chen et al., 2019). Sequencing of the internal transcribed spacer ITS2 region of ribosomal DNA has been used to determine the kinship of Syringa species in Northeast China (Li et al., 2010). The ITS2 sequence, located between the 5.8S rRNA and 25S rRNA, was chosen. The ITS2 sequence achieved a 100% amplification success rate in Syringa identification studies and demonstrated advantages in terms of variation, sequence quality, and high interspecific and intraspecific differentiation capabilities (Wang et al., 2015). Studies have shown that the ITS2 sequence has a good distinguishing effect on plants in the genus Hoya (Xia et al., 2023). In the selection of chloroplast genomic sequences, in addition to psbA-trnH and trnL-trnF, the trnL intron, which has not been used in Syringa plant studies, was also included. The trnL intron is also a commonly used barcode sequence in molecular systematics, with a variation degree greater than that of mitochondrial gene sequences but significantly lower than the evolutionary rate of nuclear genes, often used for analyzing phylogenetic relationships at the interspecific and intraspecific levels within a genus. In the identification of five species of Phoebe, the trnL sequence was able to distinguish between Machilus oreophila and Machilus pauhoi (Qiao et al., 2019). Sometimes, the selection of plant barcodes cannot be limited to a single fragment; it is necessary to supplement with additional fragments according to the requirements. The use of a combination of multiple fragment markers is often required. In Syringa species, a single psbA-trnH sequence was found to be insufficient for distinguishing 33 Syringa samples, whereas the combined barcode of psbA-trnH and trnC-petN showed a higher identification rate for these samples. These findings highlight the need for further research on both single and combined DNA barcode sequences for Syringa species. This study employs four individual sequences ITS2, psbA-trnH, trnL-trnF, and trnL as well as 11 combined sequences for analysis. The aim is to compare different analytical methods and to explore whether combined sequences can enhance the identification capability of Syringa species. Ultimately, this study seeks to identify the optimal DNA barcode combination for the accurate identification of Syringa species.
The morphological characteristics of three leaf traits (leaf shape, leaf base shape, leaf color) and four flower traits (flowering period, inflorescence shape, petal type, flower color) were statistically analyzed for nine Syringa species, as shown in Table 1.
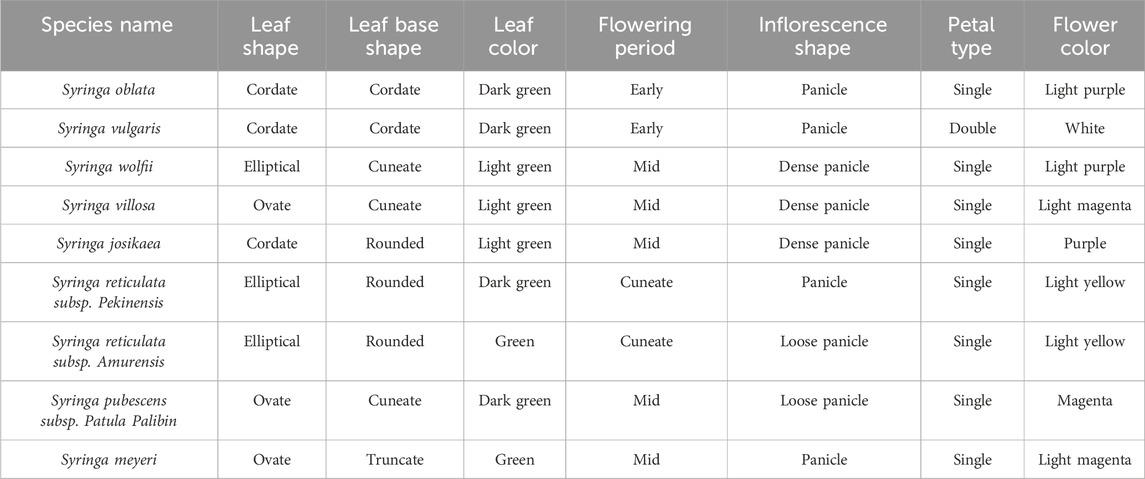
Table 1. Morphological characteristic statistics of nine syringa species.
In taxonomy, Syringa species are divided into two sections: Sect. Syringa and Sect. Ligustrina. Sect. Syringa comprises Ser. Pinnatifoliae, Ser. Pubescentes (Schneid.) Lingelsh., Ser. Syringa, and Ser. Villosae (Schneid.) Rehd, while Sect. Ligustrina consists of Syringa pekinensis and Syringa reticulata. The study found that species within the same taxonomic group exhibit more similar morphological characteristics. For instance, Syringa oblata and Syringa vulgaris, both belonging to Ser. Syringa, show similarities in leaf shape, leaf base shape, leaf color, lowering period, and Inflorescence shape. However, Syringa vulgaris exhibits a double petal morphology and a different color compared to Syringa oblata. Among Syringa wolfii, Syringa villosa, and Syringa josikaea, which are all classified under Ser. Villosae (Schneid.) Rehd., similarities are observed in leaf color, lowering period, Inflorescence shape, and Petal type, while differences exist in leaf shape, leaf base shape, and flower color. Syringa pubescens subsp. Patula Palibin and Syringa meyeri, both belonging to Ser. Pubescentes (Schneid.) Lingelsh., share similar morphological characteristics in leaf shape, lowering period, and Petal type, but exhibit differences in leaf base shape, leaf color, Inflorescence shape, and flower color. Lastly, Syringa reticulata subsp. Pekinensis and Syringa reticulata subsp. Amurensis, which are both part of Sect. Ligustrina, show high similarity in eaf shape, leaf base shape, flowering period, Petal type, and flower color, with only slight differences in leaf color and Inflorescence shape.
The single and combined sequences of nine species of Syringa were compared and the sequence information is shown in Table 2. The length of in-dividual sequences varied from 225 bp (ITS2) to 510 bp (psbA-trnH). Among the sequences, psbA-trnH had the highest proportion of informative sites (55/480 bp), fol-lowed by ITS2 (25/225 bp), trnL-trnF (12/351 bp), and trnL (8/510 bp). The combined sequences ranged in length from 576 bp (ITS2 + trnL-trnF) to 1,566 bp (ITS2 + psbA-trnH + trnL-trnF + trnL), with the number of informative sites for each combination being ITS2+psbA-trnH (54/705), ITS2+psbA-trnH + trnL-trnF (62/1,056), psbA-trnH + trnL-trnF (46/831), ITS2+psbA-trnH + trnL (56/1,215), ITS2+trnL-trnF (24/576), ITS2+psbA-trnH + trnL-trnF + trnL (64/1,566), psbA-trnH + trnL (40/990), psbA-trnH + trnL-trnF + trnL (48/1,341), trnL-trnF + trnL (24/861), ITS2+trnL (18/735), and ITS2+trnL-trnF + trnL (26/1,086).
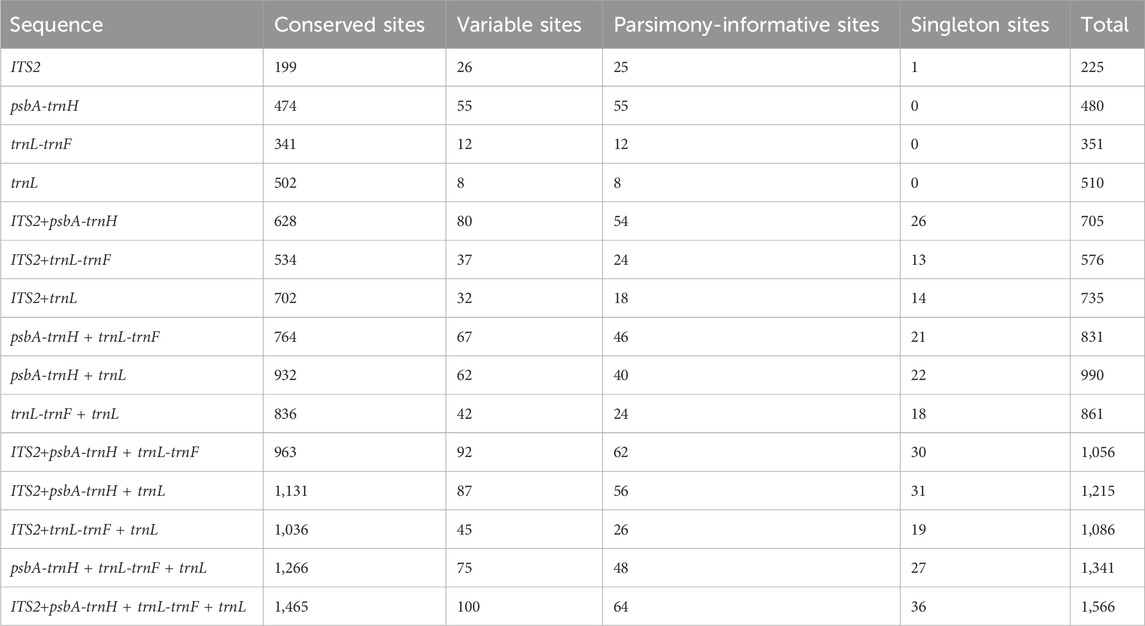
Table 2. Statistics of sequence characteristics.
The mean intraspecific and interspecific genetic distances for single and combined sequences were studied, as shown in Table 3. The interspecific distances ranged from 0.0044 to 0.0549, while the intraspecific distances ranged from 0.0004 to 0.0104. These distances can be utilized to assess the genetic variation of the sequences. Notably, psbA-trnH exhibited the highest genetic variation for both interspecific and intraspecific variations, the trnL intron showed the lowest interspecific genetic variation, and trnL-trnF had the lowest intraspecific genetic variation. In all sequences, interspecific distances were higher than intraspecific distances. The interspecific distances ranged from 0.0108 to 0.0678, and the intraspecific distances ranged from 0.0032 to 0.0126. The highest interspecific genetic variation was observed in the combined sequences ITS2 + psbA-trnH, while the lowest interspecific and intraspecific genetic variation was found in trnL-trnF + trnL.
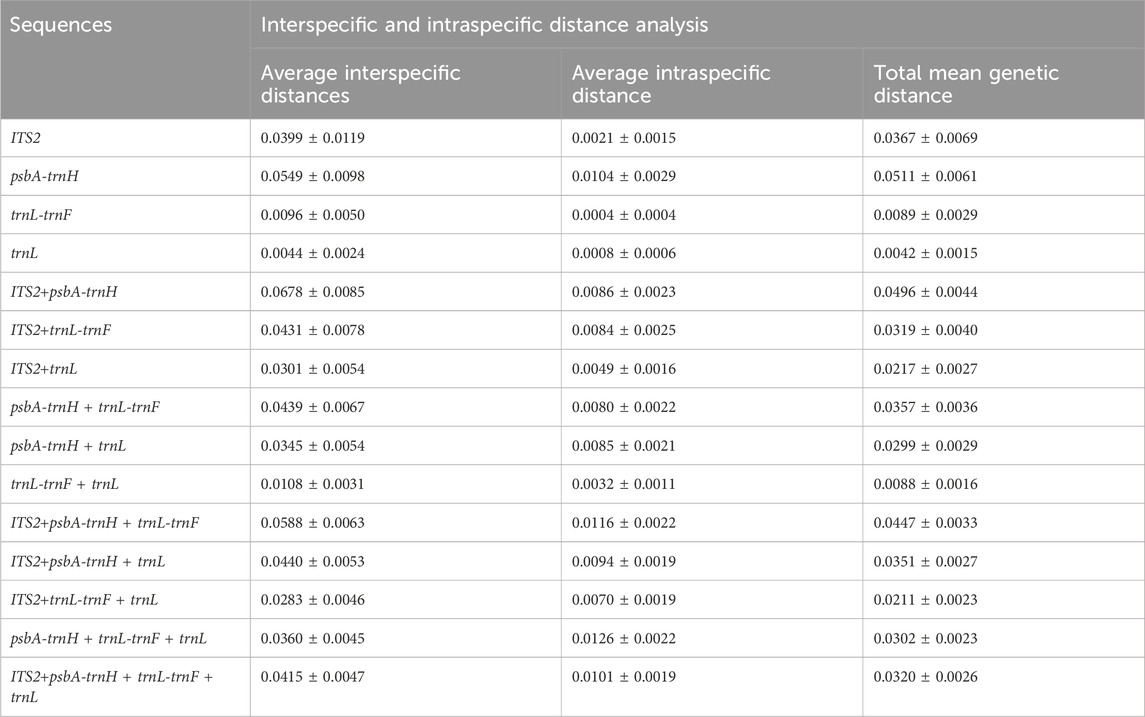
Table 3. Interspecific and intraspecific distance analysis of sequences based on the Kimura two-parameter model.
The frequency distribution of intraspecific and interspecific genetic distances is depicted in Figure 1. The results indicate that for single sequences, ITS2, trnL, and trnL-trnF showed relatively stable intraspecific variation, with overlapping distributions for both interspecific and intraspecific distances. For combined sequences, ITS2 + psbA-trnH showed a stable trend in intraspecific variation, and all combined sequences exhibited overlapping intraspecific and interspecific distances.
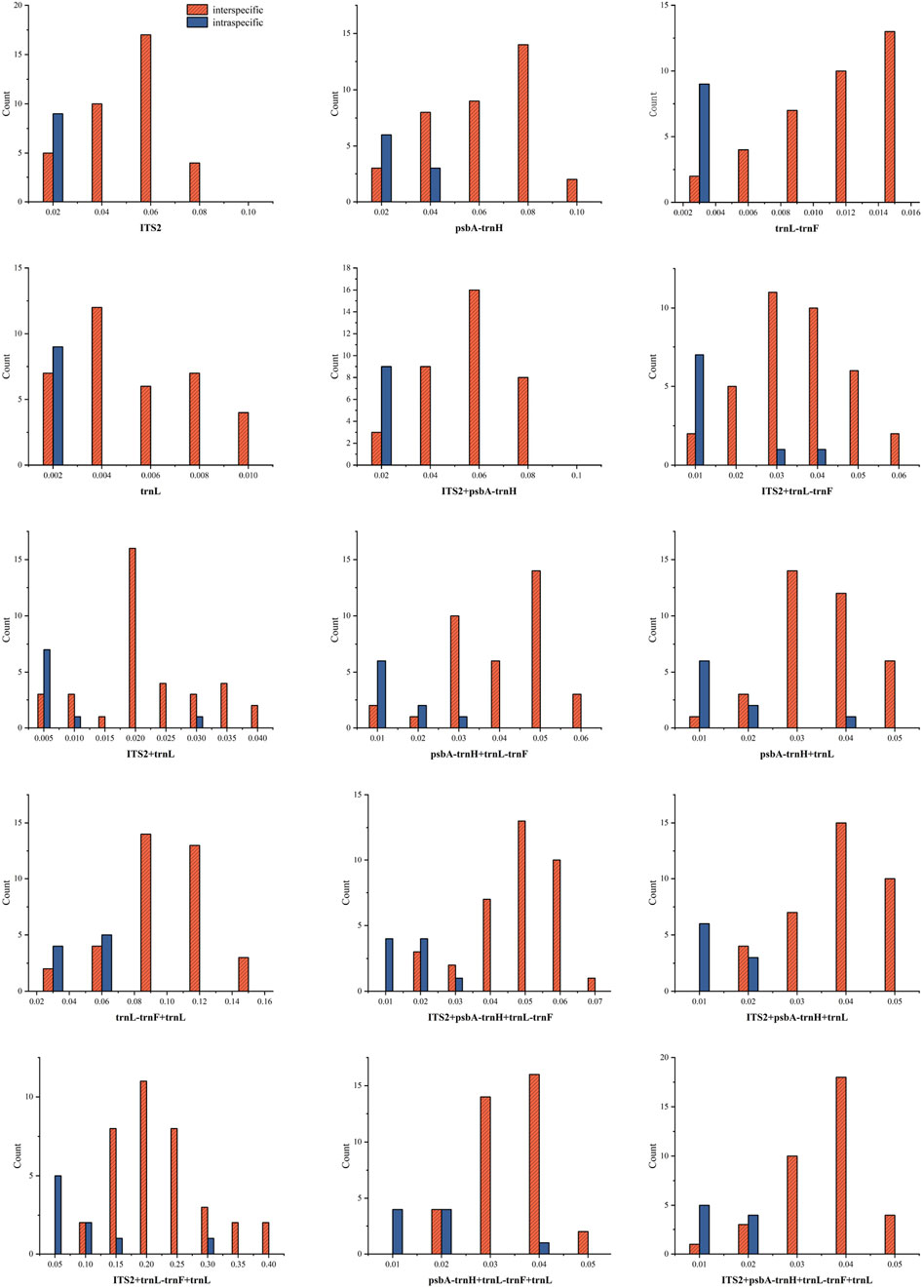
Figure 1. Distribution of interspecific and intraspecific distances of sequences (A: ITS2; B: psbA- trnH; C: trnL-trnF; D: trnL; E: ITS2 + psbA- trnH ; F: ITS2 + trnL-trnF; G: ITS2 + trnL; H: psbA- trnH + trnL-trnF; I: psbA- trnH + trnL; J: trnL-trnF + trnL; K: ITS2 + psbA- trnH + trnL-trnF; L: ITS2 + psbA- trnH + trnL; M: ITS2 + trnL-trnF + trnL; N: psbA-trnH + trnL-trnF + trnL; O: ITS2 + psbA-trnH + trnL-trnF + trnL).
The interspecific distances of single and combined sequences were subjected to Wilcoxon Signed-Rank Test analysis, with results presented in Tables 4 and 5. The order of single sequences was psbA-trnH > ITS2 > trnL-trnF > trnL. The order of combined sequences was ITS2+psbA-trnH > ITS2+psbA-trnH + trnL-trnF > ITS2+psbA-trnH + trnL, psbA-trnH + trnL-trnF > ITS2+trnL-trnF, ITS2+psbA-trnH + trnL-trnF + trnL, psbA + trnH + trnL-trnF + trnL, psbA-trnH + trnL > ITS2+trnL, ITS2+trnL-trnF + trnL > trnL-trnF + trnL. In the Wilcoxon Signed-Rank Test analysis, a p-value less than 0.05 is considered to indicate statistically significant with a meaningful difference between two sequences. Notably, comparisons could not be made between ITS2+trnL-trnF and psbA-trnH + trnL, psbA-trnH + trnL-trnF + trnL, and ITS2+psbA-trnH + trnL-trnF + trnL due to not statistically significant (p > 0.05). Additionally, comparisons could not be made between ITS2+trnL and ITS2+trnL-trnF + trnL, and between ITS2+psbA-trnH + trnL and psbA-trnH + trnL-trnF due to not statistically significant (p > 0.05). Furthermore, this study also tested the interspecific distances of single and combined sequences (Supplementary Table S1). The results indicate that the interspecific divergence of the single sequence psbA-trnH is higher than that of all combined sequences. The interspecific divergence of the single sequence ITS2 is generally greater than that of the combined sequences. The interspecific divergence of the single sequences trnL-trnF and trnL is lower than that of the combined sequences.
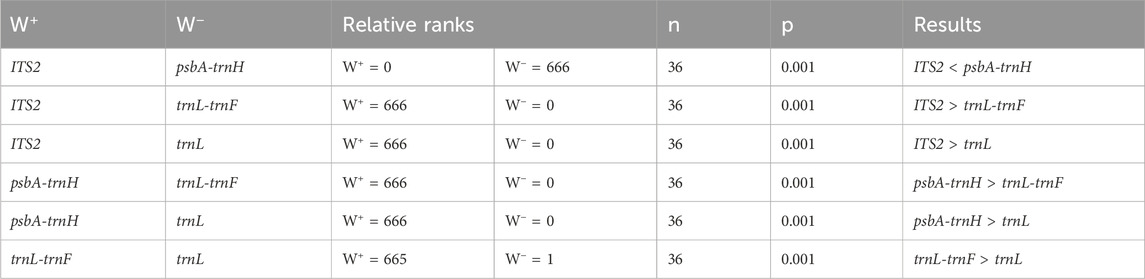
Table 4. Wilcoxon signed-rank test for the interspecies distances of the single sequences.
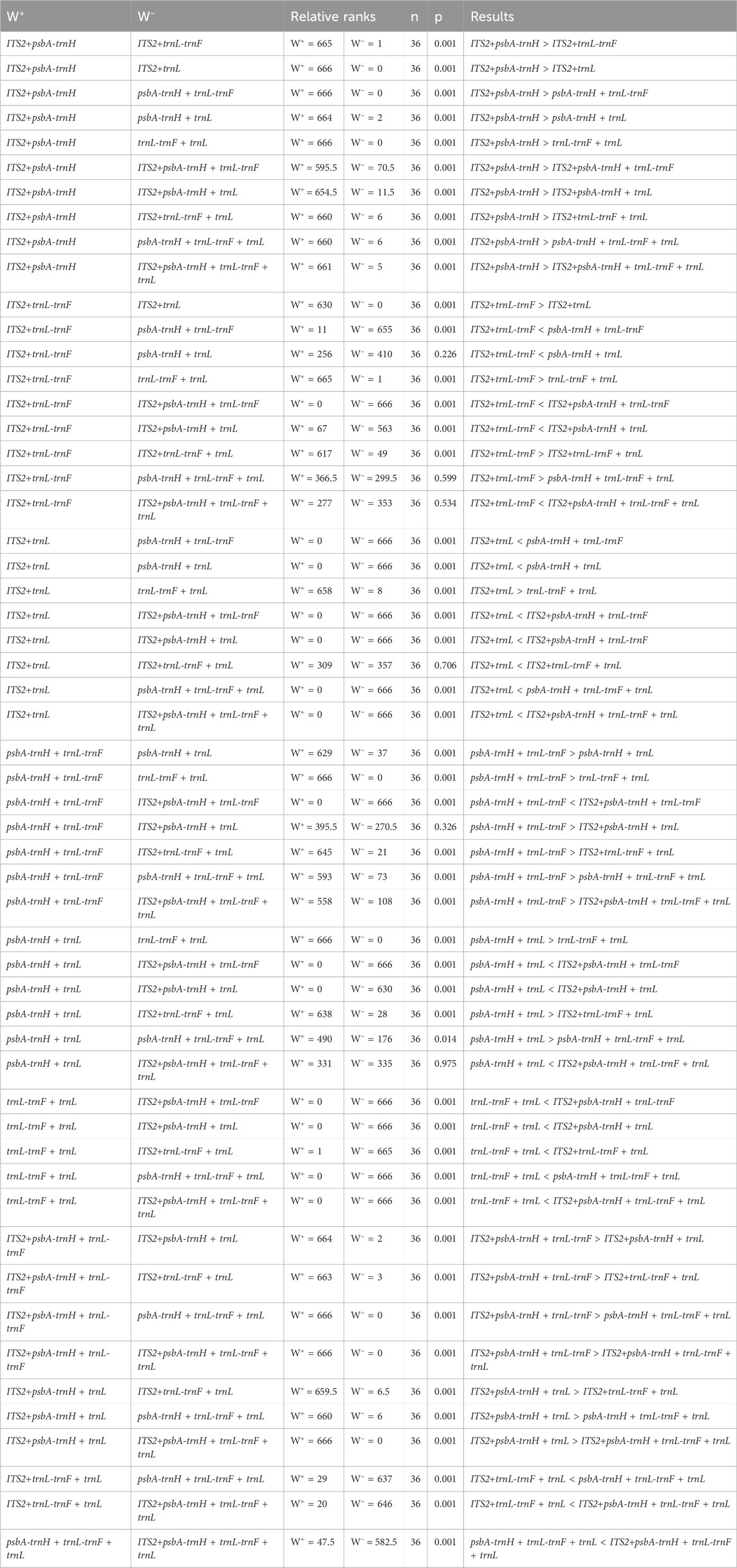
Table 5. Wilcoxon signed-rank test for the interspecies distances of the combined sequences.
The average scores, alignment rates and recognition rates of single and combined sequences of 9 Syringa species retrieved by BLAST from NCBI are shown in Supplementary Table S2. The average scores and recognition rates of different sequences are shown in Figure 2. Among the single sequences, the trnL sequence had the highest score, while the ITS2 sequence had the lowest. In the combined sequences, the psbA-trnH + trnL-trnF + trnL sequence had the highest score, and the ITS2+trnL-trnF sequence had the lowest. For single sequences, the ITS2 had the highest average identification rate at 99.80%, while the trnL had the lowest at 98.61%. The average identification rates for the ITS2 and trnL-trnF sequences ranged from 99% to 100%, and for the psbA-trnH and trnL sequences, they ranged from 98% to 99%. Among the combined sequences ITS2+psbA-trnH + trnL had the highest identification rates at 99.70%, while ITS2+psbA-trnH had the lowest at 97.51%. The average identification rates for the sequences ITS2+trnL-trnF, ITS2+trnL, psbA-trnH + trnL, trnL-trnF + trnL, ITS2+psbA-trnH + trnL, ITS2+trnL-trnF + trnL, psbA-trnH + trnL-trnF + trnL, and ITS2+psbA-trnH + trnL-trnF + trnL ranged from 99% to 100%, while the average identification rates for the ITS2+psbA-trnH, ITS2+psbA-trnH + trnL-trnF and psbA-trnH + trnL-trnF sequences ranged from 97% to 99%.
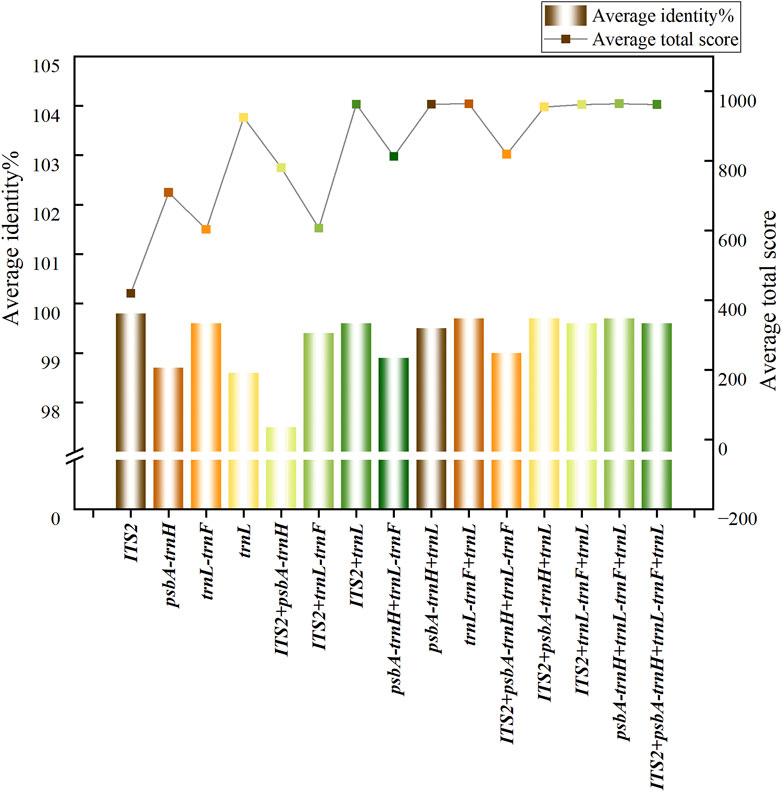
Figure 2. Trends of average score and average recognition rate in BLAST analysis.
The identification rates and logical formulas for each sequence based on the BLOG algorithm are presented in Table 6. For the nine Syringa species, the single sequence psbA-trnH and all combined sequences achieved a correct classification rate of 100%. Among the single sequences, the trnL-trnF sequence revealed specific base positions for different species: S. reticulata subsp. Amurensis has G at position 88, T at position 189, and A at position 192; S. reticulata subsp. Pekinensis has T at position 88; S. pubescens subsp. patula Palibin has A at position 192 and G at position 292; S. meyeri has G at position 189 and A at position 292; S. villosa has A at position 79, C at position 192, and A at position 292; S. wolfii has C at position 79; S. josikaea has C at position 192 and G at position 292; whereas S. oblata and S. vulgaris did not have corresponding positions to distinguish them. In the trnL sequence, S. oblata has T at position 10; S. vulgaris has T at position 10 and A at position 198; S. reticulata subsp. Amurensis has G at position 88 and T at position 189, and A at position 192; S. reticulata subsp. Pekinensis has A at position 278; S. pubescens subsp. patula Palibin has A at position 12 and G at position 278; S. meyeri has G at position 12, G at position 198, and G at position 278; S. wolfii has A at position 12 and C at position 385; while S. villosa and S. josikaea did not have corresponding positions to distinguish them. Among the combined sequences, all nine Syringa species had corresponding base positions for differentiation.
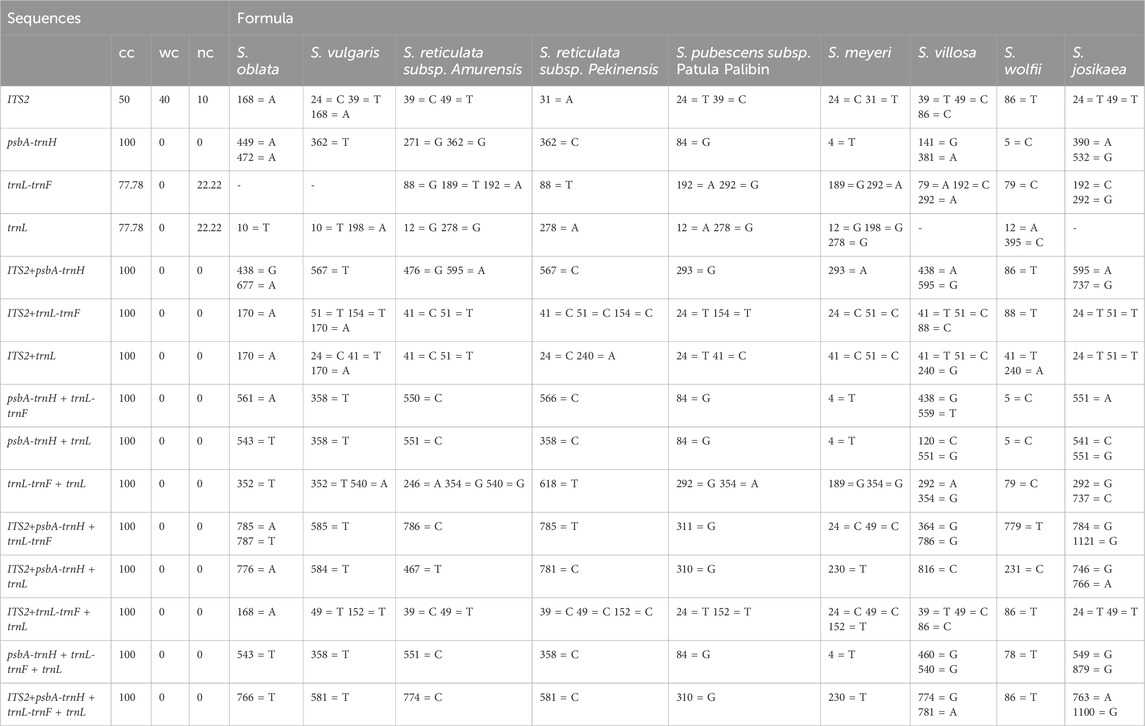
Table 6. Character-based approach for species identification.
The Forsythia suspensa ITS2 sequence was used as an outgroup to root the tree. In the single sequence NJ tree analysis of the ITS2 region, a broad distribution pattern is observed (Figure 3), with specimens from the evolutionary branches formed by the species pairs (S. oblata and S. vulgaris), (S. villosa and S. wolfii), and (S. pubescens subsp. Patula Palibin and S. meyeri) showing clear clustering. However, the ITS2 sequence differences between the two species in each of these pairs are minimal, preventing further sub-clustering. From the figure, it is evident that S. oblata and S. vulgaris form the basal branches, followed by a larger branch composed of two sub-branches: one grouping S. villosa, S. wolfii, and S. josikaea; the other sub-branch divides into two smaller branches, with S. pubescens subsp. Paulat palibin and S. meyeri forming one branch; and S. reticulata subsp. Pekinensis and S. reticulata subsp. Amurensis forming the other. The tree diagram clearly shows that S. reticulata subsp. Pekinensis, S. reticulata subsp. Amurensis, and S. josikaea can be distinctly clustered and form separate sub-clusters.
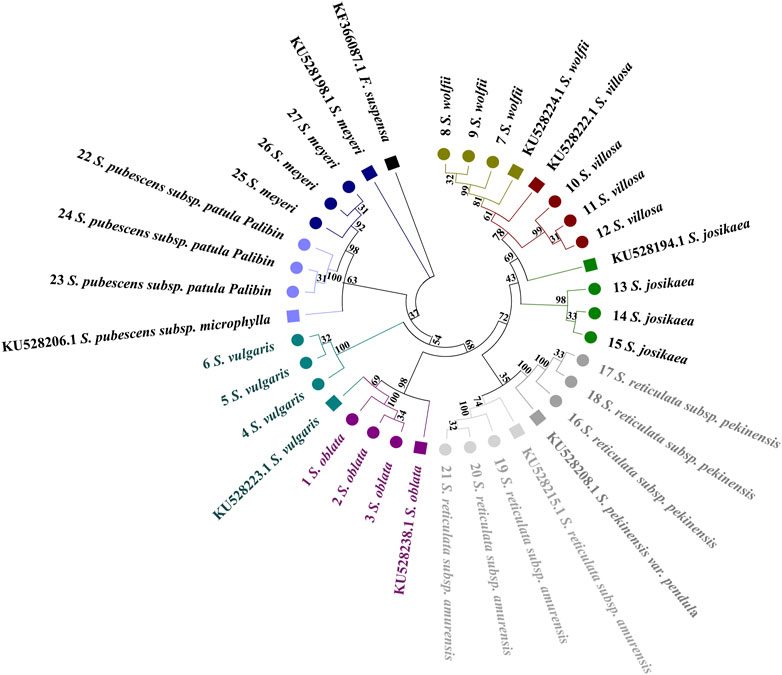
Figure 3. Phylogenetic tree based on the ITS2 alignment matrix of 27 samples from 9 Syringa species. The F. suspensa ITS2 sequence (GenBank accession number: MG219753.1) was used as an outgroup to root the tree. Taxa are color-coded at the species level for easy discrimination of each species. Syringa species analyzed in this study are marked with circles. Numbers in the species labels correspond to sample ID (Table 6). Reference sequences obtained from GenBank are marked with squares and accession numbers in the taxon labels.
Distinct distribution patterns from those observed with the ITS2 barcode are evident in the psbA-trnH tree (Figure 4). The phylogenetic tree is primarily divided into two main sections. Syringa meyeri and S. pubescens subsp. Patula Palibin form a basal clade, followed by the S. vulgaris clade. The last major branch consists of two sub-branches: S. oblata and a branch comprising S. reticulata subsp. Amurensis, S. reticulata subsp. Pekinensis, S. josikaea, S. villosa, and S. wolfii. High sequence similarity among S. josikaea, S. villosa, and S. wolfii presents challenges in further sub-clustering. Unlike the analysis of ITS2 sequences, S. oblata and S. vulgaris do not form a single clade, with the reference sequence of S. vulgaris being clustered within S. oblata. However, it is clear from the tree that species specimens of S. reticulata subsp. Pekinensis, S. reticulata subsp. Amurensis, S. oblata, and S. vulgaris can be distinctly clustered and form separate sub-clusters.
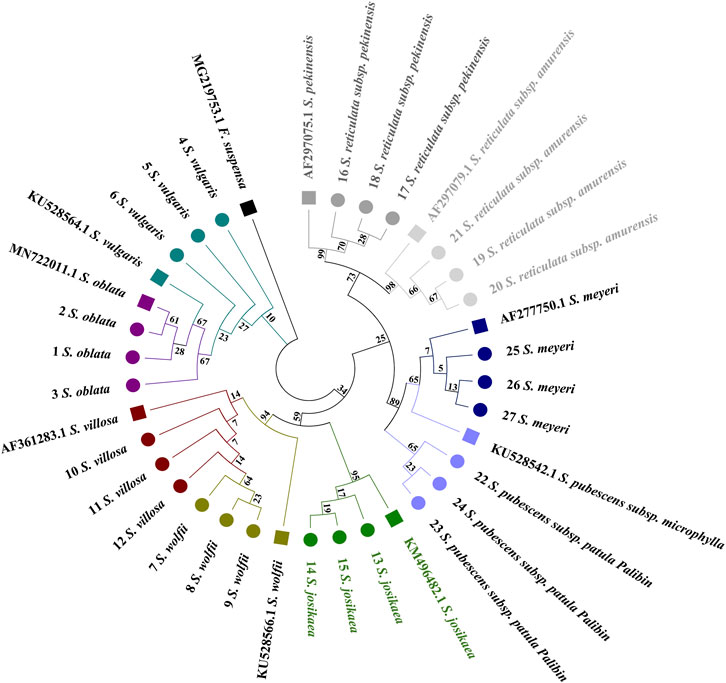
Figure 4. Phylogenetic tree generated from the psbA-trnH alignment matrix of 27 samples from 9 Syringa species. The F. suspensa psbA-trnH sequence (GenBank accession number: KF366087.1) was used as an outgroup to root the tree. Taxa are color-coded at the species level for easy discrimination of each species. Syringa species analyzed in this study are marked with circles. Numbers in the species labels correspond to sample ID (Table 6). Reference sequences obtained from GenBank are marked with squares and accession numbers in the taxon labels.
A distribution pattern similar to that observed with the ITS2 barcode is clearly discernible in the trnL-trnF tree (Figure 5). Specimens from the evolutionary branches formed by S. oblata and S. vulgaris, S. reticulata subsp. Pekinensis and S. reticulata subsp. Amurensis, and S. pubescens subsp. Patula Palibin and S. meyeri are noticeably clustered. However, the trnL-trnF sequence differences between the two species in these three pairs are minimal, preventing further sub-clustering. The tree illustrates that S. oblata and S. reticulata subsp. Pekinensis form a basal branch, followed by a larger branch composed of two sub-branches, one consisting of S. reticulata subsp. Pekinensis and S. reticulata subsp. Amurensis, and the other divided into three branches, namely, S. meyeri and S. pubescens subsp. Patula Palibin, S. josikaea as a separate branch, and a branch composed of S. villosa and S. wolfii. The reference sequence of S. pubescens subsp. Patula Palibin and the specimens of S. meyeri cluster together. However, it is clear from the tree that specimens of S. josikaea, S. wolfii, and S. villosa can be distinctly clustered and form separate sub-clusters.
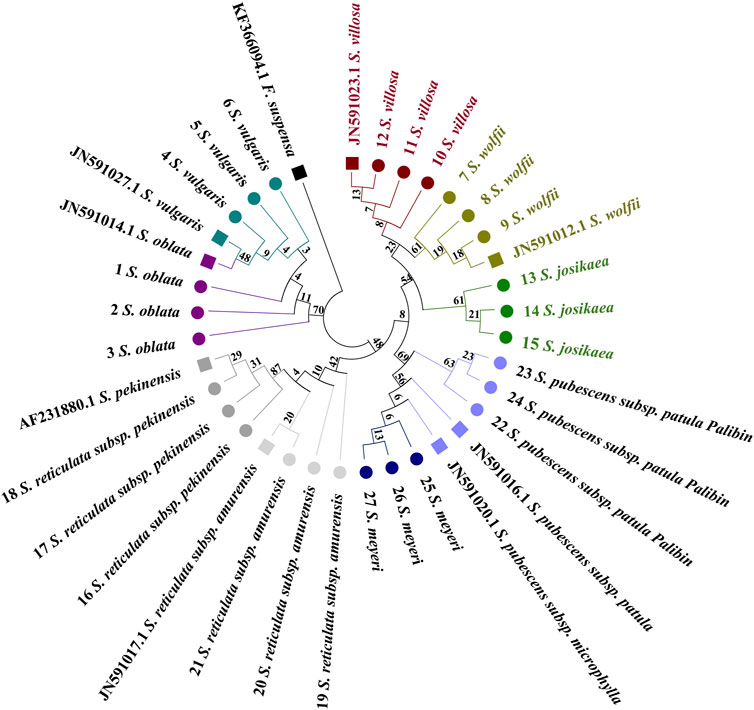
Figure 5. Phylogenetic tree generated from the trnL-trnF alignment matrix of 27 samples from 9 Syringa species. The F. suspensa trnL-trnF sequence (GenBank accession number: KF366094.1) was used as an outgroup to root the tree. Taxa are color-coded at the species level for easy discrimination of each species. Syringa species analyzed in this study are marked with circles. Numbers in the species labels correspond to sample ID (Table 6). Reference sequences obtained from GenBank are marked with squares and accession numbers in the taxon labels.
The trnL tree does not clearly exhibit a broad distribution pattern (Figure 6). The evolutionary tree is primarily divided into two sections. It can be observed that the sequences of S. reticulata subsp. Pekinensis and the reference sequence of S. reticulata subsp. Amurensis are positioned at the basal position, with the sample sequences of S. reticulata subsp. Amurensis following. Subsequently, a branch emerges, which splits into two smaller branches, one being S. meyeri, and the other branch further divides into two branches, one consisting of S. oblata and S. vulgaris forming a single branch, and the other branch splits again into two branches, one being S. pubescens subsp. Patula Palibin, and the other branch further divides into two branches, one branch being S. wolfii, and the other branch being S. villosa and S. josikaea. The reference sequence of S. reticulata subsp. Amurensis clusters with S. reticulata subsp. Pekinensis, the reference sequences of S. villosa and S. josikaea cluster with S. wolfii, and the reference sequence of S. oblata clusters with S. vulgaris, still presenting issues with high sequence similarity preventing sub-clustering. However, it is clear from the tree that the species specimens of S. pubescens subsp. Patula Palibin and S. meyeri can be distinctly clustered and form separate sub-clusters.
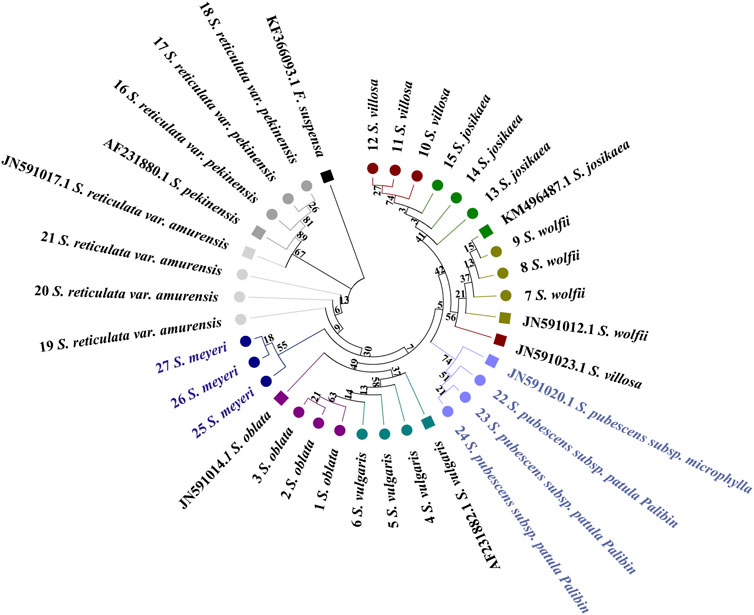
Figure 6. Phylogenetic tree generated from the trnL alignment matrix of 27 samples from 9 Syringa species. The F. suspensa trnL sequence (GenBank accession number: KF366093.1) was used as an outgroup to root the tree. Taxa are color-coded at the species level for easy discrimination of each species. Syringa species analyzed in this study are marked with circles. Numbers in the species labels correspond to sample ID (Table 6). Reference sequences obtained from GenBank are marked with squares and accession numbers in the taxon labels.
The results indicate that single sequences are unable to cluster all nine species of Syringa into a single clade on the phylogenetic tree. Instead, they can only group two or three species of Syringa together, with relatively high bootstrap values.
In the combined sequences, the NJ trees constructed withITS2+psbA-trnH + trnL, and ITS2+psbA-trnH + trnL-trnF + trnL and ITS2+psbA-trnH + trnL-trnF align with the phylogenetic analysis model for Syringa species and are capable of distinguishing the nine Syringa species, with high support rates for individual sub-clustering (Figures 7A, B, 8). The identification success rates for the three combined sequences, ITS2+psbA-trnH + trnL-trnF, ITS2+psbA-trnH + trnL, and ITS2+psbA-trnH + trnL-trnF + trnL, were 93.6%, 70%, and 81%, respectively. Among these, the combination of ITS2+psbA-trnH + trnL-trnF exhibited the highest identification success rate. Among other sequences, the NJ tree constructed with ITS2+psbA-trnH sequences clusters S. wolfii and S. villosa together, failing to distinguish between these two species (Supplementary Figure S1). In the NJ tree constructed with ITS2+trnL-trnF sequences, S. wolfii and S. villosa cluster together, and S. oblata and S. vulgaris also cluster together, preventing the distinction between these two pairs of species (Supplementary Figure S2). In the NJ trees constructed with ITS2+trnL and ITS2+trnL-trnF + trnL sequences, S. oblata and S. vulgaris cluster together, failing to distinguish between S. oblata and S. vulgaris (Supplementary Figures S3, S7). In the NJ tree constructed with trnL-trnF + trnL sequences, S. oblata and S. vulgaris cluster together, and S. reticulata subsp. Amurensis as a single species fails to cluster, making it unclear to distinguish these three species (Supplementary Figure S6). The NJ trees constructed with psbA-trnH + trnL-trnF, psbA-trnH + trnL, and psbA-trnH + trnL-trnF + trnL sequences do not conform to the phylogenetic analysis model for Syringa species but can complete individual sub-clustering (Supplementary Figures S4, S5, S8). Therefore, these three combined sequences cannot be used to identify the nine Syringa species.
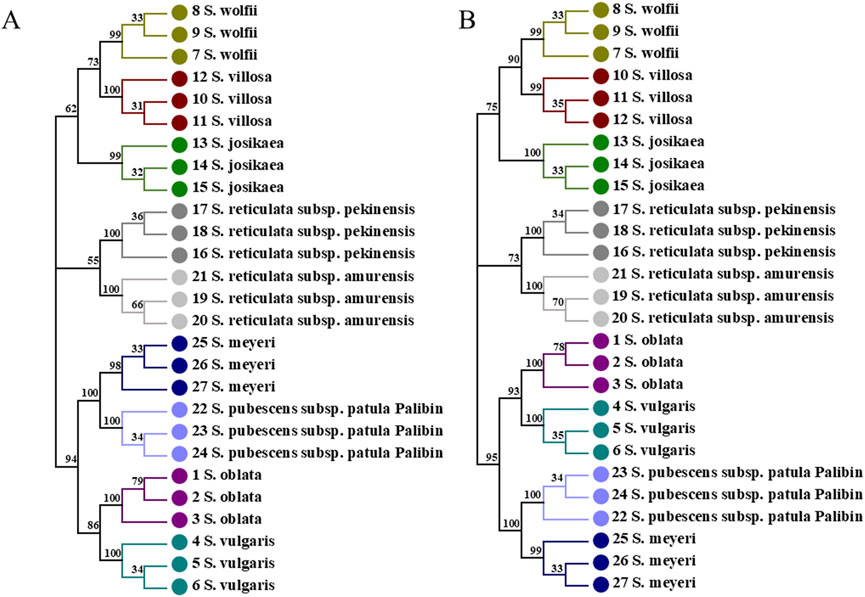
Figure 7. NJ trees constructed based on ITS2+psbA-trnH + trnL, ITS2+psbA-trnH + trnL-trnF + trnL [(A): ITS2+psbA-trnH + trnL; (B) ITS2+psbA-trnH + trnL-trnF + trnL].

Figure 8. NJ trees constructed based on ITS2+psbA-trnH + trnL-trnF.
The results demonstrated that individual sequences were inadequate for distinguishing among the nine species of the genus Syringa. Among the combinations of sequences, three specific combinations were effective in differentiating the nine species of Syringa. Notably, the sequence combination of ITS2+psbA-trnH + trnL-trnF achieved the highest identification rate, reaching up to 93.6%.
Current DNA barcode analysis methods primarily include sequence feature analysis, genetic distance analysis, BLAST search analysis in NCBI, and evolutionary tree construction analysis (Zhang et al., 2015). The genetic distance analysis and phylogenetic tree construction methods have been applied in the identification of plants in the genus Syringa to assess whether the combination of psbA-trnH and trnC-petN sequences can serve as a DNA barcode for Syringa plants (Yao et al., 2022). In this study, the analysis methods were expanded to include BLAST analysis and BLOG method analysis, and the effectiveness of these four analysis methods in the selection of DNA barcodes for nine species of Syringa was evaluated.
For genetic distances, intraspecific and interspecific genetic distances are generally assessed, with the ideal DNA barcode characterized by small intraspecific distances and a significant difference from interspecific distances (Hao et al., 2012). Wilcoxon signed-rank tests can further analyze interspecific genetic distances (Kim and Kim, 2003; Kasuya, 2010). DNA barcodes, combined with the distribution of genetic distances among sequences and the results of Wilcoxon signed-rank tests, indicate that the ITS2+psbA-trnH sequence exhibits stable intraspecific variation and significant interspecific variation. However, this method could not evaluate the identification effect of barcodes. BLAST is a commonly used method to compare sequence similarities in the NCBI database, comparing identification effects based on ratios and scores. The results indicate that the BLAST method exhibits a high identification rate, with all sequences achieving identification rates between 97% and 100%. Among the single sequences, ITS2 has the highest identification rate at 99.80%. Among the combined sequences, the ITS2 + psbA-trnH + trnL sequence has the highest identification rate at 99.70%. It shows that the BLAST-based method has a high identification rate at the genus level. Character-based analysis methods identify species through different base substitutions at specific positions in the sequence, typically using BLOG analysis software. This method was applied in this study to select DNA barcodes for Syringa genus species. The BLOG algorithm was used to analyze the DNA barcodes of the nine Syringa genus plants, with an accurate identification rate of 50% for ITS2, and 77.78% for trnL-trnF and trnL, while the remaining individual and combined sequences could accurately identify these nine species.
The construction of phylogenetic trees using UPGMA, NJ, and MP methods serves as a basis for DNA barcode identification assessment (Liu et al., 2015). The Neighbor-Joining (NJ) method is relatively accurate for constructing phylogenetic trees when the evolutionary distances are short and the number of informative sites is limited in short sequences (Li and Gao, 2009). The results of this study show that NJ trees constructed with ITS2+psbA-trnH + trnL-trn conform to the phylogenetic analysis model of Syringa genus plants and the results obtained in this study are in alignment with those reported by preceding scholars (He M., 2007). In summary, distance-based methods and BLAST methods cannot directly assess the identification effects of DNA barcodes, but character-based and tree-based methods can. In this study, the construction of phylogenetic trees showed the best identification effects.
Based on current research on the structure of the nuclear and chloroplast genomes, it is difficult to find a universal DNA barcode suitable for all plants. Since Kress proposed the idea of sequence combinations, more studies have proven that combined sequences have higher species discrimination ability than single sequences (Kress et al., 2005). In the study of Syringa genus species, two barcode sequences and their combinations were analyzed for 33 species, revealing that combined sequences have higher identification capabilities than any single sequence (Yao et al., 2022).
This study analyzed four single sequences and 11 combined sequences of Syringa genus plants. The results of distance-based analysis indicated that, except for ITS2 and psbA-trnH, the average interspecific distance of combined sequences was higher than that of other single sequences, supported by the results of Wilcoxon signed-rank tests. However, the genetic distance distribution showed that intraspecific variation was more stable for single sequences compared to combined sequences, with all sequences having overlapping regions. BLAST-based analysis results indicated that combined sequences had higher scores than single sequences, but both single and combined sequences had high identification success rates. Feature-based analysis showed that only one single sequence could accurately identify Syringa genus plants, while all sequence combinations could achieve this goal, indicating a significant improvement in identification ability compared to single sequences. In the NJ tree analysis, none of the four single sequences could cluster the nine Syringa species separately. However, among the combined sequences, six were able to cluster the nine plant species. Yet, only the three NJ trees constructed using ITS2+psbA-trnH + trnL-trnL, ITS2+psbA-trnH + trnL, and ITS2+psbA-trnH + trnL-trnF + trnL enabling the separate clustering of these nine species. Ultimately, the sequence combination of ITS2+psbA-trnH + trnL-trnF was selected for its highest accuracy in identification. Therefore, the results of this study indicate that combined sequences have higher identification capabilities for these nine Syringa genus plants compared to single sequences. However, not all combined sequences can accurately identify these nine species, which is also related to the choice of analysis method.
The results obtained in this study regarding the morphological traits of Syringa species in the northeast region are consistent with those collected by previous researchers in their cladistic analysis of the Syringa genus based on morphological traits. However, the morphological clustering analysis does not support the traditional classification results (He N., 2007). Traditional morphological markers are significantly influenced by the developmental stage of plants and environmental factors, making it difficult to effectively distinguish species with very similar morphologies.
Currently, molecular marker techniques are also frequently employed in species classification and identification due to their characteristics of being rapid, accurate, and objective. Specifically, polymorphisms such as AFLP, SSR, and ISSR are identified through the amplification of DNA fragments and the detection of changes in DNA length (Varma and Shrivastava, 2018; Gyorgy et al., 2020; Kocsisne et al., 2020; Yang et al., 2020). Study have shown that ISSR molecular marker techniques can be used for the identification of plants in the genus Syringa (Yao et al., 2021). The results indicate that there is an overlapping phenomenon in the clustering of species between the Ser. Pubescentes (Schneid.) Lingelsh and the Ser. Villosae (Schneid.) Rehd. This finding is consistent with the results of the AFLP analysis on the phylogenetic relationships of plants in the genus Syringa (Gao et al., 2011). Therefore, neither of these markers can accurately distinguish between these two groups. With the rapid development of sequencing technologies, the method of species identification using DNA sequences has been recognized as reliable and accurate. DNA barcoding technology can accurately identify species through variation sites in marker sequences. In previous studies, different researchers have utilized nuclear genomic sequences such as ITS and ETS, as well as chloroplast genomic sequences like psbA-trnH, trnL-trnF, and trnC-petN for the identification and phylogenetic analysis of plants in the genus Syringa. This study employed four analytical methods, namely, distance-based methods, BLAST-based methods, character-based methods, and tree-based methods, to evaluate whether the ITS2 + psbA-trnH + trnL-trnF sequences could serve as a barcode for nine species of Syringa. The study incorporated the trnL intron and chloroplast genome sequences, which had not been used in previous Syringa research, with the aim of screening new DNA barcodes suitable for differentiating these nine tree species from the sequences.
From the perspective of genetic distance distribution results, the inter-specific variation of the ITS2 + psbA-trnH + trnL-trnF sequence overlaps with the intra-specific variation, and the inter-specific variation distance is relatively large. The analysis based on BLAST indicates that this sequence can achieve a species-level identification rate of 98.97%. The analysis based on sequence characteristics also shows that the sequence has an accuracy of 100% for the nine tree species. In the NJ tree constructed based on ITS2 + psbA-trnH + trnL-trnF, the nine species of Syringa can be clustered into three different clades, among which Sect. Ligustrina and Ser. Villosae (Schneid.) Rehd. Cluster together, and Ser. Syringa and Ser. Pubescentes (Schneid.) Lingelsh each form a separate clade. The research results are similar to those of the NJ tree results from the identification of Syringa based on chloroplast genomes. However, in the NJ phylogenetic tree constructed using the combined sequences of psbA-trnH and trnC-petN, Ser. Villosae (Schneid.) Rehd. Clusters separately and is closer to the root. The results are also similar to the NJ phylogenetic tree constructed using the trnL-trnF single sequence in the molecular systematics study of Syringa in the Northeast region, where Ser. Syringa is closer to the root (He M., 2007). In the NJ phylogenetic tree constructed using the combined sequences of psbA-trnH and trnC-petN, Ser. Villosae (Schneid.) Rehd. Clusters separately and is closer to the root (Yao et al., 2022). The presentation of these results may be related to the selection of sequences. The experimental results obtained from multiple evaluation methods demonstrate that the ITS2 + psbA-trnH + trnL-trnF sequence has strong discriminatory power, providing strong support for the conclusion that it can serve as a DNA barcode for these nine tree species. In addition to ITS2 + psbA-trnH + trnL-trnF, in the combined sequence research, ITS2 + psbA-trnH + trnL and ITS2 + psbA-trnH + trnL-trnF + trnL can also distinguish the nine species of Syringa, but the identification rate is lower than that of the ITS2 + psbA-trnH + trnL-trnF sequence. In conclusion, it is recommended to use the ITS2 + psbA-trnH + trnL-trnF sequence as the DNA barcode for the identification of the nine species of Syringa.
Through the combined analysis of morphological methods and DNA barcoding technology, the results of the barcode ITS2 + psbA-trnH + trnL-trnF in the identification of nine species of Syringa were verified. The phylogenetic tree of the DNA barcode ITS2 + psbA-trnH + trnL-trnF shows that S. oblata and S. vulgaris, which have similar morphological features such as leaf shape, leaf base shape, leaf color, lowering period, and Inflorescence shape, cluster within Ser. Syringa. Syringa wolfii, S. villosa, and S. josikaea, which have similar features such as leaf color, lowering period, Inflorescence shape, and Petal type, cluster within Ser. Villosae (Schneid.) Rehd. Syringa oblata and S. vulgaris, which have similar features such as leaf shape, lowering period, and Petal type, cluster within Ser. Pubescentes (Schneid.) Lingelsh. Syringa reticulata subsp. Pekinensis and S. reticulata subsp. Amurensis, which have similar features such as leaf shape, leaf base shape, flowering period, and Petal type, cluster within Sect. Ligustrina. This is consistent with previous morphological and taxonomic research results. Excluding the group division, Sect. Ligustrina is included in Sect. Syringa. Therefore, currently, the use of morphological feature analysis and single or combined barcode fragments can only be applied and identified within a small range of higher plants (at the family, genus, and species levels), and the results of phylogenetic analysis may be incorrect or contradictory to traditional taxonomy. It is evident that the screening of traditional plant barcodes still has a long way to go.
The seeds of 27 specimens of nine different species of syringa were provided by the Botanical Garden of Heilongjiang University of Chinese Medicine Pharmacy Botanical Garden and The Tree Specimen Garden of the Heilongjiang Forest Botanical Garden. All plant samples were identified by researcher Ma Wei, Department of Chinese Medicine Resources, Heilongjiang University of Chinese Medicine. Species names and source information are shown in Table 7. The wax leaf specimens of nine species of Syringa are shown in Figure 9.
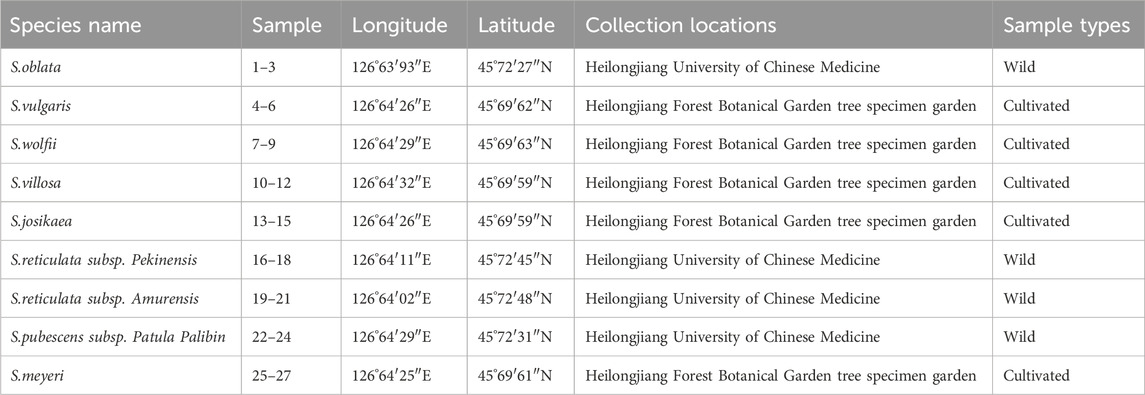
Table 7. Information of the plant samples.

Figure 9. Nine species of Syringa L. Herbarium specimens. (A) S.oblata (Serial Number:20230513X1HRB); (B) S.vulgaris (Serial Number:20230516Z5HRB); (C) S.wolfii (Serial Number:20230531Z7HRB); (D) S.villosa (Serial Number:20230602Z6HRB); (E) S.josikaea (Serial Number:20230531Z8HRB); (F) S.reticulata subsp. Pekinensis (Serial Number:20230616X17HRB); (G) S.reticulata subsp. Amurensis (Serial Number:20230606X11HRB); (H) S.pubescens subsp. Patula Palibin (Serial Number:20230519X10HRB); (I) S.meyeri (Serial Number:20230525Z1HRB).
For each sample, three mature fresh leaves of the whole plant in different orientations were taken in a pre-cooled mortar, ground and crushed with liquid nitrogen, and the DNA of the samples was extracted using the Plant Genomic DNA Extraction Kit (TIANGEN), and the extracted DNA was stored at −20°C.
PCR amplification was performed to a final volume of 25 µL in an eppendorf research thermocycler (Eppendorf AG 22331, Hamburg, Germany). The reaction mixture contained 2 µM genomic DNA, 1 µM forward and reverse primers, and 8.5 mM ddH2O, 12.5 mM 2 × MEGA Fast Taq Master Mix (Msunflowers, China). The primer sequences used for the DNA barcoding analyses are shown in Supplementary Table S4. PCR cycles consisted of an initial denaturation step for 5 min at 94°C, followed by 30 cycles of denaturation (30 s at 94°C), annealing (30 s at 58°C) and elongation (1 min at 72°C), a final elongation at 72°C for 10 min. The PCR products were sequenced in two directions with magnetic bead method in an automated ABI 3730 sequencer (PE Applied Biosystems). Sequence ambiguities were manually corrected using the Sequencing analysis software version 5.2 (Carlsbad, California, United States of America).
MEGA7 software was used to perform multiple alignment of sequences, and the basic information of each sequence was calculated after manually adjusting the sequence. In addition, in this study, ITS2 + psbA-trnH, ITS2 + trnL-trnF, ITS2 + trnL, psbA-trnH + trnL-trnF, psbA-trnH + trnL, psbA-trnH + trnL, trnL-trnF + trnL, ITS2 + psbA-trnH + trnL-trnF, ITS2 + psbA-trnH + trnL, psbA-trnH + trnL-trnF + trnL, ITS2 + psbA-trnH + trnL-trnF + trnL, ITS2 + psbA-trnH + trnL-trnF + trnL were selected as candidate DNA barcodes for further identification and analysis. The GenBank accession numbers of each sequence are shown in Supplementary Table S3. Kimura two-parameter (K2P) model in MEGA seven software was used to calculate genetic distance. Three parameters, interspecific, intraspecific and mean genetic distance, were used to evaluate the results. The distribution of genetic variation was observed by intraspecific and interspecific genetic distances. DNA barcode sequences should show independent and non-overlapping distributions of genetic variation in intraspecific and interspecific samples. The method of Wilcoxon signed rank test was used to verify the significance of the difference among the species by using IBM SPSS Statistics 27 software. In recognition ability, BLAST, character method and evolutionary tree method were selected to evaluate each sequence (Bosmali et al., 2022). Blast search was performed in NCBI database, and the most similar uploaded sequences of the same species were selected for statistical analysis, and the recognition ability of each sequence was evaluated. Blog 2.0 is based on sequence feature analysis, using classification rules to analyze the features of base sites (Weitschek et al., 2013). In this study, Blog 2.0 software was used to evaluate the discrimination rate of different sequences, and the logic rules were obtained. MEGA7 is used to build adjacent (NJ) trees (Ross et al., 2008), and the Bootstrap support option is set to 1,000 random addition replicates to determine the branch’s statistical support. When all individuals of a species can congregate in a single clade, the species is considered to have been successfully identified.
In this study, we described the morphological characteristics of nine Syringa species. Employing DNA barcoding techniques, four different methods were utilized to evaluate the identification capabilities of four single sequences and eleven combined sequences for the nine Syringa tree species. Across all methods, the sequences demonstrated the best identification performance when analyzed using the NJ tree approach. Moreover, compared to single sequences, combined sequences showed a notable enhancement in identification capabilities when employing the character-based method. Experimental results indicated that single sequences could only identify 2-3 out of the nine Syringa genus plants, whereas the combined sequence analyses, specifically ITS2+psbA-trnH + trnL-trnF accurately distinguished all nine species of Syringa genus plants using the four evaluation methods, exhibiting excellent discrimination and identification capabilities. The study ultimately selected the combination of ITS2+psbA-trnH + trnL-trnF as the optimal DNA barcode for the identification of nine species within the genus Syringa.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MZ: Formal Analysis, Methodology, Software, Validation, Writing - original draft. XZ: Resources, Writing - original draft. LH: Investigation, Software, Writing - original draft. ZW: Validation, Writing - original draft. HC: Investigation, Writing - original draft. PW: Data curation, Writing - original draft. WR: Conceptualization, Funding acquisition, Supervision, Visualization, Writing - review and editing. WM: Conceptualization, Funding acquisition, Project administration, Supervision, Writing - review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Key Research and Development Project, specifically the project on the quality resources collection and screening of genuine medicinal materials, including ginseng, as well as the breeding technology research and demonstration project. The Article Processing Charge (APC) was covered by the grant 2021YFD1600901. Source of funds: Grant number 2021YFD1600901.
The authors thank all of the above funds for their support. We would also like to thank Yongying Yu and Xiaoou Zhai (Heilongjiang Forest Botanical Garden) provides plant material support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1544062/full#supplementary-material
Bafeel, S. O., Arif, I. A., Bakir, M. A., Al Homaidan, A. A., Al Farhan, A. H., and Khan, H. A. (2012). DNA barcoding of arid wild plants using rbcL gene sequences. Genet. Mol. Res. 11, 1934–1941. doi:10.4238/2012.July.19.12
Bi, Y., Wen, X., Pan, Y., Cai, H., Zhong, H., and Wang, A. (2020). Application and research progress of chloroplast DNA barcoding in forest trees. Mol. Plant Breed. 18, 5444–5452.
Bosmali, I., Lagiotis, G., Haider, N., Osathanunkul, M., Biliaderis, C., and Madesis, P. (2022). DNA-based identification of eurasian vicia species using chloroplast and nuclear DNA barcodes. Plants 11, 947. doi:10.3390/plants11070947
Cai, Y., Dai, J., Zheng, Y., Ren, Y., Chen, H., Feng, T., et al. (2022). Screening of DNA barcoding sequences for molecular identification of Uncaria genus. Chin. Traditional Herb. Drugs 53, 1828–1837.
Chase, M. W., Cowan, R. S., Hollingsworth, P. M., Van Den Berg, C., Madrinan, S., Petersen, G., et al. (2007). A proposal for a standardised protocol to barcode all land plants. Taxon 56, 295–299. doi:10.1002/tax.562004
Chen, F., Guo, Q., Yang, F., Zhu, Z., and Wang, T. (2019). Application of ITS2 secondary structure phylogenetic information in DNA barcode identification of Chrysanthemum indicum and its related plants. China J. Chin. materia medica 44, 4813–4819. doi:10.19540/j.cnki.cjcmm.20190829.110
Chen, J. (2006). The taxonomic revision of Syringa L.(Oleaceae). master’s thesis, Institute of Botany. Chinese Academy of Sciences.
Chen, J., Zhang, Z., and Hong, D. (2009). A taxonomic revision of the syringa pubescens complex (OLEACEAE). Ann. Mo. Botanical Gard. 96, 237–250. doi:10.3417/2006072
Chen, S., Wu, Y., Xu, L., Feng, Q., Wang, B., Kang, T., et al. (2016). DNA barcoding of Mongolian oxytropis medicinal materials. J. Chin. Med. Mater. 39, 284–288.
Cheng, X., Wang, A., Gu, Z., Wang, Y., Zhan, X., and Shi, Y. (2011). Current progress of DNA barcoding. Genomics Appl. Biol. 30, 738–747.
Frigerio, J., Agostinetto, G., Mezzasalma, V., De Mattia, F., Labra, M., and Bruno, A. (2021). DNA-based herbal teas’ authentication: an ITS2 and psbA-trnH multi-marker DNA metabarcoding approach. Plants 10, 2120. doi:10.3390/plants10102120
Fu, M., Peng, J., Wang, Y., Yu, D., Wang, L., and Zhang, Y. (2010). Application and analysis of DNA barcoding. J. Henan Normal Univ. Nat. Sci. 38, 118–122.
Gao, H., Yang, K., and Liu, J. (2011). Analysis of the phylogeneticrel ationship of Syringa by AFLP technique. J. China Agric. ural Univ. 16, 50–54.
Gyorgy, Z., Incze, N., and Pluhar, Z. (2020). Differentiating Thymus vulgaris chemotypes with ISSR molecular markers. Biochem. Syst. Ecol. 92, 104118. doi:10.1016/j.bse.2020.104118
Hao, D.-H., Xiao, P., Peng, Y., Dong, J., and Liu, W. (2012). Evaluation of the chloroplast barcoding markers by mean and smallest interspecific distances. Pak. J. Bot. 44, 1271–1274.
He, M. (2007a). Study on molecular phylogenetics of the genus syringa in northeastern China. master’s thesis. Northeast Forestry University.
He, N. (2007b). Cladistics analysis of Syringa from northeast China based on morphological characters. For. Technol. 32, 60–64.
Hu, X., Zheng, X., Guo, E., Xing, Z., and Tian, C. (2009). Study on the ornamental characteristics of Syringa plants and their development and exploitation in Henan. J. Henan Agric. Univ. 43, 560–563.
Juntheikki-Palovaara, I., Antonius, K., Linden, L., and Korpelainen, H. (2013). Microsatellite markers for common lilac (Syringa vulgaris L.). Plant Genet. Resources-Characterization Util. 11, 279–282. doi:10.1017/s1479262113000166
Kasuya, E. (2010). Wilcoxon signed-ranks test: symmetry should be confirmed before the test. Anim. Behav. 79, 765–767. doi:10.1016/j.anbehav.2009.11.019
Kim, D. H., and Kim, H. G. (2003). Ranked-set sample Wilcoxon signed rank test for quantiles under equal allocation. Commun. Stat. Appl. Methods 10, 535–543. doi:10.5351/ckss.2003.10.2.535
Kocsisne, G. M., Bolla, D., Anhalt-Bruederl, U. C. M., Forneck, A., Taller, J., and Kocsis, L. (2020). Genetic diversity and similarity of pear (Pyrus communisL.) cultivars in Central Europe revealed by SSR markers. Genet. Resour. Crop Evol. 67, 1755–1763. doi:10.1007/s10722-020-00937-0
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A., and Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. U. S. A. 102, 8369–8374. doi:10.1073/pnas.0503123102
Lattier, J. D., and Contreras, R. N. (2017). Intraspecific, interspecific, and interseries cross-compatibility in lilac. J. Am. Soc. Hortic. Sci. 142, 279–288. doi:10.21273/jashs04155-17
Li, Y., and Gao, K. (2009). An improved neighbor-joining method and its applications. J. Beijing Univ. Technol. 35, 283–288.
Li, Y., Wei, J., He, M., and Zhuo, L. (2010). Phylogenetic relationships of the genus syringa in Northeast China based on nrDNA ITS sequence. J. North-East For. Univ. 38, 45–47.
Li, Y., Wu, D., and Gao, L. (2013). Highly universal DNA barcoding primers of ITS2 for gymnosperms. Plant Divers. Resourcues 35, 751–760.
Liu, C., Jiao, S., Zhou, X., Li, A., Ma, X., Shana, W., et al. (2020). An updated phytochemical and pharmacological progress on Syringa pinnatifolia. China J. Chin. materia medica 45, 4196–4204. doi:10.19540/j.cnki.cjcmm.20200608.201
Liu, J., Hu, D., Zhou, Z., Liu, S., and Wan, S. (2019). Applications of DNA barcoding in forestry. For. Res. 32, 152–159.
Liu, J., Provan, J., Gao, L., and Li, D.-Z. (2012). Sampling strategy and potential utility of indels for DNA barcoding of closely related plant species: a case study in taxus. Int. J. Mol. Sci. 13, 8740–8751. doi:10.3390/ijms13078740
Liu, Q., Guo, S., Zheng, X., Shen, X., Zhang, T., Liao, B., et al. (2021). Licorice germplasm resources identification using DNA barcodes inner-variants. Plants 10, 2036. doi:10.3390/plants10102036
Liu, S., Wang, X., Song, Y., and Guo, B. (2015). Evolutionary bayesian rose trees. Ieee Trans. Knowl. Data Eng. 27, 1533–1546. doi:10.1109/tkde.2014.2373384
Ming, J., Gu, W., Liu, C., Liu, K., and Wang, L. (2007). Advances in germplasm resources of syringa linn. Research. World For. Res. 20, 20–26.
Qiao, M., Chen, B., and Fu, Y. (2019). DNA extraction and DNA barcoding identification of 5 wood species of Phoebe spp. Machilus spp 3, 141–148.
Ross, H. A., Murugan, S., Sibon Li, W. L., and Hedin, M. (2008). Testing the reliability of genetic methods of species identification via simulation. Syst. Biol. 57, 216–230. doi:10.1080/10635150802032990
Su, G., Chen, J., Cao, Y., Bai, R., Chen, S., Tu, P., et al. (2015). Phytochemical and pharmacological progress on peeled stem of Syringa pinnatifolia, a Mongolian folk medicine. China J. Chin. materia medica 40, 4333–4338.
Varma, A., and Shrivastava, N. (2018). Genetic structuring in wild populations of two important medicinal plant species as inferred from AFLP markers. Plant Biosyst. 152, 1088–1100. doi:10.1080/11263504.2017.1418446
Wang, L., Jiao, W., Chen, X., Liao, B., Wang, X., and Han, J. (2015). Molecular identification of ilicis chinensis folium from its closely related species and adulterants based on ribosomal internal transcribed spacer 2 (ITS2) sequence. Journol Agric. Biotechnol. 23, 598–605.
Wang, Q., Huo, S., Bao, Y., and Ao, W. (2018). Chemical constituents of Syringa pinnatifolia and its chemotaxonomic study. Chem. Nat. Compd. 54, 435–438. doi:10.1007/s10600-018-2373-4
Wang, Y., Li, Y., Wang, Y., Lu, S., and Yang, Z. (2003). Quantitative determination of syringopicroside in leaves of Syringa oblata by RP-HPLC. Chin. Traditional Herb. Drugs 34, 268–269.
Wang, Y.-S., Jin, Y.-X., Liu, K.-J., Guo, C., Wang, Y.-H., Xu, C., et al. (2023). Species identification of Ligustrum lucidum. China J. Chin. materia medica 48, 2940–2948. doi:10.19540/j.cnki.cjcmm.20230315.101
Wei, L., Pacheco-Reyes, F. C., Villarreal-Quintanilla, J. A., Robledo-Torres, V., Encina-Dominguez, J. A., Lara-Ramirez, E. E., et al. (2024). Effectiveness of dna barcodes (rbcl, matk, its2) in identifying genera and species in cactaceae. Pak. J. Bot. 56, 1911–1928. doi:10.30848/pjb2024-5(11)
Weitschek, E., Van Velzen, R., Felici, G., and Bertolazzi, P. (2013). BLOG 2.0: a software system for character-based species classification with DNA Barcode sequences. What it does, how to use it. Mol. Ecol. Resour. 13, 1043–1046. doi:10.1111/1755-0998.12073
Xia, Y., Yin, J., Zeng, L., Wang, Y., Zhan, R., Lin, Y., et al. (2023). Study on interspecies identification of Hoya plants based on ITS2 sequence. J. Chin. Med. Mater. 46, 603–609.
Yang, C., Wu, K., Chuang, L., and Chang, H. (2018). Decision tree algorithm-generated single-nucleotide polymorphism barcodes of rbcL genes for 38 brassicaceae species tagging. Evol. Bioinforma. 14, 1176934318760856. doi:10.1177/1176934318760856
Yang, D., Li, J., Liang, C., Tian, L., Shi, C., Hui, N., et al. (2021). Syringa microphylla Diels: a comprehensive review of its phytochemical, pharmacological, pharmacokinetic, and toxicological characteristics and an investigation into its potential health benefits. Phytomedicine 93, 1–28. doi:10.1016/j.phymed.2021.153770
Yang, H., Dong, Y., Gu, Z., Liang, N., and Yang, J. (2012). A preliminary assessment of matK, rbcL and trnH-psbA as DNA barcodes for Calamus (Arecaceae) species in China with a note on ITS. Ann. Bot. Fenn. 49, 319–330. doi:10.5735/085.049.0603
Yang, Y., He, R., Zheng, J., Hu, Z., Wu, J., and Leng, P. (2020). Development of EST-SSR markers and association mapping with floral traits in Syringa oblata. BMC Plant Biol. 20, 1–13. doi:10.1186/s12870-020-02652-5
Yao, R., Guo, R., Liu, Y., Kou, Z., and Shi, B. (2022). Identification and phylogenetic analysis of the genus Syringa based on chloroplast genomic DNA barcoding. Plos One 17, e0271633. doi:10.1371/journal.pone.0271633
Yao, R., Wei, J., Meng, X., Jin, M., Liu, Y., Kou, Z., et al. (2021). Phylogenetic analysis of Syringa based on ISSR markers. J. HEBEI Agric. Univ. 44, 48–56.
Zhang, C., Fang, X., Qiu, H., Li, N., Li, X., and Fang, X. (2015). A review of wood identification by DNA barcoding. World For. Res. 28, 50–55.
Zhang, S., Zhang, J., and Wang, J. (2006). Chemical constituents in stem bark of Syringa oblata. Chin. Traditional Herb. Drugs 37, 1624–1626.
Zhao, M., Tang, W., Li, J., Bai, L., Wang, J., Zhang, W., et al. (2016). Two new monoterpenoids from the fresh leaves of Syringa oblata. Chem. Nat. Compd. 52, 1023–1025. doi:10.1007/s10600-016-1852-8
Zheng, R., and Guo, W. (2013). Simultaneous determination of rutin,astragalin and isoquercetin in the leaves of Syringa veutina Kom by HPCE-DAD. Chin. Tradit. Pat. Med. 35, 2457–2461.
Zhu, W., Wang, Z., Sun, Y., Yang, B., Wang, Q., and Kuang, H. (2021). Traditional uses, phytochemistry and pharmacology of genus Syringa: a comprehensive review. J. Ethnopharmacol. 266, 113465. doi:10.1016/j.jep.2020.113465
Keywords: Syringa, DNA barcoding, ITS2, PSBA-TRNH, trnL-trnF, TRNL, species identification
Citation: Zhang M, Zhai X, He L, Wang Z, Cao H, Wang P, Ren W and Ma W (2025) Morphological description and DNA barcoding research of nine Syringa species. Front. Genet. 16:1544062. doi: 10.3389/fgene.2025.1544062
Received: 12 December 2024; Accepted: 03 February 2025;
Published: 26 February 2025.
Edited by:
Hongqiang Lyu, Xi’an Jiaotong University, ChinaReviewed by:
Fuad Bahrul Ulum, University of Göttingen, GermanyCopyright © 2025 Zhang, Zhai, He, Wang, Cao, Wang, Ren and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weichao Ren, bHp5cmVud2VpY2hhb0AxMjYuY29t; Wei Ma, bWF3ZWlAaGxqdWNtLmVkdS5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.