 Qinglong Yu1†
Qinglong Yu1† Risna Begam Mohammed Nazar
Risna Begam Mohammed Nazar Junhui Wang
Junhui Wang Xueping Chen
Xueping Chen- 1Department of Neurology,Affiliated Hospital of Panzhihua University, Panzihua, China
- 2Department of Neurology, West China Hospital, Sichuan University, Chengdu, Sichuan, China
- 3Department of Neurology, Mount Sinai Hospital, Toronto, ON, Canada
- 4Sunsimiao Hospital, Beijing University of Chinese Medicine, Tongchuan, Shanxi, China
We present the case of a 16-year-old East Asian Chinese girl with a novel mutation in the SIGMAR1 gene, initially diagnosed as juvenile amyotrophic lateral sclerosis (JALS). At the age of five, she began to exhibit gait abnormalities while walking, a condition that persisted for 4 years until muscle weakness and atrophy emerged, predominantly affecting her distal muscles symmetrically. Electromyography (EMG) initially revealed early abonormal motor conduction, and subsequent examinations indicated neurogenic damage accompanied by localized denervation potentials. Whole-exome sequencing identified compound heterozygous mutations in the SIGMAR1 gene. Throughout the course of her illness, the patient exhibited slow disease progression without cognitive impairment or scoliosis development. We ultimately revised the diagnosis to distal hereditary motor neuropathy (dHMN). This study reports the case of SIGMAR1 new locus mutation leading to dHMN in China, contributing to the expansion of the dHMN genetic database. In our patient, the initial EMG findings indicated issues with neurogenic conduction, followed by a slow progression of the disease. Subsequently, EMG results revealed axonal damage and denervation potentials. These clinical features can easily lead to confusion with JALS. This insight is valuable for improving diagnostic accuracy and understanding the clinical spectrum of dHMN related to SIGMAR1 mutations.
Introduction
Distal hereditary motor neuropathy (dHMN) is a relatively new and debated condition characterized by clinical and genetic heterogeneity (Li et al., 2015). It commonly presents with progressive distal muscle weakness and atrophy, sometimes accompanied by pyramidal signs, but notably lacks sensory abnormalities (Li et al., 2015; Ma et al., 2020; Tazir and Nouioua, 2024). To date, approximately 30 genes have been identified in association with dHMN, yet together they explain fewer than one-third of the observed cases (Ververis et al., 2020). Notably, the SIGMAR1 gene is especially contentious due to significant phenotypic overlap; mutations within SIGMAR1 are frequently associated with conditions that exhibit clinical features reminiscent of Amyotrophic Lateral Sclerosis (ALS) (Al-Saif et al., 2011; Luty et al., 2010; Li et al., 2023; Izumi et al., 2018; Kim et al., 2014). ALS is a fatal neurodegenerative disorder characterized by the progressive degeneration of upper and lower motor neurons (Feldman et al., 2022). When symptom onset occurs before the age of 25, the condition is classified as Juvenile ALS (JALS) (Lehky and Grunseich, 2021). However, “JALS” cases diagnosed due to SIGMAR1 gene mutations do not typically exhibit the rapid progression and widespread neurogenic damage characteristic of ALS, leading to considerable confusion among clinicians and the families of patients.
We reported on an adolescent patient with involvement of both upper and lower motor neurons who harbors a novel locus variant in the SIGMAR1 gene.
Case presentation
At the age of 5, the patient began experiencing difficulties with walking, characterized by increased muscle tone in the legs and a tendency to fall while running quickly. Despite these symptoms being relatively mild and not significantly interfering with her daily activities, no medical evaluation was sought at that time. By the age of 9, she developed slight weakness in the distal upper limbs, reduced range of motion in her arms, and upper limb tremors when gripping objects. These symptoms progressively worsened over time, resulting in more frequent falls and diminished performance in sports activities. She did not report any symptoms related to fasciculations, choking, swallowing difficulties, respiratory challenges, and cognitive impairment. The patient went to the local hospital for medical help. Neurological examination revealed muscle atrophy in the limbs, particularly in the interosseous muscles of the hands. A postural tremor was observed in the left upper limb and muscle strength was nearly normal, with only a positive result for the Clipper Paper Test and a slight limitation in standing on tiptoes noted. Hyperactive tendon reflexes were present in the limbs, and the Babinski sign was positive. The EMG examination revealed decreased amplitudes of compound muscle action potential (CMAP) waves and motor nerve conduction abnormalities, including extended distal latencies and reduced F-wave frequencies. However, the initial EMG did not demonstrate spontaneous potentials or evidence of chronic denervation in the upper limbs (Supplementary Table S1). The patient was diagnosed with clinically “possible ALS” at that time in local hospital, although her EMG was not consistent with classic ALS manifestations.
At age 14, the subsequent EMG revealed neurogenic damage affecting both the upper and lower limbs was observed, with evident axonal involvement, and a notable finding was the presence of abundant fasciculation potentials in the bilateral anterior tibialis muscles. Both ulnar and sural nerve conduction velocities were found to be decreased upon re-examination (Supplementary Table S2). At this time, her symptoms remained stable except for a slight reduction in lower limb muscle strength to grade 4+. Whole exome sequencing identified a compound heterozygous mutation in the SIGMAR1 gene (Figure 1). The first gene mutation site [NM_005866. 2: 131A > c (p.Gln 44 Pro)] came from her father, and this mutation was not listed in all gene database, and the mutation frequency among the general populace being 0%. Analytical predictions employing multiple statistical methodologies indicated a detrimental impact on the gene and its protein products. Consequently, this mutation satisfied the classification for “possible pathogenic gene variation” (PM2+PM3+PP3+PP4). The other SIGMAR1 gene variant [NM_005866.2: 151 + 1G>T] was inherited from her mother which has been shown to impair gene function in vitro and in vivo, and therefore met the standard of “pathogenic gene variant” (Richards et al., 2015). Notably, the patient’s parents exhibited no phenotypical manifestations of the disease (Figure 2). At this point, the patient was diagnosed with “clinically probable ALS”, considering the clinical presentation of both upper and lower motor neuron involvement coupled with genetic corroboration, although her EMG was not consistent with classic ALS manifestations (Shefner et al., 2020). The treatment regimen involved administering riluzole at a dose of 50 mg twice daily, accompanied by routine follow-up appointments.
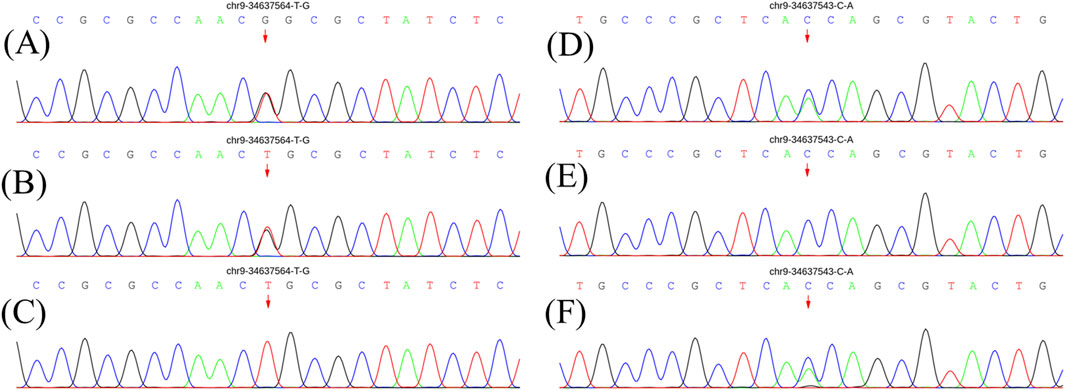
Figure 1. Genetic results for patients and parents. (A) and (D) show electrophoresis images of the proband. (B) and (E) show electrophoresis images of her father. (C) and (F) show electrophoresis images of her mother. The pathogenic gene mutation is consistent with autosomal recessive inheritance (compound heterozygous mutation pattern).

Figure 2. Diagram of family tree.
At age 16, during a follow-up visit, we found that her clinical symptoms remained stable, and no further progression of muscle atrophy was observed. Symmetrical muscle atrophy was observed in the distal extremities, with the lower limbs being more severely affected (Figure 3). We conducted a cognitive assessment, which revealed a Montreal Cognitive Assessment (MoCA) score of 23, a Mini-Mental State Examination (MMSE) score of 26, and a Frontal Assessment Battery (FAB) score of 14, indicating normal cognitive function. Furthermore, her brain magnetic resonance imaging was unremarkable, and no scoliosis was observed (Figure 4). The patient’s MRI of both lower legs exhibited noticeable bilateral calf muscle atrophy, which was symmetric, with areas showing infiltration of white adipose tissue (Figures 4M, N). Somatosensory evoked potential (SEP) and motor evoked potential (MEP) showed no significant abnormalities. Unfortunately, the patient declined the muscle biopsy due to concerns about invasive procedures. Based on the clinical features of distal symmetrical onset, slow progression, absence of fasciculations, and significantly reduced amplitudes on EMG, along with acutely denervated potentials that were localized and symmetrical, we ultimately revised the patient’s diagnosis to dHMN (Ma et al., 2020).
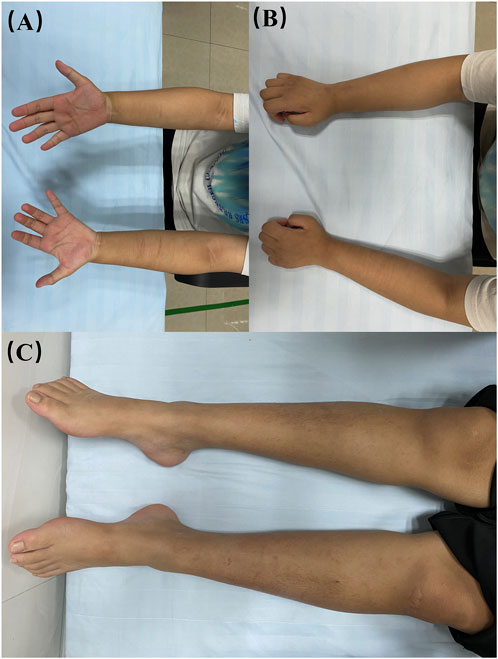
Figure 3. Visualisation of the patient’s limbs. (A, B) bilateral forearms; (C) bilateral lower limbs.
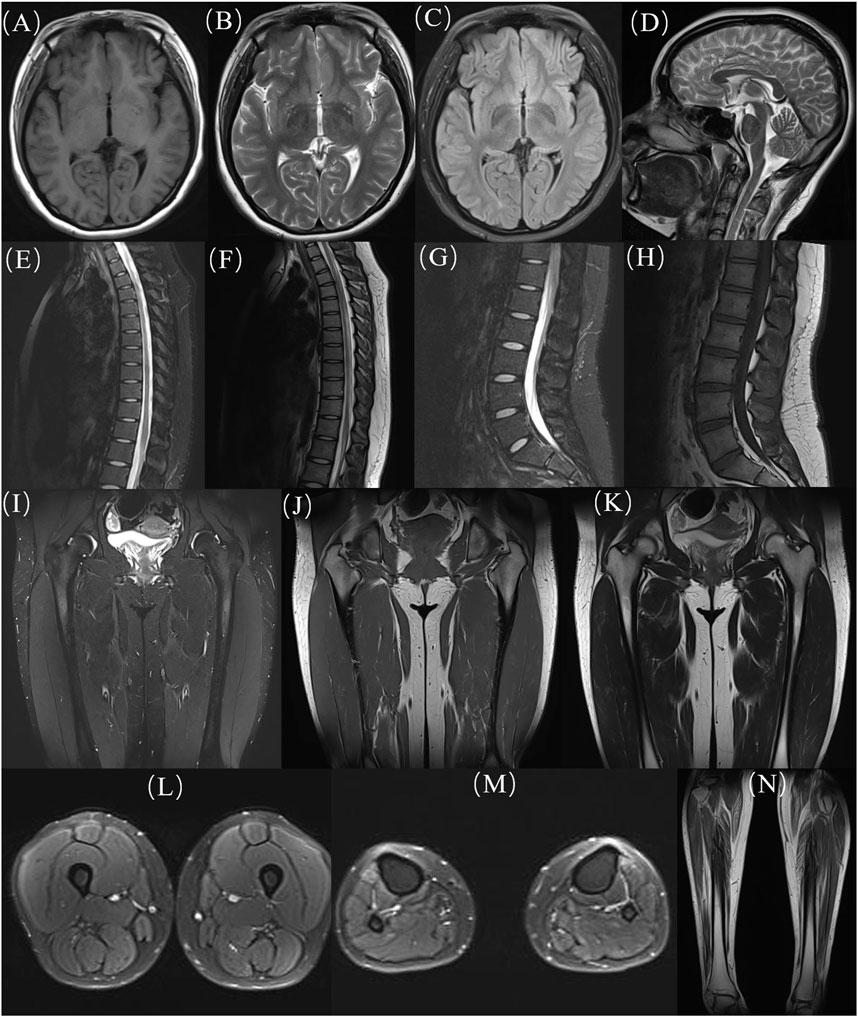
Figure 4. MRI images of the patient. (A-D) head, (E, F) thoracic segments; (G, H) lumbar segments; (H-K) hips; (L) thighs; (M, N) calves. MRI: Magnetic resonance imaging.
Discussion
In this report, we presented a case diagnosed dHMN girl harboring a novel mutation within the SIGMAR1 gene. The genetic affliction manifests an autosomal recessive inheritance pattern, congruent with the observation of a compound heterozygous mutation in our patient.
Although our patient exhibited segmental involvement of both upper and lower motor neurons, which aligns with the diagnostic criteria for ALS, the clinical course presents several atypical features. The patient’s symptoms began in childhood, with relatively rapid progression during the first 4 years, followed by a markedly slow disease progression. The pattern of muscle atrophy displayed a length-dependent distribution, and there was no report of fasciculations. Additionally, the initial EMG showed abnormalities in nerve conduction without the presence of acute denervation potentials. As the disease progressed, the EMG findings evolved to show neurogenic damage characteristic of ALS. However, these denervated potentials were very localized and symmetrically distributed. Notably, although muscle atrophy was not severe, EMG revealed significantly reduced amplitudes, suggesting predominantly axonal involvement. These findings do not support an ALS diagnosis and instead suggest the possibility of dHMN, especially given the prominence of atrophy in both lower limbs. Furthermore, previous reports indicate that ALS associated with SIGMAR gene mutations is often accompanied by cognitive impairments, which contrasts with our patient’s normal cognitive function. Research has shown that dHMN primarily affects the peripheral nerves and does not typically impact cognitive function. In summary, we believe the diagnosis of dHMN is more appropriate and aligns better with the family’s psychological expectations (Pareyson and Marchesi, 2009; Kasselimis et al., 2020).
dHMN is a disorder characterized by distal weakness and muscle atrophy. Some phenotypes, as illustrated in this case, also exhibit pyramidal tract signs, suggesting concurrent upper motor neuron dysfunction, which can mimic ALS and lead to misdiagnosis (Ma et al., 2020). A previous report described a consanguineous family with six members carrying a homozygous SIGMAR1 mutation (Al-Saif et al., 2011). The affected individuals presented with juvenile-onset, symmetrical distal limb weakness, atrophy, and spasticity, and were diagnosed as fALS based on the El Escorial criteria. However, the unusually slow progression of the disease and the absence of fasciculations in these patients are puzzling. Furthermore, it has been suggested that SIGMAR1 mutations may also cause a classic adult-onset ALS phenotype. However, subsequent findings revealed that one of these patients carried a C9ORF72 repeat expansion, which further weakens the evidence linking SIGMAR1 to ALS, suggesting that the association between SIGMAR1 mutations and familial ALS may be coincidental (Luty et al., 2010; Dobson-Stone et al., 2013; Pickering-Brown and Hardy, 2015). Therefore, we agree with recent studies that classifying SIGMAR1-related motor disorders as dHMN, rather than fALS, more accurately reflects the clinical and pathological characteristics of these cases (Ma et al., 2020).
Currently, therapeutic strategies for dHMN associated with SIGMAR1 gene mutations remain in the investigational phase, with management primarily focused on improving quality of life and slowing disease progression (Frasquet and Sevilla, 2022). As our understanding of these diseases deepens, and recognizing that they differ from ALS in terms of their rapid progression, it is believed that with ongoing advancements in gene therapy, SIGMAR-1 receptor-targeted therapies, and other molecular treatments, more effective therapeutic options may emerge in the future.
Conclusion
Our case enriches the dHMN database and highlights that adolescent onset, slow progression, length-dependent muscle atrophy, and localized, symmetrical denervation potentials should raise suspicion for dHMN.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of West China Hospital, Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
QY: Conceptualization, Formal Analysis, Methodology, Writing – original draft. RM: Formal Analysis, Investigation, Methodology, Writing – original draft. SC: Formal Analysis, Investigation, Methodology, Writing – review and editing. QQ: Investigation, Methodology, Writing – review and editing. JW: Writing – review and editing. XC: Conceptualization, Formal Analysis, Methodology, Resources, Supervision, Validation, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1477518/full#supplementary-material
References
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi:10.1002/ana.22534
Dobson-Stone, C., Hallupp, M., Loy, C. T., Thompson, E. M., Haan, E., Sue, C. M., et al. (2013). C9ORF72 repeat expansion in Australian and Spanish frontotemporal dementia patients. PLoS One 8, e56899. doi:10.1371/journal.pone.0056899
Feldman, E. L., Goutman, S. A., Petri, S., Mazzini, L., Savelieff, M. G., Shaw, P. J., et al. (2022). Amyotrophic lateral sclerosis. Lancet 400, 1363–1380. doi:10.1016/S0140-6736(22)01272-7
Frasquet, M., and Sevilla, T. (2022). Hereditary motor neuropathies. Curr. Opin. Neurol. 35, 562–570. doi:10.1097/WCO.0000000000001087
Izumi, Y., Morino, H., Miyamoto, R., Matsuda, Y., Ohsawa, R., Kurashige, T., et al. (2018). Compound heterozygote mutations in the SIGMAR1 gene in an oldest-old patient with amyotrophic lateral sclerosis. Geriatr. Gerontol. Int. 18, 1519–1520. doi:10.1111/ggi.13506
Kasselimis, D., Karadima, G., Angelopoulou, G., Breza, M., Tsolakopoulos, D., Potagas, C., et al. (2020). Evidence for cognitive deficits in X-linked charcot-marie-tooth disease. J. Int. Neuropsychol. Soc. 26, 294–302. doi:10.1017/S1355617719001188
Kim, H. J., Kwon, M. J., Choi, W. J., Oh, K. W., Oh, S. I., Ki, C. S., et al. (2014). Mutations in UBQLN2 and SIGMAR1 genes are rare in Korean patients with amyotrophic lateral sclerosis. Neurobiol. Aging 35, 1957.e7–1957.e1.957E18. doi:10.1016/j.neurobiolaging.2014.03.001
Lehky, T., and Grunseich, C. (2021). Juvenile amyotrophic lateral sclerosis: a review. Genes (Basel) 12, 1935. doi:10.3390/genes12121935
Li, H., Xuan, T., Xu, T., Yang, J., Cheng, J., and Wang, Z. (2023). SIGMAR1 variants in ALS-PD complex cases: a case report of a novel mutation and literature review. Front. Neurol. 14, 1242472. doi:10.3389/fneur.2023.1242472
Li, X., Hu, Z., Liu, L., Xie, Y., Zhan, Y., Zi, X., et al. (2015). A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 84, 2430–2437. doi:10.1212/WNL.0000000000001680
Luty, A. A., Kwok, J. B., Dobson-Stone, C., Loy, C. T., Coupland, K. G., Karlström, H., et al. (2010). Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol. 68, 639–649. doi:10.1002/ana.22274
Ma, M. T., Chen, D. H., Raskind, W. H., and Bird, T. D. (2020). Mutations in the SIGMAR1 gene cause a distal hereditary motor neuropathy phenotype mimicking ALS: report of two novel variants. Neuromuscul. Disord. 30, 572–575. doi:10.1016/j.nmd.2020.05.005
Pareyson, D., and Marchesi, C. (2009). Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 8, 654–667. doi:10.1016/S1474-4422(09)70110-3
Pickering-Brown, S., and Hardy, J. (2015). Is SIGMAR1 a confirmed FTD/MND gene? Brain 138, e393. doi:10.1093/brain/awv173
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. official J. Am. Coll. Med. Genet. 17, 405–424. doi:10.1038/gim.2015.30
Shefner, J. M., Al-Chalabi, A., Baker, M. R., Cui, L. Y., de Carvalho, M., Eisen, A., et al. (2020). A proposal for new diagnostic criteria for ALS. Clin. Neurophysiol. 131, 1975–1978. doi:10.1016/j.clinph.2020.04.005
Tazir, M., and Nouioua, S. (2024). Distal hereditary motor neuropathies. Rev. Neurol. Paris. 180, 1031–1036. doi:10.1016/j.neurol.2023.09.005
Keywords: SIGMAR, JALS, dHMN, diagnosis, case report
Citation: Yu Q, Mohammed Nazar RB, Chen S, Qian Q, Wang J and Chen X (2025) A novel SIGMAR1 missense mutation leads to distal hereditary motor neuropathy phenotype mimicking juvenile ALS: a case report of China. Front. Genet. 16:1477518. doi: 10.3389/fgene.2025.1477518
Received: 14 August 2024; Accepted: 31 March 2025;
Published: 16 April 2025.
Edited by:
Karen Muller Smith, University of Louisiana at Lafayette, United StatesReviewed by:
Partha Sarathi Sarkar, University of Texas Medical Branch at Galveston, United StatesVibha Taneja, Sir Ganga Ram Hospital, India
Stefano Carlo Previtali, San Raffaele Scientific Institute (IRCCS), Italy
Copyright © 2025 Yu, Mohammed Nazar, Chen, Qian, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueping Chen, Y2hlbnh1ZXBpbmcwNjA2QHNpbmEuY29t
†These authors have contributed equally to this work and share first authorship