
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 28 February 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1512070
 Haoyang Zheng1,2,3†
Haoyang Zheng1,2,3† Gui Chen1,2,3†
Gui Chen1,2,3† Tingting Wang4
Tingting Wang4 Weisheng Cheng5,6,7,8,9,10Jing Yuan5,6,7,8,9,10Fang Liu1,2,3*
Weisheng Cheng5,6,7,8,9,10Jing Yuan5,6,7,8,9,10Fang Liu1,2,3* Yuanhong Xu1,2,3*
Yuanhong Xu1,2,3*Lesch-Nyhan syndrome (LNS, OMIM #300322) is a rare X-linked genetic disorder caused by variants in the HPRT1 gene, which codes for the Hypoxanthine-guanine phosphoribosyltransferase (HGPRT). HPRT1 gene variants disrupt normal purine metabolism, leading to the involvement of multiple organ systems, primarily characterized by hyperuricemia, dystonia, and neurological abnormalities, which makes LNS clinically heterogeneous and diagnostically challenging. Here, we report a rare case of a 27-year-old Chinese male exhibiting severe lower limb motor disorders, hyperuricemia, and intellectual development delay. Blood tests showed hyperuricemia and whole exome sequencing (WES) identified a novel hemizygous variant in the HPRT1 (NM-000194.3) gene: c.104T > C in exon 2, respectively. Bioinformatics techniques indicated that the variant may disrupt the activity of HGPRT. According to the clinical presentation, diagnostic examination, and WES results, the patient was finally diagnosed with LNS. This study identified a previously unreported pathogenic variant in the HPRT1 gene. Although no curative therapy is currently available for HPRT1 gene variants at present, a definite diagnosis of its genetic etiology is of great significance for genetic counseling and family planning.
Lesch-Nyhan syndrome (LNS, OMIM #300322), also termed “self-destructive features syndrome,” is an autosomal recessive metabolic disorder. The typical clinical manifestations include self-injurious behavior (SIB), hyperuricemia, and a constellation of developmental delays, intellectual disability, and hypotonia. The incidence of this disease in Canada is estimated at 1/380,000, while in Spain it is approximately 1/250,000. However, it is less well-reported worldwide (Harris, 2018; Nanagiri and Shabbir, 2025). The causative gene HPRT1 encodes hypoxanthine-guanine phosphor ribosyl transferase (HGPRT), which plays a central role in the remedial synthesis pathway of purine nucleotides by catalyzing the conversion of hypoxanthine and guanine to hypoxanthine nucleotides (IMP) and guanine nucleotides (GMP) (Bell et al., 2021).
A novel variant in the HPRT1 gene (NM_000194.3: c.104T > C [p.Val35Ala]) was identified in this study by whole exome sequencing. The variant affects the function of HGPRT to some extent, and the patient exhibits typical LNS symptoms. LNS patients are less commonly reported in China (Chen et al., 2014; Huang et al., 2018; Jian et al., 2013; Li et al., 2022; Liu et al., 2015; Tong et al., 2022; Wang et al., 2023; Yang and Guo, 2022). Our research broadened the genetic and phenotypic diversity of LNS patients in China, while also offering important insights for genetic counseling.
This case report presents a Chinese patient with Lesch-Nyhan syndrome (LNS) who harbors a novel variant in the HPRT1 gene.
The patient with Lesch-Nyhan syndrome were recruited from the First Affiliated Hospital of Anhui Medical University. This study received approval from the Ethics Committee of the First Affiliated Hospital of Anhui Medical University. Informed consent was procured from the patient and his family in consonance with institutional regulations and the Declaration of Helsinki. The patient and his family members received comprehensive clinical assessments performed by skilled doctors and laboratory staff at our hospital, and they submitted blood samples for genetic testing.
The patient was a 27-year-old male with “congenital weakness of both lower limbs and inability to walk”. At the age of six, he could ambulate with the assistance of supportive devices. However, he subsequently experienced a gradual decline in his ability to walk, ultimately becoming wheelchair dependent. The patient did not show obvious self-injurious behavior during the clinical visit, but it was learned from the family that the patient is prone to emotional outbursts and may bang into walls. Additionally, the patient presented with characteristic features of developmental delay, cognitive impairment, and multiple gouty tophi. Laboratory tests showed that the concentration of uric acid in the patient’s blood was significantly higher than the normal level (Table 1).
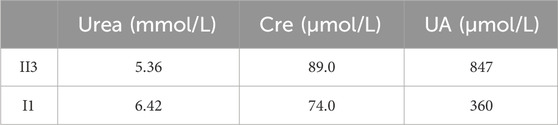
Table 1. Biochemical examination indicators of the patient and his father.
At the time of the patient’s hospital admission, the patient’s mother was deceased. The patient’s father and sister were normal, and the parents were not consanguineous. Of the three uncles of the patient, one was phenotypically normal, while the remaining two displayed clinical symptoms comparable to those observed in the patient but have since passed away. Additionally, the patient’s cousin exhibited no phenotypical abnormalities and the patient’s mother did not present with similar symptoms during her lifetime. Due to the passage of time, the photographs and clinical records of the patient’s mother and two affected uncles were not adequately preserved, thereby limiting our ability to further corroborate the association between this variant and the disease. However, based on the available information, we suspect a maternal family history of Lesch-Nyhan syndrome. (Figure 1).
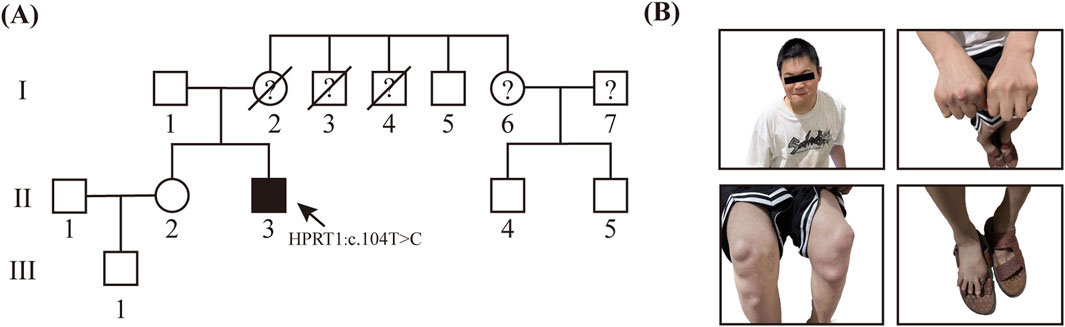
Figure 1. Genetic and Clinical Analysis of LNS in Unrelated Family (A) Precedenters’ family lineage diagram, which illustrates the inheritance pattern of LNS. (B) Hyperuricemia resulted in the occurrence of multiple gouty tophi in this patient.
The information pertaining to the primary case and his family members was obtained from the complete autopsy report. DNA extraction was performed using the TIANamp DNA Extraction Kit (TIANGEN Biotech, DP348). DNA from the patient and their family members was extracted and quantified with a NanoDrop 3,000 spectrophotometer (Allsheng, Hangzhou, China).
Enriched DNA samples were sequenced on the AmCareSeq-2000 Sequencer (AmCare Genomics Lab, Ltd., Guangzhou, China). The sequencing targeted 20,324 genes associated with clinical manifestations in pre-certified individuals, covering a total of 209,175 coding regions. The average depth of coverage of the assay was 315 ± 146×, with 98.5% of the coverage intervals having an average depth of >10× and 98.4% of the coverage intervals having an average depth of >20×.
The results of WES are analyzed and interpreted using a range of tools, including pathogenic variant databases such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (https://www.hgmd.cf.ac.uk/ac/index.php), databases of the normal population such as gnomAD (https://gnomad.broadinstitute.org/) and AllofUs (https://databrowser.researchallofus.org/), the Mendelian genetic disease database OMIM (https://www.omim.org/), and protein function prediction software PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (https://sift.bii.a-star.edu.sg/). Concurrently, the genetic variants were screened and graded within the context of the test in accordance with the American College of Medical Genetics and Genomics (ACMG) variant classification guidelines and supplementary guidelines (Richards et al., 2015). Furthermore, the clinical manifestations of the patient and the corresponding test results were considered in order to identify rare variants that may be associated with the patient’s clinical manifestations. Subsequently, Sanger sequencing was conducted on DNA samples from the patient and his family members to confirm the pathogenic variants.
The amino acid sequence of the HGPRT protein was obtained from Uniprot (https://www.uniprot.org/). The 3D structures of both the wild-type and missense mutant were modeled using SwissModel (https://swissmodel.expasy.org/). Available crystal structures of HGPRT from the RCSB Protein Data Bank (http://www.rcsb.org) were used to refine the model. The protein’s functional domains were analyzed using the InterPro database (https://www.ebi.ac.uk/interpro/). Structures were visualized and analyzed with PyMOL.
To elucidate the pathogenic variants in this patient, a whole exome sequencing (WES) study of the patient’s DNA was performed. The genome of the patient was evaluated for single nucleotide variants (SNVs) and copy number variants (CNVs) in accordance with the guidelines established by the American College of Medical Genetics and Genomics (ACMG). It is regrettable to report that no disease-causing or potentially disease-causing variants were identified. Subsequently, all variants of unknown significance (VUS) were subjected to further analysis, which revealed a hemizygous missense variant in exon two of the HPRT1 gene on the X chromosome (NM_000194.3: c.104T > C [p.Val35Ala]) (Supplementary Table S1). This finding was subsequently confirmed by Sanger sequencing. However, no relatives of the patient have been identified as carriers of this variant. When considered alongside the clinical manifestations observed in the patient’s family, the HPRT1 c.104T > C variant probably originated from the patient’s mother (Figure 2A). Furthermore, this variant was not identified in various population-based databases, including the ClinVar, the All of Us Research Program and the Genome Aggregation Database, etc.
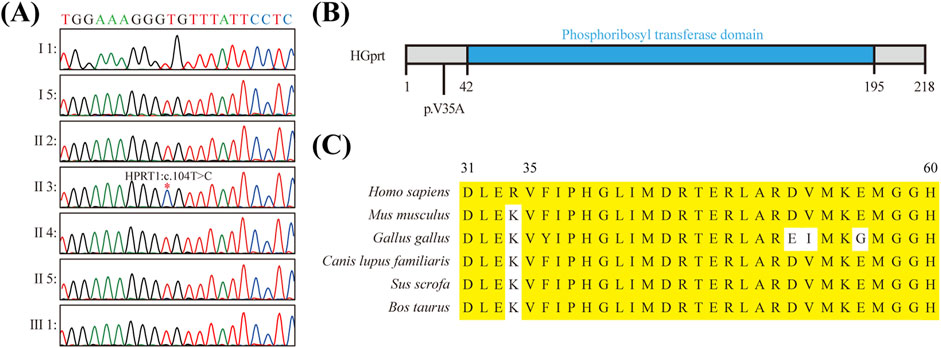
Figure 2. Molecular Characterization of HPRT1 Missense Mutation (A) Sanger sequencing results verified the presence of the c.104T > C mutation in the HPRT1 gene of the preterist. (B) Schematic representation of the structural domain of the HGPRT protein and the position of the missense mutation c.104T > C (p.Val35Ala) within the nonstructural domain. (C) Amino acid sequence conservation analysis of the p.V35A mutation site in the HGPRT protein. The following amino acid sequence comparison results illustrate the high conservation of the valine residue at position 35.
To ascertain the suspected pathogenicity of the detected variant, a series of variant prediction tools were employed to analyze its potential harm. The results of the bioinformatics analysis indicated a high probability that the variant is indeed pathogenic. Moreover, this variant has not been identified in various population-based databases, including ClinVar, Genome AD (Genome Aggregation Database dataset), and AllofUs (All of Us Research Program). It is noteworthy that this variant is situated within a gene region that exhibits high levels of conservation (Figures 2B,C; Supplementary Table S2).
The structural analysis of the p.Val35Ala mutation was conducted using SwissModel to predict the 3D structure of the HGprt protein, based on available crystal structures from the RCSB Protein Data Bank. Using InterPro to identify functional domains, we found that the mutation occurs within the Phosphoribosyltransferase domain (InterPro: IPR000836), a region located at the front of the catalytic site, potentially involved in substrate binding or enzyme activity regulation. Visualization using PyMOL revealed that the mutation causes a narrowing of a surface-exposed concave pocket, which is critical for ligand binding (Figure 3). This alteration in the pocket’s geometry could hinder substrate accessibility, thereby impairing the protein’s ability to bind its substrates. Based on this structural insight and the clinical manifestations observed in the patient, we hypothesize that this structural change may reduce the enzyme’s catalytic efficiency, supporting the pathogenicity of the p.Val35Ala variant.
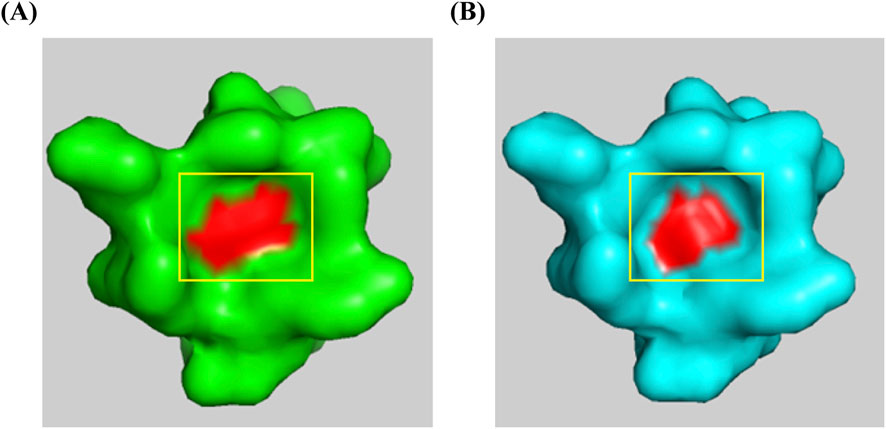
Figure 3. Structural comparison of wild-type and p.Val35Ala mutant HGPRT. The red area represents the 35th amino acid residue of the HGPRT protein. (A) Wild-type HGPRT structure showing the open concave pocket. (B) p.Val35Ala mutant HGPRT structure demonstrating the narrowing of the concave pocket due to the mutation at position 35.
LNS is a rare disorder of metabolic abnormalities that was first described by Michael Lesch and William Nyhan in 1964 (Lesch and Nyhan, 1964). Patients with LNS typically present with an early onset of disease, often within the first year of age. The clinical manifestations of LNS are variable in their severity and may include hyperuricemia, movement disorders, intellectual disability, mental and behavioral abnormalities, such as recurrent self-injury and aggressive behavior. It is an X-linked recessive genetic disease. Reported patients are mainly male, and female patients are mostly carriers. In rare cases, female LNS patients have been reported (AlBakheet et al., 2022; Cho et al., 2019; Fu et al., 2014; Li et al., 2022; Tong et al., 2022). Early identification of hyperuricemia through blood tests is essential for the prognosis of LNS and highlights the importance of early diagnosis in rare disorders. Indeed, early diagnosis not only aids in symptom management but also plays a significant role in reducing long-term neurological damage. Importantly, genetic testing can provide critical information regarding inheritance patterns, offering families the opportunity to make informed decisions regarding reproductive planning.
The HPRT1 gene is the causal gene for LNS, which is located in human Xq26.1 and has a total length of 40.5 kb. It consists of nine exons and eight introns and encodes 218 amino acids. To date, over 600 pathogenic variants have been documented, encompassing various forms of base substitutions, shifts, deletions, splices, and insertions (Nanagiri and Shabbir, 2025). Early genetic testing, particularly in families with a history of LNS, can help in understanding potential inheritance risks, reinforcing the need for genetic counseling and informed family planning.
The HGPRT protein, encoded by the HPRT1 gene, is a ubiquitous cytoplasmic enzyme within the human body. Under normal physiological conditions, it catalyzes the conversion of hypoxanthine to hypoxanthine acid, guanine to guanosine-phosphate, and the metabolism of the purine moieties of hypoxanthine and guanine to uric acid (Fu et al., 2015). The synthesis of purine nucleotides in the human body occurs through two distinct pathways: the “ab initio” and the “remedial synthesis” pathways. However, the brain lacks the enzyme system necessary for the ab initio pathway, rendering it incapable of synthesizing purine nucleotides through this route. Consequently, HGPRT is expressed at a higher level in the brain, particularly in the basal ganglia, where the concentration of HGPRT is the highest. In patients with LNS, the reduced or absent activity of the HGPRT enzyme, which is encoded by the abnormal HPRT1 gene, results in impaired purine metabolism in the body. This leads to excessive production of uric acid, which in turn causes hyperuricemia and related conditions such as gout and kidney stones (Guo et al., 2022). In addition, it has been found that HGPRT enzyme deficiency may affect the development of the dopamine system and cerebral cortex in the basal ganglia of the brain (Dinasarapu et al., 2022). The deposition of uric acid in the brain also results in the impairment of the basal ganglia, leading to cognitive deficits, neurological dysfunction, and SIB (Bell et al., 2021). The patient described exhibits significantly elevated serum uric acid levels, whereas his father’s uric acid levels are within the normal range. The patient presents with intellectual disability and severe motor impairments, characterized by congenital weakness in both lower limbs, preventing him from walking upright. Additionally, the patient demonstrates emotional instability, frequently engaging in self-injurious behaviors such as banging into walls. These metabolic disturbances highlight the importance of early diagnosis and genetic testing, which not only inform treatment decisions but also help identify at-risk family members. Genetic counseling can play an instrumental role in helping families understand inheritance patterns and potential risks.
The pathogenicity of abnormal HPRT1 genes is mainly associated with reduced or absent HGPRT enzyme activity. HGPRT activity is mainly measured using whole blood, natural cells, solutions and other specimens to determine the HGPRT-catalyzed production of inosinic acid and guanosine from hypoxanthine and guanine, respectively (Lesch and Nyhan, 1964). LNS is classified into three types according to the degree of HGPRT activity impairment (Fu et al., 2014; Fu et al., 2015; Harris, 2018): (1) A complete lack of enzymatic activity (<1.5%) leads to classical LNS, with typical clinical features including hyperuricemia and its sequelae such as gouty arthritis and urinary tract stones, movement disorders such as dystonia, involuntary movements, cognitive impairment, and SIB; (2) HGPRT activity is partially lacking (1.5%–8%), resulting in a variant LNS, also known as an intermediate type, with relatively mild clinical symptoms; (3) The mildest phenotype is HPRT-related hyperuricemia (OMIM #300323), in which patients have HGPRT activity >8.0%. Some patients exhibit hyperuricemia and its accompanying symptoms, while symptoms of movement disorders and cognitive impairment are mild and can only be detected through a professional neurological examination (Keebaugh et al., 2007). Among them, the neurological function and behavioral abnormalities between Lesch-Nyhan syndrome and the HPRT-related hyperuricemia phenotype are referred to as HPRT-related neuro dysfunction. Patients have an HGPRT activity of 1.5%–8.0%. The clinical manifestations include hyperuricemia and neurological dysfunction, but no SIB (Fang et al., 2024; Guo et al., 2022; Jinnah, 1993). Through computer prediction and protein model construction of the HPRT1:p.Val35Ala variant carried by the patient in this study, we found that this variant resides within the phosphoribosyltransferase domain (IPR000836). This structural modification suggests that the variant may impair HGPRT enzyme activity by hindering substrate binding. These findings support the hypothesis that this variant is potentially deleterious. Based on the patient’s clinical manifestations, we speculate that this case represents a classic presentation of LNS. However, our hospital currently lacks the capability to perform HGPRT enzyme activity assays, which limits our ability to definitively confirm the diagnosis. This is a limitation of our study. We plan to conduct further research to validate these findings.
Compared with other reported HPRT1 variants, the variant in our patient has its unique characteristics. For example, (Baba et al., 2017), reported a novel duplication variant (c.372_374 TTT > TTTT) in exon four of HPRT1 in a 9-month-old boy. The variant led to abnormal splicing and manifested as developmental delay, athetosis, and dystonic postures without SIB at that time. In contrast, our patient, with a different variant type, not only had hyperuricemia and developmental delay but also more severe motor-function impairment, such as the complete inability to walk due to congenital lower - limb weakness (AlBakheet et al., 2022). described a novel frameshift variant in HPRT1 in a 4-year-old girl with LNS, presenting with global developmental delay, self-injurious behavior, hyperuricemia, hypotonia, speech delay, spasticity, and seizures. Our patient’s variant-induced symptoms showed differences in the frequency and intensity of self-injurious behavior and the pattern of motor-disorder manifestations. Zhang et al. (2019) reported two hotspot variants, c.508 C > T and c.151 C > T, which led to premature translation termination. These variants are different from the one in our patient at the genetic locus. Functionally, our patient’s variant had a more profound impact on the three-dimensional structure of the HGPRT protein, as predicted by in silico analysis, which might be related to the more complex clinical manifestations. Furthermore, we have conducted a comprehensive statistical analysis of LNS patients reported in China over the past 5 years (Supplementary Table S3).
Lesch-Nyhan syndrome is difficult to treat and mostly treated with symptomatic therapy. Pharmacological treatments mainly use allopurinol to control uric acid, while baclofen or benzodiazepines are used to treat muscle tone disorders (Qurie et al., 2025; Satow et al., 2021). Nevertheless, the optimal treatment for LNS’s motor and neurological symptoms remains under investigation. The most widely studied interventions are S-adenosylmethionine (SAM) and deep brain stimulation (DBS) (Benchoua et al., 2021; Deng et al., 2023), the former is considered an effective treatment for SIB in LNS patients (Krajewski et al., 2024). Botulinum toxin can also be used to treat dystonia in LNS; however, it is incapable of affecting other symptoms and can only be used as one of several adjuvant treatments for LNS (Dressler et al., 2021; Gilbert et al., 2021). Levodopa/carbidopa is reported to be beneficial for LNS treatment, early use of adequate doses of levodopa/carbidopa in LNS may prevent the occurrence of SIB (Visser et al., 2024). While these treatments provide symptom relief, they also raise important ethical considerations, especially regarding long-term use and the potential for quality-of-life impacts. Genetic counseling and informed decision-making become critical in such cases, ensuring that families can make the best choices regarding treatment options and future care. Additionally,to achieve precise correction of LNS, a previous study has utilized CRISPR-mediated cytosine base editors (CBEs) to introduce HPRT1 c.430C > T and c.508C > T variants in the HAP1 cell line, thereby establishing LNS disease models. Adenine base editors (ABEs) were subsequently employed to correct these variants, achieving repair efficiencies of 3% and 5.2%, respectively. Furthermore, prime editors (PEs) have been used for both constructing and repairing LNS disease models, with variant correction efficiencies reaching up to 14%. (Jang et al., 2023).
The potential of induced pluripotent stem cells (iPSCs) in modeling genetic diseases has opened new avenues for understanding disease mechanisms and developing novel treatments. iPSCs have been successfully applied to model a variety of genetic disorders, such as Rett’s syndrome, Angelman syndrome, Dravet syndrome, Cockayne’s syndrome and X-fragile syndrome (Camoes et al., 2023; Haase et al., 2021; Lee et al., 2022; Schuster et al., 2019; Szepanowski et al., 2024), providing valuable insights into disease pathogenesis and therapeutic interventions. By generating patient-specific models, iPSC technology enables drug screening and personalized therapy testing. In the context of Lesch-Nyhan syndrome, iPSC-derived neural models, including neural organoids, could serve as innovative platforms for studying the effects of HPRT1 variants and evaluating potential therapies. These models, previously used for neurological disorders like Parkinson’s disease (PD) (Kim et al., 2024), could be adapted to explore the impact of HGPRT deficiency on neural development and function, while high-throughput screening in these systems could help identify novel drugs to alleviate the neurological symptoms of LNS. A total of six potential therapeutic drugs have been evaluated using iPSCs derived from LNS patients (Ruillier et al., 2020). The drugs, including SAM, all contain at least one adenosine structure, and SAM has been tested in LNS patients. Meanwhile, studies have shown that chromosome transplantation (CT) in iPSCs derived from LNS patients may represent a potential therapeutic approach for LNS (Paulis et al., 2020).In addition, the experimental models should be further optimized with the use of neural organoids cultured from iPSCs, which would enable both a high throughput drug screening and investigation of disease mechanisms (Vandana et al., 2023).
Lesch-Nyhan syndrome is clinically heterogeneous, presenting with a range of manifestations including motor retardation, extrapyramidal and pyramidal tract symptoms. It is frequently misdiagnosed as cerebral palsy. The early detection of hyperuricemia through blood uric acid testing is crucial for the prognosis of Lesch-Nyhan syndrome. Early diagnosis and appropriate treatment can significantly improve the prognosis of Lesch-Nyhan syndrome. Genetic diagnosis is the key to preventing and controlling Lesch-Nyhan syndrome. Carrier screening and prenatal diagnosis can reduce the recurrence of Lesch-Nyhan syndrome in families.
This study identifies a novel variant in the HPRT1 gene (HPRT1:c.104C > T) associated with Lesch-Nyhan Syndrome (LNS), a rare genetic disorder that leads to severe neurological symptoms. Our findings could improve the early diagnosis and genetic counseling for families affected by LNS, providing crucial insights for better management and prevention strategies.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of the First Affiliated Hospital of Anhui Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HZ: Data curation, Validation, Writing–original draft. GC: Investigation, Methodology, Software, Writing–original draft. TW: Data curation, Investigation, Resources, Writing–original draft. WC: Conceptualization, Supervision, Validation, Writing–review and editing. JY: Data curation, Formal Analysis, Investigation, Writing–review and editing. FL: Conceptualization, Funding acquisition, Investigation, Writing–review and editing. YX: Formal Analysis, Resources, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Key Research and Development Program of China (2021YFC1005303), National Natural Science Foundation of China (82101954, 82302101), China Postdoctoral Science Foundation (2023M740023), Anhui Provincial Natural Science Foundation (2308085QH244), Postdoctoral Researchers Foundation of Anhui Province (2022A574), University Natural Foundation of Anhui Educational Committee (2022AH051161), and Research Fund of Anhui Institute of Translational Medicine (2023zhyx-C23).
We extend our gratitude to the patient and their family for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1512070/full#supplementary-material
AlBakheet, A., AlQudairy, H., Alkhalifah, J., Almoaily, S., Kaya, N., and Rahbeeni, Z. (2022). Detailed genetic and clinical analysis of a novel de novo variant in HPRT1: case report of a female patient from Saudi Arabia with lesch-nyhan syndrome. Front. Genet. 13, 1044936. [Case Reports]. doi:10.3389/fgene.2022.1044936
Baba, S., Saito, T., Yamada, Y., Takeshita, E., Nomura, N., Yamada, K., et al. (2017). Novel mutation in HPRT1 causing a splicing error with multiple variations. Nucleosides Nucleotides Nucleic Acids 36 (1), 1–6. doi:10.1080/15257770.2016.1163381
Bell, S., McCarty, V., Peng, H., Jefri, M., Hettige, N., Antonyan, L., et al. (2021). Lesch-Nyhan disease causes impaired energy metabolism and reduced developmental potential in midbrain dopaminergic cells. Stem Cell Rep. 16 (7), 1749–1762. doi:10.1016/j.stemcr.2021.06.003
Benchoua, A., Lasbareilles, M., and Tournois, J. (2021). Contribution of human pluripotent stem cell-based models to drug discovery for neurological disorders. Cells 10 (12), 3290. doi:10.3390/cells10123290
Camoes, D. S. J., Appleton, C., Cazaux, M. F., Covas, R., Bekman, E. P., and Da, R. S. (2023). Stem cell models of Angelman syndrome. Front. Cell. Dev. Biol. 11, 1274040. doi:10.3389/fcell.2023.1274040
Chen, B. C., Balasubramaniam, S., McGown, I. N., O'Neill, J. P., Chng, G. S., Keng, W. T., et al. (2014). Treatment of Lesch-Nyhan disease with S-adenosylmethionine: experience with five young Malaysians, including a girl. Brain. Dev. 36 (7), 593–600. doi:10.1016/j.braindev.2013.08.013
Cho, J. H., Choi, J. H., Heo, S. H., Kim, G. H., Yum, M. S., Lee, B. H., et al. (2019). Phenotypic and molecular spectrum of Korean patients with Lesch-Nyhan syndrome and attenuated clinical variants. Metab. Brain Dis. 34 (5), 1335–1340. doi:10.1007/s11011-019-00441-0
Deng, H., Xiong, B. T., Wu, Y., and Wang, W. (2023). Deep brain stimulation in Lesch-Nyhan syndrome: a systematic review. Neurosurg. Rev. 46 (1), 40. doi:10.1007/s10143-023-01950-4
Dinasarapu, A. R., Sutcliffe, D. J., Seifar, F., Visser, J. E., and Jinnah, H. A. (2022). Abnormalities of neural stem cells in Lesch-Nyhan disease. J. Neurogenet. 36 (2-3), 81–87. doi:10.1080/01677063.2022.2129632
Dressler, D., Adib, S. F., and Rosales, R. L. (2021). Botulinum toxin therapy of dystonia. [Journal article; research support, non-U.S. Gov't; review]. J. Neural Transm. 128 (4), 531–537. doi:10.1007/s00702-020-02266-z
Fang, H. H., Lee, C. L., Chen, H. J., Chuang, C. K., Chiu, H. C., Chang, Y. H., et al. (2024). Whole exome sequencing facilitates early diagnosis of lesch-nyhan syndrome: a case series. Diagnostics 14 (24), 2809. doi:10.3390/diagnostics14242809
Fu, R., Ceballos-Picot, I., Torres, R. J., Larovere, L. E., Yamada, Y., Nguyen, K. V., et al. (2014). Genotype-phenotype correlations in neurogenetics: lesch-Nyhan disease as a model disorder. Brain. 137 (Pt 5), 1282–1303. doi:10.1093/brain/awt202
Fu, R., Sutcliffe, D., Zhao, H., Huang, X., Schretlen, D. J., Benkovic, S., et al. (2015). Clinical severity in Lesch-Nyhan disease: the role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 114 (1), 55–61. doi:10.1016/j.ymgme.2014.11.001
Gilbert, C., Sauer, M., and Cheng, J. (2021). Reduction of self-mutilating behavior and improved oromotor function in a patient with Lesch-Nyhan syndrome following botulinum toxin injection: a case report. J. Pediatr. Rehabil. Med. 14 (1), 133–136. doi:10.3233/PRM-200729
Guo, M., Chen, Y., Lin, L., Wang, Y., Wang, A., Yuan, F., et al. (2022). The study on the clinical phenotype and function of HPRT1 gene. Child. Neurol. Open 9, doi:10.1177/2329048X221108821
Haase, F. D., Coorey, B., Riley, L., Cantrill, L. C., Tam, P., and Gold, W. A. (2021). Pre-clinical investigation of rett syndrome using human stem cell-based disease models. Front. Neurosci. 15, 698812. [Journal Article; Review]. doi:10.3389/fnins.2021.698812
Harris, J. C. (2018). Lesch-Nyhan syndrome and its variants: examining the behavioral and neurocognitive phenotype. Curr. Opin. Psychiatr. 31 (2), 96–102. doi:10.1097/YCO.0000000000000388
Huang, J., Zhang, C., Guo, Q., Zhang, X., Ma, L., Zhan, Y., et al. (2018). Lesch-nyhan syndrome in a Chinese family with mutation in the hypoxanthine-guanine phosphoribosyltransferase gene. Clin. Lab. 64 (1), 197–200. [Case Reports]. doi:10.7754/Clin.Lab.2017.170813
Jang, G., Shin, H. R., Do, H. S., Kweon, J., Hwang, S., Kim, S., et al. (2023). Therapeutic gene correction for lesch-nyhan syndrome using CRISPR-mediated base and prime editing. Mol. Ther. Nucleic Acids 31, 586–595. [Journal Article]. doi:10.1016/j.omtn.2023.02.009
Jian, W., Peng, W., Li, H., Feng, Q., Wang, W., and Su, Q. (2013). Molecular characterization and structure analysis of hprt in a Chinese patient with lesch-nyhan disease. Nucleosides Nucleotides Nucleic Acids 32 (4), 189–195. doi:10.1080/15257770.2013.774013
Keebaugh, A. C., Sullivan, R. T., and Thomas, J. W. (2007). Gene duplication and inactivation in the HPRT gene family. Genomics 89 (1), 134–142. doi:10.1016/j.ygeno.2006.07.003
Kim, M. S., Kim, H., and Lee, G. (2024). Precision medicine in Parkinson's disease using induced pluripotent stem cells. Adv. Healthc. Mater. 13 (21), e2303041. doi:10.1002/adhm.202303041
Krajewski, O., Opielka, M., Urbanowicz, K., Chojnowski, K., Kochany, P., Pawlowski, K., et al. (2024). Management of neurological symptoms in lesch-nyhan disease: a systematic review. Neurosci. Biobehav. Rev. 165, 105847. [Journal Article; Review; Systematic Review]. doi:10.1016/j.neubiorev.2024.105847
Lee, A., Xu, J., Wen, Z., and Jin, P. (2022). Across dimensions: developing 2d and 3d human iPSC-based models of fragile x syndrome. J. Article; Res. Support 11 (11), 1725. doi:10.3390/cells11111725
Lesch, M., and Nyhan, W. L. (1964). A familial disorder of uric acid metabolism and central nervous system function. Am. J. Med. 36, 561–570. [Journal Article]. doi:10.1016/0002-9343(64)90104-4
Li, L., Qiao, X., Liu, F., Wang, J., Shen, H., Fu, H., et al. (2022). Description of the molecular and phenotypic spectrum of lesch-nyhan disease in eight Chinese patients. Front. Genet. 13, 868942. [Journal Article]. doi:10.3389/fgene.2022.868942
Liu, N., Zhuo, Z. H., Wang, H. L., Kong, X. D., Shi, H. R., Wu, Q. H., et al. (2015). Prenatal diagnosis based on HPRT1 gene mutation in a lesch-nyhan family. J. Obstet. Gynaecol. 35 (5), 490–493. [Case Reports; Journal Article; Research Support, Non-U.S. Gov't]. doi:10.3109/01443615.2014.969209
Paulis, M., Susani, L., Castelli, A., Suzuki, T., Hara, T., Straniero, L., et al. (2020). Chromosome transplantation: a possible approach to treat human x-linked disorders. Mol.Ther.-Methods Clin. Dev. 17, 369–377. [Journal Article]. doi:10.1016/j.omtm.2020.01.003
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. [Consensus Development Conference; Guideline; Journal Article; Research Support, N.I.H., Extramural]. doi:10.1038/gim.2015.30
Ruillier, V., Tournois, J., Boissart, C., Lasbareilles, M., Mahe, G., Chatrousse, L., et al. (2020). Rescuing compounds for Lesch-Nyhan disease identified using stem cell-based phenotypic screening. JCI Insight 5 (4), e132094. doi:10.1172/jci.insight.132094
Satow, T., Ogawa, M., and Komuro, T. (2021). Intrathecal baclofen therapy for Lesch-Nyhan disease: illustrative case. J. Neurosurg. Case Lessons 1 (1), CASE202. doi:10.3171/CASE202
Schuster, J., Fatima, A., Sobol, M., Norradin, F. H., Laan, L., and Dahl, N. (2019). Generation of three human induced pluripotent stem cell (iPSC) lines from three patients with Dravet syndrome carrying distinct SCN1A gene mutations. Stem Cell Res. 39, 101523. doi:10.1016/j.scr.2019.101523
Szepanowski, L. P., Wruck, W., Kapr, J., Rossi, A., Fritsche, E., Krutmann, J., et al. (2024). Cockayne syndrome patient iPSC-derived brain organoids and neurospheres show early transcriptional dysregulation of biological processes associated with brain development and metabolism. Cells 13 (7), 591. [Journal Article; Research Support, Non-U.S. Gov't]. doi:10.3390/cells13070591
Tong, M., Li, Q., Sun, A., Chen, C., and Hu, S. (2022). Analysis of HPRT1 gene variant and prenatal diagnosis for a Chinese pedigree with lesch-nyhan syndrome but no specimen from affected probands. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 39 (11), 1243–1246. [English Abstract; Journal Article]. doi:10.3760/cma.j.cn511374-20211029-00861
Vandana, J. J., Manrique, C., Lacko, L. A., and Chen, S. (2023). Human pluripotent-stem-cell-derived organoids for drug discovery and evaluation. Cell Stem Cell 30 (5), 571–591. doi:10.1016/j.stem.2023.04.011
Visser, J. E., Chorin, O., and Jinnah, H. A. (2024). Very early levodopa may prevent self-injury in lesch-nyhan disease. Pediatr. Neurol. 155, 156–159. doi:10.1016/j.pediatrneurol.2024.03.020
Wang, D., Zhao, J., Teng, J., Li, W., Zhao, X., and Li, L. (2023). Genetic analysis of a Chinese pedigree with lesch-nyhan syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 40 (6), 723–726. doi:10.3760/cma.j.cn511374-20220916-00629
Yang, L., and Guo, H. (2022). Case report: early-onset renal failure as presenting sign of lesch-nyhan disease in infancy. Front. Pediatr. 10, 1080486. [Case Reports]. doi:10.3389/fped.2022.1080486
Keywords: lesch-nyhan syndrome, HPRT1, X-linked disease, HGPRT, novel variant
Citation: Zheng H, Chen G, Wang T, Cheng W, Yuan J, Liu F and Xu Y (2025) Case report: Whole exome sequencing identifies a novel variant in the HPRT1 gene in a male with developmental delay. Front. Genet. 16:1512070. doi: 10.3389/fgene.2025.1512070
Received: 16 October 2024; Accepted: 10 February 2025;
Published: 28 February 2025.
Edited by:
Jeffrey Dennis Calhoun, Northwestern University, United StatesReviewed by:
Ted Han, New York Genome Center, United StatesCopyright © 2025 Zheng, Chen, Wang, Cheng, Yuan, Liu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanhong Xu, eHlob25nMTk2NEAxNjMuY29t; Fang Liu, bWF1ZC5saXVAb3V0bG9vay5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.