 Joshua T. Veluz1*
Joshua T. Veluz1* Laurence Anthony N. Mallari1,2
Laurence Anthony N. Mallari1,2 Paul Christian T. Gloria1,2
Paul Christian T. Gloria1,2 Maria Auxilia T. Siringan1
Maria Auxilia T. Siringan1- 1Natural Sciences Research Institute, College of Science, University of the Philippines Diliman, Quezon City, Philippines
- 2Institute of Biology, College of Science, University of the Philippines Diliman, Quezon City, Philippines
1 Introduction
Submarine groundwater discharge (SGD) refers to the movement of water from land to coastal waters, flowing across the land-ocean interface (Adyasari et al., 2019). SGD is ubiquitous in sandy, rocky, and muddy shorelines and may include fresh groundwater of terrestrial origin, recirculated seawater, or a combination of both (Adyasari et al., 2019; Santos et al., 2021). The presence of SGD in these areas results in physical and chemical gradients that create unique biogeochemical environments. SGD acts as a conduit for the transport of materials such as gases, nutrients, and trace metals, from land to sea (Moore, 2010; Knee and Paytan, 2011). The flux of nitrogen and phosphorus to the ocean from total SGD, which includes both fresh and recirculated seawater, is estimated to exceed riverine inputs on a global scale (Cho et al., 2018). The SGD-mediated inflow of nutrients can significantly impact coastal ecosystems and water quality, altering levels of dissolved and gaseous metabolites, including ammonium, methane, and hydrogen sulfide (Bernard et al., 2014; Santos et al., 2021; Schlüter et al., 2004). This influences microbial communities and their metabolic activities in these specific locations (Purkamo et al., 2022).
Similar to terrestrial subsurface environments, deep marine sediments are also characterized by an absence of photosynthetically produced labile organic carbon (Chen et al., 2023). Because of this, groundwater microorganisms have developed diverse strategies to ensure survival and persistence. Among these strategies is the ability to utilize ancient organic carbon from rocks, allochthonous organic carbon, or byproducts from the degradation of organic contaminants (Griebler and Lueders, 2009; Smith et al., 2015). Other groundwater microorganisms also have adaptations that enable them to fix inorganic carbon by utilizing energy from the oxidation of substrates such as nitrite, ammonium, reduced iron, and sulfur compounds (Ruiz-González et al., 2021).
Applying shotgun metagenomics and assembling microbial genomes from metagenomic data can reveal important insights into the structure-function relationships of microbial communities within complex environments (Overholt et al., 2020). Through the use of metagenome-assembled genomes (MAGs), researchers can reconstruct near-complete microbial genomes, enabling precise taxonomic identification and comprehensive functional profiling of these assembled genomes (Mangoma et al., 2024).
For instance, Mangoma et al. (2024) successfully recovered taxonomically diverse MAGs from Buhera soda pans in Zimbabwe. These genomes revealed metabolic pathways associated with nitrogen fixation and sulfur cycling, highlighting the ecological roles of microbial communities in such extreme environments. Similarly, a study by He et al. (2016) leveraged metagenomic sequencing of sediment samples from deep-sea hydrothermal vents in the Guaymas Basin, Mexico, to identify Bathyarchaeota MAGs. These genomes were found to contain genes encoding the Wood-Ljungdahl (WL) pathway, an acetogenic process crucial for carbon fixation and acetate production. This finding underscores the pivotal role of Bathyarchaeota in the carbon cycle within deep-sea vent ecosystems.
Building on the work of Mangoma et al. (2024) and He et al. (2016), this data report seeks to deepen our understanding of specific environments, such as SGDs, from a microbial perspective. This report expands our knowledge of microbial species thriving in such environments, sheds light on their functional roles, and reveals untapped resources for industrial and medical applications through the analysis of these assembled genomes.
In this study, high-quality MAGs were generated from shotgun metagenome data derived from water samples from an SGD vent in Mabini, Batangas, Philippines. The presence of SGD in the collection site was documented by Cardenas et al. (2020) using Radon (222Rn) concentrations, a natural tracer for SGD (Burnett et al., 2003; Burnett et al., 2006; Cardenas et al., 2020). The generated MAGs were annotated to identify their potential environmental roles and subsequently compared to existing genomes for further insights. This data report offers preliminary findings that can serve as a foundation for future studies. It provides valuable insights into the limited research on the microbial dimension in SGD-associated areas within the Philippines.
2 Methods
Water samples (4 L) were collected from an SGD vent in the Sea Spring site in a coastal area in Mabini, Batangas, Philippines (13.68701 N, 120.89573 E). Divers brought a 4 L-Nalgene bottle filled with surface water to the floor of the Sea Spring site, about 7 m in depth. The surface water was purged from the bottle using pressurized air from the divers’ tanks. After purging, the bottles were titled towards the vent to collect the SGD water. Using a vacuum pump filtration system, the water samples were filtered through a 3 µm polycarbonate track-etched (PCTE) membrane (Sterlitech Corp., United States). After filtration, the membrane filter was placed in a cryovial and stored in a portable cryotank containing liquid nitrogen before transporting to the laboratory.
Total DNA was extracted from the membrane filter using the DNeasy PowerSoil® Pro Kit (QIAGEN), following the manufacturer’s protocol with minimal modifications. Specifically, three sample replicates were prepared, each subjected to the lysis step, and combined in a single column to ensure sufficient DNA concentration. The concentration of the extracted DNA was determined using the Denovix QFX Fluorometer and dsDNA broad-range kit following the manufacturer’s instructions. The DNA extract was sent to Macrogen, Inc. (South Korea) for shotgun metagenomic sequencing, at 25 Gb throughput, and 150 bp paired-end setting using the Illumina NovaSeq™ 6,000 system.
De novo metagenome assembly was performed using various bioinformatics tools in the KBase v2.7.11 platform (Arkin et al., 2018). After merging the forward and reverse reads during the importing stage, the resulting merged raw reads were analyzed with FastQC v0.12.1 (Andrews, 2010) and yielded 77,717,154 total sequences, 11.7 Gbp total bases, 45% G+C content, and no poor-quality sequences. Consequently, the merged raw reads were trimmed using Trimmomatic v0.36 (Bolger et al., 2014) with a sliding window size of 4 and a minimum quality set to 30. The Q30-trimmed reads were assembled using three different assemblers namely: metaSPAdes v3.15.3 (Nurk et al., 2017; Prjibelski et al., 2020), MEGAHIT v1.2.9 (Li et al., 2016), and IDBA-UD v1.1.3 (Peng et al., 2012), all using default parameters. The assemblers metaSPAdes, MEGAHIT, and IDBA-UD showed a total DNA length of 187,749,451 bp, 178,126,656 bp, and 108,098,378 bp, respectively. The metaSPAdes assembly, which had the longest total sequence length, was selected for downstream processing. The metagenomic contigs from the assembly chosen were grouped into bins using three binning tools–CONCOCT v1.1 (Alneberg et al., 2014), MaxBin2 v2.2.4 (Wu et al., 2015), and MetaBAT2 v1.7 (Kang et al., 2019), following default parameters (minimum contig length of 300 ≤ 2000). The generated bins from these three binning tools were consolidated and optimized using the DAS Tool v1.1.2 (Sieber et al., 2018) using default parameters (diamond, 0.5 score threshold, 0.6 duplicate penalty, and 0.5 megabin penalty). The generated bins from DAS Tool optimization were quality-filtered using CheckM v1.0.18 (Parks et al., 2015) to have ≥90% completeness and ≤5% contamination according to the high-quality MiMAG standards (Bowers et al., 2017). The completeness of the genomes was also determined using BUSCO v5.4.6 (Simão et al., 2015). MAGs’ information and taxonomic identities were visualized using the circlize package (Gu et al., 2014) in RStudio (RStudio Team, 2024).
The filtered bins were compared to the top 10 closest known genomes using the SpeciesTreeBuilder v0.1.4 (Arkin et al., 2018), which analyzes 49 clusters of orthologous groups (COGs) to determine phylogenetic relationships. In this report, tree reconstruction was conducted without an outgroup to maintain focus on the relationships among the GenBank genomes, allowing for a more refined understanding of their phylogenetic proximity. Consequently, all genomes underwent general annotation using Rapid Annotation using Subsystem Technology (RAST) through the SEED viewer v2.0 platform (Aziz et al., 2008; Overbeek et al., 2013) using the RASTtk annotation scheme. This report specifically highlights genes with critical functions in biogeochemical cycles focusing on those involved in sulfur metabolism, iron acquisition and metabolism, potassium metabolism, phosphorus metabolism, and nitrogen metabolism. The phylogenetic trees and RAST results were visualized using ggtree (v1.14.6; Yu et al., 2016) and ggplot (v3.5.1; Wickham, 2016) packages through RStudio (RStudio Team, 2024). The bins were also subjected to Distilled and Refined Annotation of Metabolism (DRAM) v0.1.2 (Shaffer et al., 2020) using default settings. To determine the functional COGs, eggNOG-mapper v2 (Cantalapiedra et al., 2021) with eggNOG v5.0 (Huerta-Cepas et al., 2018) were utilized using default settings. Afterward, the resulting .csv files were manipulated in RStudio (RStudio Team, 2024) using the packages dplyr (v1.1.4; Wickham et al., 2023), readr (v2.1.5; Wickham et al., 2024), and stringr (v1.5.1; Wickham, 2023) to generate relative frequencies of COG categories for each MAG and across all MAGs.
Lastly, genome mining of biosynthetic gene clusters (BGCs) was also performed using antiSMASH v7.0 (Blin et al., 2023) with a relaxed detection strictness. The predicted BGCs from the seven high-quality MAGs were recorded and tallied.
3 Data and analysis
From the Q30-trimmed metagenomic reads, seven bins were filtered and optimized. Figure 1A provides a summary of their genome characteristics. These seven MAGs exhibited CheckM completeness of over 90% and contamination of under 5%, aligning with MIMAG standards (Bowers et al., 2017). Notably, bins 002, 023, and 027 surpass 98% in CheckM completeness, while bins 010, 023, 024, and 027 reported less than 1% contamination. Additionally, using BUSCO, four of the seven bins scored above 90% completeness. Further quality assessment with QUAST v5.2.0 (Mikheenko et al., 2018) revealed genome sizes ranging from 1.37 to 2.17 Mbp across the bins. Bins 002, 010, 023, and 027 contain fewer than 75 contigs, with bin 027 having the least number at 55 contigs. All bins maintained an L50 value below 50, with bins 002 and 010 presenting the lowest at 13. The N50 values for all bins were above 14,000, with bin 010 having the highest at 48,078. The GC content of all seven bins ranged from 39.6% to 62.88%. The comprehensive results for CheckM, BUSCO, and QUAST assessments of the seven MAGs are detailed in Supplementary File 1.
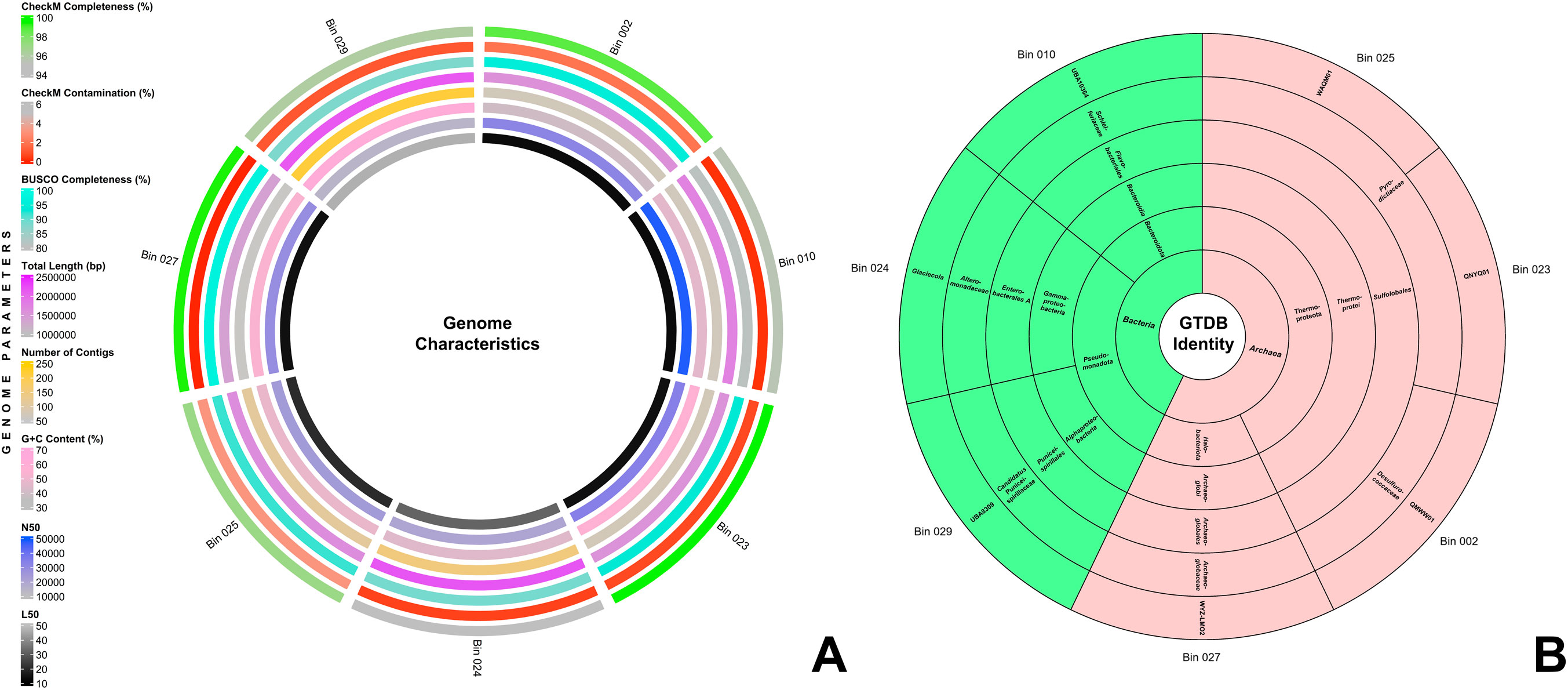
Figure 1. (A) Genome characteristics of the seven MAGs. From the innermost to the outermost circle, the rings display the following information: L50, N50, G+C content, contig count, total genome length, BUSCO completeness, CheckM contamination, and CheckM completeness. (B) Sunburst chart depicting the GTDB taxonomic identities of the seven MAGs. The innermost ring represents the domain, followed by successive rings for phylum, class, order, family, and genus in the outermost ring.
The seven bins were taxonomically classified using the Genome Taxonomy Database toolkit (GTDB-tk) v2.3.2 (Chaumeil et al., 2019), and their taxonomic identities are shown in Figure 1B. Four of the seven MAGs were classified under Archaea, and three were under Bacteria. The MAGs under Archaea were divided into two phyla: Thermoproteota (Bins 002, 023, 025) and Halobacteriota (Bin 027), while the MAGs under Bacteria were divided into Pseudomonadota (Bins 024, 029) and Bacteroidota (Bin 010). Detailed taxonomic identities of all MAGs up to genus level are presented in Supplementary File 2.
Figure 2A presents the top 10 closest genomes to the bins classified under the domain Archaea. Figure 2B illustrates the phylogenetic relationships for bins identified under Bacteria and their respective top 10 closest genomes.
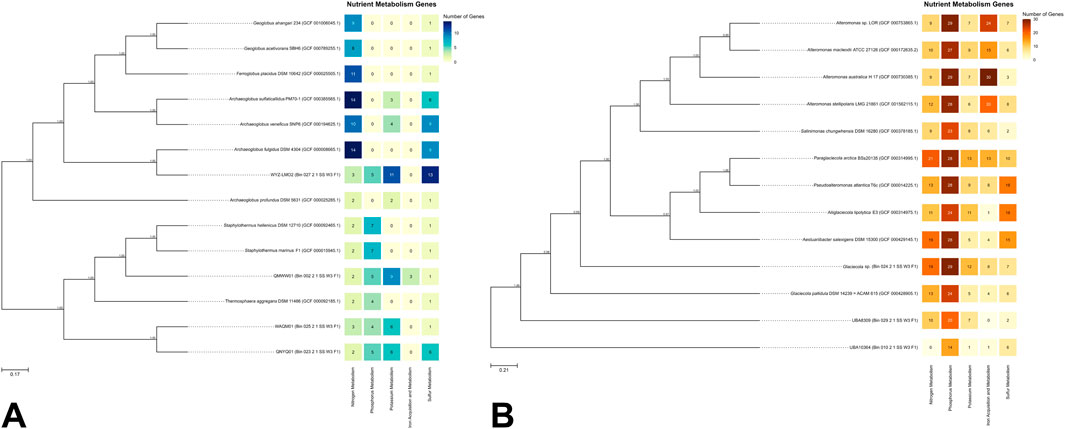
Figure 2. (A) Phylogenetic trees of archaeal and (B) bacterial MAGs with associated nutrient metabolism gene heatmaps. Node values indicate bootstrap values based on 1,000 replicates. Scale bars represent (A) 17% and (B) 21% genetic variation per unit of scale length.
For archaeal MAGs (Figure 2A), QMWW01 (Bin 002) was found to cluster near the Staphylothermus clade, suggesting it may belong to this genus. This is supported by GTDB results (Figure 1B), which identified QMWW01 within the family Desulfurococcaceae, the same family as Staphylothermus. QNYQ01 (Bin 023) and WAQM01 (Bin 025) formed a monophyletic clade near to Thermosphaera aggregans, also within Desulfurococcaceae (Anderson et al., 2009). QMWW01, QNYQ01, and WAQM01 clustered with other GenBank genomes, including Staphylothermus and Thermosphaera, all classified under the order Sulfolobales. The Sulfolobales genomes displayed genes linked to nitrogen, phosphorus, and sulfur metabolism. Sulfolobales is a group of thermoacidophilic Archaea where the majority of which are facultatively or obligately chemolithoautotrophic. The ability to metabolize sulfur, whether in its elemental form or reduced inorganic sulfur compounds, enables Sulfolobales to grow autotrophically. As a result, it is their most important physiological characteristic (Liu et al., 2021). Additionally, only the Sulfolobales MAGs were observed to possess genes associated with potassium metabolism. Notably, QMWW01 uniquely exhibited genes involved in iron acquisition and metabolism within this clade.
On the opposite branch of the tree, WYZ-LMO2 (Bin 027) formed a monophyletic clade with Archaeoglobus fulgidus, along with other members of the genera Ferroglobus, Geoglobus, and other Archaeoglobus, all belonging to the family Archaeoglobaceae (Brileya and Reysenbach, 2014). All Archaeoglobaceae genomes exhibited genes associated with nitrogen and sulfur metabolism. This thermophilic and obligate anaerobic family, found in marine and terrestrial environments, is known for their diverse metabolic capabilities including nitrate and sulfate reduction (Brileya and Reysenbach, 2014). Notably, WYZ-LMO2 was the only genome within the clade to possess genes associated with phosphorus metabolism. Lastly, no genome within the clade exhibited genes for iron acquisition and metabolism.
For bacterial MAGs (Figure 2B), Glacieola sp. (Bin 024) and UBA8309 (Bin 029) were identified as closely related to the GenBank genome Glaciecola pallidula DSM 14239. All three genomes were classified under the phylum Pseudomonadota (syn. Proteobacteria). Both Glaciecola pallidula DSM 14239 and Glaciecola sp. contained genes associated with all the types of metabolism genes analyzed in this report. Members of the genus Glaciecola are known to inhabit various marine environments (Bian et al., 2011; Qin et al., 2014), including seawater similar to the sample source in this study. This genus was also recognized for its ability to break down biopolymers such as cellulose, chitin, and xylan (Qin et al., 2014), suggesting a significant role in organic matter degradation and carbon cycling in marine environments. For UBA8309, the MAG was classified under the family Candidatus Puniceispirillaceae. Puniceispirillum marinum IMCC1322, which belongs to the same family, is known for its metabolic generalism in oceanic nutrient cycling and possesses genes associated with dimethylsulfoniopropionate (DMSP), which may play a role in marine sulfur cycling (Oh et al., 2010). This suggests that UBA8309 may also play a role in nutrient cycling in SGD areas.
UBA10364 (Bin 010) was identified as the only MAG classified under Bacteroidota and was the most distantly related bin compared to the GenBank bacterial genomes. The MAG falls under the order Flavobacteriales. Members of this group are known to metabolize sulfur-containing substances and participate in the sulfur cycle (Wang et al., 2023). They also produce extracellular hydrolases to degrade macromolecular organic substances (Wang et al., 2023). The presence of sulfur metabolism genes in UBA10364 suggests a role in sulfur cycling, and the detection of genes related to phosphorus, potassium, and iron metabolism indicates potential involvement in cycling these elements as well.
The nutrient metabolism genes identified in the bins were further analyzed (Supplementary File 3). For nitrogen metabolism, the bins were found to contain genes associated with nitrate and nitrite ammonification, as well as ammonia assimilation. In the case of phosphorus metabolism, genes involved in phosphate metabolism, high-affinity phosphate transport, phosphate regulon, and polyphosphate synthesis were detected. For potassium metabolism, the identified genes included those related to the glutathione-regulated potassium-efflux system, hyperosmotic potassium uptake, and potassium homeostasis. For sulfur metabolism, genes linked to galactosylceramide and sulfatide metabolism, sulfite reduction, and thioredoxin-disulfide reductase were observed. Lastly, within the iron acquisition and metabolism category, genes encoding encapsulating proteins for peroxidase enzymes, iron acquisition mechanisms, and the hemin transport system were identified. In addition to the RAST results, the DRAM analysis (Supplementary File 4) of the bins revealed a diverse array of nutrient metabolism genes. These included genes involved in carbon, nitrogen, and sulfur metabolism, as well as genes associated with arsenate and mercury reduction, methanogenesis, and alcohol production.
The diverse metabolic capabilities of the recovered bacterial and archaeal MAGs, supported by annotated genes and their taxonomic identities, suggest that these putative microorganisms may play a crucial role in biogeochemical cycling in SGD areas of Mabini, Batangas. This conclusion is further supported by various studies highlighting the metabolic functions of similar taxa in nutrient cycling.
Functional COGs were identified across all MAGs using eggNOG. The analysis revealed that the category with the highest relative frequency of COGs was “Unknown function” (S) (Supplementary File 5; Supplementary Figure S1). This observation aligns with individual MAG results, where the most frequently assigned COG in all bins also belonged to the “Unknown function” category (Supplementary File 5; Supplementary Figures S2–S8). This finding suggests that many COGs within the bins require additional data for proper classification. In addition to the “Unknown function” category, several functional COGs were found to have relative frequencies exceeding 5%, including J (translation, ribosomal structure, and biogenesis), C (energy production and conversion), E (amino acid transport and metabolism), H (coenzyme transport and metabolism), and L (replication, recombination, and repair). The abundance of these functional genes suggests that the microorganisms in the vent water heavily rely on these processes for survival, particularly in the SGD environment.
In addition to the analysis of the nutrient metabolism genes and COGs, the presence of BGCs within the MAGs was assessed, as detailed in Supplementary File 6. Using antiSMASH, it was found that only QNYQ01 (Bin 023) and WYZ-LMO2 (Bin 027) lacked detectable BGCs. In contrast, the remaining MAGs exhibited BGCs associated with ectoine, ribosomally synthesized, and post-translationally modified peptides (RiPP)-like compounds, terpene, and redox-cofactors. Among these, Glacieola sp. (Bin 024) contained the highest number of BGCs, including those related to ectoine, RiPP-like compounds, and terpene. Notably, RiPP-like and terpene-related BGCs were the most prevalent across all MAGs. RiPP-like compounds, previously identified as bacteriocin-encoding genes (Blin et al., 2021), have been studied for their anticancer and antibiotic applications (Negash and Tsehai, 2020; Thapar and Kumar Salooja, 2023). Similarly, terpenes are renowned for their diverse biological activities, including antiplasmodial, antiviral, anticancer, and antidiabetic properties (Cox-Georgian et al., 2019).
The identification of these BGCs in putative microbes from vent water at SGD sites in Mabini, Batangas, suggests their potential biotechnological and pharmaceutical applications. Although these microorganisms remain unculturable, future advances in cultivation and isolation techniques may enable us to unlock their potential, leading to the discovery of novel therapeutic compounds and other valuable bioactive agents.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/1111281 https://zenodo.org/records/14490404 https://narrative.kbase.us/narrative/202183.
Author contributions
JV: Data curation, Formal Analysis, Project administration, Writing–original draft, Writing–review and editing, Visualization. LM: Conceptualization, Data curation, Investigation, Methodology, Project administration, Writing–original draft, Writing–review and editing. PG: Conceptualization, Data curation, Investigation, Methodology, Project administration, Writing–review and editing. MS: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This data report was an output of the SGD Project 4: Probing Microbial Diversity in Submarine Groundwater Discharge (SGD) Areas. The project was implemented under the Biodiversity and Resilience of Coral Reefs and Associated Ecosystems in Submarine Groundwater Discharge Areas (BioRe CoARE SGD) program, with funding support from the Department of Science and Technology-Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (DOST-PCAARRD).
Acknowledgments
The authors extend their gratitude to the Microbiological Research and Services Laboratory of the Natural Sciences Research Institute, University of the Philippines Diliman, for hosting the SGD Project 4. Special thanks also go to Mr. Edwin E. Dumalagan Jr., Mr. Peter Paul P. Bucsit, and Dr. Fernando P. Siringan, who collected the SGD vent water samples featured in this data report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that Gen AI was used in the creation of this manuscript. OpenAI’s ChatGPT-4o was utilized to improve the writing of the data report.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1522253/full#supplementary-material
References
Adyasari, D., Hassenrück, C., Oehler, T., Sabdaningsih, A., and Moosdorf, N. (2019). Microbial community structure associated with submarine groundwater discharge in Northern Java (Indonesia). Sci. Total Environ. 689, 590–601. doi:10.1016/j.scitotenv.2019.06.193
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11 (11), 1144–1146. doi:10.1038/nmeth.3103
Anderson, I. J., Sun, H., Lapidus, A., Copeland, A., Glavina Del Rio, T., Tice, H., et al. (2009). Complete genome sequence of Staphylothermus marinus Stetter and Fiala 1986 type strain F1. Stand. Genomic Sci. 1 (2), 183–188. doi:10.4056/sigs.30527
Arkin, A. P., Cottingham, R. W., Henry, C. S., Harris, N. L., Stevens, R. L., Maslov, S., et al. (2018). KBase: the United States department of energy systems biology knowledgebase. Nat. Biotechnol. 36 (7), 566–569. doi:10.1038/nbt.4163
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9 (1), 75. doi:10.1186/1471-2164-9-75
Bernard, R., Mortazavi, B., Wang, L., Ortmann, A., MacIntyre, H., and Burnett, W. (2014). Benthic nutrient fluxes and limited denitrification in a sub-tropical groundwater-influenced Coastal Lagoon. Mar. Ecol. Prog. Ser. 504, 13–26. doi:10.3354/meps10783
Bian, F., Qin, Q.-L., Xie, B.-B., Shu, Y.-L., Zhang, X.-Y., Yu, Y., et al. (2011). Complete genome sequence of seawater bacterium Glaciecola nitratireducens FR1064T. J. Bacteriol. 193 (24), 7006–7007. doi:10.1128/jb.06296-11
Blin, K., Shaw, S., Augustijn, H. E., Reitz, Z. L., Biermann, F., Alanjary, M., et al. (2023). antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51 (W1), W46–W50. doi:10.1093/nar/gkad344
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49 (W1), W29–W35. doi:10.1093/nar/gkab335
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35 (8), 725–731. doi:10.1038/nbt.3893
Brileya, K., and Reysenbach, A.-L. (2014). The class Archaeoglobi. Prokaryotes, 15–23. doi:10.1007/978-3-642-38954-2_323
Burnett, W. C., Aggarwal, P. K., Aureli, A., Bokuniewicz, H., Cable, J. E., Charette, M. A., et al. (2006). Quantifying submarine groundwater discharge in the coastal zone via multiple methods. Sci. Total Environ. 367 (2–3), 498–543. doi:10.1016/j.scitotenv.2006.05.009
Burnett, W. C., Cable, J. E., and Corbett, D. R. (2003). Radon tracing of submarine groundwater discharge in coastal environments. Land Mar. Hydrogeology, 25–43. doi:10.1016/b978-044451479-0/50015-7
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). EggNOG-mapper V2: functional annotation, Orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38 (12), 5825–5829. doi:10.1093/molbev/msab293
Cardenas, M. B., Rodolfo, R. S., Lapus, M. R., Cabria, H. B., Fullon, J., Gojunco, G. R., et al. (2020). Submarine groundwater and vent discharge in a volcanic area associated with coastal acidification. Geophys. Res. Lett. 47 (1). doi:10.1029/2019gl085730
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-TK: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36 (6), 1925–1927. doi:10.1093/bioinformatics/btz848
Chen, X., Cai, R., Zhuo, X., Chen, Q., He, C., Sun, J., et al. (2023). Niche differentiation of microbial community shapes vertical distribution of recalcitrant dissolved organic matter in deep-sea sediments. Environ. Int. 178, 108080. doi:10.1016/j.envint.2023.108080
Cho, H.-M., Kim, G., Kwon, E. Y., Moosdorf, N., Garcia-Orellana, J., and Santos, I. R. (2018). Radium tracing nutrient inputs through submarine groundwater discharge in the Global Ocean. Sci. Rep. 8 (1), 2439. doi:10.1038/s41598-018-20806-2
Cox-Georgian, D., Ramadoss, N., Dona, C., and Basu, C. (2019). Therapeutic and medicinal uses of Terpenes. Med. Plants, 333–359. doi:10.1007/978-3-030-31269-5_15
Griebler, C., and Lueders, T. (2009). Microbial Biodiversity in groundwater ecosystems. Freshw. Biol. 54 (4), 649–677. doi:10.1111/j.1365-2427.2008.02013.x
Gu, Z., Gu, L., Eils, R., Schlesner, M., and Brors, B. (2014). Circlize implements and enhances circular visualization in R. Bioinformatics 30 (19), 2811–2812. doi:10.1093/bioinformatics/btu393
He, Y., Li, M., Perumal, V., Feng, X., Fang, J., Xie, J., et al. (2016). Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat. Microbiol. 1 (4), 16035. doi:10.1038/nmicrobiol.2016.35
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2018). Eggnog 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47 (D1), D309–D314. doi:10.1093/nar/gky1085
Kang, D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from Metagenome Assemblies. PeerJ 7, e7359. doi:10.7717/peerj.7359
Knee, K. L., and Paytan, A. (2011). 4.08 - submarine groundwater discharge: a source of nutrients, metals, and pollutants to the coastal ocean. Treatise Estuar. Coast. Sci., 205–233. doi:10.1016/b978-0-12-374711-2.00410-1
Li, D., Luo, R., Liu, C.-M., Leung, C.-M., Ting, H.-F., Sadakane, K., et al. (2016). Megahit v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi:10.1016/j.ymeth.2016.02.020
Liu, L.-J., Jiang, Z., Wang, P., Qin, Y.-L., Xu, W., Wang, Y., et al. (2021). Physiology, taxonomy, and sulfur metabolism of the Sulfolobales, an order of thermoacidophilic archaea. Front. Microbiol. 12, 768283. doi:10.3389/fmicb.2021.768283
Mangoma, N., Zhou, N., and Ncube, T. (2024). Metagenome-assembled genomes provide insight into the microbial taxonomy and ecology of the Buhera soda pans, Zimbabwe. PLOS ONE 19 (12), e0299620. doi:10.1371/journal.pone.0299620
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D., and Gurevich, A. (2018). Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34 (13), i142–i150. doi:10.1093/bioinformatics/bty266
Moore, W. S. (2010). The effect of submarine groundwater discharge on the Ocean. Annu. Rev. Mar. Sci. 2 (1), 59–88. doi:10.1146/annurev-marine-120308-081019
Negash, A. W., and Tsehai, B. A. (2020). Current applications of bacteriocin. Int. J. Microbiol. 2020, 4374891–4374897. doi:10.1155/2020/4374891
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27 (5), 824–834. doi:10.1101/gr.213959.116
Oh, H.-M., Kwon, K. K., Kang, I., Kang, S. G., Lee, J.-H., Kim, S.-J., et al. (2010). Complete genome sequence of “Candidatus Puniceispirillum marinum” IMCC1322, a representative of the SAR116 clade in the Alphaproteobacteria. J. Bacteriol. 192 (12), 3240–3241. doi:10.1128/jb.00347-10
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2013). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42 (D1), D206–D214. doi:10.1093/nar/gkt1226
Overholt, W. A., Hölzer, M., Geesink, P., Diezel, C., Marz, M., and Küsel, K. (2020). Inclusion of oxford nanopore long reads improves all microbial and viral metagenome-assembled genomes from a complex aquifer system. Environ. Microbiol. 22 (9), 4000–4013. doi:10.1111/1462-2920.15186
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). Checkm: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25 (7), 1043–1055. doi:10.1101/gr.186072.114
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28 (11), 1420–1428. doi:10.1093/bioinformatics/bts174
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., and Korobeynikov, A. (2020). Using SPAdes de novo assembler. Curr. Protoc. Bioinforma. 70 (1), e102. doi:10.1002/cpbi.102
Purkamo, L., von Ahn, C. M., Jilbert, T., Muniruzzaman, M., Bange, H. W., Jenner, A.-K., et al. (2022). Impact of submarine groundwater discharge on biogeochemistry and microbial communities in Pockmarks. Geochimica Cosmochimica Acta 334, 14–44. doi:10.1016/j.gca.2022.06.040
Qin, Q.-L., Xie, B.-B., Yu, Y., Shu, Y.-L., Rong, J.-C., Zhang, Y.-J., et al. (2014). Comparative genomics of the marine bacterial genus Glaciecola reveals the high degree of genomic diversity and genomic characteristic for cold adaptation. Environ. Microbiol. 16 (6), 1642–1653. doi:10.1111/1462-2920.12318
RStudio Team (2024). RStudio: Integrated development environment for R. RStudio. PBC. Available at: https://www.rstudio.com.
Ruiz-González, C., Rodellas, V., and Garcia-Orellana, J. (2021). The microbial dimension of submarine groundwater discharge: current challenges and Future Directions. FEMS Microbiol. Rev. 45 (5), fuab010. doi:10.1093/femsre/fuab010
Santos, I. R., Chen, X., Lecher, A. L., Sawyer, A. H., Moosdorf, N., Rodellas, V., et al. (2021). Submarine groundwater discharge impacts on coastal nutrient biogeochemistry. Nat. Rev. Earth and Environ. 2 (5), 307–323. doi:10.1038/s43017-021-00152-0
Schlüter, M., Sauter, E. J., Andersen, C. E., Dahlgaard, H., and Dando, P. R. (2004). Spatial distribution and budget for submarine groundwater discharge in eckernförde bay (western baltic sea). Limnol. Oceanogr. 49 (1), 157–167. doi:10.4319/lo.2004.49.1.0157
Shaffer, M., Borton, M. A., McGivern, B. B., Zayed, A. A., La Rosa, S. L., Solden, L. M., et al. (2020). Dram for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 48 (16), 8883–8900. doi:10.1093/nar/gkaa621
Sieber, C. M., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3 (7), 836–843. doi:10.1038/s41564-018-0171-1
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Smith, M. B., Rocha, A. M., Smillie, C. S., Olesen, S. W., Paradis, C., Wu, L., et al. (2015). Natural bacterial communities serve as quantitative geochemical biosensors. mBio 6 (3), e00326. doi:10.1128/mbio.00326-15
Thapar, P., and Kumar Salooja, M. (2023). Bacteriocins: applications in food preservation and therapeutics. Lact. - A Multifunct. Genus. doi:10.5772/intechopen.106871
Wang, Y., Wang, L., Liu, Y., Su, S., and Hao, W. (2023). Marine bacterial communities in the xisha islands, South China sea. Diversity 15 (7), 865. doi:10.3390/d15070865
Wickham, H. (2016). ggplot2: elegant graphics for data analysis. New York: Springer-Verlag. Available at: https://ggplot2.tidyverse.org (ISBN 978-3-319-24277-4.
Wickham, H. (2023). stringr: simple, consistent wrappers for common string operations (R package version 1.5.1). Available at: https://stringr.tidyverse.org.
Wickham, H., François, R., Henry, L., Müller, K., and Vaughan, D. (2023). dplyr: a grammar of data manipulation (R package version 1.1.4). Available at: https://github.com/tidyverse/dplyr.
Wickham, H., Hester, J., and Bryan, J. (2024). readr: read rectangular text data (R package) version 2.1.5. Available at: https://readr.tidyverse.org.
Wu, Y.-W., Simmons, B. A., and Singer, S. W. (2015). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32 (4), 605–607. doi:10.1093/bioinformatics/btv638
Keywords: biosynthethic gene clusters, metagenome-assembled genomes, nutrient metabolism genes, shotgun sequencing, submarine groundwater discharge, vent water
Citation: Veluz JT, Mallari LAN, Gloria PCT and Siringan MAT (2025) Exploring the taxonomical and functional profiles of marine microorganisms in Submarine Groundwater Discharge vent water from Mabini, Batangas, Philippines through metagenome-assembled genomes. Front. Genet. 16:1522253. doi: 10.3389/fgene.2025.1522253
Received: 04 November 2024; Accepted: 13 January 2025;
Published: 10 February 2025.
Edited by:
Christopher Fields, University of Illinois at Urbana-Champaign, United StatesReviewed by:
Etienne Z. Gnimpieba, University of South Dakota Biomedical Engineering, United StatesHilal Ay, Yıldız Technical University, Türkiye
Copyright © 2025 Veluz, Mallari, Gloria and Siringan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua T. Veluz, anR2ZWx1ekB1cC5lZHUucGg=