 Yao Wang
Yao Wang Jing Guo1
Jing Guo1 Hua Li
Hua Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 28 February 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1528563
Objectives: To describe a case of Progressive Encephalopathy with or without Lipodystrophy (PELD), characterized by a late onset of neurological regression at 9 years old, due to a homozygous c.974dupG variant in the BSCL2 gene.
Methods: An 11-year, 9-month-old girl with repeated seizures over 2 years underwent clinical assessment and genetic investigation. We also reviewed the published literature.
Results: The patient exhibited mild intellectual disability, a lipodystrophic appearance, precocious puberty, voracious appetite, elevated transaminase levels, hyperlipidemia, hypercortisolism, hepatomegaly, fatty liver, and splenomegaly. Motor and cognitive regression occurred at 9 years. A homozygous pathogenic variant c.974dup (p.Ile326HisfsTer12) in exon 7 of BSCL2 (NM_001122955.4) was identified. Despite multiple antiseizure medications, seizures were refractory, leading to status epilepticus and rapid death after genetic diagnosis.
Conclusion: We confirm that the BSCL2 c.974dupG variant is a cause of PELD. Regression may occur later than previously reported. Literature review suggests that the c.974dupG variant may present a milder phenotype compared to the classic c.985C>T variant. Early genetic testing and diagnosis are crucial for improving outcomes in rare neurodegenerative disorders like PELD.
The BSCL2 gene is located on the long arm of chromosome 11 at position 11q13. It encodes the protein seipin, an integral membrane protein of the endoplasmic reticulum (ER). The gene is highly expressed in the brain (Magré et al., 2001), encoding three main seipin isoforms: 462 (BSCL2-203), 398 (BSCL2-205/207/210) and 287 (BSCL2-201) amino acids long, respectively (Guillén-Navarro et al., 2013). Biallelic mutation in the BSCL2 gene can cause progressive encephalopathy with or without lipodystrophy (PELD; MIM: #615924) and congenital generalized lipodystrophy type 2 (CGL2; MIM: #269700). Heterozygous mutations in the BSCL2 gene can cause distal hereditary motor neuronopathy type VC(HMND13; MIM: #619112) and Silver syndrome (SPG17; MIM: #270685). PELD, also known as Celia’s encephalopathy, is a rare and severe neurodegenerative disorder. It is characterized by developmental regression of motor and cognitive skills before the age of 5, often leading to death within the first decade. Patients may exhibit a mild or typical lipodystrophic appearance (Guillén-Navarro et al., 2013). The variant c.985C>T is considered the classic genotype (Guillén-Navarro et al., 2013). However, other variants in BSCL2 associated with severe neurodegenerative manifestations have been reported, some of which resemble PELD. We report a patient with PELD due to a homozygous c. 974dupG variant in the BSCL2 gene, who experienced motor and cognitive regression at 9 years old. We also review the features of all published PELD patients.
The study was approved by the ethics review committee of Guangdong Sanjiu Brain Hospital and conducted according to the Helsinki Declaration’s ethical guidelines. Consent for disclosure was obtained from the patient’s parents. We also reviewed published literature.
An 11-year, 9-month-old girl was hospitalized in June 2024, due to repeated seizures over 2 years. She was born to non-consanguineous parents in Guangdong Province, China. The pregnancy and delivery were uneventful. Mild intellectual disability was noted at 30 months. In infancy, she exhibited a lipodystrophic appearance and acanthosis nigricans. She experienced precocious puberty, voracious appetite, elevated transaminase levels, hyperlipidemia, hypercortisolism, hepatomegaly, fatty liver, and splenomegaly. At 9 years old, the patient experienced epileptic seizures characterized by generalized tonic-clonic seizures (GTCS) and myoclonus. The seizures were refractory despite treatment with levetiracetam and topiramate. She experienced a status epilepticus (SE) episode lasting approximately 10 h. At that time, the patient demonstrated psychomotor regression. She exhibited motor deterioration, ataxia, action myoclonus, tremors, reduced speech, dysarthria, and sleep disturbances. By 11 years and 9 months, the patient could only speak single words and had no independent gait, requiring assistance for eating, bathing, and toileting. Physical examination revealed marked generalized lipoatrophy, hypotonia, ataxia, and action myoclonus. Video electroencephalography (VEEG) indicated a slow background and diffuse epileptiform discharges. Frequent myoclonus was recorded (Figure 1). Brain MRI indicated diffuse brain atrophy, particularly in the caudate nucleus (Figure 2). Gynecological ultrasound indicated a small uterine volume. Blood glucose, lipids, lactic acid, urine organic acids, electrocardiogram, echocardiogram, and abdominal ultrasound were unremarkable. After obtaining informed consent from the parents, whole exome sequencing was conducted. Genetic analysis revealed a homozygous pathogenic frameshift variant in BSCL2 (NM_001122955.4:c.974dup; p.Ile326HisfsTer12), classified as pathogenic according to ACMG guidelines, with both parents identified as heterozygous carriers. Clonazepam and perampanel were successively prescribed, providing transient seizure control. Seizure occurrences gradually increased and progressed to SE on July 23rd. She died at the age of 11 years and 11 months from SE on July 25th.

Figure 1. VEEG findings of the PELD patient: (A) Slow background; (B) Diffuse epileptiform discharges (indicated by the arrow); (C) Rhythmic myoclonus; (D) Irregular myoclonus (action myoclonus).
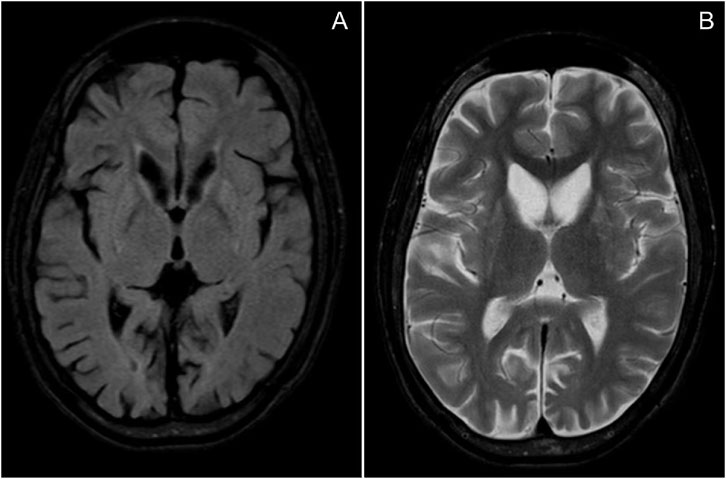
Figure 2. MRI of the patient: (A) T2 FLAIR; (B) T2-weighted imaging (T2WI), indicating diffuse brain atrophy, particularly in the caudate nucleus.
Clinical, imaging and genetic findings of our patient and the other 23 reported PELD patients are summarized in Tables 1, 2 (Guillén-Navarro et al., 2013; Alaei et al., 2016; Zhang et al., 2019; Wu et al., 2009; Sánchez-Iglesias et al., 2019; Fernández-Marmiesse et al., 2019; Opri et al., 2016; Ferranti et al., 2020; Serino et al., 2019; Pedicelli et al., 2020; Araújo-Vilar et al., 2018; Poisson et al., 2019; Stanley et al., 2022; Ding et al., 2021).
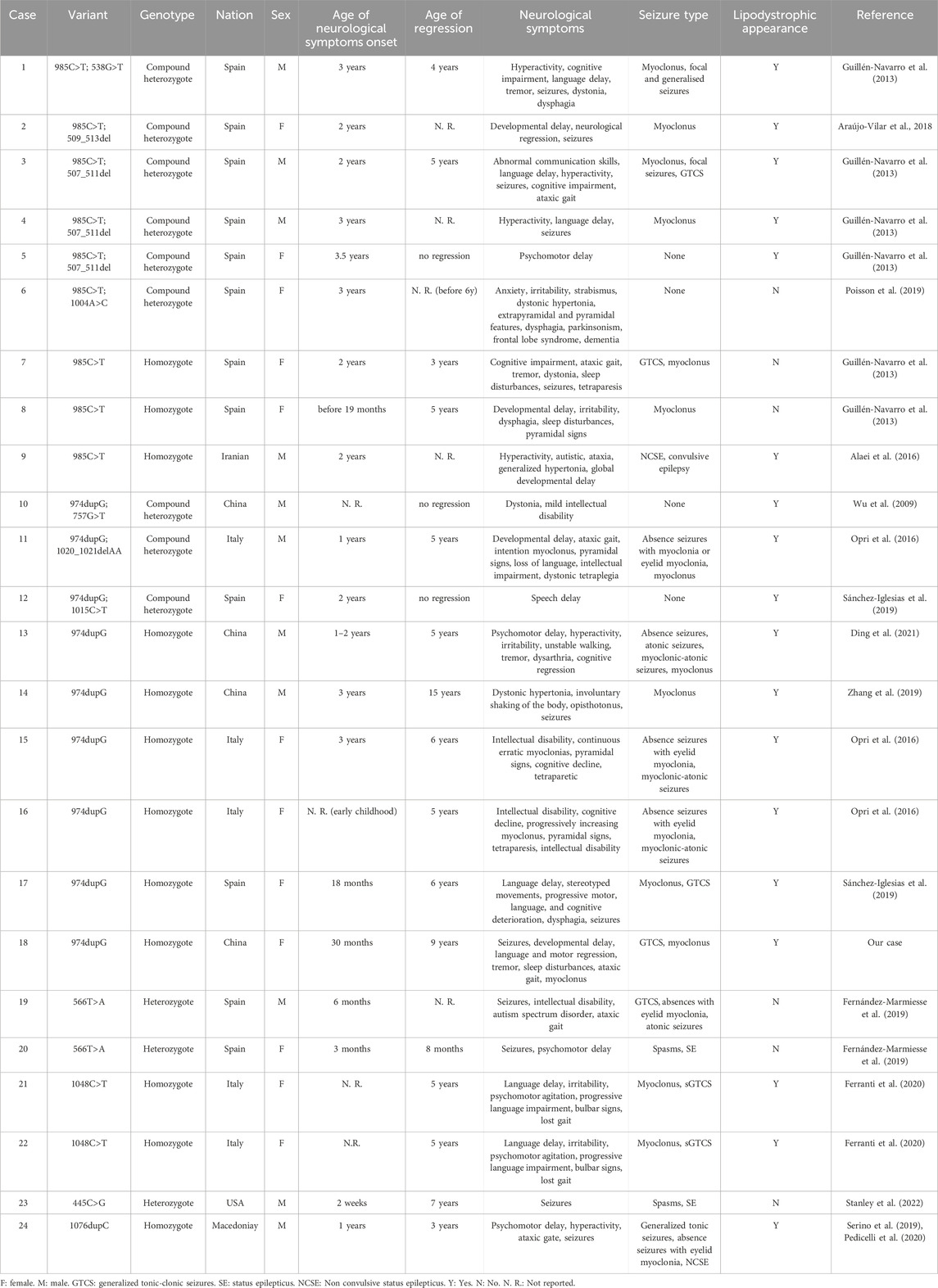
Table 1. Clinical features of PELD patients (part 1).
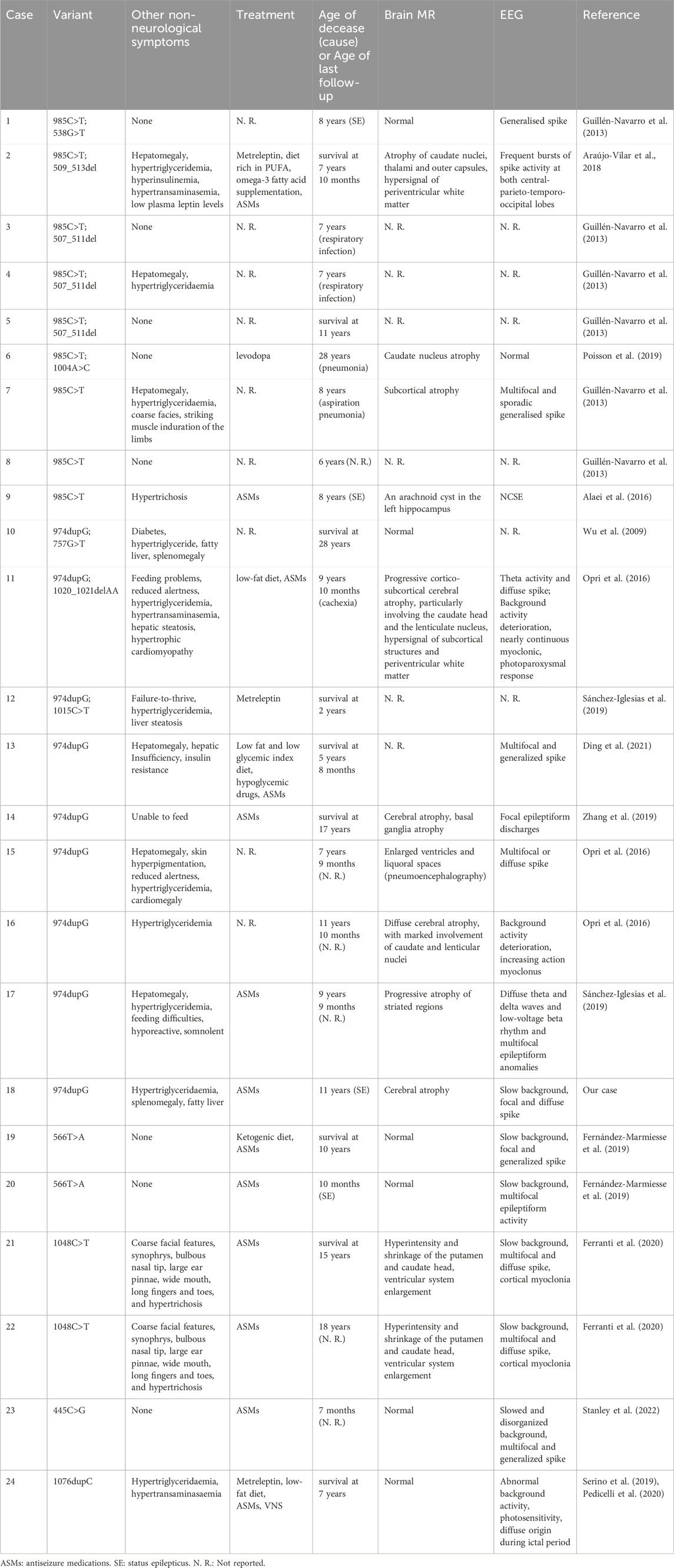
Table 2. Clinical features of PELD patients (part 2).
The patient cohort consisted of 11 males and 13 females, with cases distributed as follows: 12 from Spain, 5 from Italy, 4 from China, 1 from the United States, 1 from Iran, and 1 from Macedonia. 79.2% (19/24) of patients had neurological symptoms before the age of 3. (87.5%) 21 of cases experienced regression, with 14 regressing before the age of 6. 75% (18/24) of cases exhibited a lipodystrophic appearance. The cohort included homozygous (12/24), compound heterozygous (9/24), and heterozygous cases (3/24), with the c.985C>T and c.974dupG variants being the most common mutations. 15 patients died prematurely, with 11 dying before the age of 10. Nine patients were alive, with the oldest reported being 28 years old.
This study reports a girl with PELD caused by a c.974dupG biallelic mutation in the BSCL2 gene. A literature review revealed no gender differences among individuals with PELD. These cases were commonly reported in European and Asian countries. Most patients exhibited neurological symptoms before the age of 3 and experienced regression before the age of 6. Developmental delay and neurological regression were the most common neurological symptoms. The prognosis was poor, with most patients dying prematurely. Typical imaging showed brain atrophy, primarily in the caudate nucleus, and EEG findings indicated a slow background with multifocal or generalized epileptiform discharges.
Our case presents unique features including late-onset regression at 9 years old, rapid progression to death within 2 years after regression, and the presence of both lipodystrophic appearance and PME symptoms, which distinguishes it from previously reported cases. Among the documented cases, a notable c.974dupG mutation carrier manifested neurological regression at 15 years of age, representing the latest reported onset in the literature (Zhang et al., 2019). Remarkably, the longest documented survival was observed in a 28-year-old patient harboring the same mutation (Wu et al., 2009). These clinical observations collectively suggest that individuals with c.974dupG mutation exhibit delayed disease progression and extended survival compared to c.985C>T mutation carriers, potentially reflecting the differential impact of these variants on BSCL2 function.
The pathogenesis of Celia’s encephalopathy involves abnormal splicing of BSCL2 gene, leading to exon 7 skipping. This results in increased expression of BSCL2-201 transcript, which generates truncated seipin protein. The accumulation of aberrant seipin induces endoplasmic reticulum (ER) stress and the formation of intranuclear aggregates, ultimately triggering neuronal apoptosis (Sánchez-Iglesias et al., 2021). Notably, patients with c.974dupG mutation exhibit lower expression of BSCL2-201 transcript compared to c.985C>T carriers (Sánchez-Iglesias et al., 2019), correlating with relatively milder neurological manifestations. In this case, the progression from regression to death was rapid, with the cause of death being SE. SE was common in PELD and always difficult to treat, having been the cause of death in at least four reported cases (Guillén-Navarro et al., 2013; Alaei et al., 2016; Fernández-Marmiesse et al., 2019).
Epilepsy was common in PELD, affecting 20 out of 24 patients. Among these, myoclonic seizures were the most common type. The high expression of the BSCL2 gene in the brain and the widespread deposition of aberrant protein may contribute to this phenomenon. The presence of myoclonic seizures, ataxia, and progressive neurological deterioration in this patient supports the diagnosis of Progressive Myoclonus Epilepsy (PME). This finding strengthens the evidence for the coexistence of PME and BSCL2 mutations, as previously suggested in the literature (Zhang et al., 2019; Opri et al., 2016; Ferranti et al., 2020; Serino et al., 2019). In the literature review, we found that PME is underdiagnosed in PELD. Therefore, we propose that PELD should be considered as one of the PMEs. The BSCL2 gene should be considered in gene panels for PME diagnostics.
All patients with 974dupG, 1048C>T, and 1076dupC variants displayed a lipodystrophic appearance. In contrast, those with the 445C>G and 566T>A heterozygous variants did not, while the phenotype for patients with the 985C>T variant remains uncertain. This suggests a potential link between genotype and lipodystrophic appearance. Given the small number of cases, this link still needs further investigation. We found that both compound heterozygotes and homozygotes may or may not display a lipodystrophic appearance, indicating that these features are independent of zygosity type, which is inconsistent with previous studies (Guillén-Navarro et al., 2013).
It has been reported that leptin-replacement therapy delayed neurological regression or allowed better seizure control in patients with PELD (Sánchez-Iglesias et al., 2019; Pedicelli et al., 2020; Araújo-Vilar et al., 2018). Unfortunately, the patient died 23 days after genetic cause confirmation, highlighting the importance of early diagnosis. Early genetic testing and diagnosis are crucial for improving outcomes in rare neurodegenerative disorders like PELD.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving humans were approved by the ethics review committee of Guangdong Sanjiu Brain Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YW: Conceptualization, Data curation, Investigation, Methodology, Software, Writing–original draft, Writing–review and editing. JG: Supervision, Writing–review and editing. PZ: Data curation, Writing–review and editing. FL: Investigation, Writing–review and editing. HL: Conceptualization, Supervision, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We would like to thank the patient for permission of using her clinical data shown in this report.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alaei, M. R., Talebi, S., Ghofrani, M., Taghizadeh, M., and Keramatipour, M. (2016). Whole exome sequencing reveals a BSCL2 mutation causing progressive encephalopathy with lipodystrophy (PELD) in an Iranian pediatric patient. Iran. Biomed. J. 20, 295–301. doi:10.22045/ibj.2016.07
Araújo-Vilar, D., Domingo-Jiménez, R., Ruibal, Á., Aguiar, P., Ibáñez-Micó, S., Garrido-Pumar, M., et al. (2018). Association of metreleptin treatment and dietary intervention with neurological outcomes in Celia's encephalopathy. Eur. J. Hum. Genet. 26, 396–406. doi:10.1038/s41431-017-0052-8
Ding, J., Ma, M., and Qiu, Z. (2021). A congenital generalized lipodystrophy patient complicated with critical central neurological involvement due to BSCL2 mutations. Basic Clin. Med. 41, 1333–1337. doi:10.16352/j.issn.1001-6325.2021.09.009
Fernández-Marmiesse, A., Sánchez-Iglesias, S., Darling, A., O'Callaghan, M. M., Tonda, R., Jou, C., et al. (2019). A de novo heterozygous missense BSCL2 variant in 2 siblings with intractable developmental and epileptic encephalopathy. Seizure 71, 161–165. doi:10.1016/j.seizure.2019.07.019
Ferranti, S., Lo Rizzo, C., Renieri, A., Galluzzi, P., and Grosso, S. (2020). Focus on progressive myoclonic epilepsy in Berardinelli-Seip syndrome. Neurol. Sci. 41, 3345–3348. doi:10.1007/s10072-020-04418-1
Guillén-Navarro, E., Sánchez-Iglesias, S., Domingo-Jiménez, R., Victoria, B., Ruiz-Riquelme, A., Rábano, A., et al. (2013). A new seipin-associated neurodegenerative syndrome. J. Med. Genet. 50, 401–409. doi:10.1136/jmedgenet-2013-101525
Magré, J., Delépine, M., Khallouf, E., Gedde-Dahl, T., Van Maldergem, L., Sobel, E., et al. (2001). Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 28, 365–370. doi:10.1038/ng585
Opri, R., Fabrizi, G. M., Cantalupo, G., Ferrarini, M., Simonati, A., Dalla Bernardina, B., et al. (2016). Progressive myoclonus epilepsy in congenital generalized lipodystrophy type 2: report of 3 cases and literature review. Seizure 42, 1–6. doi:10.1016/j.seizure.2016.08.008
Pedicelli, S., de Palma, L., Pelosini, C., and Cappa, M. (2020). Metreleptin for the treatment of progressive encephalopathy with/without lipodystrophy (PELD) in a child with progressive myoclonic epilepsy: a case report. Ital. J. Pediatr. 46, 158. doi:10.1186/s13052-020-00916-2
Poisson, A., Chatron, N., Labalme, A., Till, M., Broussolle, E., Sanlaville, D., et al. (2019). Regressive autism spectrum disorder expands the phenotype of BSCL2/seipin-associated neurodegeneration. Biol. Psychiatry 85, e17–e19. doi:10.1016/j.biopsych.2018.05.010
Sánchez-Iglesias, S., Crocker, M., O'Callaghan, M., Darling, A., García-Cazorla, A., Domingo-Jiménez, R., et al. (2019). Celia’s encephalopathy and c.974dupG in BSCL2 gene: a hidden change in a known variant. Neurogenetics 20, 73–82. doi:10.1007/s10048-019-00574-5
Sánchez-Iglesias, S., Fernández-Pombo, A., Cobelo-Gómez, S., Hermida-Ameijeiras, Á., Alarcón-Martínez, H., Domingo-Jiménez, R., et al. (2021). Celia's encephalopathy (BSCL2-Gene-Related): current understanding. J. Clin. Med. 10, 1435. doi:10.3390/jcm10071435
Serino, D., Davico, C., Specchio, N., Marras, C. E., and Fioretto, F. (2019). Berardinelli-Seip syndrome and progressive myoclonus epilepsy. Epileptic Disord. 21, 117–121. doi:10.1684/epd.2019.1038
Stanley, N. E., Robinson, L. J., and Mao, Q. (2022). A novel, heterozygous BSCL2 variant in association with early-onset epileptic encephalopathy. J. Neuropathol. Exp. Neurol. 81, 377–380. doi:10.1093/jnen/nlac013
Wu, Y. R., Hung, S. I., Chang, Y. C., Chen, S. T., Lin, Y. L., and Chung, W. H. (2009). Complementary mutations in seipin gene in a patient with Berardinelli-Seip congenital lipodystrophy and dystonia: phenotype variability suggests multiple roles of seipin gene. J. Neurol. Neurosurg. Psychiatry 80, 1180–1181. doi:10.1136/jnnp.2008.165977
Keywords: BSCL2 gene, progressive encephalopathy with or without lipodystrophy, celia’s encephalopathy, progressive myoclonus epilepsy, neurological regression
Citation: Wang Y, Guo J, Zhang PQ, Liu F and Li H (2025) Case Report: A case of progressive encephalopathy with or without lipodystrophy caused by BSCL2 variant and literature review. Front. Genet. 16:1528563. doi: 10.3389/fgene.2025.1528563
Received: 15 November 2024; Accepted: 10 February 2025;
Published: 28 February 2025.
Edited by:
David Araujo-Vilar, University of Santiago de Compostela, SpainReviewed by:
Lourdes Loidi, Galician Public Foundation of Genomic Medicine, SpainCopyright © 2025 Wang, Guo, Zhang, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Li, bGlodWExMDUxQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.