
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 25 March 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1497915
This article is part of the Research TopicInsights in Genetics of Common and Rare Diseases 2024View all 11 articles
 Qinghong Lin1,2,3,4†
Qinghong Lin1,2,3,4† Xuejun Wang1,2,3†Xiaoliao Peng1,2,3†Tian Han1,2,3
Xuejun Wang1,2,3†Xiaoliao Peng1,2,3†Tian Han1,2,3 Ling Sun1,2,3Xiaoyu Zhang1,2,3
Ling Sun1,2,3Xiaoyu Zhang1,2,3 Xingtao Zhou1,2,3*
Xingtao Zhou1,2,3*Purpose: To investigate the genetic characteristics of four Chinese families affected by keratoconus (KC).
Methods: In the four families affected by KC, medical records, clinical observations, and blood samples were collected from all individuals. One hundred subjects without KC served as healthy controls. All controls and subjects in the four families underwent whole exome sequencing of their genomic DNA and polymerase chain reaction to confirm the variants. All variants were analyzed using online software; and in silico predictions of three-dimensional protein structures were performed.
Results: The clinical manifestations in those first-degree family members of the probands were atypical. The following four variants were identified in the four probands and other family members with KC: heterozygous missense variation c.109G>A (p.Glu37Lys, rs369263247) in the procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1) gene; heterozygous missense variation c.3766G>A (p.Ala1256Thr, rs148216434) in the collagen type I alpha 1 (COL1A1) gene; heterozygous missense variant c.4364G>A (p.Gly1455Glu) in the collagen type V alpha 2 (COL5A2) gene; and missense variation c.976G>A (p.Glu326Ser) in the collagen type IV alpha 1 (COL4A1) gene. The above genotypes were co-segregated with corresponding phenotypes. All variations in these families appeared to be pathogenic.
Conclusion: Four variants in the PLOD1, COL1A1, COL5A2, and COL4A1 genes were identified in this study, which are collagen-coding genes and collagen crosslink regulatory genes and may be associated with the origin and development of KC. This study updates the knowledge of genes related to KC and the biomedical implications.
Keratoconus (KC) is a corneal disorder characterized by thinning of the cornea, conical protrusion, irregular astigmatism, and impaired vision, typically emerging during adolescence (Rabinowitz, 1998). KC is genetically characterized by a complex polygenic inheritance involving multiple susceptibility loci and environmental interactions, such as eye rubbing. About 5%–14% of KC patients have a family history (Bykhovskaya et al., 2016; Loukovitis et al., 2018). Collagens are the major component of the cornea stroma. The interaction between collagens and proteoglycans maintains the normal optical function of the cornea (Egrilmez et al., 2004; Legare et al., 2013). However, in the cornea of a patient with KC, there is decreased total collagen and the crosslinking of collagen molecules is defective (Farjadnia and Naderan, 2015). This can cause the cornea to thin and protrude forward due to reduced mechanical resistance. Therapies such as ultraviolet-A (UVA) crosslinking with riboflavin treat KC by increasing the mechanical resistance of the cornea involving anti-collagenase activity (Wu et al., 2021).
In the last two decades, accompanied by the advancement and prevalence of myopia correction surgery, young patients with myopia come in great numbers to the clinic. Through comprehensive pre-operative ocular examinations, an increasing number of corneal morphological changes have been identified and prompted further exploration of KC, especially the incipient stage of the disease. Therefore, identifying genetic susceptibility to KC is very important in the early diagnosis of these myopia correction surgery candidates. Numerous genes code for various collagens in the cornea stroma; their association with the central cornea thickness (CCT) and KC have been studied such as COL1A1, COLIA2, COL4A3, COL4A4, COL5A1, and COL8A2 (Dimasi et al., 2010; Vithana et al., 2011; Gao et al., 2013; Li et al., 2013; Abu-Amero et al., 2015; Saravani et al., 2015; Hosoda et al., 2020). Thus, further exploring genetic alterations in collagen-related genes is meaningful and will assist in the early detection and management of KC.
In the present study, we recruited four Chinese families with KC to evaluate their genetic characteristics. Four variants were identified and analyzed, and their biomedical implications are discussed.
This study enrolled 124 people including 24 family members from four Chinese families with KC and 100 healthy Chinese individuals without KC. Among the 24 family members, 4 were from family 1 (2 patients and 2 healthy members), 7 from family 2 (4 patients and 3 healthy members), 10 from family 3 (5 patients and 5 healthy members), and 3 from family 4 (2 patients and 1 healthy member). The 100 unrelated healthy individuals served as controls; they were diagnosed with refractive errors but without KC or any other corneal disease.
This study adhered to the Chinese Expert Consensus on the Diagnosis and Treatment of Keratoconus (2019) for the clinical diagnostic criteria and stages of KC (Chinese Medical Association Ophthalmology Branch Corneal Disease Group, 2019; Shi et al., 2019). Written informed consent was provided by all subjects. All participants received an examination, which included VA, anterior and posterior segments of the eye, and corneal evaluation using the Scheimpflug Camera System (Pentacam; Oculus Optikgeräte GmbH, Wetzlar, Germany). Approval for this study was obtained from the institutional review board of Fudan University (Shanghai, China) (Approval No. 2022128), and all procedures were carried out in accordance with the principles of the Declaration of Helsinki.
Exome sequencing (ES) was performed for 15 participants (all 4 members of family 1; Ⅲ1, Ⅲ2, Ⅱ3, and Ⅱ4 in family 2; Ⅳ1, Ⅲ1, Ⅲ2, and Ⅲ3 in family 3; all 3 members of family 4) using the method described in our previous study (Lin et al., 2023). Leukocyte-derived DNA was extracted, followed by ES and enrichment of exonic sequences from the genomic DNA of KC subjects. Subsequently, the loci that met the criteria for co-segregation in the four pedigrees were listed in the Supplementary Table S1. Considering population frequencies, genes related to the cornea, functional mutations in exonic regions or splice site regions, and conducting first-generation sequencing verification for all members of the pedigrees at the listed loci in Supplementary Table S1, the loci identified were ultimately selected as suspected mutations causing the diseases in the mentioned pedigrees. Reported variants (e.g., transforming growth factor beta-induced) and those occurring in corneal disorder, especially KC subjects at frequencies ≤1%, were assessed using the 1000 Genomes Project. Only variants co-segregating with KC in affected family members were considered candidate variants.
Variant validation and analyses were conducted following the methodology outlined in our previous work (Lin et al., 2023). All variants underwent analysis using online software tools as detailed in our published report (Lin et al., 2023) including Polyphen2, Sorting Intolerant from Tolerant, Protein Variation Effect Analyzer, fathmmMKL, and Mutation Taster. In addition, CADD v1.4 (http://cadd.gs.washington.edu) and VarSite datebase (www.ebi.ac.uk), with scoring in accordance with the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). Genomic Evolutionary Rate Profiling ++ (GERP++) (https://bio.tools/gerp) was used to predict the conservation of the variants.
To validate the candidate variants, polymerase chain reaction (PCR) and Sanger sequencing were performed on the other family members in the study and healthy controls. Primer3 was utilized for PCR primer design. The NCBI VARIANT, NCBI HomoloGene, and 1000 Genomes Project databases were used for validation and analysis. Three-dimensional (3D) protein structures of the variants were generated utilizing the online server Iterative Threading ASSEmbly Refinement (I-TASSER; https://zhanggroup.org/I-TASSER/).
Analyses of the protein–protein interaction (PPI) network were performed using the online Search Tool for the Retrieval Interacting Genes 11.5 (STRING) (https://cn.string-db.org). Then Cytoscape (v3.9.0) was applied to study the network of interactions. The filter of crucial proteins was set at a degree ≥5.
Figure 1 displays the genograms (1a: family 1; 1b: family 2; 1c: family 3; and 1d: family 4) and shows autosomal dominant inheritance of KC in all families. Two subjects in family 1 were diagnosed with KC. An 18-year-old female in this family was the proband (II.2); she had impaired vision in both eyes and was diagnosed as KC grade 1 in the right eye and grade 2 in the left eye; another subject in family 1 was 41 years old and had latent stage KC in her right eye and grade 1 in her left eye. In family 2, a 22-year-old young man was the proband (III.2) and his right eye was KC grade 1 and left eye was KC grade 2; the second subject in family 2 was a 23-year-old male with KC grade 2 in his right eye and incipient stage in his left eye; the third one was a 43-year-old female who had the same diagnosis as the second subject; the fourth subject in family 2 was a 45-year-old female with KC grade 2 in her right eye and grade 1 in her left eye. The proband in family 3 was a 17-year-old male (Ⅳ1) with KC grade 2 in both eyes; the second subject in family 3 was a 40-year-old male with KC grade 1 in his right eye and grade 2 in his left eye; the third subject in family 3 was a 22-year-old male with KC incipient stage in the right eye and latent stage in the left eye; the fourth subject was a 59-year-old female with KC grade 2 in both eyes; the fifth subject in family 3 was a 55-year-old female with KC incipient stage in her right eye and grade 1 in her left eye. In family 4, the proband was a 23-year-old male (II.1) with KC latent stage in his right eye and grade 1 in his left eye; another subject was a 45-year-old female with KC grade 2 in her right eye and incipient stage in her left eye. Corneal topography (Pentacam) reports of these probands are present in Figure 2 to show the mean values of posterior elevation of the cornea (PEC), maximum anterior surface curvatures (MASCs), and CCTs. The clinical data and grades of all KC patients in these four families are summarized in Table 1.
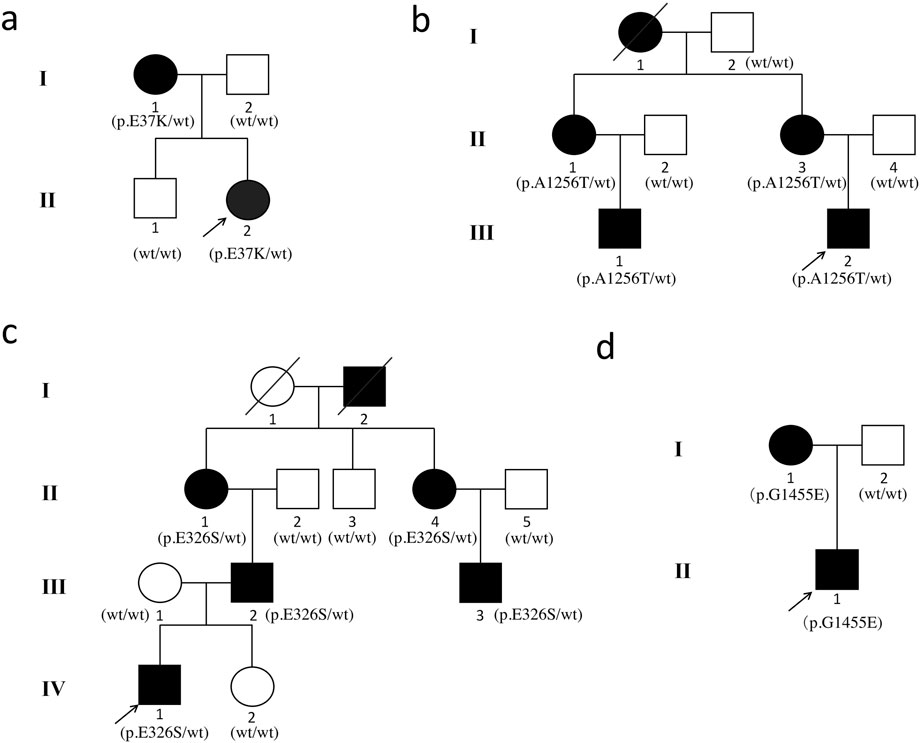
Figure 1. Pedigrees of the Chinese families with keratoconus. (a) family 1; (b) family 2; (c) family 3; and (d) family 4. The squares represent the males, and the circles represent the females. The open symbols indicate the unaffected family members. The solid symbols indicate the infected individuals. The diagonal line indicates the family member who passed away. The arrows indicate the probands.
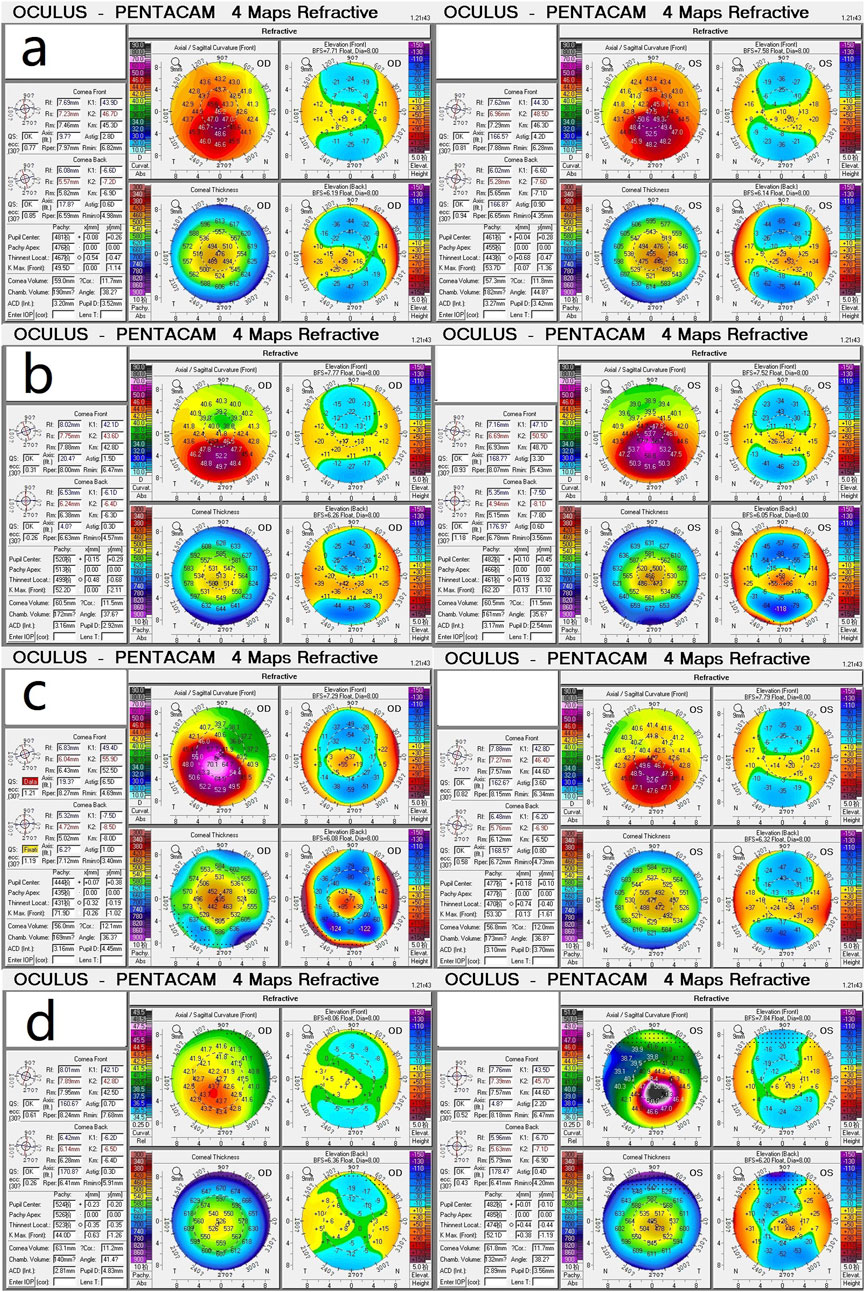
Figure 2. Corneal topography (Pentacam) reports show the mean values of posterior elevation of the cornea, maximum anterior surface curvatures, and central corneal thicknesses. (a) Proband (II.2) in family 1; (b) Proband (III.2) in family 2; (c) Proband (Ⅳ1) in family 3; (d) Proband (II.1) in family 4.
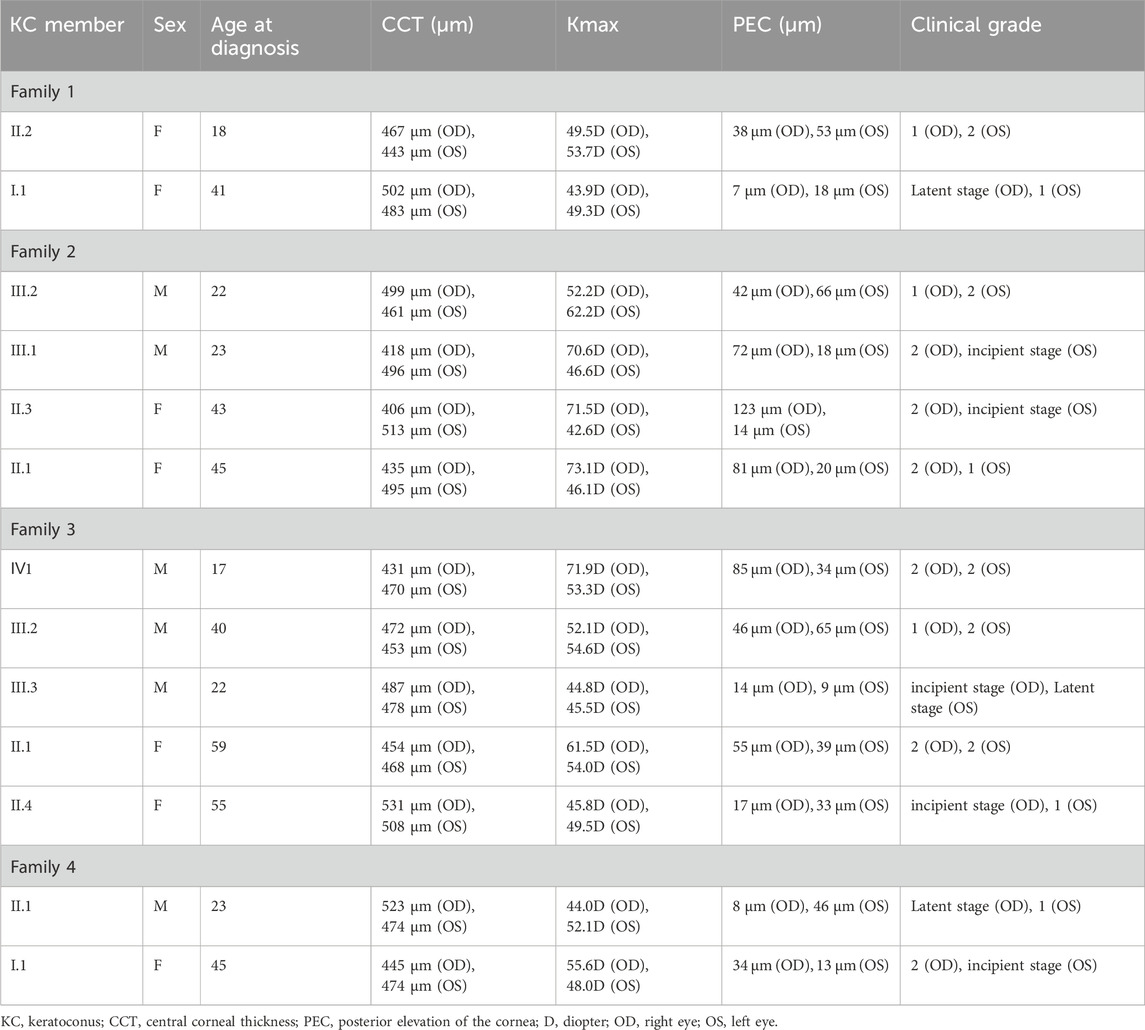
Table 1. Clinical data of the KC patients.
The following four variants were detected in the four families and were absent in the healthy controls: 1) a heterozygous missense variation c.109G>A (p.Glu37Lys, rs369263247) in the exon 2 of the procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1) gene in family 1 (Figure 3A). According to the VarSite database, a transition from a Glu to a Lys side chain is quite substantial, and could likely lead to a modification in the protein’s function. The variant residue is a lysine, possessing a positively charged side chain, thus enhancing its hydrophilicity. The amino acid alteration from a Glu to a Lys has a high “disease propensity” value of 1.14, and its occurrence in the gnomAD population database is exceedingly rare (PM2); 2) a heterozygous missense variation c.3766G>A(p.Ala1256Thr, rs148216434) in the exon 18 of the collagen type I alpha 1 (COL1A1) gene in family 2 (Figure 3B). Following the ACMG guidelines, its occurrence in the gnomAD population database is exceptionally rare (PM2) and is likely to have a deleterious effect (PP3); 3) a missense variation c.976G>A (p.Glu326Ser) in the collagen type IV alpha 1 (COL4A1) gene in family 3 (Figure 3C). Its occurence in the gnomAD population database is exceedingly rare (PM2), and it is likely to have a deleterious effect (PP3). In the VarSite database, residue alteration from a Gly to a Ser has a high ‘disease propensity’ value of 1.34; and 4) a heterozygous missense variant c.4364G>A (p.Gly1455Glu) in exon 54 of the collagen type V alpha 2 (COL5A2) gene in family 4 (Figure 3D). This variant is also located in the COLFI domain, close to the C-terminal end (1,265–1,499 aa). The VarSite database also showed that a transition from a Gly to Glu side chain is substantial, and could likely lead to a modification in the protein’s function. Its occurrence in the gnomAD population database is exceedingly rare (PM2) and is likely to have a deleterious effect (PP3).
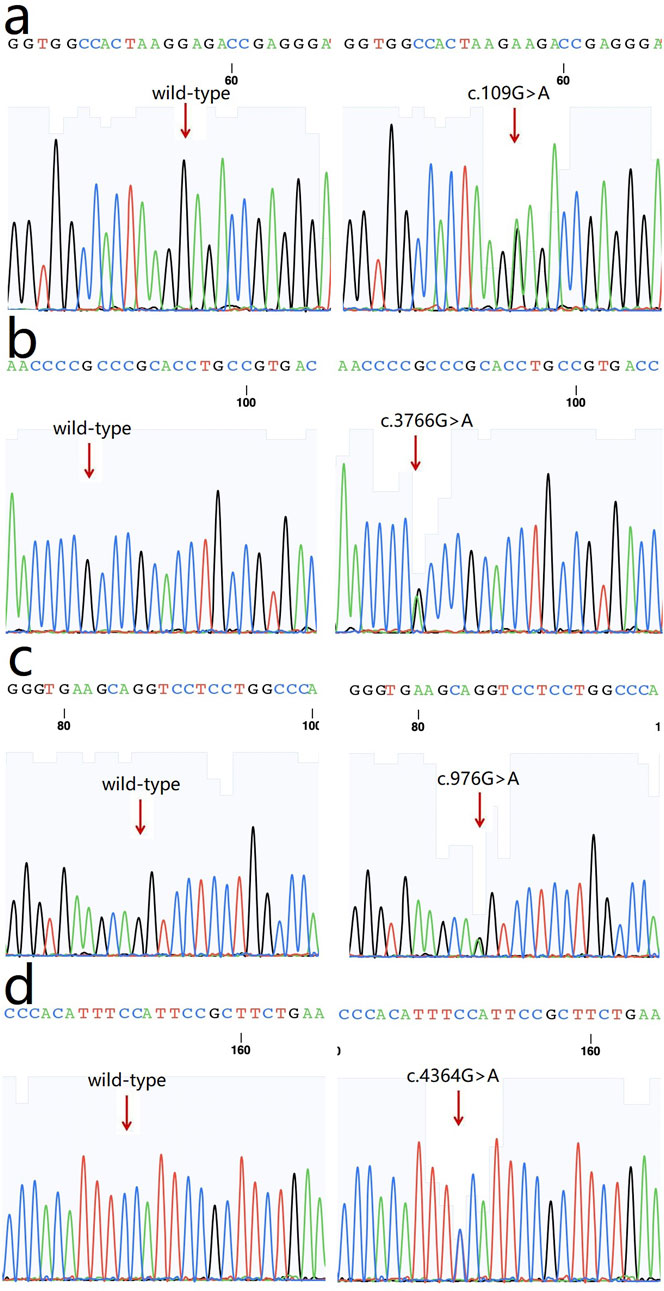
Figure 3. Sequence chromatograms of the variants. (a) c.109G>A (p.Glu37Lys, rs369263247) in PLOD1 gene (arrow); (b) a heterozygous missense variation c.3766G>A (p.Ala1256Thr, rs148216434) in COL1A1 gene (arrow); (c) a missense variation c.976G>A (p.Glu326Ser) in COL4A1 gene; (d) a heterozygous missense variant c.4364G>A (p.Gly1455Glu) in COL5A2 gene.
The above genotypes were co-segregated with the corresponding phenotypes. All of the missense variations were predicted to be damaging and highly conserved using software programs (Table 2). The conformational alteration of the proteins caused by these variations was revealed by 3D modeling and was compared with their wild-type proteins (Figures 4A–D). GERP++ scores indicated that these four variants were located in a highly conservative region. Based on the ACMG guidelines, these four variations were predicted to be variants of uncertain significance (VUS) (Table 2; Supplementary Table S1).
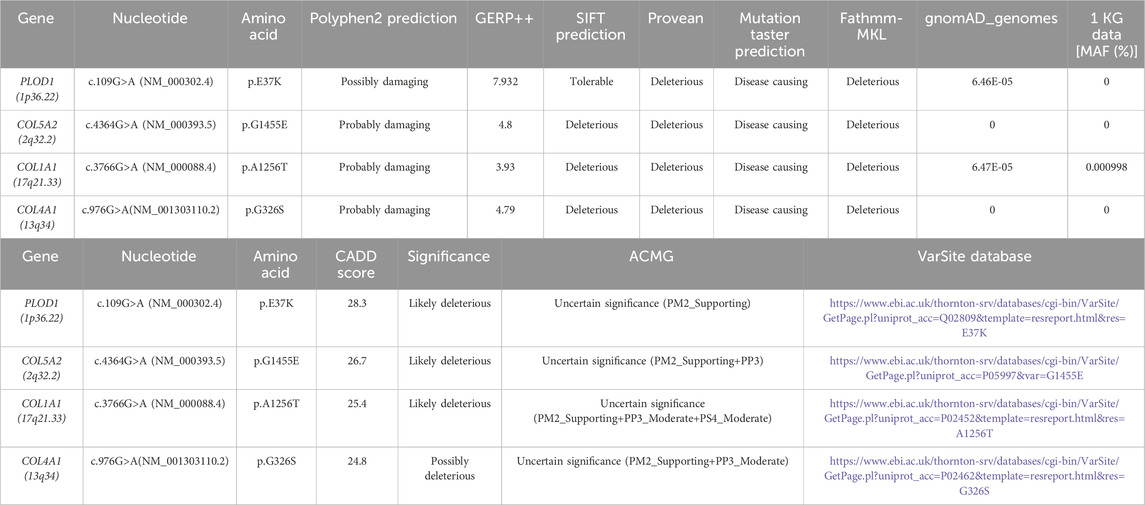
Table 2. Computational predictions and ACMG classification of all identified variants and their frequency in the gnomAD genomes.
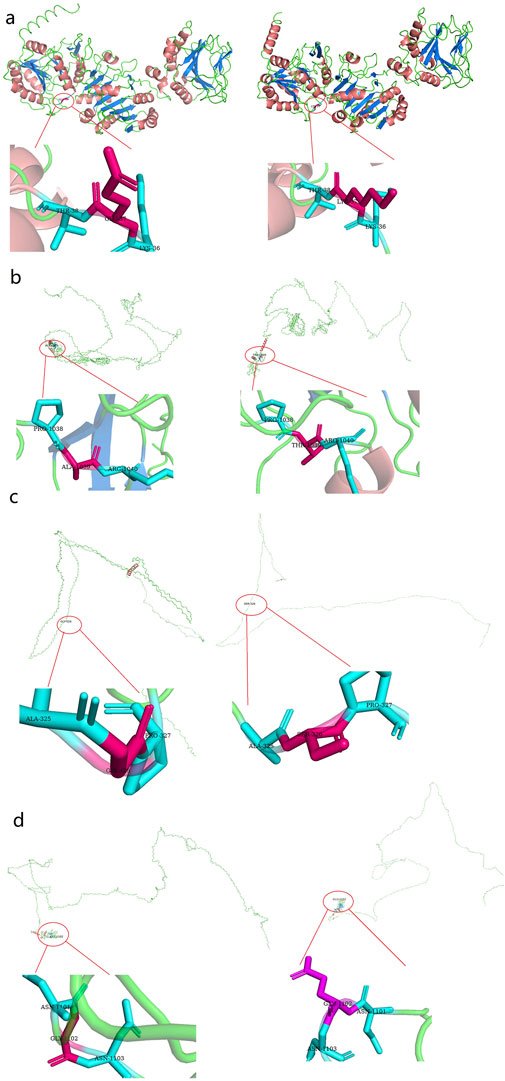
Figure 4. Three dimensional (3D) structures of the proteins show the sites of variants. The insert pictures are regional enlargements of the variants. (a) 3D modeling of wild-type PLOD1 and p.Glu37Lys variant. (b) 3D modeling of wild-type COL1A1 and p.Ala1256Ter variant. (c) 3D modeling of wild-type COL4A1 and p.Glu326Ser variant; (d) 3D modeling of wild-type COL5A2 and p.Gly1455Glu variant.
Using STRING, 11 significantly upregulated genes were updated to build the PPI networks of PLOD1, COL4A1, COL1A1, and COL5A2 (Figure 5A). Then the subnetworks were constructed using Cytoscape. In Figure 5B, the interaction of COL4A1 with integrin subunit alpha (ITGA) and ITGB are shown. Integrin is an important component of the extracellular matrix (ECM) involving in cytoskeleton remodeling, cell adhesion and migration, and integrin outside-in signaling, etc., which are involved in corneal development (https://www.wikipathways.org/pathways). In Figure 5C, the interaction among COL1A1, COL1A2 and COL5A1, and their interaction with ITGB are shown, which involves the assembly of collagen fibrils and other multimeric structures, integrin inside-out signaling, integrin-mediated cell adhesion and migration, ECM remodeling, and cytoskeleton remodeling, etc. (https://pubchem.ncbi.nlm.nih.gov/pathway;and https://www.genecards.org)
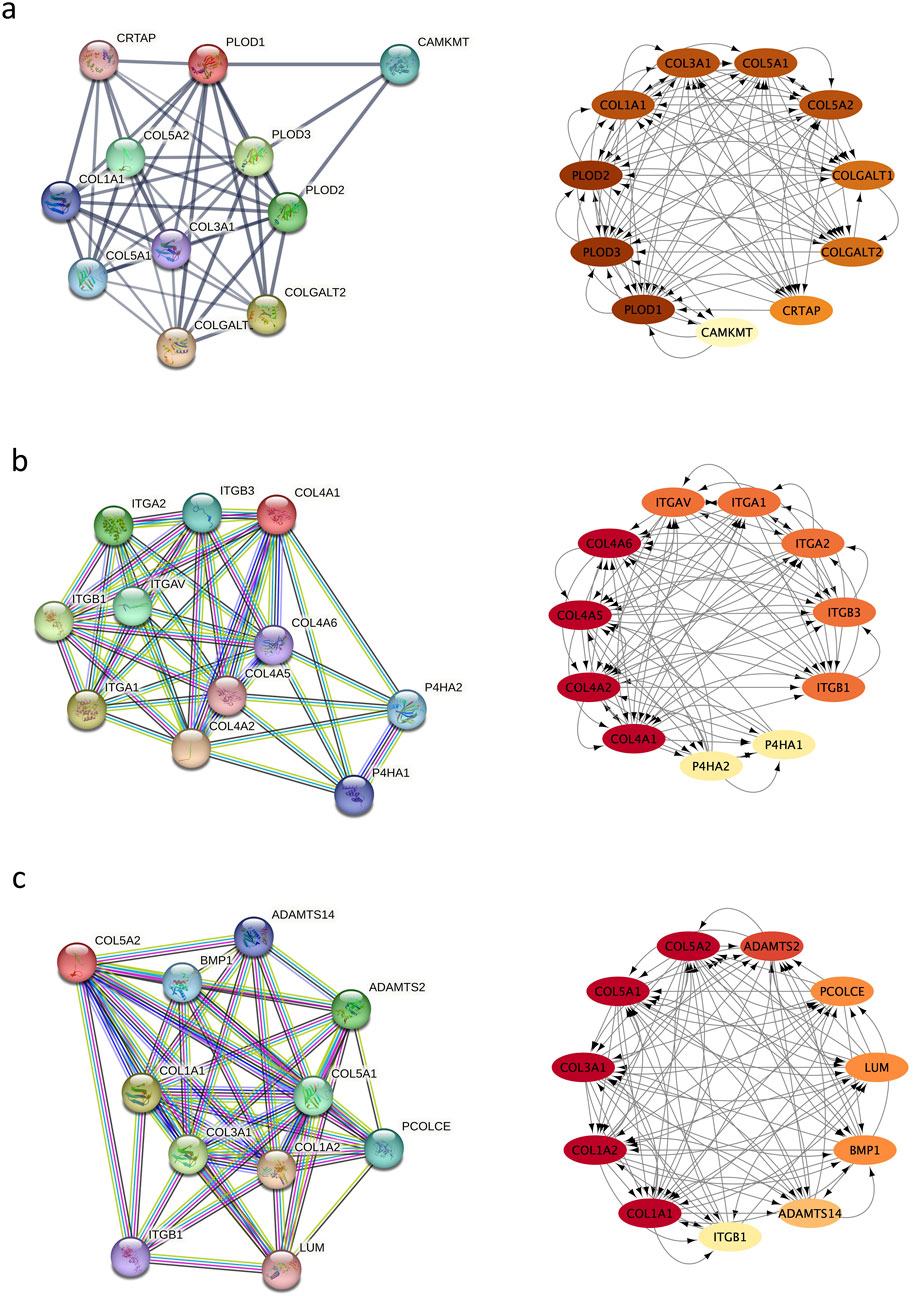
Figure 5. Protein–protein interaction (PPI) network. (a) the interaction of PLOD1 with COL1A1, COL4A1, and COL5A2; (b) the interaction of COL4A1 with ITGA and ITGB; (c) the interaction among COL1A1, COL1A2 and CO5A1, and their interaction with ITGB.
The etiology of KC is complex and involves environmental and genetic factors. Reduced corneal collagen or abnormal distribution of the collagen fibers are pathologic features of KC. In this study, four variants in COL1A1, COL5A2, COL4A1, and PLOD1 genes were identified in four Chinese families with KC. COL1A1, COL5A2, and COL4A1 are collagen genes and the PLOD1 gene is associated with collagen crosslinking. The variants detected in the present study were all computationally predicted to be damaging and pathogenic for KC; however, further verification is warranted.
Before discussing the results of this study, a review of key activities related to corneal collagen and its crosslinkage should provide an understanding of the biomedical implication of the findings. In collagens, lysyl hydroxylase (LH) is important for catalyzing the hydroxylation of lysine residues in -X-LysGly- triplets (Kivirikko and Pihlajaniemi, 1998). LH has three isoenzymes: LH1, LH2, and LH3 (Ruotsalainen et al., 1999). The hydroxylysine residues are crucial for stability of the intermolecular crosslinks to ensure the mechanical stability and tensile strength of collagen fibrils, as well as the attachment sites for carbohydrate units (Gao et al., 2013). The formation of collagen crosslink takes place in the extracellular matrix and begins with the transformation of certain lysine or hydroxylysine residues in the telopeptides, catalyzed by lysyloxidase (Kagan and Li, 2003). All three primary fibril-forming collagens (types I, II, and III) possess four crosslinking sites, each in its own telopeptide and two within the triple helical region, near the N- and C-terminal ends (Eyre and Wu, 2005). Pyridinoline crosslinks are abundant in bone, tendons, ligaments, and cornea (Eyre and Wu, 2005). The hydroxylysine-linked carbohydrate units play a crucial role in controlling the formation and morphology of collagen fibrils(Notbohm et al., 1999) as well as type IV collagen-mediated crosslinking (Rautavuoma et al., 2004; Ruotsalainen et al., 2006). In Ehlers-Danlos syndrome (EDS), an inherited connective tissue disorder, LH1 deficiency caused by variants in the PLOD1 gene is associated with the kyphoscoliotic subtype of EDS (Yeowell and Walker, 2000; Wilcox, 2003; Myllyharju and Kivirikko, 2004). In this subtype, ocular fragility cases and KC development have been reported (Wilcox, 2003; Myllyharju and Kivirikko, 2004).
COL1A1, COL5A2, and COL4A1 encode type I, IV, and V collagens, which are the primary constituents of the corneal stroma and basement membranes (Chan and Snibson, 2013). The composition of collagens in the stroma is 80%–90% type I and 10%–20% type V, whereas type IV collagen is one of the main components of basement membranes. The quantities of collagen type I and type V are vital for preserving the organization of the stroma as well as the shape and robustness of the cornea (Chakravarti et al., 1998; Quantock et al., 2003). Collagen type I forms collagen type V fibrils. Then the fibril growth and diameter are determined by the protrusion of nonhelical terminal extensions of collagen type V (Linsenmayer et al., 1993). Thus the variants in COL1A1 and COL5A2 could cause the abnormalities of collagen synthesis. In this research, a heterozygous variant c.3766G>A in COL1A1 was identified in family 2 and it caused alteration in amino acid (p.Ala1256Thr), which is located in the fibrillar collagen C-terminal (COLFI) domain, 1,228–1,464 aa, one of the crosslinking sites (Figure 6A). Amino acid change in this region may cause protein dysfunction and deformation. Besides, in VarSite database, for wildtype, at sequence position 1,256 in this protein, the residue is an alanine which has an aliphatic side chain (i.e., containing only carbon and hydrogen atoms), which is hydrophobic (i.e., tending to avoid water and preferring to interact with other hydrophobic side chains), but the variant residue is a threonine which has a neutral side chain, this change may affect the structure stability.
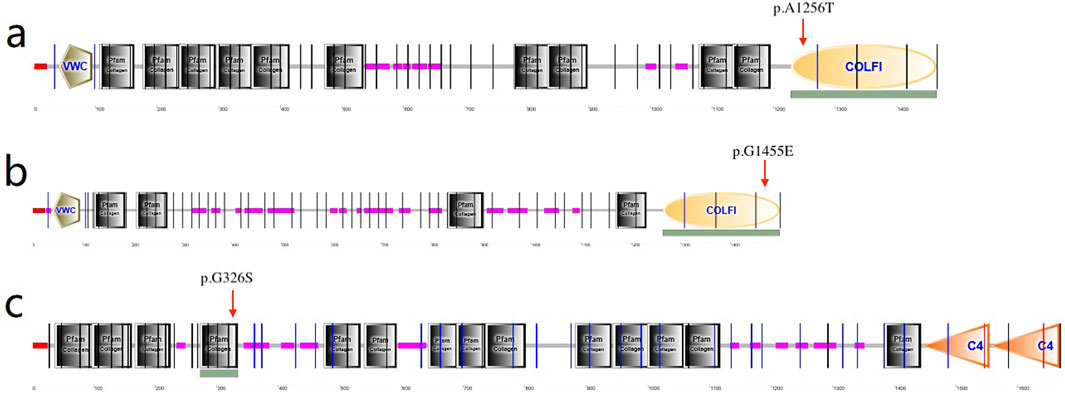
Figure 6. (a) p.Ala1256Ter amino acid change in COL1A1 located in the COLFI domain (arrow indicates 1,228–1,464aa); (b) p.Gly1455Glu in COL5A2 located in the COLFI domain (arrow indicates 1,265–1,499aa); (c) p.Gly326Ser in COL4A1 located in the Pfarm collagen domain (arrow indicates 273–334aa).
In family 4, the variant in COL5A2 (c.4364G>A, p.Gly1455Glu) is within the fourth cross-linking site (Figure 6B), and is expected to be highly conserved across species and causes potential harm. For wild-type, at sequence position 1,455 in this protein, the residue is a glycine which has no side chain, giving the protein increased flexibility at this location; the variant residue is a glutamic acid, possessing a negatively charged side chain, making it hydrophilic (i.e., favoring the protein’s surface over its interior).
Corneal basement membranes are essential structures in tissue development and maintenance. The main protein in the basement membranes, type IV collagen, is a family of six proteins encoded by six distinct genes: COL4A1, COL4A2, COL4A3, COL4A4, COL4A5, and COL4A6 (Bai et al., 2009). COL4A3, COL4A4, and COL4A5 have been identified as KC candidate genes (Bochert et al., 2003; Stabuc-Silih et al., 2009; Krolo et al., 2021). In 2020, Tian et al. (2020) investigated the role of the XIST-miR-181a-COL4A1 axis in the development of KC and suggested that COL4A1 might be correlated with KC. However, to date, COL4A1 variants in KC families have not been reported. In this study, missense variation c.976G>A (p.Gly326Ser) was detected in the COL4A1 gene and was predicted to be damaging. The substitution p.Gly326Ser was in the Pfam collagen domain (273–334 aa) (Figure 6C), which is predominantly repeats of the G-X-Y and polypeptide chain-formed triple helix and is considered highly conserved. This is the first time a COL4A1 variation in a family with KC has been identified.
In this study, another variation detected was in the PLOD1 gene. PLOD1 is located on chromosome 1, which consists of 727 aa and is expressed in corneal stroma. PLOD1 is expressed in corneal collagens and is associated with collagen crosslinking (Burdon et al., 2008). In 2017, Hudson et al. also confirmed a common lysine under-hydroxylation effect at the helical domain crosslinking sites in the skin, bone, tendon, aorta, scleral, and cornea (Hudson et al., 2017). In our study, a heterozygous PLOD1 mutation c.109G>A was identified in family 1. The structural change in the protein induced by this variant may disturb the crosslinking of corneal fibrils. In addition, PPI network showed the interaction between PLOD1 and COL1A1 or PLOD1 and COL5A1. Therefore, misfolding and/or aggregate formation of PLOD1 likely cause the development of KC by disturbing corneal crosslinking. To date, this is the first report to identify a PLOD1 variation in a family with KC.
Clinically, the corneal crosslinking (CXL) technique uses riboflavin and UVA light to treat KC. The mechanism of this treatment is that UVA, in conjunction with riboflavin, initiates oxidative and glycosylation processes, resulting in the generation of collagen crosslinks and strengthening the corneal stroma (Gomes et al., 2015; Jacob et al., 2018). However, corneal structures such as endothelial cells can potentially be damaged after CXL, leading to corneal edema and vision loss (Bagga et al., 2012; Sharma et al., 2012). Moreover, transient or persistent corneal haze after CXL and post-CXL infectious keratitis have been reported (Kim et al., 2016; Kodavoor et al., 2020). CXL also cannot be performed in patients who are pregnant or breastfeeding or have eye inflammation, pronounced corneal scarring, autoimmune diseases, or corneal thickness <400 µm. In view of the limitations of CXL surgery and the risk of complications, treatment with collagen crosslinking without these issues should be developed. Since the PLOD1 gene plays an important role in regulating collagen crosslinking, gene therapy for the variations in PLOD1 could be a treatment strategy to be considered.
In conclusion, through investigating four Chinese families with KC, four variants in COL1A1, COL5A2, COL4A1, and PLOD1 genes were identified in this study. COL1A1, COL5A2, and COL4A1 are collagen genes and PLOD1 is a collagen crosslinking regulatory gene. These discoveries broaden the range of recognized gene variants in KC and warrant additional studies on these variants.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SCV004030248, SCV004030250, SCV004030251 and SCV004030252.
The studies involving humans were approved by institutional review board of Fudan University (Shanghai, China) (approval no. 2022128). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. No potentially identifiable images or data are presented in this study.
XW: Data curation, Formal Analysis, Software, Writing–original draft. XP: Data curation, Formal Analysis, Writing–original draft. TH: Data curation, Formal Analysis, Writing–original draft. LS: Data curation, Formal Analysis, Writing–original draft. XaZ: Data curation, Formal Analysis, Writing–original draft. XnZ: Conceptualization, Funding acquisition, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. The present research received partial support from the following fundings. The National Natural Science Foundation of China (Grant No. 82305379); the China National Postdoctoral Program for Innovative Talents (Grant No. BX20230095); the China Postdoctoral Science Foundation (Grant No. 2023M740740); the Project of Shanghai Science and Technology (No. 20410710100); the Clinical Research Plan of SHDC (No. SHDC2020CR1043B); the Project of Shanghai Xuhui District Science and Technology (No. 2020-015); the Project of Shanghai Xuhui District Science and Technology (No. XHLHGG202104); the Shanghai Engineering Research Center of Laser and Autostereoscopic 3D for Vision Care (No. 20DZ2255000); and the construction of a 3D digital intelligent prevention and control platform for the whole life cycle of highly myopic patients in the Yangtze River Delta (No. 21002411600).
The authors wish to extend their appreciation to all study participants for their collaboration. We thank Medjaden Inc. for scientific editing of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1497915/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Conservation analysis diagram of the variants. (a) Conservation analysis of the amino acid position where the p.Glu37Lys of the PLOD1 gene is located among various species; (b) Conservation analysis of the amino acid position where p.Ala1256Thr of the COL1A1 gene is located among various species; (c) Conservation analysis of the amino acid position where p.Glu326Ser of the COL4A1 gene is located among various species; (d) Conservation analysis of the amino acid position where p.Gly1455Glu of the COL5A2 gene is located among various species.
SUPPLEMENTARY TABLE S1 | Variants evaluated in the four families with KC.
KC, Keratoconus; UVA, ultraviolet-A; UCVA, uncorrected visual acuity; BSCVA, best spectacle corrected visual acuity; CC, corneal curvature; CTTP, corneal thickness at the thinnest point; ES, exome sequencing; SIFT, Solar Instant Feasibility Tool; PCR, polymerase chain reaction; 3D, three-dimensional; PPI, protein-protein interaction; PEC, posterior elevation of the cornea; MASCs, maximum anterior surface curvatures; CCTs, central corneal thicknesses; PLOD1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1; COL1A1, collagen type I alpha 1; COL4A1, collagen type IV alpha 1; COL5A2, collagen type V alpha 2; LH, lysyl hydroxylase; EDS, Ehlers-Danlos syndrome; COLFI, fibrillar collagen C-terminal; CXL, corneal crosslinking.
Abu-Amero, K. K., Helwa, I., Al-Muammar, A., Strickland, S., Hauser, M. A., Allingham, R. R., et al. (2015). Case-control association between CCT-associated variants and keratoconus in a Saudi Arabian population. J. Negat. Results Biomed. 14, 10. doi:10.1186/s12952-015-0029-5
Bagga, B., Pahuja, S., Murthy, S., and Sangwan, V. S. (2012). Endothelial failure after collagen cross-linking with riboflavin and UV-A: case report with literature review. Cornea 31 (10), 1197–1200. doi:10.1097/ICO.0b013e31823cbeb1
Bai, X., Dilworth, D. J., Weng, Y. C., and Gould, D. B. (2009). Developmental distribution of collagen IV isoforms and relevance to ocular diseases. Matrix Biol. 28 (4), 194–201. doi:10.1016/j.matbio.2009.02.004
Bochert, A., Berlau, J., Koczan, D., Seitz, B., Thiessen, H. J., and Guthoff, R. (2003). Genexpression bei Keratokonus. Ophthalmologe 100 (7), 545–549. doi:10.1007/s00347-003-0808-0
Burdon, K. P., Coster, D. J., Charlesworth, J. C., Mills, R. A., Laurie, K. J., Giunta, C., et al. (2008). Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum. Genet. 124 (4), 379–386. doi:10.1007/s00439-008-0555-z
Bykhovskaya, Y., Margines, B., and Rabinowitz, Y. S. (2016). Genetics in Keratoconus: where are we? Eye Vis. (Lond) 3, 16. doi:10.1186/s40662-016-0047-5
Chakravarti, S., Magnuson, T., Lass, J. H., Jepsen, K. J., LaMantia, C., and Carroll, H. (1998). Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J. Cell Biol. 141 (5), 1277–1286. doi:10.1083/jcb.141.5.1277
Chan, E., and Snibson, G. R. (2013). Current status of corneal collagen cross-linking for keratoconus: a review. Clin. Exp. Optom. 96 (2), 155–164. doi:10.1111/cxo.12020
Chinese Medical Association Ophthalmology Branch Corneal Disease Group (2019). Expert consensus on the diagnosis and treatment of keratoconus in China (2019). Chin. J. Ophthalmol. 55 (12), 891–895. doi:10.3760/cma.j.issn.0412-4081.2019.12.004
Dimasi, D. P., Chen, J. Y., Hewitt, A. W., Klebe, S., Davey, R., Stirling, J., et al. (2010). Novel quantitative trait loci for central corneal thickness identified by candidate gene analysis of osteogenesis imperfecta genes. Hum. Genet. 127 (1), 33–44. doi:10.1007/s00439-009-0729-3
Egrilmez, S., Sahin, S., and Yagci, A. (2004). The effect of vernal keratoconjunctivitis on clinical outcomes of penetrating keratoplasty for keratoconus. Can. J. Ophthalmol. 39 (7), 772–777. doi:10.1016/s0008-4182(04)80072-3
Eyre, D. R., and Wu, J. J. (2005). Collagen cross-links. Top. Curr. Chem. 247, 207–229. doi:10.1007/b103828
Farjadnia, M., and Naderan, M. (2015). Corneal cross-linking treatment of keratoconus. Oman J. Ophthalmol. 8 (2), 86–91. doi:10.4103/0974-620x.159105
Gao, X., Gauderman, W. J., Liu, Y., Marjoram, P., Torres, M., Haritunians, T., et al. (2013). A genome-wide association study of central corneal thickness in Latinos. Invest Ophthalmol. Vis. Sci. 54 (4), 2435–2443. doi:10.1167/iovs.13-11692
Gomes, J. A., Tan, D., Rapuano, C. J., Belin, M. W., Ambrósio, R., Guell, J. L., et al. (2015). Global consensus on keratoconus and ectatic diseases. Cornea 34 (4), 359–369. doi:10.1097/ico.0000000000000408
Hosoda, Y., Miyake, M., Meguro, A., Tabara, Y., Iwai, S., Ueda-Arakawa, N., et al. (2020). Keratoconus-susceptibility gene identification by corneal thickness genome-wide association study and artificial intelligence IBM Watson. Commun. Biol. 3 (1), 410. doi:10.1038/s42003-020-01137-3
Hudson, D. M., Weis, M., Rai, J., Joeng, K. S., Dimori, M., Lee, B. H., et al. (2017). P3h3-null and Sc65-null mice phenocopy the collagen lysine under-hydroxylation and cross-linking abnormality of ehlers-danlos syndrome type VIA. J. Biol. Chem. 292 (9), 3877–3887. doi:10.1074/jbc.M116.762245
Jacob, S., Patel, S. R., Agarwal, A., Ramalingam, A., Saijimol, A. I., and Raj, J. M. (2018). Corneal allogenic intrastromal ring segments (CAIRS) combined with corneal cross-linking for keratoconus. J. Refract Surg. 34 (5), 296–303. doi:10.3928/1081597x-20180223-01
Kagan, H. M., and Li, W. (2003). Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J. Cell Biochem. 88 (4), 660–672. doi:10.1002/jcb.10413
Kim, B. Z., Jordan, C. A., McGhee, C. N., and Patel, D. V. (2016). Natural history of corneal haze after corneal collagen crosslinking in keratoconus using Scheimpflug analysis. J. Cataract. Refract Surg. 42 (7), 1053–1059. doi:10.1016/j.jcrs.2016.04.019
Kivirikko, K. I., and Pihlajaniemi, T. (1998). Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv. Enzymol. Relat. Areas Mol. Biol. 72, 325–398. doi:10.1002/9780470123188.ch9
Kodavoor, S. K., Tiwari, N. N., and Ramamurthy, D. (2020). Profile of infectious and sterile keratitis after accelerated corneal collagen cross-linking for keratoconus. Oman J. Ophthalmol. 13 (1), 18–23. doi:10.4103/ojo.OJO_115_2018
Krolo, I., Kasumović, A., Radman, I., and Pavić, P. (2021). Corneal cross-linking for treatment of progressive keratoconus in a patient with Alport syndrome: a case report. Eur. J. Ophthalmol. 31 (4), 1584–1587. doi:10.1177/1120672121997672
Legare, M. E., Iovieno, A., Yeung, S. N., Kim, P., Lichtinger, A., Hollands, S., et al. (2013). Corneal collagen cross-linking using riboflavin and ultraviolet A for the treatment of mild to moderate keratoconus: 2-year follow-up. Can. J. Ophthalmol. 48 (1), 63–68. doi:10.1016/j.jcjo.2012.11.007
Li, X., Bykhovskaya, Y., Canedo, A. L., Haritunians, T., Siscovick, D., Aldave, A. J., et al. (2013). Genetic association of COL5A1 variants in keratoconus patients suggests a complex connection between corneal thinning and keratoconus. Invest Ophthalmol. Vis. Sci. 54 (4), 2696–2704. doi:10.1167/iovs.13-11601
Lin, Q., Wang, X., Han, T., and Zhou, X. (2023). Identification of genetic variants in two families with Keratoconus. BMC Med. Genomics 16 (1), 299. doi:10.1186/s12920-023-01738-x
Linsenmayer, T. F., Gibney, E., Igoe, F., Gordon, M. K., Fitch, J. M., Fessler, L. I., et al. (1993). Type V collagen: molecular structure and fibrillar organization of the chicken alpha 1(V) NH2-terminal domain, a putative regulator of corneal fibrillogenesis. J. Cell Biol. 121 (5), 1181–1189. doi:10.1083/jcb.121.5.1181
Loukovitis, E., Sfakianakis, K., Syrmakesi, P., Tsotridou, E., Orfanidou, M., Bakaloudi, D. R., et al. (2018). Genetic aspects of keratoconus: a literature review exploring potential genetic contributions and possible genetic relationships with comorbidities. Ophthalmol. Ther. 7 (2), 263–292. doi:10.1007/s40123-018-0144-8
Myllyharju, J., and Kivirikko, K. I. (2004). Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20 (1), 33–43. doi:10.1016/j.tig.2003.11.004
Notbohm, H., Nokelainen, M., Myllyharju, J., Fietzek, P. P., Müller, P. K., and Kivirikko, K. I. (1999). Recombinant human type II collagens with low and high levels of hydroxylysine and its glycosylated forms show marked differences in fibrillogenesis in vitro. J. Biol. Chem. 274 (13), 8988–8992. doi:10.1074/jbc.274.13.8988
Quantock, A. J., Dennis, S., Adachi, W., Kinoshita, S., Boote, C., Meek, K. M., et al. (2003). Annulus of collagen fibrils in mouse cornea and structural matrix alterations in a murine-specific keratopathy. Invest Ophthalmol. Vis. Sci. 44 (5), 1906–1911. doi:10.1167/iovs.02-0884
Rabinowitz, Y. S. (1998). Keratoconus. Surv. Ophthalmol. 42 (4), 297–319. doi:10.1016/s0039-6257(97)00119-7
Rautavuoma, K., Takaluoma, K., Sormunen, R., Myllyharju, J., Kivirikko, K. I., and Soininen, R. (2004). Premature aggregation of type IV collagen and early lethality in lysyl hydroxylase 3 null mice. Proc. Natl. Acad. Sci. U. S. A. 101 (39), 14120–14125. doi:10.1073/pnas.0404966101
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ruotsalainen, H., Sipilä, L., Kerkelä, E., Pospiech, H., and Myllylä, R. (1999). Characterization of cDNAs for mouse lysyl hydroxylase 1, 2 and 3, their phylogenetic analysis and tissue-specific expression in the mouse. Matrix Biol. 18 (3), 325–329. doi:10.1016/s0945-053x(99)00016-5
Ruotsalainen, H., Sipilä, L., Vapola, M., Sormunen, R., Salo, A. M., Uitto, L., et al. (2006). Glycosylation catalyzed by lysyl hydroxylase 3 is essential for basement membranes. J. Cell Sci. 119 (Pt 4), 625–635. doi:10.1242/jcs.02780
Saravani, R., Hasanian-Langroudi, F., Validad, M. H., Yari, D., Bahari, G., Faramarzi, M., et al. (2015). Evaluation of possible relationship between COL4A4 gene polymorphisms and risk of keratoconus. Cornea 34 (3), 318–322. doi:10.1097/ico.0000000000000356
Sharma, A., Nottage, J. M., Mirchia, K., Sharma, R., Mohan, K., and Nirankari, V. S. (2012). Persistent corneal edema after collagen cross-linking for keratoconus. Am. J. Ophthalmol. 154 (6), 922–926.e1. doi:10.1016/j.ajo.2012.06.005
Shi, W. Y., Gao, H., and Li, Y. (2019). Standardizing the clinical diagnosis and treatment of keratoconus in China (In Chinese). Zhonghua Yan Ke Za Zhi 55 (6), 401–404. doi:10.3760/cma.j.issn.0412-4081.2019.06.001
Stabuc-Silih, M., Ravnik-Glavac, M., Glavac, D., Hawlina, M., and Strazisar, M. (2009). Polymorphisms in COL4A3 and COL4A4 genes associated with keratoconus. Mol. Vis. 15, 2848–2860.
Tian, R., Wang, L., Zou, H., Song, M., Liu, L., and Zhang, H. (2020). Role of the XIST-miR-181a-COL4A1 axis in the development and progression of keratoconus. Mol. Vis. 26, 1–13.
Vithana, E. N., Aung, T., Khor, C. C., Cornes, B. K., Tay, W. T., Sim, X., et al. (2011). Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum. Mol. Genet. 20 (4), 649–658. doi:10.1093/hmg/ddq511
Wilcox, W. R. (2003). Connective tissue and its heritable disorders: molecular, genetic, and medical aspects. Am. J. Hum. Genet. 72, 503–504. doi:10.1086/345996
Wu, D., Lim, D. K., Lim, B. X. H., Wong, N., Hafezi, F., Manotosh, R., et al. (2021). Corneal cross-linking: the evolution of treatment for corneal diseases. Front. Pharmacol. 12, 686630. doi:10.3389/fphar.2021.686630
Keywords: keratoconus, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1), collagen type V alpha 2, collagen type I alpha 1, collagen type IV alpha 1, early diagnosis
Citation: Lin Q, Wang X, Peng X, Han T, Sun L, Zhang X and Zhou X (2025) Novel variations in the PLOD1, COL1A1, COL5A2 and COL4A1 genes related to keratoconus. Front. Genet. 16:1497915. doi: 10.3389/fgene.2025.1497915
Received: 18 September 2024; Accepted: 07 March 2025;
Published: 25 March 2025.
Edited by:
Mara Marongiu, National Research Council (CNR), ItalyReviewed by:
Muzammil Ahmad Khan, Gomal University, PakistanCopyright © 2025 Lin, Wang, Peng, Han, Sun, Zhang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingtao Zhou, WmhvdXhpbmd0YW9kb2N0QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.