
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 07 March 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1523228
 Wen Li1*†
Wen Li1*† Mengjie Lin1†Jinwei Dao2Li Shi3Wei Yi4Jia Lei5Yaxian Song3Jiaolou Dong6Meiwei Zhao7
Mengjie Lin1†Jinwei Dao2Li Shi3Wei Yi4Jia Lei5Yaxian Song3Jiaolou Dong6Meiwei Zhao7 Yushan Xu3*
Yushan Xu3* Lulu Chen8*
Lulu Chen8*Background: Kabuki syndrome (KS) is a rare autosomal dominant genetic disorder. The full understanding of KS remains elusive due to the heterogeneity of gene mutations, clinical phenotypes, and the associations and mechanisms linking genotypes to phenotypes. This study reports on a 16-year-old male patient diagnosed with type I Kabuki syndrome following the identification of a de novo mutation, c.15535C>T (p.Arg5179Cys), in the KMT2D gene.
Case Report: A 16-year-old male presented with bilateral breast enlargement persisting for over 1 month. Historically, the patient exhibited intellectual disability. Both parents are healthy with no similar family history. The patient’s father had a history of heroin use for 8 years prior to the patient’s birth. On examination, the patient had unclear speech and slow speech rate, with diminished reading comprehension and calculation abilities. Characteristic facial features of KS were noted. Breast development was observed (Tanner stage II on the right and III on the left), with pain upon deep palpation of the left nipple. Molecular genetic testing identified a heterozygous missense mutation, c.15535C>T (p.Arg5179Cys), in theKMT2Dgene, confirming the diagnosis of type I Kabuki syndrome.
Discussion: KS is characterized by distinctive facial features: arched eyebrows, eversion of the eyelids, long palpebral fissures, a short nasal septum, a flat nasal tip, auricular deformities, a small mandible, a high palatal arch, or cleft palate. The patient exhibited a heterozygous missense mutation in the coding region of the KMT2D gene, identified as a de novo mutation. Currently, KS management primarily involves symptomatic and rehabilitative therapies.
Kabuki syndrome (KS) has an incidence of approximately 1:30,000 (Del Cerro et al., 2020) and was first described by Niikawa et al. (1981) and Kuroki et al. (1981) in 1981. Patients with KS often present with distinctive facial features resembling those of Kabuki actors, . KS is a rare autosomal dominant genetic disorder caused by mutations in the lysine (K)-specific methyltransferase 2D (KMT2D) gene or the X-linked lysine-specific demethylase 6A (UTX/KDM6A) gene. The first case of KS in China was reported in 1998 (Jiao et al., 1998). The typical clinical manifestations of KS include distinctive facial features: arched eyebrows, eyelid eversion, long palpebral fissures, short nasal septum, flat nasal tip, auricular deformities, small mandible, high palatal arch, or cleft palate. These features are crucial for distinguishing KS from other multiple malformation syndromes (Xu, 2007). Other common manifestations include short stature, brachydactyly, joint laxity, hypotonia, skeletal anomalies, intellectual disability, speech impairments, hearing loss, inner ear malformations, congenital heart defects, urogenital malformations, dental anomalies, epilepsy (Li, 2018), and dermatoglyphic anomalies, with persistent fetal fingertip pads being characteristic symptoms of KS. Endocrine abnormalities such as precocious puberty and isolated premature thelarche, athelia, polythelia, hypoglycemia, hormonal imbalances are more common in patients with KMT2D mutations. The etiology of these features has not been definitively established, but Pescowitz et al. suggest it may be due to premature activation of hypothalamic gonadotropin-releasing hormone (LHRH) neurons. Male breast development has not been previously reported. This study reports on a 16-year-old male patient found to have a mutation in the KMT2D gene, c.15535C>T (p.Arg5179Cys), leading to a diagnosis of type I Kabuki syndrome.
The patient, a 16-year-old male, was delivered at home with no history of asphyxia or birth trauma. Birth weight was 2.8 kg. The mother (G2P2) underwent regular prenatal check-ups, remained healthy during pregnancy, and was not of advanced maternal age (25 years), with no significant medication history. Both the father and the patient’s older sister are in good health. The child was breastfed for the first year of life, with timely introduction of complementary foods. The child could stand independently at 1 year but did not begin walking independently until the age of two and a half, with an unsteady gait. At 3 years old, the child could only speak simple repeated words like “Papa” and “Mama,” unable to form complete sentences or engage in communication effectively. He often faced bullying during play and preferred solitary play. At the age of three, parents noticed delays in speech, walking, and height development compared to peers and sought medical attention, resulting in a diagnosis of developmental delay. A laryngeal examination was recommended but not performed. There is no history of epilepsy, hypoglycemia, constipation, celiac disease, malnutrition, or heart defects. The child has not taken any medication for chronic diseases and has been vaccinated as per the schedule. The child began receiving compulsory education on time but transferred to a special school in second grade due to poor academic performance. Height was below average for their age group. Height growth accelerated at the age of 15, increasing by 6 cm last year. Secondary sexual characteristics (voice change, ejaculation, or clear signs of erection) were not observed. Appetite remains good, with no nausea, vomiting, or diarrhea. Weight increased by 3 kg over the past year. He was admitted after noticing a bilateral breast hypertrophy for over a month. The enlargement was more pronounced on the left side and was painful upon palpation. No nipple discharge was observed. The patient had not used anti-androgens, estrogens, diuretics, or glucocorticoids. Initial evaluation by the general surgery department at our hospital, followed by a color ultrasound of the breasts, indicated male breast development. The parents are nonconsanguineous, and there is no similar family history. The patient’s father had an 8-year history of heroin use prior to the patient’s birth. Physical examination revealed unclear and slow speech, moderate intellectual disability (Using the Wechsler Intelligence Scale for Children, the child scored an IQ of 45),and reduced reading comprehension and calculation abilities. Distinctive facial and physical features (see Figure 1) included a flat expression, small mandible, arched eyebrows with sparse outer thirds, high palatal arch, ptosis of the upper eyelids, long palpebral fissures with mild eversion of the lower eyelids, a short nose with a flat tip and flared nostrils, a thin upper lip and thick, indented lower lip. Fetal fingertip pads were visible on all digits; the palmar lines were numerous, fine, and had many folds and transverse palmar creases. The fifth digits of both hands were short and bent outward. Bilateral breast development was noted (Tanner stage II on the right and III on the left), with pain on deep palpation of the left nipple. There was no axillary hair growth, and the posterior hairline was low. Pubic hair was sparse, curly, and limited to the base of the penis; the penis measured 2.2 cm, with right testicular volume of 3 mL and left of 4 mL. The patient’s developmental timeline in Table 1.
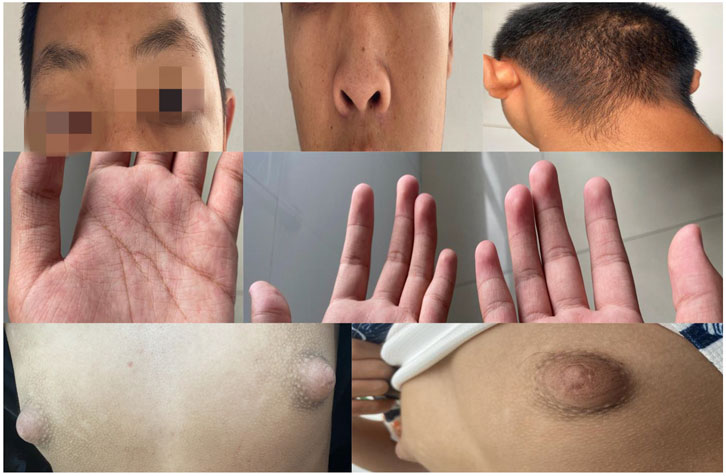
Figure 1. Distinctive Facial and Physical Features: Arched eyebrows with sparse outer thirds; long palpebral fissures with mild eversion of the lower eyelids; a short nose with a flat nasal tip; low posterior hairline. Dermatoglyphic Anomalies: Increased number of palmar lines, fine and multiple creases; visible fetal fingertip pads on all digits; breast hypertrophy.
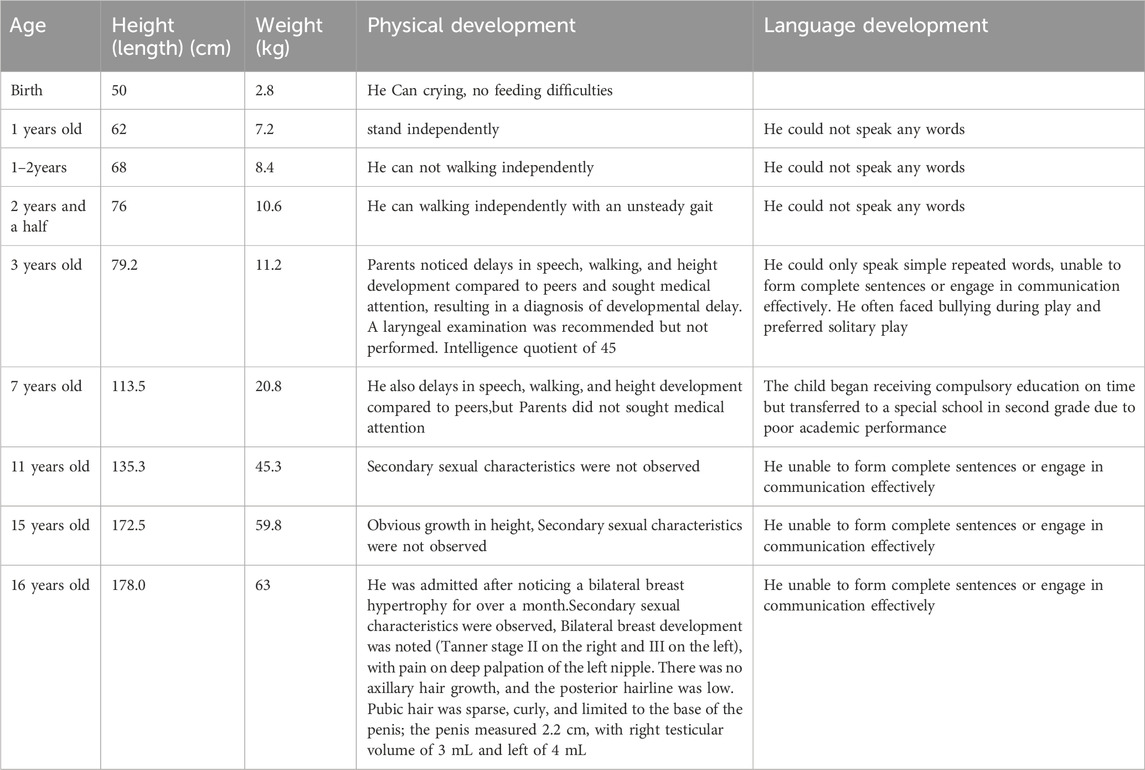
Table 1. The patient’s developmental timeline.
Breast ultrasound revealed male breast development (right 4.5 cm × 1.1 cm, left 5.3 cm × 1.6 cm). Electrocardiogram, thyroid ultrasound, cardiac echocardiogram, urinary system ultrasound, MRI of the brain and pituitary, X-rays of the pelvis and spine showed no abnormalities; Bone age assessment corresponded to approximately 17 years in a male. Bone density indicated reduced bone mass; chromosomal karyotype analysis revealed a 46,XY karyotype. Blood test results are presented in Table 2.
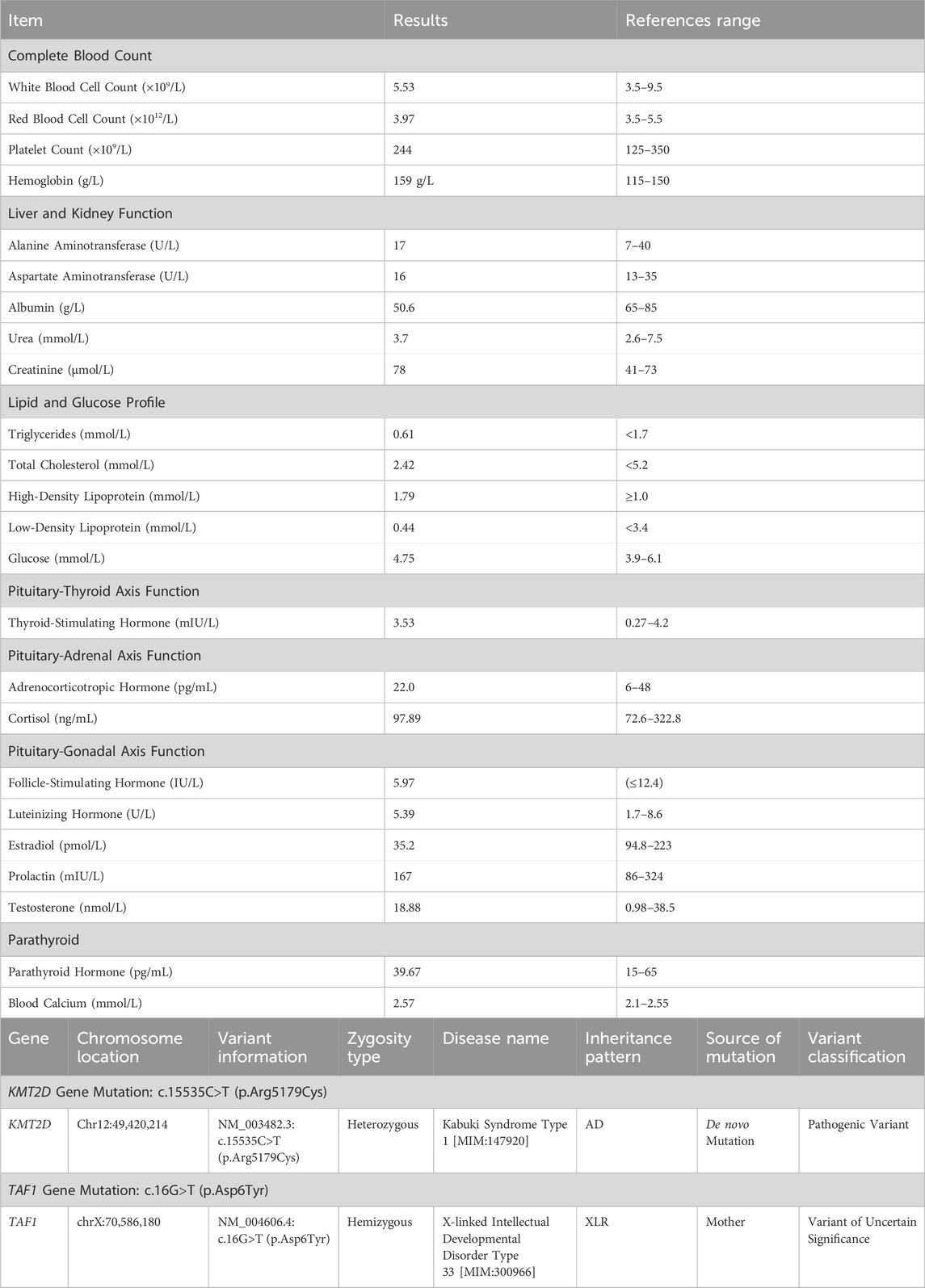
Table 2. Laboratory test results.
Genetic testing was performed using next-generation sequencing data for single nucleotide variations, small segment insertions and deletions, and large segment copy number variations. Real-time quantitative PCR and Western blot analyses were employed to evaluate the transcriptional and protein expression levels of the KMT2D gene (also called MLL2 gene, OMMI:147920) respectively.
A pathogenic variant that explains the patient’s phenotype and another variant correlating with the patient’s main clinical features were detected, though the latter’s clinical significance remains uncertain. The results indicated the presence of a heterozygous missense mutation in the coding region of the KMT2D gene: c.15535C>T (p.Arg5179Cys).
Additionally, genetic testing identified a hemizygous missense mutation in the TAF1gene: c.16G>T (p.Asp6Tyr). This variant can cause X-linked syndromic intellectual developmental disorder 33 (MRXS33) [MIM:300966], inherited in an X-linked manner. The c.16G>T (p.Asp6Tyr) variant, inherited from the patient’s mother, represents a missense mutation in the TAF1 gene coding region. This variant has not been reported in large-scale population frequency databases or literature. Based on available evidence, this variant is classified as a variant of uncertain clinical significance (see Figure 2). No clinical manifestations related to TAF1 gene mutations were observed in the patient.

Figure 2. Electropherogram of KMT2D Sequencing/Electropherogram of TAF1 Sequencing. Comparison of the 15535th base with the patient’s parents reveals that the cytosine (C) in the patient has mutated to thymine (T).
To validate the expression levels of the KMT2D gene at both transcriptional and protein levels in the proband, skin-derived fibroblasts were isolated and cultured in primary cell culture, with normal fibroblasts serving as controls. Real-time quantitative PCR results indicated a higher expression trend of the KMT2D gene in the proband’s cells compared to controls. Western blot analysis (SDS-PAGE) also showed increased protein expression levels of KMT2D in the proband’s cells compared to controls, consistent with the transcriptional level results. These findings suggest that the KMT2D gene harboring the c.15535C>T (p.Arg5179Cys) mutation exhibits upregulated expression at both transcriptional and protein levels (see Figure 3).
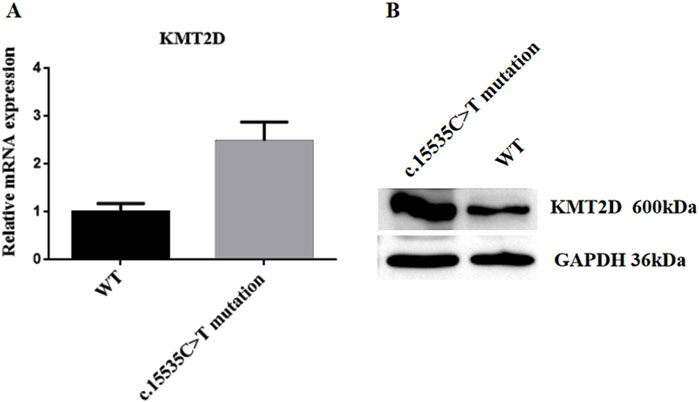
Figure 3. KMT2D Expression Analysis. The back skin fibroblasts of the patient were isolated and cultured to the third generation, and the normal human fibroblasts (BJ cell No. FH0191) were used as the control group. Real-time fluorescence quantitative PCR and protein Western blot detection were performed. (A) Real-Time Quantitative PCR Analysis. (B) Western blot Analysis. c.15535C>T mutation: Proband’s fibroblasts; WT (Wild Type): Normal fibroblasts.
The patient has been diagnosed with Type I KS.
There is no specific treatment for KS; management primarily involves symptomatic and rehabilitative therapies, with systematic follow-up of growth and development indices and symptomatic treatment tailored to the clinical manifestations at different stages. The patient’s gonadal development is slightly delayed, but pituitary-gonadal axis hormones are currently normal, and he is still in puberty, thus continuing observation is recommended. The development of male breast tissue will continue to be monitored, and post-puberty, surgical intervention may be considered if aesthetically or psychologically necessary (Thiruchelvam et al., 2016). By evaluating the pathogenic gene causing KS, targeted therapy may be considered. Gene therapy or histone methylation-modulating molecules for Kabuki Syndrome (KS) remain in the research stage. Early identification, diagnosis, and personalized treatment for KS can improve prognosis. Genetic counseling can reduce the birth of children with genetic disorders and effectively lower birth defects.
The parents expressed relief that their child received an accurate diagnosis and treatment, resolving a long-standing doubt about why their child differed from peers. They expressed gratitude to the doctors and agreed to continue monitoring for breast tissue overgrowth until 18 years old, consenting to surgery if necessary. Moving forward, they plan to provide more care and focus on their child’s psychological health and nutrition.
This article reports on a 16-year-old case of KS, in which comprehensive exome sequencing revealed a heterozygous missense mutation in the coding region of the KMT2Dgene, c.15535C>T (p.Arg5179Cys), ultimately leading to a diagnosis of Type I Kabuki Syndrome. There is a heterozygous missense mutation in the coding region of the KMT2Dgene:c.15535C>T (p.Arg5179Cys). At the 15535th base of the KMT2D gene, a cytosine (C) was replaced by a thymine (T), resulting in the substitution of the amino acid at position 5,179 from arginine to cysteine, altering the amino acid’s polarity. This mutation was not detected in peripheral blood samples from the patient’s parents, suggesting it is a de novo mutation. The mutation is extremely rare, with no reports in HGMD databases. Base on current evidence, this variant is classified as pathogenic, and the c.15535C>T (p.Arg5179Cys) mutation is likely the cause of the patient’s condition (see Figure 2). The particularity of this case lies in the fact that molecular genetic testing found a mutation at the locus of KMT2D gene C.15535C>T (P.Arg5179cys), which is a new mutation and has not been reported before. In addition, the patient presented with bilateral breast enlargement, and the father had a history of drug use for 8 years prior to birth. In 2010, Ng et al. (2010) reported that the KMT2D gene is the primary causative gene for Kabuki Syndrome Type I, accounting for 56%–75% of KS cases, characterized by autosomal dominant inheritance. Additionally, heterozygous deletions in the Kabuki Syndrome due to a KDM6A gene mutation (Bögershausen and Wollnik, 2013),Currently, Detailed distinctive clinical pictures have not yet been fully characterized, But,KDM6A mutations are typically associated with more severe phenotypes, including higher risks of epilepsy, developmental delays, and feeding difficulties. Dai Xiaowei et al. reported a case of a 3-year-old child, who presented with developmental delays, found to have a heterozygous mutation in the MLL2 gene coding region: c.2546 > T (p.Ser849Leu), leading to KS (Dai et al., 2018). Lu Huimin et al. performed genetic testing on a child with characteristic facial features, developmental delays, and cardiovascular abnormalities, identifying a frameshift mutation in the KMT2D gene, c.16028delC (p.Pro5343LeufsTer13) (Lu, 2022). Xin et al. (2018) detected two novel mutations in the KMT2D gene (c.5235delA, p.(A1746Lfs39) and c.7048G>A, p.(Q2350)) in two patients, confirmed by Sanger sequencing-based family pedigree analysis. These 62 patients with the Kabuki syndrome were collected in a collaborative study among 33 institutions and analyzed clinically, Many other inconsistent anomalies were observed. Important among them were early breast development in infant girls (23%), (Niikawa et al., 1988). Wu et al. described a 10-month-old female infant diagnosed with KS due to a c.12697C>T mutation, presenting developmental delays and early breast development (Wu and Su, 2017).The clinical manifestations in this study align with previous literature, such as developmental delays and unique facial features. Although rare breast tissue overgrowth was observed, the patient had no significant life-threatening organ malformations. The comparison between the performance of this patient and the previous literature are shown in Table 3. Barry et al.'s meta-analysis of 1,369 KS cases suggested that KMT2D mutations were predominantly truncation mutations, followed by missense mutations (Barry et al., 2022), consistent with this case’s mutation type.
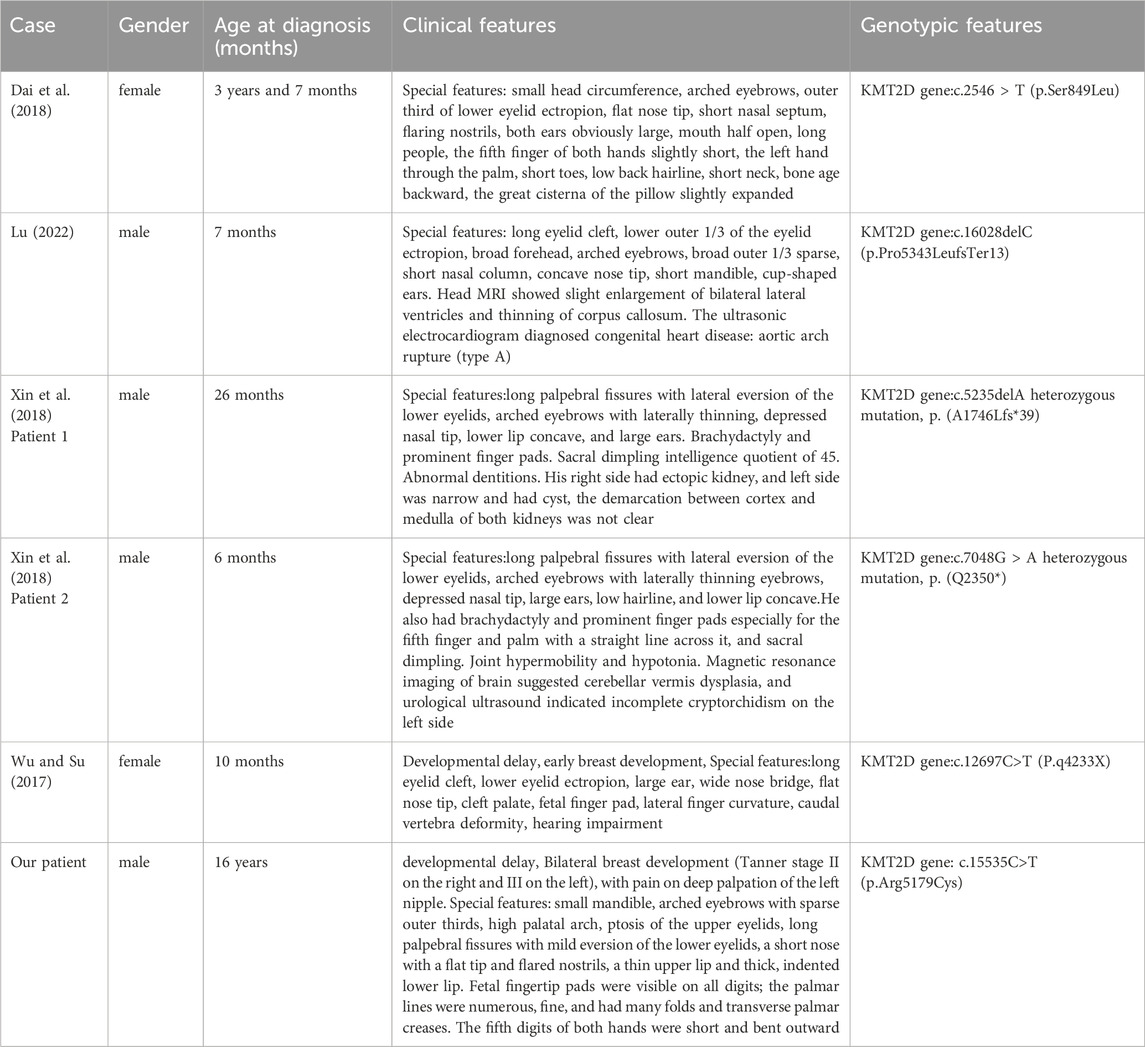
Table 3. The comparison between the performance of this patient and the previous literature.
KS patients often have multiple endocrine system diseases, such as developmental delay, hypoglycemia, hypothyroidism, precocious puberty, and diabetes insipidus. The underlying mechanisms remain unclear but may involve estrogen receptor dysfunction due to KMT2D gene mutations. Our study identified a rare heterozygous missense mutation, c.15535C>T (p.Arg5179Cys), in the coding region of the KMT2D gene, not reported in large population frequency databases and not detected in the peripheral blood samples of the patient’s parents, indicating a de novo mutation.
Not only did we identify the pathogenic gene mutation, but we also validated the expression levels of the KMT2D gene at both transcriptional and protein levels in the proband. The results indicated that compared to normal fibroblasts, the proband’s cells showed increased transcriptional expression of the KMT2D gene and elevated protein expression, consistent with the transcriptional data. These findings suggest that the KMT2D gene, harboring the c.15535C>T (p.Arg5179Cys)mutation, exhibits upregulated expression at both transcriptional and protein levels, confirming that this gene mutation is likely the cause of the patient’s condition. This case further confirms the impact of KMT2D gene mutations on protein levels in KS patients, clarifying the diagnosis and enriching the evidence for the gene’s influence. It contributes to future research on gene therapy and epigenetic modifications, enhancing understanding of the relationship between epigenetic changes and human disease development.
Kabuki syndrome is a rare disease, many doctors lack sufficient understanding of this disease, the most prominent feature of this disease is the special face, because there is no thorough treatment method at present, early prevention, early diagnosis and early treatment are very important, which can reduce the burden of patients and their families to the greatest extent, so further epidemiological investigation and research of this disease are needed. Modern molecular biology diagnostic technology, especially second-generation sequencing, is helpful to find the pathogenic genes of complex diseases, so as to make clear diagnosis, judge prognosis, guide treatment, and truly achieve the purpose of eugenics and fertility.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by the Ethics Committee of Dehong People’s Hospital (No. DYLL-KY2023032). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
WL: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Writing–original draft, Writing–review and editing. ML: Conceptualization, Data curation, Investigation, Methodology, Writing–original draft, Writing–review and editing. JDa: Conceptualization, Formal Analysis, Writing–review and editing. LS: Conceptualization, Formal Analysis, Writing–review and editing. WY: Conceptualization, Formal Analysis, Writing–review and editing. JL: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. YS: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. JDo: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. MZ: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. YX: Project administration, Writing–original draft, Writing–review and editing. LC: Project administration, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by Yunnan Province Clinical Research Center for Metabolic diseases (202102AA100056),Yunnan Clinical Medical Center for Endocrine and Metabolic Diseases (YWLCYXZXXYS20221005), and “Xingbian Talents” Support Project of Dehong Prefecture.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Barry, K. K., Tsaparlis, M., Hoffman, D., Hartman, D., Adam, M. P., Hung, C., et al. (2022). From genotype to phenotype-a review of Kabuki syndrome. Genes 13 (10), 1761. doi:10.3390/genes13101761
Bögershausen, N., and Wollnik, B. (2013). Unmasking Kabuki syndrome. Clin. Genet. 83 (3), 201–211. doi:10.1111/cge.12051
Dai, X., Zheng, L., Xu, Y., and Wu, G. (2018). A case and genetic analysis of a child with Kabuki syndrome. Chin. J. Eugen. Genet. 26, 85–86+70. doi:10.13404/j.cnki.cjbhh.2018.02.033
Del, C. I., Merino, P., Gómez de Liaño, P., and Alan, G. (2020). Changes in ocular motility in Kabuki syndrome. Arch. Soc. Espanola Oftalmol. 95 (1), 38–41. doi:10.1016/j.oftal.2019.09.016
Del Cerro, I., Merino, P., Gómez de Liaño, P., and Alan, G. (2020). Changesin ocularmotility in Kabuki syndrome. Arch. Soc. Esp. Oftalmol. (EnglEd) 95 (1), 38–41. doi:10.1016/j.oftal.2019.09.016
Jiao, X. H., Yasushi, O., and Nobuyuki, L. (1998). Kabuki facial spectrum syndrome. Chin. J. Stomatol. (03), 26.
Kuroki, Y., Suzuki, Y., Chyo, H., Hata, A., Matsui, I., and Ears, L. (1981). A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 99 (4), 570–573. doi:10.1016/s0022-3476(81)80256-9
Li, J. C. (2018). Kabuki syndrome: a report of 2 cases. J. Clin. Pediatr. 36, 53–56. doi:10.3969/j.issn.1000-3606.2018.01.012
Lu, H. (2022). Phenotypic analysis of Kabuki syndrome caused by new pathogenic variation of Kmt2d gen. Chin. J. Eugen. Genet. 30, 803–807. doi:10.13404/j.cnki.cjbhh.2022.05.032
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., McMillin, M. J., Gildersleeve, H. I., et al. (2010). Exome sequencing identifies Mll2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42 (9), 790–793. doi:10.1038/ng.646
Niikawa, N., Kuroki, Y., Kajii, T., Matsuura, N., Ishikiriyama, S., Tonoki, H., et al. (1988). Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am. J. Med. Genet. 31 (3), 565–589. doi:10.1002/ajmg.1320310312
Niikawa, N., Matsuura, N., Fukushima, Y., Ohsawa, T., and Kajii, T. (1981). Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 99 (4), 565–569. doi:10.1016/s0022-3476(81)80255-7
Thiruchelvam, P., Walker, J. N., Rose, K., Lewis, J., and Al-Mufti, R. (2016). Gynaecomastia. BMJ Clin. Res. ed 354, i4833. doi:10.1136/bmj.i4833
Wu, B. B., and Su, Y. J. (2017). Etc. Kabuki syndrome caused by Kmt2d gene mutation: report of 6 cases and literature review. Chin. J. Evidence-Based Pediatr. 12 (2), 135–139. doi:10.3969/j.issn.1673-5501.2017.02.011
Xin, C., Wang, C., Wang, Y., Zhao, J., Wang, L., Li, R., et al. (2018). Identification of novel Kmt2d mutations in two Chinese children with Kabuki syndrome: a case report and systematic literature review. BMC Med. Genet. 19 (1), 31. doi:10.1186/s12881-018-0545-5
Keywords: Kabuki syndrome, male breast development, KMT2D gene, mutation, clinical presentation
Citation: Li W, Lin M, Dao J, Shi L, Yi W, Lei J, Song Y, Dong J, Zhao M, Xu Y and Chen L (2025) Case Report: Area of focus clinical presentation and KMT2D gene mutation at the c.15535C>T site in a case of Kabuki syndrome. Front. Genet. 16:1523228. doi: 10.3389/fgene.2025.1523228
Received: 05 November 2024; Accepted: 20 February 2025;
Published: 07 March 2025.
Edited by:
Marc Morgan, Northwestern University, United StatesReviewed by:
Lacramioara Butnariu, Grigore T. Popa University of Medicine and Pharmacy, RomaniaCopyright © 2025 Li, Lin, Dao, Shi, Yi, Lei, Song, Dong, Zhao, Xu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Li, ZGhsdzY5MDJAMTI2LmNvbQ==; Lulu Chen, Y2hlcmlhX2NoZW5AMTI2LmNvbQ==; Yushan Xu, eHV5dXNoYW4xMDE5QDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.