 A. Alakkas
A. Alakkas H. Shinawi1
H. Shinawi1 J. A. Bajwa
J. A. Bajwa
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 11 March 2025
Sec. Applied Genetic Epidemiology
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1504744
Dystonia prevalence and presentation varies both ethnically and geographically. There is a paucity of data on the clinical presentation of dystonia patients in Saudi Arabia and among Arabs. In this study we provide the largest description of dystonia patients in Saudi Arabia. In our population, majority, 42% of all patients with dystonia had an inherited dystonia, while 34.8% had idiopathic dystonia. In addition, we found 3 patients with homozygous GCH1 variants who displayed the classic phenotype of dopa-responsive dystonia. Two had Variant of Uncertain Significance that has been recently reclassified as likely pathogenic, and another novel homozygous Asp119Asn variant, not previously reported in ClinVar. It is the hope that this paper would be the first step for future prospective studies.
Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal postures, and/or repetitive movements (Albanese et al., 2013). Dystonia, is a highly heterogenous disorder with an ever-evolving definition and classification scheme (Albanese et al., 2013; Fahn, 2011). In 2013, an international panel of experts reviewed the definition and classification of dystonia (Albanese et al., 2013). The report classifies dystonia by a combination of two axis clinical features and etiology. Clinical features include age of onset, body distribution, disease course and associated features. Etiology is divided into inherited, acquired or idiopathic (Albanese et al., 2013; Albanese et al., 2019). The pathophysiology of dystonia is highly complex; however, genetics plays a significant role in both inherited and idiopathic dystonia (Charlesworth et al., 2013). Dystonia is a highly complex disorder with considerable genotype-phenotype variability even within members of the same family. Genetics also has a role in idiopathic dystonia where focal idiopathic dystonia tends to run in families (Charlesworth et al., 2013; Lange et al., 2021). Next-generation sequencing has resulted in the discovery of several genes causing inherited dystonia. Genetics also has a role in idiopathic dystonia where focal idiopathic dystonia tends to run in families (Charlesworth et al., 2013).
Dystonia prevalence and presentation varies both ethnically and geographically (Bailey et al., 2022). The phenotypic heterogeneity of dystonia complicates the understanding of the natural history of dystonic disorders and their response to treatment. To combat this, the dystonia collation has created a multicenter network for clinical and translational studies, unfortunately, almost all the centers included are from north American and western Europe (Kilic-Berkmen et al., 2021). To date, there is very little data on the prevalence, and clinical features of dystonia outside these regions. For example, little is known about the clinical and genetic features of patients with dystonia in Kingdom of Saudi Arabia (KSA) and the Arab world. Only one abstract described the genetic features of dystonia in Saudi Arabia (Bohlega AA et al., 2016). This paper suggested that myoclonus-dystonia might be the most common type of non-acquired dystonia in Saudi Arabia (Bohlega AA et al., 2016). Given the paucity of information on dystonic disorders in Saudi Arabia; additional studies are needed to guide clinical practice and the allocation of resources. The aim of this study is to evaluate the clinical and genetic features of patients with dystonia in a large movement disorder center. It is the hope that this paper would be the first step for future prospective studies.
Retrospective observational study using chart review at King Fahad medical city (KFMC), Riyadh.
Patients of all ages with primary dystonia who received care at KFMC from January 2003 to February 2023.
People with inherited or idiopathic dystonia (with or without a hereditary pattern) of all ages.
Diagnosed with acquired dystonia.
Data was collected from KFMC’s epic medical record system. Following Institutional Review Board (IRB) approval, we extracted the Medical Record Numbers of patients who have dystonia documented in notes, problem list or listed as a diagnosis. All charts of patients with history of dystonia were reviewed.
The following data was collected from each chart:
1- Demographic data.
a. date of birth, gender, nationality and city of residence.
2- Age of onset of dystonia.
a. Infancy (birth to 2 years).
b. Childhood (3–12 years).
c. Adolescence (13–20 years).
d. Adulthood (21–and older).
3- Anatomical onset of dystonia.
a. craniocervical dystonia (Blepharospasm/Oromandibular/cervical), Larynx (Laryngeal) or Limbs (Limb dystonia).
4- Classification of dystonia.
a. Focal (one body part) or segmental (more than 2 contiguous body parts) or multifocal (more than two non-contiguous body parts) or Hemidystonia (Ipsilateral arm and leg are involved) or generalized (more than 3 body parts).
b. Isolated (Dystonia is the only motor feature, with the exception of tremor) or combined (Dystonia is combined with other movement disorders) or complex (Dystonia accompanied by neurologic or systemic manifestations beyond movement disorders).
c. Inherited with proven gene origin or idiopathic.
5- Genetic disorder if identified.
6- Family history of dystonia or other psychiatric or neurological disorder.
7- Brain Magnetic resonance imaging (MRI) findings.
8- Electromyography & Nerve Conduction Studies (NCS/EMG) findings.
9- Treatment of dystonia.
a. Current and past anti-dystonic medications.
b. Botox injections.
c. Surgical interventions such as Deep brain stimulation (DBS).
10- Hospitalization Data.
11- Mortality Data.
Study was approved by KFMC IRB, protocol number 23-168.
Statistics done using SPSS software. Categorical variables are presented as frequencies and percentages, while continuous variables are presented as mean and standard deviation. To determine factors associated with the development of idiopathic and inherited dystonia in this population logistic regression and Chi-Square Tests are used. T-Test used for continues variables. A p-value of <0.05 was regarded as statistically significant.
Demographics and clinical characteristics are summarized in Table 1. A total of 86 participants met the including and exclusion criteria. Participants are mostly male 57% with a mean age of 26.5 years (SD 19.1). They are mostly Saudi nationals 95.3% with the majority residing in the central 67% followed by southern 18.2% regions.

Table 1. Demographics and classification of dystonia.
20.9% had a family history of dystonia. More than a third of patients had some degree of consanguinity. Of those with consanguineous parents, 88.1% had parents who are first cousins. Consanguinity was highest in the southern region where 66.7% had consanguineous parents, but this was not statistically significantly different to other regions. Patients with inherited dystonia had a higher percentage of consanguineous parents as compared with idiopathic dystonia (63.8% vs 7.7%). In addition, having parents who are first cousins was a predictor of developing inherited dystonia as compared with idiopathic dystonia [OR 16.5 (4.3- 63.1) P < 0.001].
More than half (55.8%) of patients had dystonia onset before adulthood (Table 1). Majority, 54.7% of all patients who met our inclusion and exclusion criteria had an inherited dystonia, while 45.3% had idiopathic dystonia (Table 1). Acquired dystonia made up 24.4% of original cohort, with cerebral palsy 65.4% followed by stroke 15.4% being the most common etiologies (Supplementary Table 1).
Overall, 45.3% of all patients had generalized dystonia. Body distribution was statistically significantly different between inherited and idiopathic dystonia. (P < 0.001) The Majority, 79.5% of generalized dystonia is inherited, while the majority of focal dystonia 88.9% is idiopathic in etiology. Patients with generalized dystonia were 31 times more likely to have inherited dystonia as compared with focal dystonia. (P < 0.001).
Isolated and complex dystonia had roughly equal occurrence 44.2% and 41.9% respectively (Table 1).
Forty-seven individuals had inherited dystonia (Tables 1, 2). Patients with inherited dystonia were more likely to have a younger age of onset as compared with idiopathic dystonia with a mean 14.13 [(CI 10.8- 17.4) SD 11.2] and 41.4 [(CI 36.3 – 46.5), SD 15.8] respectively [Mean Difference 27.2 (CI 33 -21.4), P < 0.001] (Supplementary Figure 2). Concerning the genetic basis of inherited dystonia, 78.7% is associated with an autosomal recessive pattern of disease, whereas only 14.8% is linked to autosomal dominant inheritance, and 4.2% is attributed to X-linked recessive inheritance.
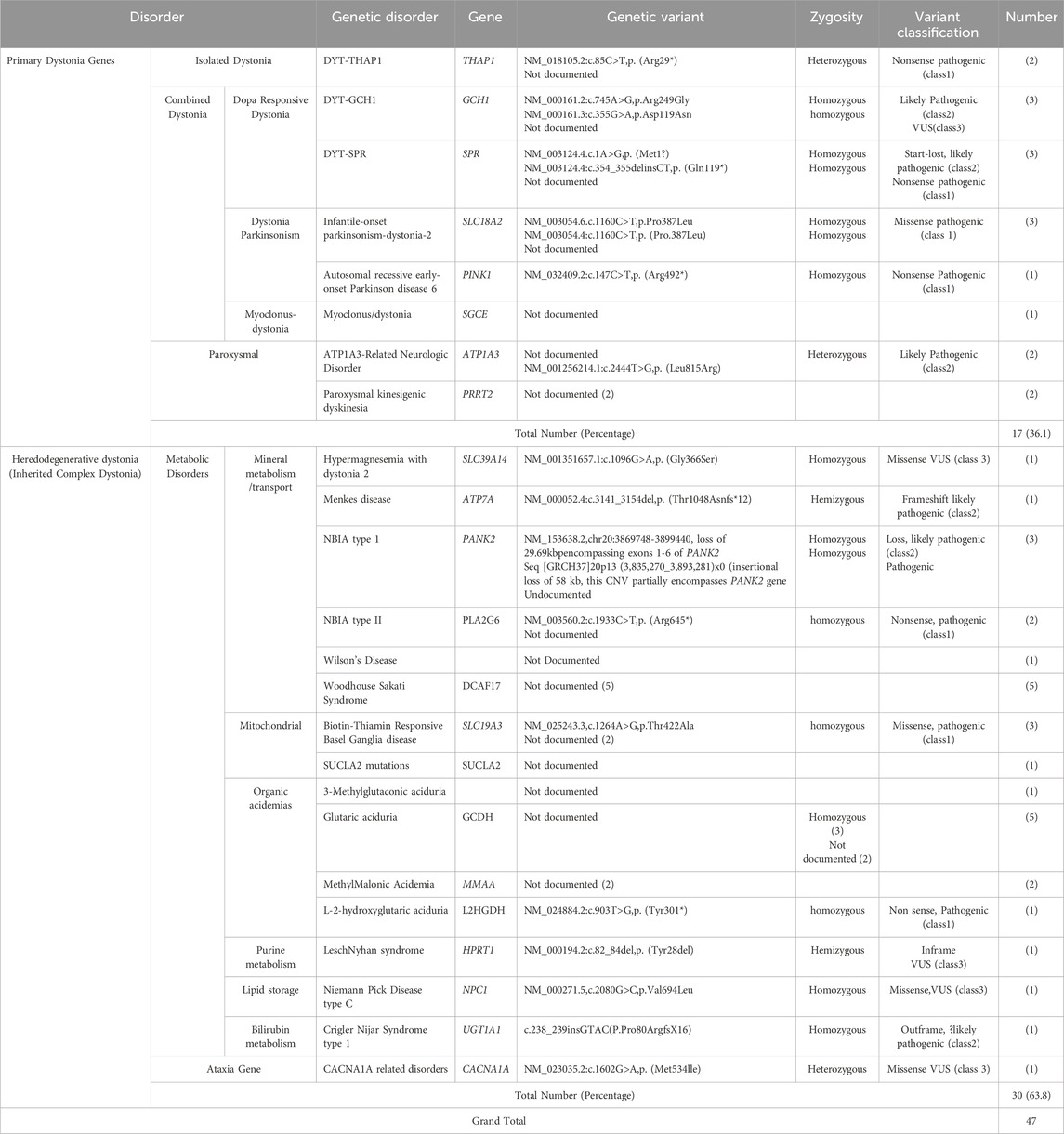
Table 2. Inherited dystonias.
Regarding genetic etiology, majority of patients 63.8% had a heredodegenerative dystonia (inherited complex dystonia). Majority 46.7% of heredodegenerative dystonia had onset in infancy. However, most 47% of primary genetic dystonia had onset in childhood. Of the heredodegenerative dystonias, mineral metabolism disorders were most common followed by organic acidemias in our cohort. Woodhouse-Sakati Syndrome was the most common mineral metabolism disorder encountered and glutaric aciduria was the most common organic academia (Table 2). Regarding primary isolated dystonia DYT-THAP1 occurred in two individuals, we had no genetically confirmed cases of DYT-TOR1A. However, one patient had childhood onset lower limb dystonia that later generalized, this patient did not undergo genetic testing (Table 2).
Majority of patients with idiopathic dystonia are male 61.5% but gender was not predictive of etiology (P 0.514). Idiopathic dystonias are mostly isolated 84.6%, of craniocervical anatomical onset 66.7% and focal 61.5%. Cervical dystonia is the most common form of idiopathic dystonia followed by Blepharospasm (Supplementary Table 2).
46 (41.8%) of individuals had a MRI brain done, 58.3% of them had an abnormal MRI. None of the patients had a documented EMG/NCS study.
94.1% of individuals are on treatment, most on benzodiazepines 45.3% followed by baclofen and anticholinergics (Supplementary Table 3). Botox was given to 37.2%, majority of which have focal dystonia 62.5%.
Bilateral Gpi DBS was performed in 5 (5.8%) individuals, current mean age is 37.6 (SD 15.3). Mean age at time of surgery is 31.2 (Range 21–59 years). All individuals who underwent DBS had generalized dystonia, 4 isolated and one combined with myoclonus. All patients had age of onset of dystonia before adulthood. The devices implanted included 4 Medtronic systems and one Abbot Medical Device.
56.9% were hospitalized at least once. Two deaths observed one due to pneumonia and another unknown. Both had NBIA type 1. The mean age of the patients who died was 8 (SD 4.2) (Supplementary Table 3).
Dystonia prevalence and presentation varies both ethnically and geographically (Bailey et al., 2022). It is highly dependent on age of the population and gender (Defazio et al., 2004; Nutt et al., 1988). In this study we present the largest description of dystonia patients from Saudi Arabia. Our population is young (mean 26.5) and male predominant 57%. This is in keeping with the Saudi population which is slightly male predominant (50.2 percent) and has a mean age of 25 years (The General Authority for Statistics, Saudi Census, 2022).
Overall, generalized dystonia was the most common type encountered in our cohort. It is important to note that the results are unlike those of prior studies, where focal dystonia was the most common form (Meoni et al., 2020; Dressler et al., 2022; Defazio et al., 2004). For example, in the Hanover study that included 316 participants with dystonia, generalized dystonia made up only 4% of all dystonia (Dressler et al., 2022). The primary reason for the discrepancy is that our study looks at the expertise of one single center, whereas others were epidemiological studies. Thus, referral bias plays a very significant role where patients with isolated and mild dystonia might not be referred to our center. In a retrospective study done in the United Arab Emeritus they also had a predominance of focal dystonia (Waqar et al., 2024).They had equal occurrence of Males to Females and majority had dystonia onset in adulthood (Waqar et al., 2024). Their cohort only included those who were aged 12 and older. Our cohort however included individuals of all age groups and was male predominant 57%.
Regarding the classification of dystonia in our cohort, this was similar to prior studies (Albanese et al., 2013; de Carvalho Aguiar and Ozelius, 2002). We found that Idiopathic dystonia is mostly isolated, of craniocervical anatomical onset and focal. While inherited dystonia was mostly generalized and had a younger age of onset. Indicating as others have noted that genetic testing in latter group is more likely to reveal positive results.
Regarding etiology, inherited dystonia was the most common. As noted above referral bias likely plays a large role. In addition to a young population and a high degree of consanguinity. Indeed, more than a third of patients had some degree of consanguinity. Not surprisingly we found that having parents who are first cousins was a predictor of developing inherited dystonia as compared with idiopathic dystonia [OR 16.5 (4.3- 63.1) P < 0.001]. This social custom would explain that glutaric aciduria and Woodhouse Sakati syndrome, both autosomal recessive disorders were the most predominant inherited disorders. It is important to note that this is unlike a previous abstract published by our collogues at King Faisal Specialist Hospital, another tertiary care center where myoclonus dystonia was the most common inherited dystonia in their cohort (Bohlega AA et al., 2016). The total population, mean age and detailed inclusion/exclusion criteria of their data remains to be published as such one could only speculate about the discrepancy. One scenario could involve KFMC operating as both a secondary and tertiary level hospital for both adults and pediatric patients, accommodating a broader patient demographic compared to a strictly tertiary care facility.
Another distinctive aspect of our data is the predominance of autosomal recessive disorders, contrasting with the predominant autosomal dominant pattern described in the literature. In a study by Zech et al., they reported 51.9% of variants being inherited through autosomal dominant disorder and 41.6% through autosomal recessive inheritance. This difference is attributed to the high level of consanguinity within our cohort (Zech et al., 2020).
Regarding early onset inherited dystonia, in our cohort, we report three pediatric patients with homozygous GCH1 variants who displayed the classic phenotype of dopa-responsive dystonia and demonstrated a significant and sustained response to low doses of L-dopa. Among these patients, two are twin sisters, both exhibiting the homozygous Arg249Gly variant. This variant has been classified as a Variant of Uncertain Significance (VUS); however, recent classification in ClinVar suggests its likely pathogenic, particularly in a child presenting with foot dystonia. The third patient, a male, experienced symptom onset at the age of 9. Whole exome analysis revealed a novel homozygous Asp119Asn variant, not previously reported in ClinVar. Reviewing the literature, we found another patient with similar presentation to our patient, 12 years old girl experiencing dystonia, diurnal fluctuations, and consistently positive response to L-dopa treatment. She exhibited a homozygous Arg249Ser mutation, with normal levels of GCH-1 mRNA but diminished GCH-1 activity (Hwu et al., 1999).
Furukawa documented 2 cases that fell between DRD and AR GCH-1 deficient HPA. In both instances, individuals had compound heterozygous mutations in GCH-1 and exhibited significantly lower levels of BH4 and neopterin in the cerebrospinal fluid compared to those with DRD. Patient 1 exhibited both frame shift and missense mutations and he had severe phenotype, while Patient 2 had two missense mutations and his phenotype was milder. They hypothesized that the mutation in Patient 1 was more severe than that in Patient 2, leading to a more pronounced biochemical deficiency and clinical phenotype (Furukawa et al., 1998). While it was previously understood that individuals with homozygous GCH1 variants exhibit a severe neurological phenotype emerging early in life, and those with heterozygous variants present a milder phenotype known as DRD, this categorization is now less accurate, particularly due to the discovery of new variants with mild effects and the clinical finding in our patients support this notion. GCH-1 serves as a remarkable illustration demonstrating how variations in a single gene can lead to distinct phenotypes like classic dopa responsive dystonia (DRD) and atypical DRD, contingent upon the degree of mutation severity and enzyme dysfunction rather than the quantity of mutations (Lee and Jeon, 2014).
Regarding the distribution of dystonia within the various Saudi regions, it is not unexpected that the percentage of dystonia was most common in the central most populous region. However, the Southern region is second to last in terms of overall population. However, it is second in terms of the prevalence of dystonia. This could be due to higher consanguinity rates in this area, genetic makeup, and of course referral basis. Given that kinship plays a significant role, an educational campaign should emphasize the importance of premarital counseling and educate the public on the availability of preimplantation genetic testing. Indeed, prevention of this disorder will likely be cost effective as at least 58.1% had at least one hospitalization.
Regarding treatment, the majority were treated with benzodiazepines, followed by anticholinergic and baclofen. Whether the preference for benzodiazepines is due to efficacy cannot be determined by our study. The reason for the preference for benzodiazepines might be due to physician comfort and availability. Future prospective studies must be performed to determine medication efficacy in a more homogeneous population.
Our paper has several areas for improvement; as noted above, we cannot determine efficacy in our retrospective study. We are also unable to know the severity of the disease. In addition, we had some missing data. In addition, we cannot determine the incidence and prevalence of dystonia in Saudi Arabia. On the other hand, this is the first and largest dystonia cohort in Saudi Arabia. It is the hope that future larger national and prospective studies will be performed to better understand the predominate phenotype and response to medications.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the King Fahad Medical City (KFMC) Institutional Review Board (IRB; study protocol number: 23-168). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
AA: Conceptualization, Data curation, Methodology, Writing–original draft, Writing–review and editing. HS: Writing–original draft, Writing–review and editing. JB: Writing–original draft, Writing–review and editing. OA: Writing–review and editing. AA-H: Data curation, Formal Analysis, Supervision, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank KFMC Research Center, Faculty of Medicine for their support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1504744/full#supplementary-material
Albanese, A., Bhatia, K., Bressman, S. B., Delong, M. R., Fahn, S., Fung, V. S., et al. (2013). Phenomenology and classification of dystonia: a consensus update. Mov. Disord. 28 (7), 863–873. doi:10.1002/mds.25475
Albanese, A., Di Giovanni, M., and Lalli, S. (2019). Dystonia: diagnosis and management. Eur. J. Neurol. 26 (1), 5–17. doi:10.1111/ene.13762
Bailey, G. A., Rawlings, A., Torabi, F., Pickrell, O., and Peall, K. J. (2022). Adult-onset idiopathic dystonia: a national data-linkage study to determine epidemiological, social deprivation, and mortality characteristics. Eur. J. Neurol. 29 (1), 91–104. doi:10.1111/ene.15114
Bohlega, S. A., Aldakheel, A., Alkhairallah, T., Alkhani, A., Almuhaizea, M., and Alotaibi, F. (2016). The phenotypic and genetic features of myoclonus-dystonia in six Saudi Arabian kindreds [abstract]. Mov. Disord. 31 (Suppl. 2), 2016. Available at: https://www.mdsabstracts.org/abstract/the-phenotypic-and-genetic-features-of-myoclonus-dystonia-in-six-saudi-arabian-kindreds (Accessed on March 01, 2025).
Charlesworth, G., Bhatia, K. P., and Wood, N. W. (2013). The genetics of dystonia: new twists in an old tale. Brain 136 (Pt 7), 2017–2037. doi:10.1093/brain/awt138
de Carvalho Aguiar, P. M., and Ozelius, L. J. (2002). Classification and genetics of dystonia. Lancet Neurol. 1 (5), 316–325. doi:10.1016/s1474-4422(02)00137-0
Defazio, G., Abbruzzese, G., Livrea, P., and Berardelli, A. (2004). Epidemiology of primary dystonia. Lancet Neurol. 3 (11), 673–678. doi:10.1016/S1474-4422(04)00907-X
Dressler, D., Altenmüller, E., Giess, R., Krauss, J. K., and Adib Saberi, F. (2022). The epidemiology of dystonia: the Hannover epidemiology study. J. Neurol. 269 (12), 6483–6493. doi:10.1007/s00415-022-11310-9
Fahn, S. (2011). Classification of movement disorders. Mov. Disord. 26 (6), 947–957. doi:10.1002/mds.23759
Furukawa, Y., Lang, A. E., Trugman, J. M., Bird, T. D., Hunter, A., Sadeh, M., et al. (1998). Gender-related penetrance and de novo GTP-cyclohydrolase I gene mutations in dopa-responsive dystonia. Neurology 50 (4), 1015–1020. doi:10.1212/wnl.50.4.1015
Hwu, W. L., Wang, P. J., Hsiao, K. J., Wang, T. R., Chiou, Y. W., and Lee, Y. M. (1999). Dopa-responsive dystonia induced by a recessive GTP cyclohydrolase I mutation. Hum. Genet. 105 (3), 226–230. doi:10.1007/s004390051093
Kilic-Berkmen, G., Wright, L. J., Perlmutter, J. S., Comella, C., Hallett, M., Teller, J., et al. (2021). The dystonia coalition: a multicenter network for clinical and translational studies. Front. Neurol. 12, 660909. doi:10.3389/fneur.2021.660909
Lange, L. M., Junker, J., Loens, S., Baumann, H., Olschewski, L., Schaake, S., et al. (2021). Genotype–phenotype relations for isolated dystonia genes: MDSGene systematic review. Mov. Disord. 36 (5), 1086–1103. doi:10.1002/mds.28485
Lee, W. W., and Jeon, B. S. (2014). Clinical spectrum of dopa-responsive dystonia and related disorders. Curr. Neurol. Neurosci. Rep. 14 (7), 461. doi:10.1007/s11910-014-0461-9
Meoni, S., Macerollo, A., and Moro, E. (2020). Sex differences in movement disorders. Nat. Rev. Neurol. 16 (2), 84–96. doi:10.1038/s41582-019-0294-x
Nutt, J. G., Muenter, M. D., Aronson, A., Kurland, L. T., and Melton, L. J. (1988). Epidemiology of focal and generalized dystonia in Rochester, Minnesota. Mov. Disord. 3 (3), 188–194. doi:10.1002/mds.870030302
Waqar, K., Thomas, M., Maiti, T., Defrietas, E., and Mittal, S. (2024). Dystonia in the Middle East - the Cleveland clinic abu dhabi experience, 122, 106808.
Keywords: dystonia, genetics, inherited, GCH1 variants, middle east
Citation: Alakkas A, Shinawi H, Bajwa JA, Alsinaidi O and Al-Hashim A (2025) Retrospective chart review of inherited and idiopathic dystonia. Front. Genet. 16:1504744. doi: 10.3389/fgene.2025.1504744
Received: 05 December 2024; Accepted: 19 February 2025;
Published: 11 March 2025.
Edited by:
Hui-Qi Qu, Children’s Hospital of Philadelphia, United StatesReviewed by:
Félix Javier Jiménez-Jiménez, Hospital Universitario del Sureste, SpainCopyright © 2025 Alakkas, Shinawi, Bajwa, Alsinaidi and Al-Hashim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Alakkas, YWxha2thcy5hbGpvaGFyYWhAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.