
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 26 March 2025
Sec. Genomics of Plants and the Phytoecosystem
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1530910
 Yujuan Tian1†
Yujuan Tian1† Jinxin Wang2†
Jinxin Wang2† Xiangyu Wang3
Xiangyu Wang3 Dongsheng Wang1
Dongsheng Wang1 Xuan Wang1
Xuan Wang1 Jing Liu1
Jing Liu1 Haie Zhang1
Haie Zhang1 Jingzheng Zhang1
Jingzheng Zhang1 Liyang Yu1*
Liyang Yu1*The phosphatidylethanolamine binding protein (PEBP) family plays an important part in growth and development of plants. Castanea mollissima is an economic plant with significant financial value and has become an important food source in the Northern Hemisphere. However, the PEBP genes in C. mollissima have not been studied yet. In this study, six PEBP genes (CmPEBP1 ∼ CmPEBP6) were identified in C. mollissima and comprehensively analyzed in terms of physicochemical properties, phylogeny, gene structures, cis-regulatory elements (CREs), transcription factor interaction, and expression profiles. The six CmPEBP genes were categorized into three subfamilies according to the phylogeny analysis, and all of them share extremely similar gene and protein structures. A total of 136 CREs were identified in the promoter regions of the CmPEBP genes, mainly related to growth and development, environmental stress, hormone response, and light response. Comparative genomic analysis indicated that the expansion of the CmPEBP genes was mainly driven by dispersed duplication, and the CmPEBP3/CmPEBP5 derived from eudicot common hexaploidization (ECH) events retained orthologous genes in all species studied. A total of 259 transcription factors (TFs) belonging to 39 families were predicted to be regulators of CmPEBP genes, and CmPEBP4 was predicted to interact with the most TFs. The RNA-seq data analysis indicated the potential roles of CmPEBP genes in the ovule, bud, and flower development of C. mollissima, as well as in the response to temperature stress, drought stress, and the gall wasp Dryocosmus kuriphilus (GWDK) infestation. Additionally, the expression of CmPEBP genes in C. mollissima seed kernel development and their response to temperature stress were confirmed by RT-qPCR assays. This study gives references and directions for future in-depth studies of PEBP genes.
The phosphatidylethanolamine binding protein (PEBP) family is a highly conserved protein family that is widely presented in all three domains (Eukaryota, Bacteria, and Archaea) of the phylogenetic tree (Chautard et al., 2004; Dong et al., 2020). PEBP genes in plants were first discovered in Arabidopsis thaliana (Xu et al., 2022). By now, the presence of PEBP gene family has been extensively reported in a large number of plants, such as Picea abies (Liu Y. Y. et al., 2016), Solanum lycopersicum (Cao et al., 2015; Sun et al., 2023c), Actinidia chinensis (Varkonyi-Gasic et al., 2013), Vitis vinifera (Carmona et al., 2007), Populus tremula (Mohamed et al., 2010), Zea mays (Danilevskaya et al., 2008), Oryza sativa (Tamaki et al., 2007; Song et al., 2018). In general, PEBP genes can be divided into three subfamilies: FLOWERING LOCUS T-like (FT-like), TFL1 TERMINAL FLOWER1-like (TFL1-like), and MOTHER OF FT AND TFL1-like (MFT-like) (Jin et al., 2021; Sun et al., 2023c). Moreover, recent studies on more species have found the existence of the PEBP-like subfamily in the PEBP family (Zhang et al., 2016; Dong et al., 2020).
Due to their diverse functions concerning the growth and development of plants, the PEBP genes in plants participate in a wide variety of biological processes, such as hormone signal transduction, flower bud differentiation, and reproductive development (Zheng et al., 2016; Huang et al., 2024; Wu et al., 2024). For instances, the overexpression of OsMFT1 delays the heading date of O. sativa and leads to remarkably increased number of spikelets and branches per panicle, while its knockout mutant leads to an earlier heading period and decreased number of spikelets per panicle (Song et al., 2018); four FT-like genes SlSP5G, SlSP5G2, SlSP5G3, and SlSP3D in S. lycopersicum are involved in the photoperiod effect of tomato flowering (Cao et al., 2015). ZCN8 interacts with DLF1 to regulate Z. mays inflorescence development (Meng et al., 2011); the overexpression of the CorfloTFL1 gene in A. thaliana, a PEBP gene in Cornus florida delays plant flowering (Liu X. et al., 2016); it has been reported that TaPEBP1, TaPEBP3 and TaPEBP5 play essential roles in response of Triticum aestivum to drought, cold stress and heat stress (Dong et al., 2020); Aradu80YRY, AraduYY72S, and AraduEHZ9Y in Arachis duranensis, along with AraipVEP8T in Arachis ipaensis, are potentially crucial regulators of flowering time (Jin et al., 2019); the overexpression of the FtFT1 and FtFT3 genes promotes flowering and yield in Fagopyrum tataricum (Nie et al., 2024).
Castanea mollissima is a plant of Castanea Mill in the family Fagaceae, known for its tasty nuts (Huang et al., 2023; Zhang et al., 2023). The nutritional value of C. mollissima has received widespread attention due to its abundance in many varieties of nutrients such as starch, protein, fat, vitamins, minerals (calcium, iron, zinc, potassium), and bioactive substances (Wang et al., 2022). The abundance of nutrients in C. mollissima nuts gives them many health benefits, including but not limited to enhancing anti-inflammatory and antioxidant effects, preventing heart disease and stress, lowering blood lipids and blood sugar, preventing cardiovascular diseases, and improving digestive function (Chang et al., 2020; Wang et al., 2022; Zhang et al., 2022). Additionally, the antioxidant substances contained in C. mollissima also have specific effects on anti-aging and cancer prevention (Zhang et al., 2014). Due to the good adaptability to the environment, developed root system, and tall tree body of C. mollissima, it can grow under relatively harsh conditions, empowering its excellent abilities in windbreak, sand stabilization, and soil and water conservation. However, there are few reports on the gene families in C. mollissima associated with its growth, development, and stress resistance, which undoubtedly limits our understanding of this miraculous plant.
The vital roles of the PEBP genes in growth and development, metabolic regulation, and response to stress factors of plants have been extensively demonstrated, but have not been systematically studied for C. mollissima. In this study, we systematically analyzed PEBP genes in the C. mollissima genome. The analysis covered the chromosome locations, phylogenetic relationships, gene structures, conserved motifs, cis-regulatory elements (CREs), collinearity, interacting transcription factors (TFs), and protein three-dimensional structures. Additionally, the expression levels of CmPEBP genes in different tissues of C. mollissima and under various environmental stresses were analyzed using RNA-seq data. The RT-qPCR results validated the differential expression of CmPEBP genes during C. mollissima seed development and temperature stress. The present study provides information for future in-depth characterization of the potential roles of PEBP genes in C. mollissima in its growth, development, and response to stress factors.
According to the results of HMM and BlastP searches, six PEBP genes were identified in the C. mollissima genome, renamed as CmPEBP1 - CmPEBP6 by their relative positions on the chromosome. The information of these CmPEBP genes and the physicochemical properties of the proteins encoded by them can be found in Supplementary Table S1. The CmPEBPs contain amino acid residues of 172 (CmPEBP4 and CmPEBP5) - 189 (CmPEBP3), with a molecular weight within the range of 18.88 kDa (CmPEBP4) to 21.34 kDa (CmPEBP3). They have maximum and minimum aliphatic indexes of 88.90 (CmPEBP4) and 74.66 (CmPEBP3), respectively. The grand average of hydropathicity of all these CmPEBPs is negative, suggesting their hydrophilic nature. All the CmPEBPs have a theoretical isoelectric point (pI) greater than seven; therefore, they are considered as alkaline. In addition, their instability index ranges from 37.14 (CmPEBP1) to 52.00 (CmPEBP5). The subcellular localization prediction results of the CmPEBP proteins indicate that they are all cytoplasmic proteins. The prediction results of the secondary structure of the CmPEBP proteins showed that all of them mainly contain random coils, accounting for more than 58% of the protein’s amino acid composition, while extended strands and alpha helices have a lower proportion, accounting for less than 42% (Supplementary Figure S1; Supplementary Table S2). In addition, no beta-turn was found in the CmPEBPs. Since the three-dimensional structure of proteins is closely related to their functions (Yang, 2008; Bhattacharya et al., 2017), the three-dimensional structures of the CmPEBPs were constructed based on SWISS-MODEL and AlphaFold3, and the results indicated that all of them share a similar three-dimensional structure, suggesting that they may serve as structural foundations for similar functions (Supplementary Figure S1).
To investigate the classification and evolution of the CmPEBP genes, the sequences of 117 PEBPs from A. thaliana (6), Malus domestica (8), O. sativa (19), Sorghum bicolor (19), Brachypodium distachyon (18), S. lycopersicum (13), V. vinifera (5), Z. mays (23), and C. mollissima (6) were used to construct a phylogenetic tree. As a result, the 117 PEBPs were classified into MFT-like, TFL1-like, and FT-like subfamilies (Figure 1). FT-like subfamilies have more PEBP members, and monocotyledonous plants tend to have more PEBP gene family members. For C. mollissima, the six CmPEBPs show an approximately uniform distribution in the three subfamilies; specifically, three of TFL1-like (CmPEBP3, CmPEBP5, CmPEBP6), two of MFT-like (CmPEBP1 and CmPEBP4), and one of FT-like (CmPEBP2).
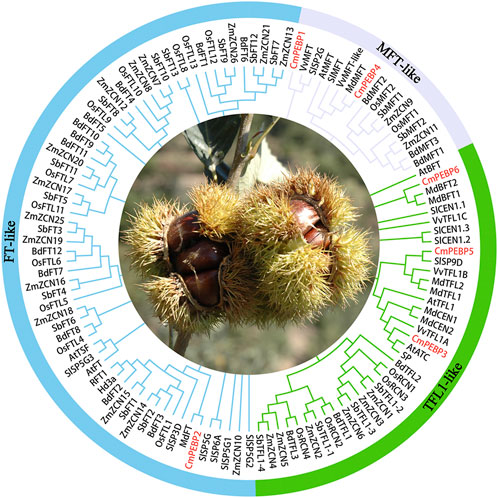
Figure 1. The phylogenetic tree of 117 PEBP proteins of Arabidopsis thaliana (6), Malus domestica (8), Oryza sativa (19), Sorghum bicolor (19), Brachypodium distachyon (18), Solanum lycopersicum (13), Vitis vinifera (5), Zea mays (23) and C. mollissima (6). MEGA 7.0 was used to construct the phylogenetic tree based on the protein sequences with the maximum likelihood method. The proteins were clustered into three groups.
The six CmPEBP genes are unevenly distributed on 5 C. mollissima chromosomes. Specifically, CmPEBP2 and CmPEBP3 are located on chromosome 6, and the other four CmPEBP genes are located on chromosomes 2, 7, 9, and 10, respectively (Figure 2A). Phylogenetic analysis classified the six CmPEBP genes into three subfamilies (Figure 2B). Furthermore, an analysis on the exon-intron structure of the CmPEBP genes was performed to investigate their gene structure. Interestingly, all the CmPEBP genes contain four exons and three introns for each, indicating the strong conservation gene structure of the CmPEBP genes (Figure 2B). Similarly, the conserved structural motifs of the proteins encoded by the CmPEBP genes were further investigated to understand the structural and functional characteristics of these genes (Figures 2C, D). As a result, eight conserved structural motifs (motif 1–8) were identified in the CmPEBPs, with five to seven conserved structural motifs in each CmPEBP. Specifically, motifs 1–4 are distributed in all the CmPEBPs and are arranged in the same order, suggesting that these four motifs are strongly conserved in the CmPEBPs; motif six is presented only in the MFT-like subfamily, while motifs 7–8 are distributed in both MFT-like and TFL1-like subfamilies. Overall, the CmPEBPs of the same subfamily tend to have the same conserved structural motifs.
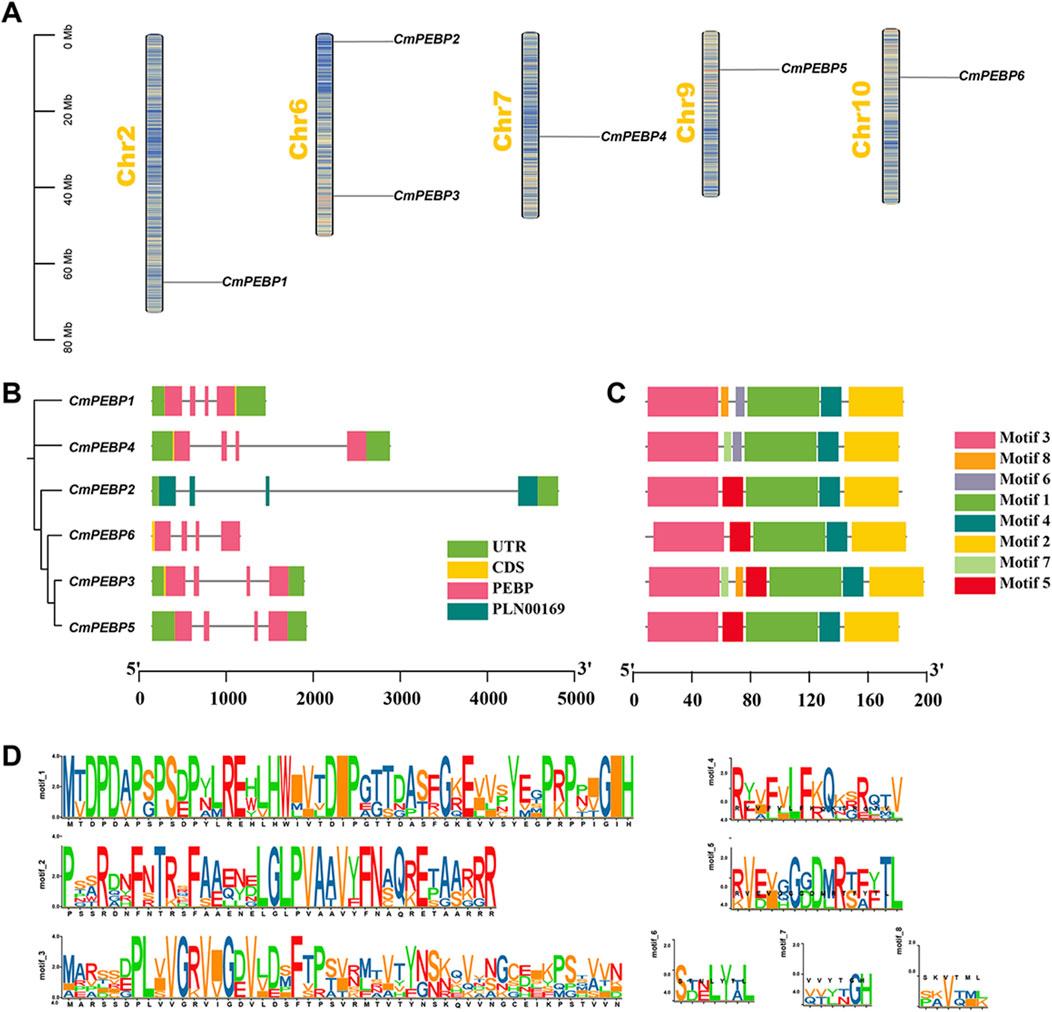
Figure 2. Chromosome distribution, gene structure, and conserved motifs of CmPEBP genes. (A) Chromosome distribution of CmPEBP genes. The color of segments in the chromosomes shows the gene density of the corresponding region. (B) Intron exon structure of CmPEBP genes. The phylogenetic tree containing only six CmPEBP genes is placed on the left side, constructed by MEGA 7.0 based on the maximum likelihood method. (C) Distribution of conserved motifs in CmPEBP proteins. (D) The sequence of eight conserved motifs in CmPEBP proteins.
The analysis of CREs in a gene’s promoter regions helps to understand a gene’s potential functions (Wittkopp and Kalay, 2012). PlantCARE (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to analyze the upstream DNA region of 2000 bp of the ATG (methionine) start codon of the CmPEBP genes to detect potential CREs (Figure 3A; Supplementary Table S3). As a result, 136 CREs in total were identified in the promoter regions of the six CmPEBP genes, which can be categorized into four types: 15 development-related CREs, 24 environmental stress-related CREs, 35 hormone-responsive CREs, and 62 photoresponsive CREs (Figures 3B, C). Notably, some CREs were distributed in almost all promoter regions of the CmPEBP genes (Figure 3). For example, Box four and G-box elements associated with light response were identified in the promoter region of six CmPEBP genes. ARE elements associated with environmental stress were identified in the promoter regions of five CmPEBP genes. CAT-box elements associated with growth and development were identified in the promoter regions of five CmPEBP genes. ABRE elements associated with hormonal responses were identified in the promoter regions of six CmPEBP genes.The analysis of promoter CREs suggested the vital roles of the PEBP gene family in hormone regulation, response to light signals, and resistance to abiotic stress.
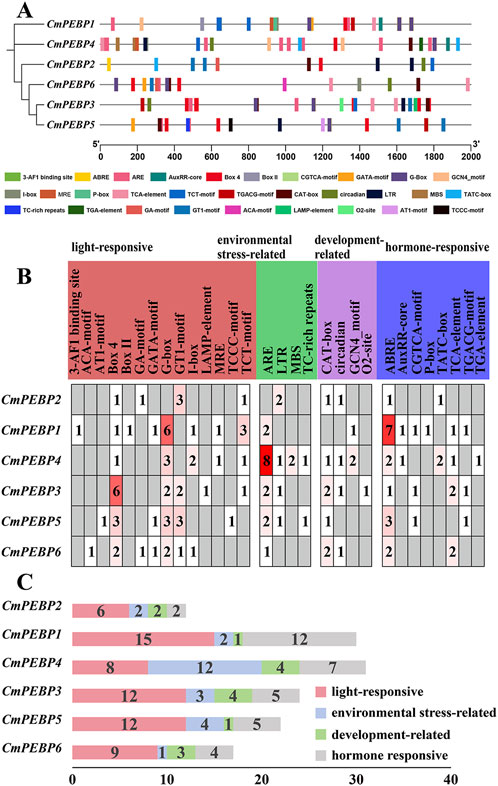
Figure 3. Prediction of cis-regulatory elements in the promoters of PEBP genes in C. mollissima. (A) Cis-regulatory elements in the promoters of six CmPEBP genes. Various color symbols present different elements, and their position in the figure indicates their relative position on the promoter. (B) The relative proportions of different cis-regulatory elements in the promoters of six CmPEBP genes are indicated in the chart. The same color represents cis-regulatory elements sharing identical or similar functions. (C) The number of various cis-regulatory elements in the promoters of each CmPEBP genes.
The collinearity between C. mollissima and seven representative species (five dicotyledonous plants: A. thaliana, Quercus, V. vinifera, Pyrus, and S. lycopersicum; and two monocotyledonous plants: O. sativa and Z. mays) was analyzed to understand the collinearity relationship of PEBP genes among different species (Figure 4). Four of the CmPEBP genes were identified in the collinear regions between C. mollissima and V. vinifera, Pyrus, Quercus, and A. thaliana, while all the six CmPEBP genes were identified in the collinear regions between C. mollissima and S. lycopersicum (Supplementary Tables S4–10). CmPEBP3, CmPEBP4, and CmPEBP5 exist in the collinear region between C. mollissima and all five dicotyledonous plants. CmPEBP3 and CmPEBP5 were also found in the collinear regions of C. mollissima with O. sativa and Zea may. As indicated by these results, the orthologous genes of CmPEBP3 and CmPEBP5 are preserved in all these species, demonstrating their conservation in the evolution of the PEBP gene family. CmPEBP3 and CmPEBP5 both have three or more orthologous genes in Pyrus and O. sativa, suggesting their potentially essential roles in the evolution of the PEBP gene family based on the gene balance hypothesis (Birchler and Veitia, 2007). We further analyzed the length of collinear blocks containing the CmPEBP genes between C. mollissima and the seven representative species (Supplementary Tables S11–17). The median length of collinear blocks containing the CmPEBP genes was 48 (C. mollissima vs. V. vinifera), 51 (C. mollissima vs. Pyrus), 10.5 (C. mollissima vs. Quercus), 19 (C. mollissima vs. A. thaliana), 17.5 (C. mollissima vs. O. sativa), 16 (C. mollissima vs. S. lycopersicum) and 11.5 (C. mollissima vs. Z. mays) gene pairs, respectively. These data indicated that the PEBP genes are better preserved in the collinear regions of C. mollissima, V. vinifera, and Pyrus genomes, regardless of genome assembly quality.
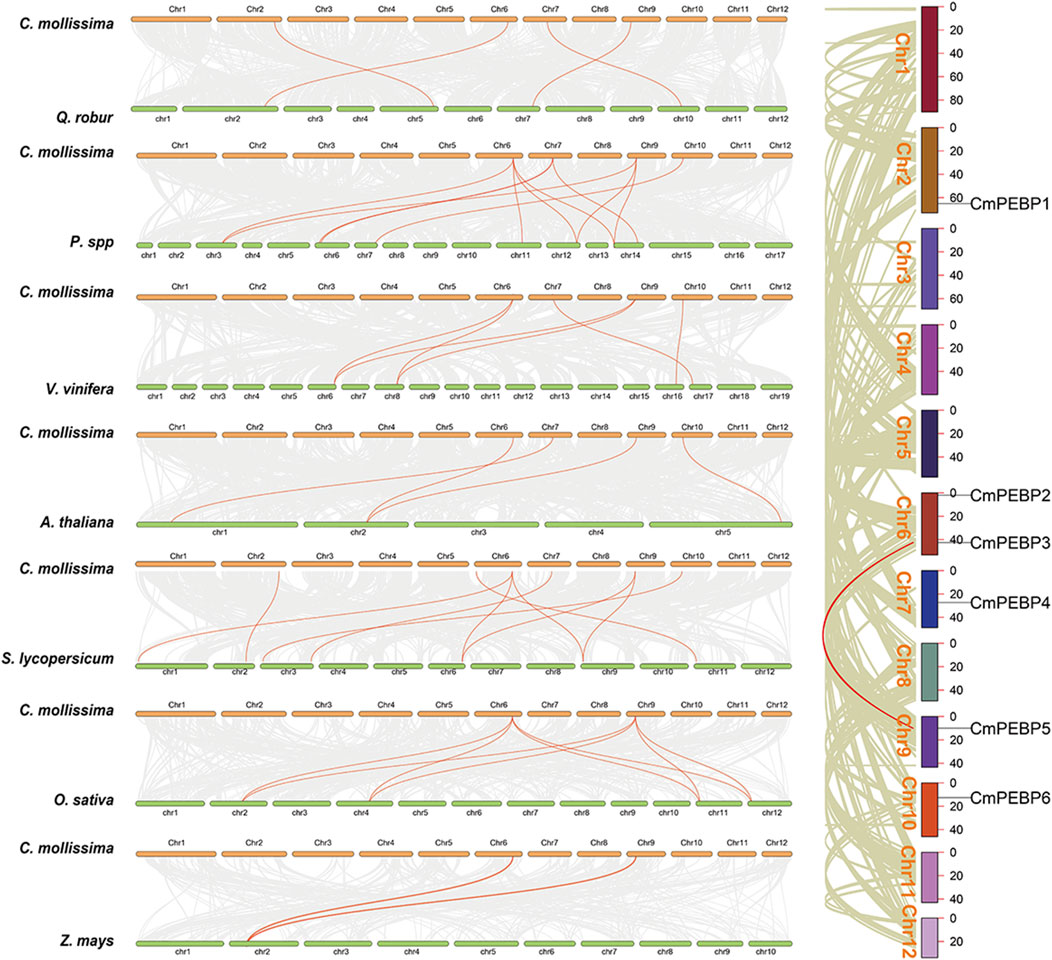
Figure 4. Collinearity analyses of PEBP genes within C. mollissima genome, and between the PEBP genes of C. mollissima and seven representative plant species (Quercus, Pyrus, V. vinifera, A. thaliana, S. lycopersicum, O. sativa, and Z. mays).
The important roles of gene duplication in gene family expansion and functional differentiation of genes have been widely reported (Liu et al., 2018; Quan et al., 2019). Therefore, the collinearity within C. mollissima genome was analyzed to explore the duplication type of the CmPEBP genes (Figure 4). The results indicated that CmPEBP1, CmPEBP2, CmPEBP4, and CmPEBP6 originate from dispersed duplication, while CmPEBP3 and CmPEBP5 are believed to originate from whole genome duplication (WGD) or segmental duplication. CmPEBP3 and CmPEBP5 were identified to originate from the eudicot common hexaploidization (ECH) events through an analysis on the collinear homologous gene dot plot of C. mollissima genome, as previously demonstrated (Figure 5) (Yu et al., 2022a; Yu et al., 2023b).
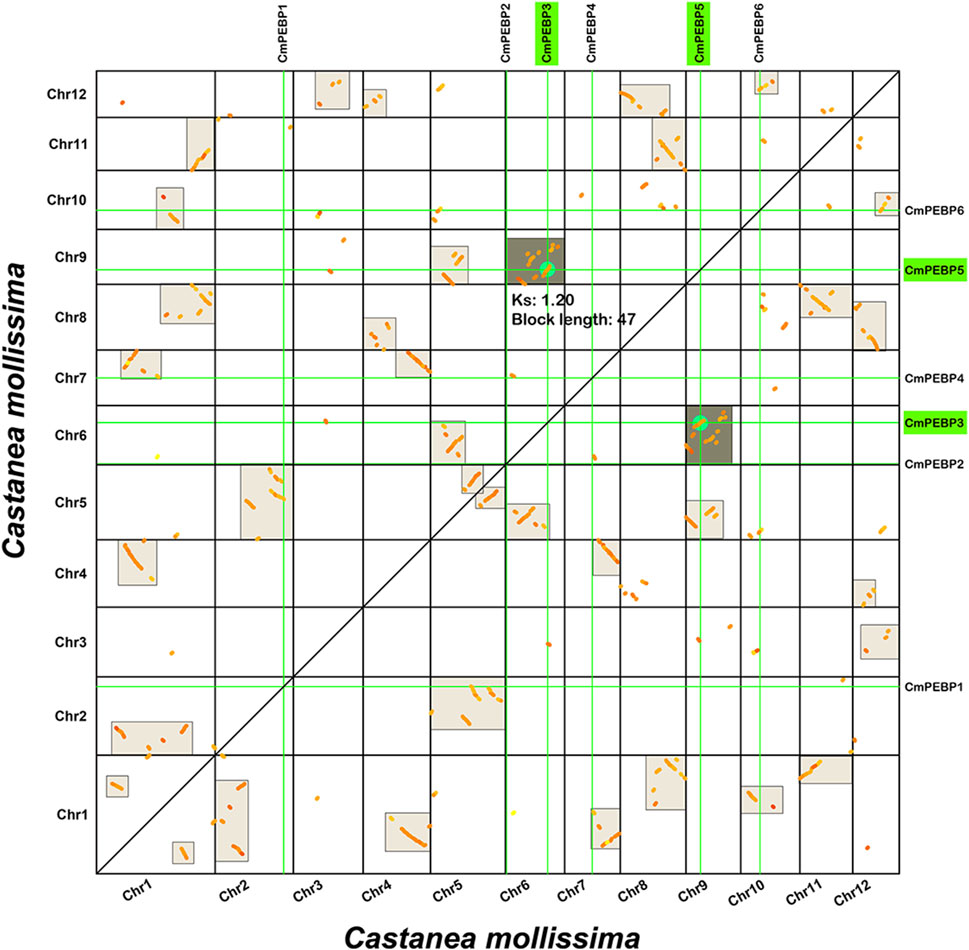
Figure 5. Homologous collinear dot-plot within the C. mollissima genome. The boxes in the figure represent collinear regions within the C. mollissima genome, in which the dark or light highlighted boxes indicate regions formed by WGD event containing CmPEBP homologous gene pairs and complementary fragments forming more significant homologous regions, respectively.
Seven proteins were predicted to interact with the CmPEBP proteins (Supplementary Figure S2). Specifically, two bZIP transcription factors interact with CmPEBP2, while three MADS-box proteins act on CmPEBP5; additionally, an unknown protein GWHTANWH026364 was predicted to interact with CmPEBP1, CmPEBP3, CmPEBP4, and CmPEBP6. The collinearity analysis showed that CmPEBP5 retained three and four orthologous genes in the Pyrus and O. sativa genomes, respectively, significantly higher than other CmPEBP members (Supplementary Table S5; Supplementary Table S8). The gene balance hypothesis suggests that genes participating in macromolecular complexes or signaling networks are more likely to be preserved in evolution (Birchler and Veitia, 2007; Xu et al., 2016). Interestingly, CmPEBP5 was predicted to interact with more other protein members, which is consistent with the interpretation of collinearity analysis results based on the gene balance hypothesis. In addition, numerous studies have demonstrated the interaction between bZIP transcription factors and FT-like proteins (Abe et al., 2005; Nan et al., 2014; Collani et al., 2019), MADS-box proteins act on TFL1-like proteins to regulate plant flowering (Liu et al., 2020). Interestingly, CmPEBP2 and CmPEBP5 belong to the FT-like and TFL1-like subfamilies, respectively (Figure 1), and have been predicted to interact with bZIP transcription factors and MADS-box proteins, respectively (Supplementary Figure S2A). These results further indicated the existence of interactions between CmPEBP proteins and the predicted proteins. Furthermore, the TFs that may interact upstream with the CmPEBP genes were predicted for further understanding of the potential functions of the CmPEBP genes (Figure 2B). As a result, 259 TFs were predicted to potentially act on the promoter regions of the CmPEBP genes, belonging to 39 TF families, such as MYB, NAC, SRS, SBP, and ERF (Supplementary Table S18). Among these predicted TF families, ERF has the most members (38), followed by MYB (25), WRKY (23), NAC (21), and bHLH (18); and the least abundant member in the TFs gene family is only one, such as FAR1, CAMTA, GRAS, ARR-B, SBP. Among the CmPEBP genes, CmPEBP4 interacts with 156 of the TFs, followed by CmPEBP1 (152), CmPEBP5 (97), CmPEBP2 (86), CmPEBP6 (81), and CmPEBP3 (69). MYB, WRKY, NAC, and bHLH are significantly enriched in the TFs regulatory network of the CmPEBP gene family, indicating their potential vital roles in regulating the driving biological functions of the CmPEBP genes. These findings provide useful information for understanding the genes that interact with the CmPEBP genes.
To explore the potential functions of CmPEBP genes, the RNA-seq data of C. mollissima from NCBI was analyzed, including ovules (three periods of development for fertile and abortive ovules), flowers (primary and secondary, male and female flowers), seed kernels (five periods of development for two varieties), and buds (three periods of development) (Figure 6; Supplementary Tables S19–22). Five of the CmPEBP genes showed almost no expression at all three stages of development of ovules (fertile and aborted ovules) (FPKM < 2), with only CmPEBP1 showing a significant decrease in expression levels during the late development of abortive ovules and in primary male flowers (Figures 6A, B; Supplementary Tables S19, 20). The expression level of CmPEBP1 significantly increased during kernel maturation in both 2 C. mollissima varieties (‘Yanshanzaofeng’ and ‘Yanlong’); the expression of CmPEBP4 was increased considerably (FPKM > 450) during the middle stage of seed kernel development (80 and 90 days after anthesis) and significantly decreased at the end of development (100 days after anthesis) (Figure 6C; Supplementary Table S21). These results indicated the potential roles of CmPEBP1 and CmPEBP4 in developing C. mollissima seed kernels. During the bud development C. mollissima, only CmPEBP1 and CmPEBP3 showed a significant decrease in expression levels, while others showed almost no expression (FPKM < 2) (Figure 6D; Supplementary Table S22). Overall, the CmPEBP genes exhibited different expression levels in various tissues of C. mollissima or at different stages of development within the same tissue, suggesting that the CmPEBP functions in other tissues of C. mollissima.
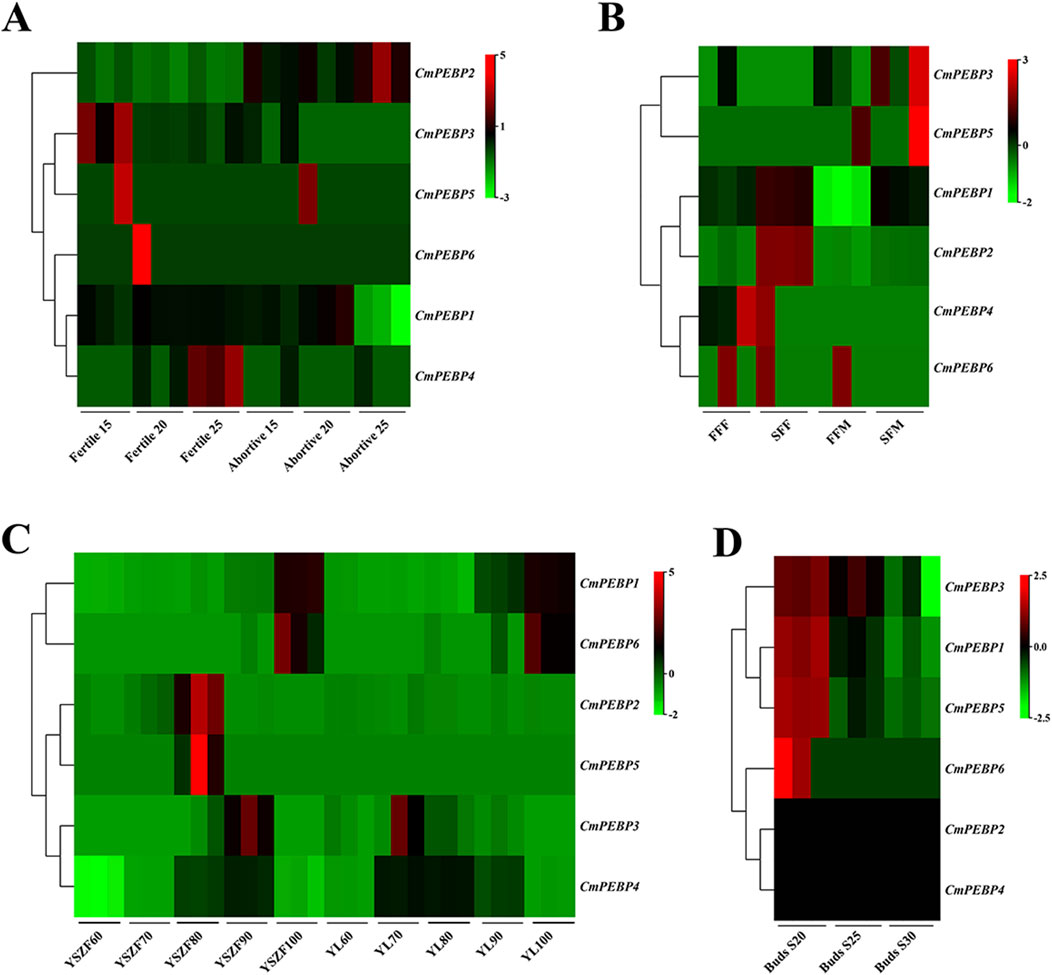
Figure 6. Genes expression of CmPEBP genes in different tissues of C. mollissima. (A) Genes expression in fertile and abortive ovules on 15-July, 20-July and 25-July. (B) Genes expression in first and second female flowering, first and second male flowering. FFF: First flowering (female), SFF: Secondary flowering (female), FFM: First flowering (male), SFM: Secondary flowering (male). (C) Genes expression in nuts of the cultivar “Yanshanzaofeng” and “Yanlong” 60, 70, 80, 90, and 100 days after flowering. (D) Genes expression in buds 20, 25, and 30 days after flowering.
RNA-seq analysis was conducted on C. mollissima under the stress factors of low-temperature, high-temperature, drought, and GWDK infestation to understand the potential functions of the CmPEBP genes in coping with environmental stresses (Figure 7; Supplementary Tables S23–27). Under low-temperature stress, CmPEBP1 showed a significantly increased expression at the beginning of the stress and always showed a high expression level throughout the stress, suggesting that it may be related to the resistance of C. mollissima resistance to low-temperature (Figure 7A; Supplementary Table S23). Under high-temperature stress, CmPEBP1 showed a continuously increasing expression level and peaked at the end (Figure 7A; Supplementary Table S23). Under drought stress, CmPEBP1 showed a significantly fluctuant expression, while the expression levels of all others of the CmPEBP genes showed almost no fluctuations, indicating that CmPEBP1 may be involved in the resistance response of the plant to drought (Figure 7B; Supplementary Table S24). Additionally, the chestnut gall wasp, Dryocosmus kuriphilus (Hymenoptera Cynipidae), is a significant pest of cultivated C. mollissima, and therefore, the RNA-seq data of the CmPEBP genes in the galls formed by GWDK at different stages were analyzed. Compared with CK, the expression of CmPEBP1 was remarkably upregulated in the galls of the initiation stage (7 April) formed by GWDK (Figure 7C; Supplementary Table S25). Furthermore, the expression level of CmPEBP1 in C. mollissima variety ‘HongLi’ (susceptible to GWDK infestation) was significantly higher than that in variety ‘Shuhe-Wuyingli’ (partially resistant to GWDK infestation) in the galls (7 April) of the initiation stage formed by GWDK (Figure 7D; Supplementary Table S26); and notably, the expression level of CmPEBP1 in galls formed by GWDK was significantly higher than that in C. mollissima leaves invaded by GWDK (Figure 7E; Supplementary Table S27). These results indicated the potential involvement of CmPEBP1 in the development of galls formed by GWDK infestation. In summary, the CmPEBP gene family may be related to the response of C. mollissima to environmental stresses, but their specific functions in coping with environmental changes need further research.
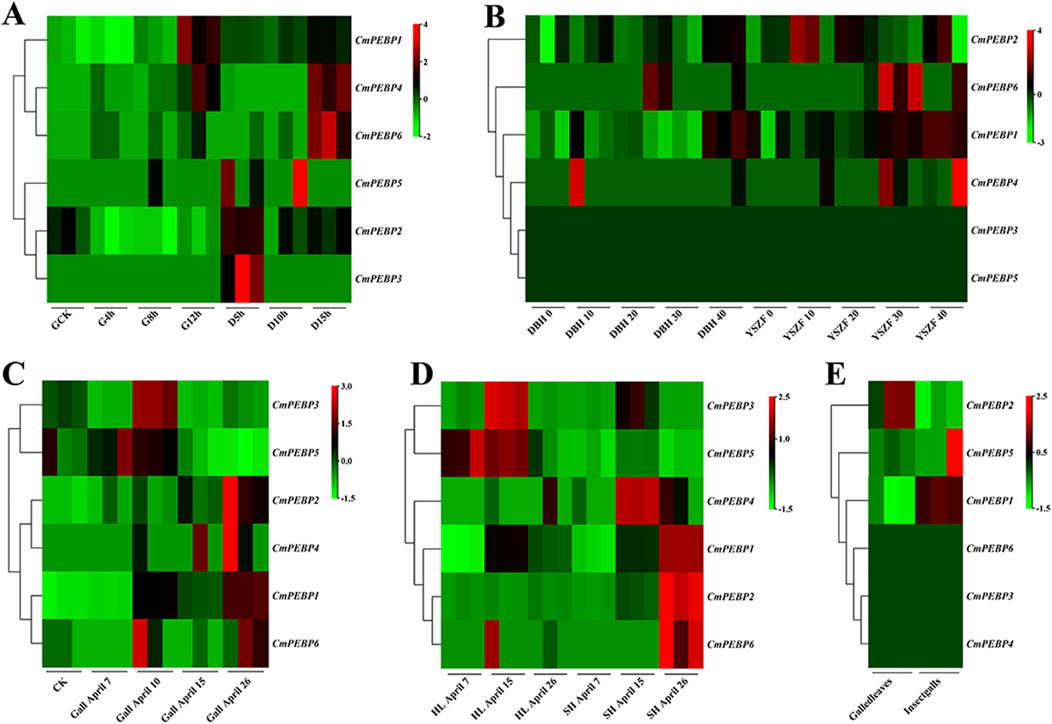
Figure 7. Genes expression of CmPEBP genes under different stresses in C. mollissima. (A) The expression profiles of CmPEBP genes under low (−15°C) and high (45°C) temperature stress at various periods. CK: control sample. G4h, G8h, G12h: high-temperature stress treatment for 4, 8, and 12 h, respectively. D5h, D10h, D15h: low-temperature stress treatment for 5, 10, and 15 h, respectively. (B) Genes expression in leaves of cultivar “Dabanhong” (DBH) and “Yanshanzaofeng” (YSZF) treated with drought for 0, 10, 20, 30, and 40 days. (C) Genes expression in leaves together with gall on 7-April, 10-April, 15-April, 26-April. (D) Genes expression in leaves with galls of cultivar “HongLi” (HL) (susceptible to GWDK infestation) and “Shuhe_Wuyingli” (SH) (partially resistant to GWDK infestation) infested with GWDK on 7-April, 15-April, 26-April. (E) Genes expression in leaves and insect galls induced by GWDK.
For further verification of the confidence of RNA-seq data, RT-qPCR assays on the CmPEBP genes in leaves of C. mollissima plant at different stages of development of seed kernels and under varying degrees of temperature stress (Figure 8). The results showed that the expression level of CmPEBP1 showed a trend of increasing with the development of C. mollissima seed kernels and peaked at 100 days after flowering. The expression level of CmPEBP4 gradually increased during the early stage of development of C. mollissima seed kernels, peaked at 90 days after flowering, and was significantly downregulated in samples collected 100 days after flowering. The expression levels of all other four CmPEBP genes did not show significant changes during the development of C. mollissima seed kernels. In addition, CmPEBP1 showed a significantly upregulated expression level under low-temperature stress, with a peak expression at D15h; CmPEBP1 presented a significantly upregulated expression level under high-temperature stress, with a peak expression at G12h. There are strong correlation between RNA-seq and RT-qPCR results of six CmPEBP genes from kernel development, low-temperature stress, and high-temperature stress. Specifically, there is a significant positive correlation between the 12 group in the 18 sets of RNA-seq and RT-qPCR results (Supplementary Figure S3). Overall, RT-qPCR assays have demonstrated the expression of the CmPEBP genes in the development of C. mollissima seed kernels under temperature stress.
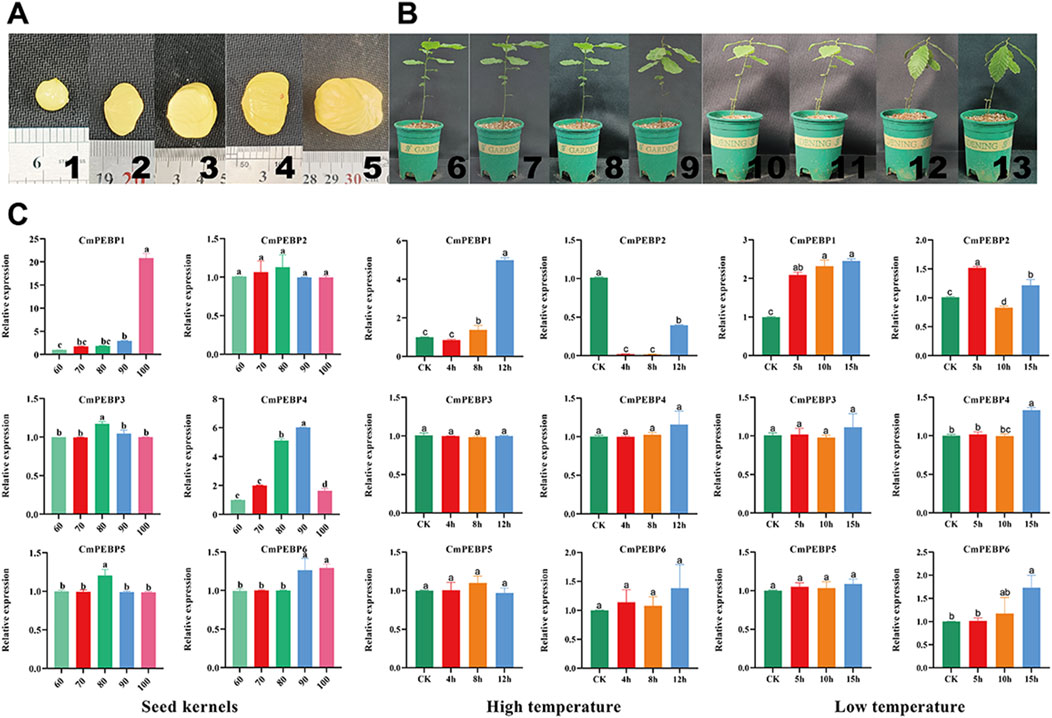
Figure 8. RT-qPCR of CmPEBP genes. (A) “Yanshanzaofeng” C. mollissima seed kernels, 1–5: seed kernels at 60, 70, 80, 90, and 100 days after flowering, respectively. (B) C. mollissima plants subjected to temperature stress treatment, 6∼9: C. mollissima plants were subjected to high-temperature stress for 0, 4, 8, and 12 h, respectively. 10∼13: C. mollissima plants were subjected to low-temperature stress for 0, 4, 8, and 12 h, respectively. (C) RT-qPCR of CmPEBP genes in C. mollissima leaves under high- and low-temperature stresses. Lowercase letter(s) above the bars indicate significant differences (α = 0.05, LSD) among the treatments.
C. mollissima is an economically and ecologically important nut that plays important roles in food supply and ecosystem maintenance, especially in the Northern Hemisphere (Nie et al., 2021; Zhou et al., 2021; Hu et al., 2022). The diverse functions of the PEBP gene family in the growth and development of plants have been widely reported, such as regulating flowering time, controlling bud development and dormancy, and affecting plant light signal transduction (Danilevskaya et al., 2008; Liu Y. Y. et al., 2016; Yang et al., 2023). Conducting a systematic analysis of the PEBP gene family in C. mollissima PEBP can offer valuable information for disclosing the critical role of PEBP genes in the biological traits of C. mollissima. The PEBP gene family has been systematically characterized in a lot of plants, including A. thaliana, O. sativa, Z. mays, S. lycopersicum, M. domestica. (Chardon and Damerval, 2005; Danilevskaya et al., 2008; Karlgren et al., 2011; Yang et al., 2023). There have also been reports of the PEBP gene family in gymnosperms (Liu Y. Y. et al., 2016). Herein, six PEBP genes were identified in the C. mollissima genome, which can be classified into three subfamilies: MFT-like, TFL1-like, and FT-like (Figure 1). The number of members of the PEBP gene family is different from species, and this may be related to the duplication and retention of the PEBP gene family in species, which is ultimately reflected in related functions. All six identified CmPEBP genes contain four exons and three introns (Figure 2). Interestingly, all thirteen PEBP genes in A. chinensis and all five PEBP genes in V. vinifera also contain four exons and three introns (Carmona et al., 2007; Voogd et al., 2017). In addition, exon two and exon three of the CmPEBP genes identified in C. mollissima are shorter, while exon one and exon four are more extended, which is similar to the distribution of exon length of the PEBP genes found in some other species (Tsaftaris et al., 2013; Li et al., 2015). The three-dimensional structures of the six CmPEBPs are similar, and the two-dimensional structures are in similar proportions (Supplementary Figure S1). The collinearity analysis between C. mollissima and seven representative species showed that the orthologous genes of CmPEBP3 and CmPEBP5 were preserved in all eight species (Figure 4), suggesting the potential conservation of their role in the course of evolution.
The important roles of gene duplication in gene family expansion and functional differentiation have been widely reported (Panchy et al., 2016; De Smet et al., 2017; Pasquier et al., 2017). The wide variation in the number of members across species suggests that the PEBP gene family may have experienced complex gene duplication and gene loss during evolution (Liu Y. Y. et al., 2016; Sun et al., 2023c; Wu et al., 2024). The duplication patterns of the CmPEBP genes were investigated to enhance the understanding of the driving forces behind the expansion of this important gene family. The analysis results based on MCScanX indicated that four of six identified CmPEBP genes (CmPEBP1, CmPEBP2, CmPEBP4, and CmPEBP6) originated from dispersed duplication. Through duplication and dispersed insertion, dispersed duplication allows similar gene copies to expand in the genome, making it one of the main mechanisms for forming gene families (Innan and Kondrashov, 2010; Qiao et al., 2019; Chen et al., 2023). Members of gene families formed by dispersed duplication often undergo functional differentiation during evolution, which is also one of the ways to increase functional genomic diversity (Innan and Kondrashov, 2010; Qiao et al., 2019). Indeed, the four CmPEBP genes thought to originate from dispersed duplication showed tissue-specific expression and response to different stresses in transcriptome data analysis, suggesting their functional differentiation (Figure 6; Figure 7). In addition, CmPEBP3 and CmPEBP6 are thought to originate from WGD or segmental duplication. Since plant genomes are usually accompanied by fusion and recombination of chromosome segments after WGD, the types of WGD and segmental duplication are not distinguished (Yu et al., 2022a). Herein, CmPEBP3 and CmPEBP5 were identified as originating from WGD using a method that we have expertly used before, based on the complementarity of the collinear blocks formed by WGDs as well as their Ks values (Yu et al., 2022a; Yu et al., 2022b; Yu et al., 2023b) (Figure 6). The C. mollissima genome has not experienced additional WGD events after the ECH event (Sun et al., 2020; Sun W. et al., 2023), which also confirmed that CmPEBP3 and CmPEBP5 originate from the ECH event. In addition, in the collinearity analysis of C. mollissima genome with seven other representative species (five dicotyledonous and two monocotyledonous plants), it was found that the orthologous genes of CmPEBP3 and CmPEBP5 were preserved in the genomes of all the eight plant species, providing evidence that CmPEBP3 and CmPEBP5 have been well preserved in the evolution of angiosperms. Considering the above results, combined with the gene balance hypothesis, which suggests that genes involved in the formation of macromolecular complexes or signaling networks are more likely to be preserved in evolution (Birchler and Veitia, 2007), it is speculated that CmPEBP3 and CmPEBP5 play a vital part in the evolution of the PEBP gene family. The PEBP genes typically function through complex regulatory mechanisms to participate in plant growth, development, and response to environmental stresses (Karlgren et al., 2011; Zhao et al., 2020). Many CREs have been predicted in the promoter regions of the CmPEBP genes, including light-responsive, environmental stress-related, development-associated, and hormone-responsive elements (Figure 3). The abundance and quantity of light- and hormone-responsive elements indicate that the promoters of the CmPEPB genes are mostly inducible promoters, which can be regulated by light and hormone signals. In addition, CREs that respond to growth, development, and environmental stress have been predicted. For example, LTR (involved in response to low temperature), MBS (involved in inducibility in response to drought), and CAT-box (related to meristem expression). These results indicated the roles of the CmPEBP genes in the growth, development, and response to environmental stresses of C. mollissima. The RNA-seq data of the CmPEBP genes in multiple tissues of C. mollissima and under different stresses were analyzed to explore their potential functions (Figure 6; Figure 7). The expression level of CmPEBP1 was significantly reduced during the late development of aborted ovules and in primary male flowers (Figures 6A, B). It was significantly upregulated during the maturation of C. mollissima seed kernels. In contrast, the expression level of CmPEBP4 was significantly upregulated during the middle stage of development of seed kernels, suggesting that both CmPEBP1 and CmPEBP4 are involved in the maturation of C. mollissima seed kernels (Figure 6C). Moreover, the expression level of CmPEBP1 was significantly decreased with the development of C. mollissima buds, suggesting that CmPEBP1 may be associated with such development (Figure 6D). Both CmPEBP1 and CmPEBP4 are of the MFT-like subfamily (Figure 1). Current research has found that the main function of genes of the MFT-like subfamily is the regulation of flowering (Yoo et al., 2004; Yuan et al., 2022; Wu et al., 2024), regulation of germination of seed kernels (Danilevskaya et al., 2008; Karlgren et al., 2011), and control of flower bud differentiation (Zhao et al., 2020). For example, ZCN8 and DLF1 have been shown to interact with and regulate the development of Z. mays flowers (Meng et al., 2011); AtMFT can regulate the germination and normal growth of A. thaliana seeds by modulating ABA and GA signals (Yoo et al., 2004; Xi et al., 2010; Vaistij et al., 2013).
CmPEBP1 showed a remarkably upregulated expression level in samples collected under low-temperature and drought stresses (Figure 7B), while the expression level of CmPEBP1 was significantly upregulated under high-temperature stress (Figure 7A). This finding indicated the potential role of CmPEBP1 in adapting C. mollissima to low-temperature and drought stresses. Furthermore, CmPEBP1 is suggested to be related to the GWDK infestation on C. mollissima. For example, compared to CK, CmPEBP1 showed an upregulated in the galls of the initiation stage (7 April) formed by GWDK (Figure 7C); the expression level of CmPEBP1 in C. mollissima variety ‘HongLi’ (susceptible to GWDK infestation) was significantly higher than that in the variety ‘Shuhe-Wuyingli’ (partially resistant to GWDK infestation), in the galls (7 April) of the initiation stage formed by GWDK (Figure 7D). All these findings justify the inference that CmPEBP1 may participate in the response of C. mollissima to GWDK infestation. In addition, temperature stress-related CREs (LTR) were identified in the promoter regions of CmPEBP1 and CmPEBP4 (Figure 3). Furthermore, the expression levels of CmPEBP1 and CmPEBP4 under both high and low-temperature stresses were verified by RT-qPCR assays, which were confirmed to be consistent with the results of RNA-seq data analysis (Figure 8). Considering the results of RNA-seq analysis and RT-qPCR assays of CmPEBP1 under temperature stresses, as well as the fact that CREs are identified in their promoter regions, it is believed that it is an important candidate gene for the response of C. mollissima to temperature stresses.
Herein, six PEBP genes were identified in the C. mollissima genome and then comprehensively characterized in terms of physicochemical properties, chromosome distribution, phylogenetics, gene structure, conserved motifs, conserved domains, collinearity relationships, CREs, and TFs regulatory networks. The results indicate that the CmPEBP genes exhibited strong conservation in gene structure, conserved motifs, and protein structure. The expression profile of the CmPEBP genes indicate their potential roles in the development of ovules, buds, seed kernels, and flowers, as well as in response to low- and high-temperatures, and GWDK infestation of C. mollissima. The results of RT-qPCR assays confirmed the expression patterns at different stages of development of seed kernels and the response to temperature stresses of the CmPEBP genes. The study offers valuable information from a theoretical point of view for future in-depth research on the functions of the PEBP gene family in C. mollissima.
The sequences of PEBPs in A. thaliana and the conserved domain of PEBP (PF01161) were sourced from the Arabidopsis Information Resource (https://www.arabidopsis.org/) and Pfam databases (https://www.ebi.ac.uk/interpro/entry/pfam/#table) (Xu et al., 2022), respectively. The genome data and annotation files of chestnut (N11-1) were downloaded from the Castanea Genome Database (http://castaneadb.net/) (Wang et al., 2020). Using the sequences of PEBPs in A. thaliana as the query sequences, the candidate genes in all protein sequences of C. mollissima were searched with BlastP (E-value ≤1.0 × e−5). The sequences of proteins in C. mollissima were searched using the HMMER3.0 software, and then the candidate genes were screened (Finn et al., 2011). All candidate sequences identified were submitted to Batch-CD to ensure the existence of PEBP conserved domains and ultimately confirm the CmPEBP genes (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi). The prediction of the physicochemical properties and subcellular localization of CmPEBPs were performed using ExPASy and Cell-PLoc, respectively (Chou and Shen, 2008), and the prediction of the secondary structure of CmPEBPs were performed using SOPMA (Geourjon and Deléage, 1995); and then the three-dimensional structures of the CmPEBPs were constructed using the Swiss model and AlphaFold 3 (Waterhouse et al., 2018; Abramson et al., 2024).
Based on publicly published articles, there are 111 PEBP genes have been identified in eight species, including A. thaliana (6), M. domestica (8), O. sativa (19), S. bicolor (19), B. distachyon (18), S. lycopersicum (13), V. vinifera (5), and Z. mays (23) (Yang et al., 2023). By combining the sequences of the above PEBPs with the sequences of six PEBPs from C. mollissima a total of 117 protein sequences were obtained. The ClustalW program was used to conduct multiple alignment of the full-length sequences of CmPEBP proteins. We used the “Find Best DNA/Protein Models (ML)” function in MEGA7.0 to obtain the best amino acid substitution model (partial deletion 95%). Finally, a phylogenetic tree was constructed with the maximum likelihood estimation and MEGA 7.0 software, with the follow parameters: Jones–Taylor–Thornton (JTT) model; Gamma Distributed (G); Partial deletion 95%; 1,000 bootstrap replications (Kumar et al., 2016). Using the genomes of seven representative species, including Quercus, Pyrus, V. vinifera, A. thaliana, S. lycopersicum, O. sativa, and Z. mays, downloaded from Phytozome database, and then the collinearity relationships between C. mollissima and these representative species were analyzed with MCScanX (Wang et al., 2012). “File Merge For MCScanX” function in TBtools was used to transfer the origin gff3 file to a format suitable for MCScanX operation that only contains chromosome, gene ID, gene start and end position information. The “duplicate_gene_classifier” in MCScanX software was used to obtain the duplication type of CmPEBP genes with default parameters, such as WGD or segmental, proximal, tandem, and dispersed. Furthermore, CmPEBP members formed by WGD events were identified, as we did before (Yu et al., 2023a; Cao et al., 2024). Specifically, the homologous collinear gene dot-plot within the C. mollissima genome was generated using TBtools. The non-synonymous (Ka) and synonymous substitution sites (Ks) values of homologous gene pairs was generated using the “add_ka_and_ks_to collinearity” function in MCScanX software. The median Ks values of collinear blocks were calculated by writing the script (Yu et al., 2022b). The collinear blocks in the homologous gene dot-plot were colored differently based on different median Ks values. Combined with the distribution of Ks corresponding to the WGD event that occurred in the C. mollissima genome before (Yu et al., 2022b), the complementarity of the collinear blocks, the CmPEBP genes formed by the WGD event were identified.
The gene structure information of CmPEBP genes was obtained based on the GFF3 file of C. mollissima, and the CmPEBPs were submitted to the online tool MEME for conservative motif prediction (Bailey et al., 2009). For each of the CmPEBP genes, the upstream sequence of 2000 bp was extracted as the promoter region by TBtools, which was then submitted to PlantCARE to predict CREs in the promoter region. Then, the gene structure, conserved motifs, and CREs were visualized with TBtools (Chen et al., 2020).
The Plant Transcriptional Regulatory Map (https://plantregmap.gao-lab.org/) was used to predict the TFs that may act on the 2000 bp upstream regions of CmPEBP genes (P-value ≤1e−6) (Hu X. et al., 2023; Yu et al., 2025). Since the C. mollissima genomic information is not yet available in the STRING (https://cn.string-db.org/) database, we conducted a protein-protein interaction network analysis to explore proteins that interact with CmPEBP proteins based on the homologs of the CmPEBP genes in A. thaliana. These homologous proteins were subjected to the STRING to obtain the interacting proteins with default parameters (Szklarczyk et al., 2019). Then, these protein sequences were aligned to obtain the homologous protein sequences in C. mollissima. This method of obtaining protein interaction networks has been widely reported and used (Chen et al., 2021; Yu et al., 2021; Hu M. et al., 2023; Sun et al., 2023b). TFs and protein interaction analysis results were visualized using the Cytoscape 3.9.1 (Kohl et al., 2011).
Transcriptomic data of C. mollissima in different tissues (ovules, flowers, seed kernels) and under various stress factors (high temperature, low temperature, drought, and GWDK infestation) were obtained from the NCBI database (Supplementary Table S28). Sratolkit 3.0 and Tophat2 software were used to align the reads to the reference genome (Chinese chestnut (N11-1) downloaded from Castanea Genome Database) and confirm the expression levels of CmPEBP genes, respectively (Goldberg et al., 2009; Kim et al., 2013). TBtools was used to standardize FPKM values (“Normalize” function in TBtools) and generate heatmaps based on log2 (FPKM+1) value conversion for comparison purposes.
The seed kernels of C. mollissima cv.Yanshanzaofeng at different stages of development were collected for RT-qPCR assays in 2024. Specifically, the fruits of ‘Yanshanzaofeng’ were collected at 60, 70, 80, 90, and 100 days after flowering, and after removing spines and shells, the seed kernels were rapidly frozen with liquid nitrogen and finally stored in the freezer at −80°C before RT-qPCR assay of the CmPEBP genes. In addition, we planted the seeds of C. mollissima cv. Yanshanzaofeng in April 2024 and subjected the plants to temperature stress treatment 60 days after sowing. Specifically, 9 C. mollissima trees were subjected to high-temperature treatment at 45°C, and leaf samples were collected after 4, 8, and 12 h. Similarly, 9 C. mollissima trees were subjected to low-temperature treatment at −15°C, and leaf samples were collected after 5, 10, and 15 h, respectively. Three C. mollissima trees grown at 25°C were used as the control. After collection, all samples were rapidly frozen with liquid nitrogen and stored at −80°C before further use. Before RT-qPCR assays, RNA extraction and reverse transcription of RNA into single-stranded cDNA were performed using the RNAprep pure Plant Kit (Tiengen, Beijing, China) and the PrimeScript RT Master Mix (Takara Biotechnology Co., Beijing, China), respectively. The RT-qPCR assays were conducted on the ABI 7500 Real-Time PCR system (Applied Biosystems Inc., Foster City, CA, USA) using TB Green Premix Ex Taq (Takara). The instrument settings were: 95°C for 300 s; 40 PCR cycles, with each cycle set at 95°C for 10 s and 60°C for 30 s. The specific primer information was shown in Supplementary Table S29, in which the 18S gene of C. mollissima was used as the reference gene. The RT-PCR primer efficiency was determined using the standard curve method (Svec et al., 2015; Zhang et al., 2021). Briefly, serial dilutions of template were used to generate a standard curve, and primer efficiency was calculated based on the resulting cycle threshold (CT) values. The calculation of relative gene expression values was completed using the comparative 2-△△CT method and three biological replicates were performed.
The data presented in the study can be found in the NCBI Sequence Read Archive (SRA) repository. The accession number(s) can be found in the article/Supplementary Material.
YT: Data curation, Formal Analysis, Methodology, Software, Writing - original draft. JW: Data curation, Formal Analysis, Methodology, Software, Visualization, Writing - original draft. XiW: Data curation, Formal Analysis, Software, Validation, Writing - original draft. DW: Formal Analysis, Methodology, Validation, Writing - original draft. XuW: Methodology, Validation, Writing - review and editing. JL: Resources, Validation, Writing - review and editing. HZ: Resources, Validation, Writing - review and editing. JZ: Resources, Validation, Writing - review and editing. LY: Conceptualization, Funding acquisition, Investigation, Supervision, Validation, Writing - original draft, Writing - review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Scientific Research Foundation of Hebei Normal University of Science and Technology (2023YB027), the Science and Technology Research Project of Higher Education in Hebei Province (2023JK01), the Natural Science Foundation of Hebei Province (C2024407040).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1530910/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | The predicted three-dimensional structure (based on SWISS-MODEL and AlphaFold3) and secondary structure analysis of CmPEBP proteins.
SUPPLEMENTARY FIGURE S2 | CmPEBP protein interaction network and TFs regulatory network analysis. (A) CmPEBP protein interaction network analysis. (B) TFs regulatory network analysis of CmPEBP genes.
SUPPLEMENTARY FIGURE S3 | The correlation between RNA-seq and RT-qPCR results of six CmPEBP genes from kernel development, low-temperature stress, and high-temperature stress.
Abe, M., Kobayashi, Y., Yamamoto, S., Daimon, Y., Yamaguchi, A., Ikeda, Y., et al. (2005). FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309 (5737), 1052–1056. doi:10.1126/science.1115983
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630 (8016), 493–500. doi:10.1038/s41586-024-07487-w
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi:10.1093/nar/gkp335
Bhattacharya, R., Rose, P. W., Burley, S. K., and Prlić, A. (2017). Impact of genetic variation on three dimensional structure and function of proteins. PLoS One 12 (3), e0171355. doi:10.1371/journal.pone.0171355
Birchler, J., and Veitia, R. (2007). The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19, 395–402. doi:10.1105/tpc.106.049338
Cao, F., Guo, C., Wang, X., Wang, X., Yu, L., Zhang, H., et al. (2024). Genome-wide identification, evolution, and expression analysis of the NAC gene family in chestnut (Castanea mollissima). Front. Genet. 15, 1337578. doi:10.3389/fgene.2024.1337578
Cao, K., Cui, L., Zhou, X., Ye, L., Zou, Z., and Deng, S. (2015). Four tomato FLOWERING LOCUS T-like proteins act antagonistically to regulate floral initiation. Front. Plant Sci. 6, 1213. doi:10.3389/fpls.2015.01213
Carmona, M. J., Calonje, M., and Martínez-Zapater, J. M. (2007). The FT/TFL1 gene family in grapevine. Plant Mol. Biol. 63 (5), 637–650. doi:10.1007/s11103-006-9113-z
Chang, X., Liu, F., Lin, Z., Qiu, J., Peng, C., Lu, Y., et al. (2020). Phytochemical profiles and cellular antioxidant activities in chestnut (Castanea mollissima BL.) kernels of five different cultivars. Molecules 25 (1), 502. doi:10.3390/molecules25030502
Chardon, F., and Damerval, C. (2005). Phylogenomic analysis of the PEBP gene family in cereals. J. Mol. Evol. 61 (5), 579–590. doi:10.1007/s00239-004-0179-4
Chautard, H., Jacquet, M., Schoentgen, F., Bureaud, N., and Bénédetti, H. (2004). Tfs1p, a member of the PEBP family, inhibits the Ira2p but not the Ira1p Ras GTPase-activating protein in Saccharomyces cerevisiae. Eukaryot. Cell 3 (2), 459–470. doi:10.1128/ec.3.2.459-470.2004
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi:10.1016/j.molp.2020.06.009
Chen, H., Zhang, Y., and Feng, S. (2023). Whole-genome and dispersed duplication, including transposed duplication, jointly advance the evolution of TLP genes in seven representative Poaceae lineages. BMC Genomics 24 (1), 290. doi:10.1186/s12864-023-09389-z
Chen, X., Liu, H., Wang, S., Zhang, C., Liu, L., Yang, M., et al. (2021). Combined transcriptome and proteome analysis provides insights into anthocyanin accumulation in the leaves of red-leaved poplars. Plant Mol. Biol. 106 (6), 491–503. doi:10.1007/s11103-021-01166-4
Chou, K.-C., and Shen, H.-B. (2008). Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 3 (2), 153–162. doi:10.1038/nprot.2007.494
Collani, S., Neumann, M., Yant, L., and Schmid, M. (2019). FT modulates genome-wide DNA-binding of the bZIP transcription factor FD. Plant Physiol. 180 (1), 367–380. doi:10.1104/pp.18.01505
Danilevskaya, O. N., Meng, X., Hou, Z., Ananiev, E. V., and Simmons, C. R. (2008). A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 146 (1), 250–264. doi:10.1104/pp.107.109538
De Smet, R., Sabaghian, E., Li, Z., Saeys, Y., and Van de Peer, Y. (2017). Coordinated functional divergence of genes after genome duplication in Arabidopsis thaliana. Plant Cell 29 (11), 2786–2800. doi:10.1105/tpc.17.00531
Dong, L., Lu, Y., and Liu, S. (2020). Genome-wide member identification, phylogeny and expression analysis of PEBP gene family in wheat and its progenitors. PeerJ 8, e10483. doi:10.7717/peerj.10483
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi:10.1093/nar/gkr367
Geourjon, C., and Deléage, G. (1995). SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11 (6), 681–684. doi:10.1093/bioinformatics/11.6.681
Goldberg, D. H., Victor, J. D., Gardner, E. P., and Gardner, D. (2009). Spike train analysis toolkit: enabling wider application of information-theoretic techniques to neurophysiology. Neuroinformatics 7 (3), 165–178. doi:10.1007/s12021-009-9049-y
Hu, G., Cheng, L., Cheng, Y., Mao, W., Qiao, Y., and Lan, Y. (2022). Pan-genome analysis of three main Chinese chestnut varieties. Front. Plant Sci. 13, 916550. doi:10.3389/fpls.2022.916550
Hu, M., Xie, M., Cui, X., Huang, J., Cheng, X., Liu, L., et al. (2023a). Characterization and potential function analysis of the SRS gene family in Brassica napus. Genes (Basel) 14 (7), 1421. doi:10.3390/genes14071421
Hu, X., Liang, J., Wang, W., Cai, C., Ye, S., Wang, N., et al. (2023b). Comprehensive genome-wide analysis of the DREB gene family in Moso bamboo (Phyllostachys edulis): evidence for the role of PeDREB28 in plant abiotic stress response. Plant J. 116 (5), 1248–1270. doi:10.1111/tpj.16420
Huang, R., Peng, F., Wang, D., Cao, F., Guo, C., Yu, L., et al. (2023). Transcriptome analysis of differential sugar accumulation in the developing embryo of contrasting two Castanea mollissima cultivars. Front. Plant Sci. 14, 1206585. doi:10.3389/fpls.2023.1206585
Huang, X., Liu, H., Wu, F., Wei, W., Zeng, Z., Xu, J., et al. (2024). Diversification of FT-like genes in the PEBP family contributes to the variation of flowering traits in Sapindaceae species. Mol. Hortic. 4 (1), 28. doi:10.1186/s43897-024-00104-4
Innan, H., and Kondrashov, F. (2010). The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11 (2), 97–108. doi:10.1038/nrg2689
Jin, H., Tang, X., Xing, M., Zhu, H., Sui, J., Cai, C., et al. (2019). Molecular and transcriptional characterization of phosphatidyl ethanolamine-binding proteins in wild peanuts Arachis duranensis and Arachis ipaensis. BMC Plant Biol. 19 (1), 484. doi:10.1186/s12870-019-2113-3
Jin, S., Nasim, Z., Susila, H., and Ahn, J. H. (2021). Evolution and functional diversification of FLOWERING LOCUS T/TERMINAL FLOWER 1 family genes in plants. Semin. Cell Dev. Biol. 109, 20–30. doi:10.1016/j.semcdb.2020.05.007
Karlgren, A., Gyllenstrand, N., Källman, T., Sundström, J. F., Moore, D., Lascoux, M., et al. (2011). Evolution of the PEBP gene family in plants: functional diversification in seed plant evolution. Plant Physiol. 156 (4), 1967–1977. doi:10.1104/pp.111.176206
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14 (4), R36. doi:10.1186/gb-2013-14-4-r36
Kohl, M., Wiese, S., and Warscheid, B. (2011). Cytoscape: software for visualization and analysis of biological networks. Methods Mol. Biol. 696, 291–303. doi:10.1007/978-1-60761-987-1_18
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi:10.1093/molbev/msw054
Li, C., Luo, L., Fu, Q., Niu, L., and Xu, Z.-F. (2015). Identification and characterization of the FT/TFL1 gene family in the biofuel plant jatropha curcas. Plant Mol. Biol. Report. 33 (2), 326–333. doi:10.1007/s11105-014-0747-8
Liu, B., Sun, Y., Xue, J., Jia, X., and Li, R. (2018). Genome-wide characterization and expression analysis of GRAS gene family in pepper (Capsicum annuum L.). PeerJ 6, e4796. doi:10.7717/peerj.4796
Liu, L., Xuan, L., Jiang, Y., and Yu, H. (2020). Regulation by FLOWERING LOCUS T and TERMINAL FLOWER 1 in flowering time and plant architecture. Small Struct. 2, 2000125. doi:10.1002/sstr.202000125
Liu, X., Zhang, J., Abuahmad, A., Franks, R. G., Xie, D.-Y., and Xiang, Q.-Y. (2016a). Analysis of two TFL1 homologs of dogwood species (Cornus L.) indicates functional conservation in control of transition to flowering. Planta 243 (5), 1129–1141. doi:10.1007/s00425-016-2466-x
Liu, Y. Y., Yang, K. Z., Wei, X. X., and Wang, X. Q. (2016b). Revisiting the phosphatidylethanolamine-binding protein (PEBP) gene family reveals cryptic FLOWERING LOCUS T gene homologs in gymnosperms and sheds new light on functional evolution. New Phytol. 212 (3), 730–744. doi:10.1111/nph.14066
Meng, X., Muszynski, M. G., and Danilevskaya, O. N. (2011). The FT-like ZCN8 gene functions as a floral activator and is involved in photoperiod sensitivity in maize. Plant Cell 23 (3), 942–960. doi:10.1105/tpc.110.081406
Mohamed, R., Wang, C. T., Ma, C., Shevchenko, O., Dye, S. J., Puzey, J. R., et al. (2010). Populus CEN/TFL1 regulates first onset of flowering, axillary meristem identity and dormancy release in Populus. Plant J. 62 (4), 674–688. doi:10.1111/j.1365-313X.2010.04185.x
Nan, H., Cao, D., Zhang, D., Li, Y., Lu, S., Tang, L., et al. (2014). GmFT2a and GmFT5a redundantly and differentially regulate flowering through interaction with and upregulation of the bZIP transcription factor GmFDL19 in soybean. PLoS One 9 (5), e97669. doi:10.1371/journal.pone.0097669
Nie, M., Li, L., He, C., Lu, J., Guo, H., Li, X. a., et al. (2024). Genome-wide identification, subcellular localization, and expression analysis of the phosphatidyl ethanolamine-binding protein family reveals the candidates involved in flowering and yield regulation of Tartary buckwheat (Fagopyrum tataricum). PeerJ 12, e17183. doi:10.7717/peerj.17183
Nie, X.-h., Wang, Z.-h., Liu, N.-w., Song, L., Yan, B.-q., Xing, Y., et al. (2021). Fingerprinting 146 Chinese chestnut (Castanea mollissima Blume) accessions and selecting a core collection using SSR markers. J. Integr. Agric. 20 (5), 1277–1286. doi:10.1016/S2095-3119(20)63400-1
Panchy, N., Lehti-Shiu, M., and Shiu, S.-H. (2016). Evolution of gene duplication in plants. Plant Physiol. 171 (4), 2294–2316. doi:10.1104/pp.16.00523
Pasquier, J., Braasch, I., Batzel, P., Cabau, C., Montfort, J., Nguyen, T., et al. (2017). Evolution of gene expression after whole-genome duplication: new insights from the spotted gar genome. J. Exp. Zool. B Mol. Dev. Evol. 328 (7), 709–721. doi:10.1002/jez.b.22770
Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., et al. (2019). Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 20 (1), 38. doi:10.1186/s13059-019-1650-2
Quan, S., Niu, J., Zhou, L., Xu, H., Ma, L., and Qin, Y. (2019). Genome-wide identification, classification, expression and duplication analysis of GRAS family genes in Juglans regia. L. Sci. Rep. 9 (1), 11643. doi:10.1038/s41598-019-48287-x
Song, S., Wang, G., Hu, Y., Liu, H., Bai, X., Qin, R., et al. (2018). OsMFT1 increases spikelets per panicle and delays heading date in rice by suppressing Ehd1, FZP and SEPALLATA-like genes. J. Exp. Bot. 69 (18), 4283–4293. doi:10.1093/jxb/ery232
Sun, W., Yin, Q., Wan, H., Gao, R., Xiong, C., Xie, C., et al. (2023a). Characterization of the horse chestnut genome reveals the evolution of aescin and aesculin biosynthesis. Nat. Commun. 14 (1), 6470. doi:10.1038/s41467-023-42253-y
Sun, Y., Jia, X., Chen, D., Fu, Q., Chen, J., Yang, W., et al. (2023b). Genome-wide identification and expression analysis of cysteine-rich polycomb-like protein (CPP) gene family in tomato. Int. J. Mol. Sci. 24 (6), 5762. doi:10.3390/ijms24065762
Sun, Y., Jia, X., Yang, Z., Fu, Q., Yang, H., and Xu, X. (2023c). Genome-wide identification of PEBP gene family in Solanum lycopersicum. Int. J. Mol. Sci. 24 (11), 9185. doi:10.3390/ijms24119185
Sun, Y., Lu, Z., Zhu, X., and Ma, H. (2020). Genomic basis of homoploid hybrid speciation within chestnut trees. Nat. Commun. 11 (1), 3375. doi:10.1038/s41467-020-17111-w
Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., and Kubista, M. (2015). How good is a PCR efficiency estimate: recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect Quantif. 3, 9–16. doi:10.1016/j.bdq.2015.01.005
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607–d613. doi:10.1093/nar/gky1131
Tamaki, S., Matsuo, S., Wong, H. L., Yokoi, S., and Shimamoto, K. (2007). Hd3a protein is a mobile flowering signal in rice. Science 316 (5827), 1033–1036. doi:10.1126/science.1141753
Tsaftaris, A., Pasentsis, K., and Argiriou, A. (2013). Cloning and characterization of FLOWERING LOCUS T-like genes from the perennial geophyte saffron crocus (Crocus sativus). Plant Mol. Biol. Report. 31 (6), 1558–1568. doi:10.1007/s11105-013-0608-x
Vaistij, F. E., Gan, Y., Penfield, S., Gilday, A. D., Dave, A., He, Z., et al. (2013). Differential control of seed primary dormancy in Arabidopsis ecotypes by the transcription factor SPATULA. Proc. Natl. Acad. Sci. U. S. A. 110 (26), 10866–10871. doi:10.1073/pnas.1301647110
Varkonyi-Gasic, E., Moss, S., Voogd, C., Wang, T., Putterill, J., and Hellens, R. (2013). Homologs of FT, CEN and FD respond to developmental and environmental signals affecting growth and flowering in the perennial vine kiwifruit. New Phytologist 198, 732–746. doi:10.1111/nph.12162
Voogd, C., Brian, L. A., Wang, T., Allan, A. C., and Varkonyi-Gasic, E. (2017). Three FT and multiple CEN and BFT genes regulate maturity, flowering, and vegetative phenology in kiwifruit. J. Exp. Bot. 68 (7), 1539–1553. doi:10.1093/jxb/erx044
Wang, J., Tian, S., Sun, X., Cheng, X., Duan, N., Tao, J., et al. (2020). Construction of pseudomolecules for the Chinese chestnut (Castanea mollissima) genome. G3 (Bethesda) 10 (10), 3565–3574. doi:10.1534/g3.120.401532
Wang, Y., Liu, C., Fang, Z., Wu, Q., Xu, Y., Gong, B., et al. (2022). A review of the stress resistance, molecular breeding, health benefits, potential food products, and ecological value of Castanea mollissima. Plants (Basel) 11 (16), 2111. doi:10.3390/plants11162111
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–w303. doi:10.1093/nar/gky427
Wittkopp, P. J., and Kalay, G. (2012). Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13 (1), 59–69. doi:10.1038/nrg3095
Wu, X., Gan, Z., Xu, F., Qian, J., Qian, M., Ai, H., et al. (2024). Molecular characterization of pepper PEBP genes reveals the diverse functions of CaFTs in flowering and plant architecture. Sci. Hortic. 335, 113345. doi:10.1016/j.scienta.2024.113345
Xi, W., Liu, C., Hou, X., and Yu, H. (2010). MOTHER OF FT AND TFL1 regulates seed germination through a negative feedback loop modulating ABA signaling in Arabidopsis. Plant Cell 22 (6), 1733–1748. doi:10.1105/tpc.109.073072
Xu, H., Guo, X., Hao, Y., Lu, G., Li, D., Lu, J., et al. (2022). Genome-wide characterization of PEBP gene family in Perilla frutescens and PfFT1 promotes flowering time in Arabidopsis thaliana. Front. Plant Sci. 13, 1026696. doi:10.3389/fpls.2022.1026696
Xu, W., Chen, Z., Ahmed, N., Han, B., Cui, Q., and Liu, A. (2016). Genome-wide identification, evolutionary analysis, and stress responses of the GRAS gene family in Castor beans. Int. J. Mol. Sci. 17 (7), 1004. doi:10.3390/ijms17071004
Yang, J. (2008). Comprehensive description of protein structures using protein folding shape code. Proteins 71 (3), 1497–1518. doi:10.1002/prot.21932
Yang, J., Ning, C., Liu, Z., Zheng, C., Mao, Y., Wu, Q., et al. (2023). Genome-wide characterization of PEBP gene family and functional analysis of TERMINAL FLOWER 1 homologs in macadamia integrifolia. Plants (Basel) 12 (14), 2692. doi:10.3390/plants12142692
Yoo, S. Y., Kardailsky, I., Lee, J. S., Weigel, D., and Ahn, J. H. (2004). Acceleration of flowering by overexpression of MFT (MOTHER OF FT AND TFL1). Mol. Cells 17 (1), 95–101. doi:10.1016/s1016-8478(23)13012-3
Yu, L., Diao, S., Zhang, G., Yu, J., Zhang, T., Luo, H., et al. (2022a). Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides. Plant Biotechnol. J. 20 (7), 1257–1273. doi:10.1111/pbi.13802
Yu, L., Fei, C., Wang, D., Huang, R., Xuan, W., Guo, C., et al. (2023a). Genome-wide identification, evolution and expression profiles analysis of bHLH gene family in Castanea mollissima. Front. Genet. 14, 1193953. doi:10.3389/fgene.2023.1193953
Yu, L., Fei, C., Wang, D., Huang, R., Xuan, W., Guo, C., et al. (2023b). Genome-wide identification, evolution and expression profiles analysis of bHLH gene family in Castanea mollissima. Front. Genet. 14, 1193953. doi:10.3389/fgene.2023.1193953
Yu, L., Hui, C., Huang, R., Wang, D., Fei, C., Guo, C., et al. (2022b). Genome-wide identification, evolution and transcriptome analysis of GRAS gene family in Chinese chestnut (Castanea mollissima). Front. Genet. 13, 1080759. doi:10.3389/fgene.2022.1080759
Yu, L., Tian, Y., Wang, X., Cao, F., Wang, H., Huang, R., et al. (2025). Genome-wide identification, phylogeny, evolutionary expansion, and expression analyses of ABC gene family in Castanea mollissima under temperature stress. Plant Physiology Biochem. 219, 109450. doi:10.1016/j.plaphy.2024.109450
Yu, L., Zhang, G., Lyu, Z., He, C., and Zhang, J. (2021). Genome-wide analysis of the GRAS gene family exhibited expansion model and functional differentiation in sea buckthorn (Hippophae rhamnoides L.). Plant Biotechnol. Rep. 15 (4), 513–525. doi:10.1007/s11816-021-00694-1
Yuan, X., Quan, S., Liu, J., Guo, C., Zhang, Z., Kang, C., et al. (2022). Evolution of the PEBP gene family in Juglandaceae and their regulation of flowering pathway under the synergistic effect of JrCO and JrNF-Y proteins. Int. J. Biol. Macromol. 223 (Pt A), 202–212. doi:10.1016/j.ijbiomac.2022.11.004
Zhang, L., Gao, H. Y., Baba, M., Okada, Y., Okuyama, T., Wu, L. J., et al. (2014). Extracts and compounds with anti-diabetic complications and anti-cancer activity from Castanea mollissina Blume (Chinese chestnut). BMC Complement. Altern. Med. 14, 422. doi:10.1186/1472-6882-14-422
Zhang, P., Liu, J., Jia, N., Wang, M., Lu, Y., Wang, D., et al. (2023). Genome-wide identification and characterization of the bZIP gene family and their function in starch accumulation in Chinese chestnut (Castanea mollissima Blume). Front. Plant Sci. 14, 1166717. doi:10.3389/fpls.2023.1166717
Zhang, S., Wang, L., Fu, Y., and Jiang, J. C. (2022). Bioactive constituents, nutritional benefits and woody food applications of Castanea mollissima: a comprehensive review. Food Chem. 393, 133380. doi:10.1016/j.foodchem.2022.133380
Zhang, X., Wang, C., Pang, C., Wei, H., Wang, H., Song, M., et al. (2016). Characterization and functional analysis of PEBP family genes in upland cotton (Gossypium hirsutum L.). PLoS One 11 (8), e0161080. doi:10.1371/journal.pone.0161080
Zhang, Y., Li, H., Shang, S., Meng, S., Lin, T., Zhang, Y., et al. (2021). Evaluation validation of a qPCR curve analysis method and conventional approaches. BMC Genomics 22 (5), 680. doi:10.1186/s12864-021-07986-4
Zhao, S., Wei, Y., Pang, H., Xu, J., Li, Y., Zhang, H., et al. (2020). Genome-wide identification of the PEBP genes in pears and the putative role of PbFT in flower bud differentiation. PeerJ 8, e8928. doi:10.7717/peerj.8928
Zheng, X.-M., Fqing, w., Zhang, X., Lin, Q., Wang, J., Guo, X.-P., et al. (2016). Evolution of PEBP family and selective signature on FT -like clade. J. Syst. Evol. 54, n/a. doi:10.1111/jse.12199
Keywords: PEBP gene family, Castanea mollissima, phylogeny, expression analysis, RT-qPCR
Citation: Tian Y, Wang J, Wang X, Wang D, Wang X, Liu J, Zhang H, Zhang J and Yu L (2025) Genome-wide identification, phylogeny, and expression analysis of PEBP gene family in Castanea mollissima. Front. Genet. 16:1530910. doi: 10.3389/fgene.2025.1530910
Received: 19 November 2024; Accepted: 06 March 2025;
Published: 26 March 2025.
Edited by:
Ertugrul Filiz, Duzce University, TürkiyeReviewed by:
Diaga Diouf, Cheikh Anta Diop University, SenegalCopyright © 2025 Tian, Wang, Wang, Wang, Wang, Liu, Zhang, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyang Yu, eXVsaXlhbmd5b3V4aWFuZ0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.