 Tingmei Wang
Tingmei Wang Dong Li
Dong Li Yunhua Deng*
Yunhua Deng*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 06 March 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1414129
Dyschromatosis, a group of pigmentary dermatoses, accompany both hyper- and hypo-pigmentation, including dyschromatosis symmetrica hereditaria (DSH), dyschromatosis universalis hereditaria (DUH), and familial progressive hyper- and hypo-pigmentation (FPHH). A peculiar phenotype of dyschromatosis presented as multiple lentigines and hypopigmentation with various sizes and shapes was found to be associated with SASH1 mutations and has recently been reported frequently. The current study evaluated the clinical manifestation, pathological pattern, and genetic basis of dyschromatosis in a five-generation family. This research also presents a case study of a sporadic patient with dyschromatosis caused by SASH1 mutations and shows different clinicopathological characteristics form DSH, DUH and FPHH. SASH1 (SAM and SH3 Domain Containing 1) gene, located on chromosome 6q24.3, encodes a tumor suppressor protein involved in cell signaling, migration, and adhesion. Additionally, the SASH1 mutations could also lead to another pigmentary phenotype: multiple lentigines. High consistency in clinicopathological features and genetic basis in these two SASH1-related pigmentary disorders suggests that SASH1 mutations cause multiple lentigines and dyschromatosis which might belong to a disease spectrum. Overall, it is expected the current study results could help enhance a more comprehensive understanding of SASH1-related pigmentary dermatoses.
The term “dyschromatosis” refers to a group of pigmentary disorders that present with both hyper- and hypo-pigmentation. Three known subtypes of this disease are dyschromatosis symmetrica hereditaria (DSH), dyschromatosis universalis hereditaria (DUH), and familial progressive hyper- and hypopigmentation (FPHH); they result from autosomal dominant inheritance without systemic involvement; DSH, DUH, and FPHH are caused by adenosine deaminase acting on RNA1 gene (ADAR1) (Miyamura et al., 2003), ATP binding cassette subfamily B member 6 gene (ABCB6) (Zhang et al., 2013), and KIT ligand gene (KITLG) (Amyere et al., 2011), respectively. Recently, several studies (Zhou et al., 2013; Zhong et al., 2019; Wu et al., 2020; Liu et al., 2021; Cao et al., 2021; Yang et al., 2023) have documented a distinct group of dyschromatosis in an autosomal dominant inherence pattern caused by mutations of SAM (sterile alpha motif) and SH3 (Src homology domain 3) domain-containing protein 1 gene (SASH1). The dyschromatosis caused by SASH1 mutations is sometimes considered to be a subtype of DUH. However, the objective of the present study is to focus on the clinical manifestation and genetic basis behind SASH1-related dyschromatosis despite the lack of adequate pathological analysis. In this study, the clinicopathologic features of the dyschromatosis caused by SASH1 mutations are analyzed to gain a better understanding of this disorder and make a clearer classification of dyschromatosis without systemic involvement.
The proband, a 20-year-old woman from central China, presents with cutaneous hyper- and hypopigmentation lasting 19 years. The condition started as multiple lentigo-like lesions on her face when the patient was 1 year old. Later on, some hypopigmented macules developed on the dorsa of interphalangeal and metacarpophalangeal joints of her hands. Over time, these macules extended to her feet, hands, trunk, face, and other limbs. Slight canities appeared in her hair at the age of 19. Her palms, soles, and nails were spared. She displayed normal intellectual ability and no other systemic or developmental defects. Dermatological examination revealed multiple ill-demarcated hypopigmented macules and patches mixed with well-circumscribed, lentigo-like hyperpigmentation on the dorsa of hands, feet, arms, legs, buttocks, trunk, neck, and face (Figures 1A–D). The most pronounced features of this proband were the symmetrical hypopigmentation on the joint prominences and the hyperpigmentation presented as multiple lentigines (Figures 1A–C).
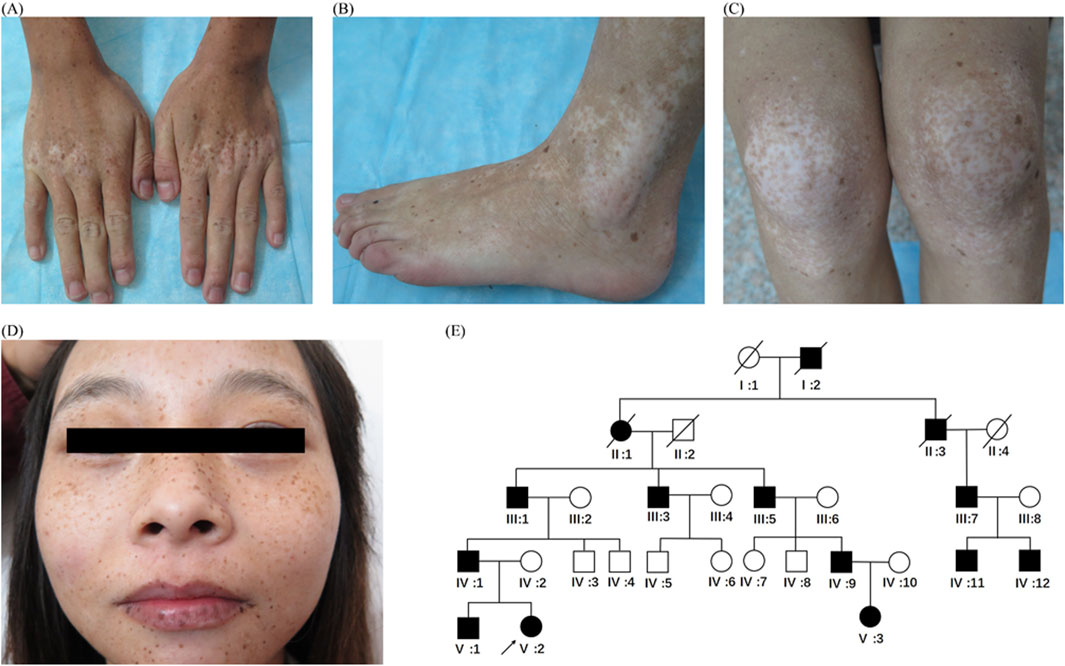
Figure 1. Clinical features and pedigree (A–D) Clinical features of the proband (V:2). Multiple hypopigmented macules or patches mixed with lentigo-like macules on the dorsa of the hands (A), feet (B) extension of knee joints, (C) and face and lips (D). (E) Pedigree chart. Black symbols represent affected individuals, and open symbols represent unaffected individuals. Circles and squares indicate women and men, respectively. The arrow indicates the proband.
All living members of the patient’s pedigree underwent a thorough medical evaluation. An autosomal dominant pattern was observed (Figure 1E). Fourteen individuals (11 men and 3 women) were affected, and the 11 surviving affected members showed similar manifestations and clinical course. Hypopigmentation was apparent on the bilateral dorsa of their extremities (Figures 2A, B, E, F, I, J) and extensor surfaces of the knees and elbows (Figures 2C, G, K) of the individuals. Diffuse hypopigmentation with some hyperpigmented macules often developed on their buttocks during their thirties (Figures 2D, H, L). Canities usually began around age 20 and gradually worsened (Supplementary Figure S1).
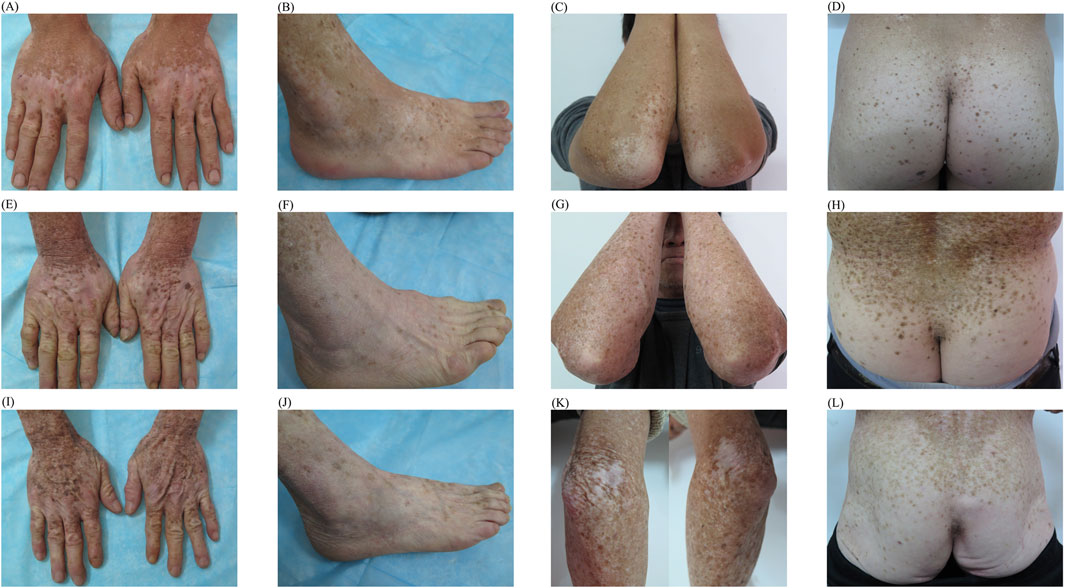
Figure 2. Typical manifestations of individuals of different ages within family (A–D) Diffuse hypopigmentation with hyperpigmented macules on the dorsa of the extremities, extensor surfaces of elbows, and buttocks of individual IV 9 (E–H) Similar manifestations on individual III 5 (I–L) Similar manifestations on individual III 3.
The sporadic patient studied here is a 24-year-old man from central China; he presented with cutaneous hyper-and hypopigmentation lasting 23 years. The clinical manifestations of this patient closely resembled those of the collected dyschromatosis family. His dermatological examination revealed multiple lentigo-like hyperpigmentation, and intermixed or confluent hypopigmented patches over the whole body. The hypopigmentation on the buttocks and lower limbs was diffuse and homogeneous (Supplementary Figure S2). Contrastively, his elder sister and both parents had no similar pigmentation defects.
Pathogenic mutations of ADAR1, ABCB6, and KITLG, the respective causative genes for DSH, DUH, and FPHH, were screened using Sanger sequencing in the proband of dyschromatosis pedigree. No causative mutation was identified in ADAR1, ABCB6, or KITLG genes. Thereafter, the mutations of SASH1 in this patient were screened, which resulted in discovery of missense mutation c.1556G>A p. Ser519Asn (S519N)(Figure 3A) in this dyschromatosis pedigree. S519N mutation was confirmed in the patients of this family, though it was absent in other unaffected individuals. This variant was co-segregated perfectly with the phenotype in this family.
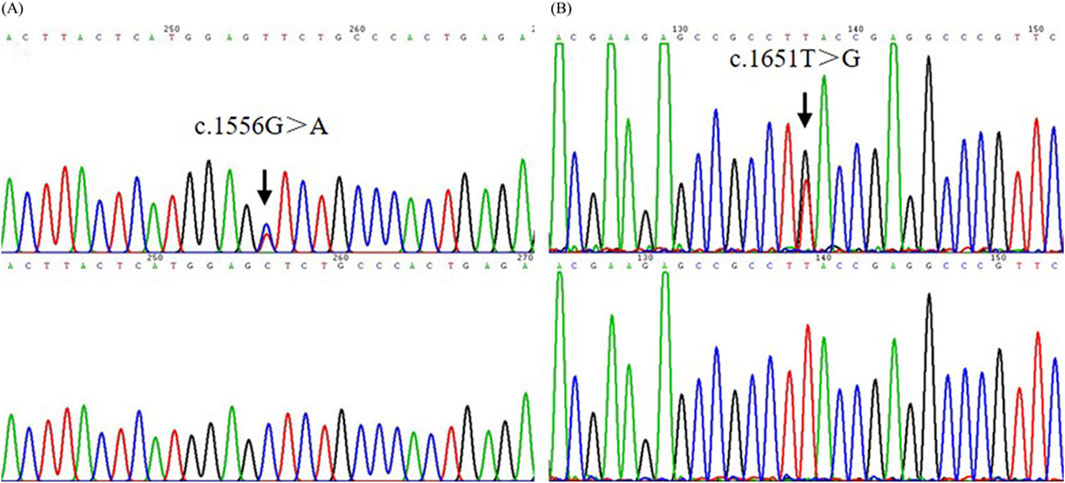
Figure 3. The mutation analysis of SASH1 in the pedigree and sporadic patient. (A) A heterozygous missense mutation c.1556G>A (S519N) in SASH1 was identified in the pedigree. (B) A heterozygous missense mutation c.1651T>G (Y551D) in SASH1 was identified in an additional individual.
The other missense mutation c.1651T>G p. Tyr551Asp (Y551D) in SASH1 was detected in the sporadic patient of dyschromatosis and was not observed in his parents and elder sister (Figure 3B). Further direct DNA sequence analysis with a panel of 300 unaffected control individuals matched for the geographical location could not detect these two mutations.
Microphthalmia-associated Transcription Factor (MITF) was used to examine if a change in the number of melanocytes participates in the occurrence and development of this dyschromatosis and to label the melanocytes in the specimens from the normally, hyper- and hypo-pigmented areas and from the skin of the normal control. The cells stained positively by the melanocyte marker MITF were counted under three different fields of ×200 magnification: 13.0 ± 0.7 (mean ± CD) in normally pigmented areas (Figure 4A), 10.3 ± 2.1 (mean ± CD) in hyperpigmented lesions (Figure 4B), and 5 ± 0.82 (mean ± CD) in the skin of unaffected controls (Figure 4D). However, only hyperpigmented lesions demonstrated a pronounced increase in melanin (Figure 4B), although both the normally pigmented and hyperpigmented skin showed increased melanocytes. Furthermore, only one MITF-positive cell was detected in any of the hypopigmented lesions on the specimen (Figure 4C).
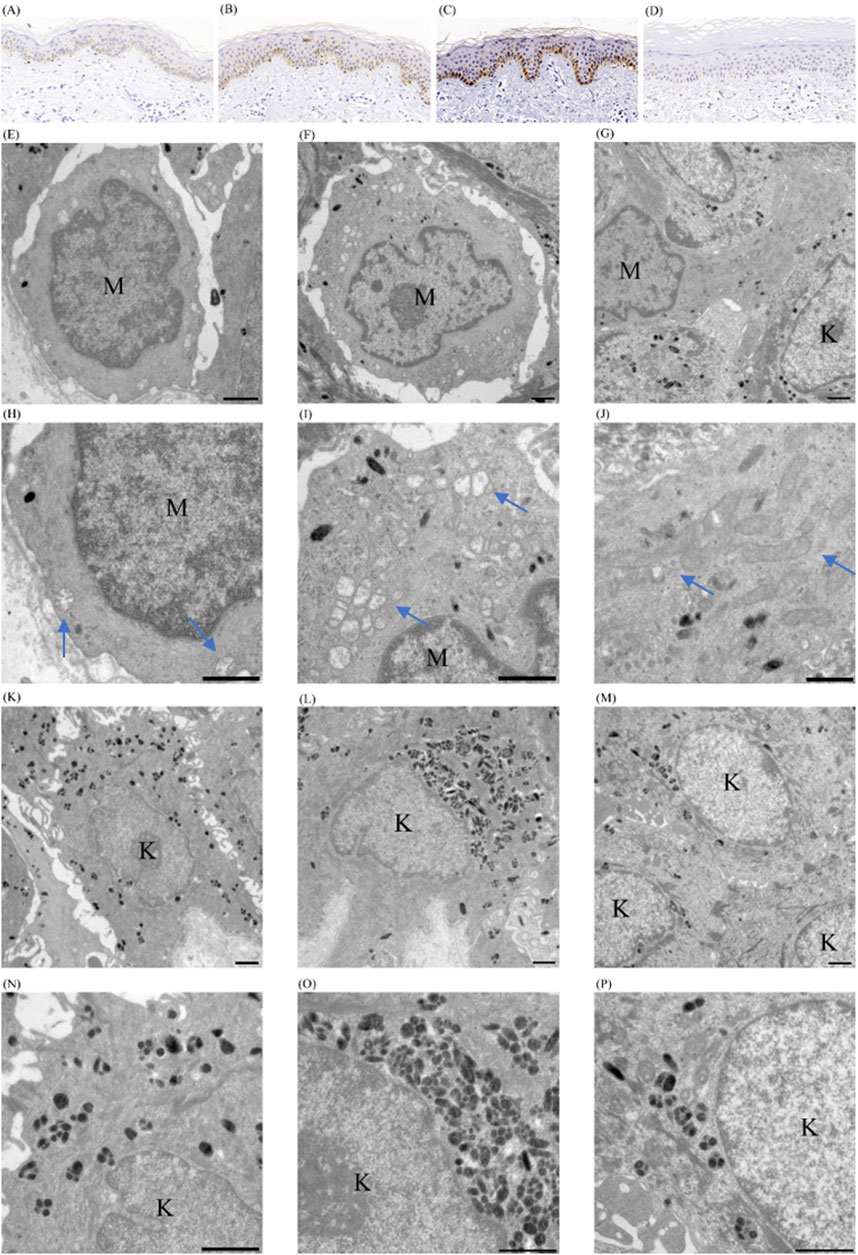
Figure 4. Pathological findings of cutaneous tissues of patient with SASH1 mutation related dyschromatosis. (A–D) Immunohistochemical analyses on the cutaneous tissues. MITF immunohistochemical staining of normally (A), hyper-(B), and hypo-(C) pigmented cutaneous tissues of patient with SASH1 related dyschromatosis, and of cutaneous tissues of normally controls (D), original magnification ×200 (E–P) Ultrastructural analyses on the cutaneous lesions. The melanocytes in normally pigmented areas showed smaller cell bodies and poor dendritic formation, loose connection with their surroundings and contained fewer melanosomes and mitochondria (E). The melanocytes in hyperpigmented areas showed poor dendritic formation, loose connection with their surroundings, and contain moderate melanosomes and mitochondria (F). The melanocyte in hypopigmented lesions was large in size with elongated dendrites and contained numerous melanosomes and mitochondria (G). Fragmentation and swelling with obscure cristae and vacuolization of mitochondria were observed in both normal- and hyper-pigmented areas. The morphology abnormalities of mitochondria were significantly more serious in melanocytes from normally pigmented areas (H, I). The mitochondria in hypopigmented lesions were normal in morphology and interconnected presenting as intracellular networks (J). More and fewer melanosomes within keratinocytes of hyper- (L) and hypopigmented (M) than in normally pigmented skin (K). The melanosomes were primarily distributed individually with a few non-membrane-bound melanosome complexes in normally pigmented keratinocytes (N). The non-membrane-bound melanosome complexes predominated in hyperpigmented keratinocytes (O). The membrane-bound melanosome complexes predominated in hypopigmented keratinocytes (P). The bar indicates 1 μm; the arrow indicates mitochondria. (MC melanocyte; KC keratinocyte).
At an ultrastructural level, the melanocytes in normally pigmented areas showed smaller cell bodies with poor dendritic formation and contained fewer melanosomes and mitochondria-related to energy generation and protein synthesis (Figures 4E, H). The poorly activated morphology of melanocytes in normally pigmented areas shows an absence of any obvious hyperpigmentation despite an increased number of melanocytes in this area. Fragmentation and swelling with obscure cristae and vacuolization were observed in the mitochondria of both normally and hyper-pigmented areas (Figures 4E, F, H, I) in the patient. However, the fragmentation and swelling of mitochondria were significantly more serious in melanocytes from normally pigmented skin (Figures 4E, F, H, I) which might be the reason for the scarce melanosomes and mitochondria in normally pigmented melanocytes. Contrastively, the remaining melanocytes in hypopigmented lesions were large in size with elongated dendrites and contained numerous melanosomes and mitochondria (Figures 4G, J). The hypopigmented lesions showed normal mitochondria in morphology and interconnected building intracellular networks (Figure 4J). The overactived state is pointed to melannocytes rahter than lesions might be associated with the functional compensatory enhancement of the remanent melanocytes. In addition, both melanocytes from normally pigmented areas and hyperpigmented lesions were loosely connected with their surroundings (Figures 4E, F).
A higher number of melanosomes were observed within keratinocytes from hyperpigmented lesions than normally pigmented areas (Figures 4K, L), and fewer melanosomes were found within keratinocytes in hypopigmented lesions (Figure 4M). The distribution of melanosomes within keratinocytes from different sources was irregular. The melanosomes were predominantly distributed individually with a few non-membrane-bound melanosome complexes documented in normally pigmented keratinocytes (Figure 4N). The non-membrane-bound melanosome complexes were predominant in hyperpigmented keratinocytes (Figure 4O), whereas membrane-bound melanosome complexes predominated in hypopigmented keratinocytes (Figure 4P). This distribution pattern supports previously reported distribution of melanosomes in keratinocytes from different skin types (Hurbain et al., 2018).
In the form of dyschromatosis related to SASH1 mutations, the lentigo-like macules and hypopigmented macules or patches started on the face or on the dorsa of interphalangeal and metacarpophalangeal joints of hands in infancy or early childhood and extended progressively onto the body with age. The most prominent feature of SASH1-related dyschromatosis is multiple lentigo-like macules coexisting with variable and symmetrical hypopigmentation which was prominent on the joint protuberances. The hypopigmented patches could progress to be diffuse and homogeneous. To our knowledge, premature canities were first observed in the present study. No other systemic or developmental defects appeared (Zhou et al., 2013; Zhong et al., 2019; Wu et al., 2020; Liu et al., 2021; Cao et al., 2021; Yang et al., 2023).
DSH, DUH, and FPHH are documented dyschromatosis subtypes of autosomal dominant inheritance without systemic involvement. A majority of SASH1-related dyschromatosis was reported as a subtype of DUH. ABCB6 gene was first reported as the disease-causing gene of DUH in a five-generation Chinese family in 2013 (Zhang et al., 2013); other studies (Cui et al., 2013; Liu et al., 2014)also corroborated this finding subsequently. However, the SASH1-related dyschromatosis presented as a distinct clinical phenotype different from that in patients with DUH. DUH, a generalized dyschromatosis, is characterized by similar-sized small hyper- and hypo-pigmented macules intermixed in a reticular or mottled pattern (Zhang et al., 2013; Cui et al., 2013; Liu et al., 2014). Though, the multiple lentigo-like macules and the symmetrical hypopigmented patches, especially on the joint prominences, are the clinical characteristics of SASH1-related dyschromatosis but they are not commonly seen in patients with DUH (Zhang et al., 2013; Cui et al., 2013; Liu et al., 2014). Moreover, patients with DUH lack diffuse or homogenous hypopigmentation (Zhang et al., 2013; Cui et al., 2013; Liu et al., 2014).
To elucidate whether SASH1-related dyschromatosis is a subtype of DUH, we analyzed the pathological manifestations of SASH1-related dyschromatosis. The pathological results also indicated that SASH1-related dyschromatosis and DUH may be different entities. In the current study, changes in the number of melanocytes were found to be involved in SASH1-related dyschromatosis: the number of melanocytes showed approximately a 2-fold increase in both normally pigmented and hyperpigmented skin than normal controls and a remarkable decrease in hypopigmented skin. Also, the mitochondrial abnormalities within melanocytes were another pathological feature of SASH1-related dyschromatosis. The more serious fragmentation and vacuolization of mitochondria within normally pigmented melanocytes might be related to the decreased number of mitochondria, and might ultimately influence the melanogenesis and transport of melanin resulting in no obvious pigment deposit despite the increased number of melanocytes in normally pigmented skin of SASH1 related dyschromatosis. DUH is not a pigmentary disorder related to the changes in melanocyte numbers (Cui et al., 2013). In addition, the abnormalities of mitochondria are not found in the melanocytes of the skin of patients with DUH (Gupta et al., 2015). Therefore, the clinicopathological features do not support the idea that DUH and SASH1-related dyschromatosis are identical entities.
DSH, caused by mutations in the ADAR1 gene (Miyamura et al., 2003), is characterized by a mixture of hyperpigmented and hypopigmented macules distributed on the dorsal aspects of the hands and feet (Miyamura et al., 2003; Kondo et al., 2008). There are some similarities in their early age of onset, intermixture of hyper- and hypopigmentation on the dorsa of hands and feet, freckle-like lesions on the face, and histological feature of the loss of melanocytes in hypopigmented lesions. However, the confluent patches of hypopigmentation, even the diffuse hypopigmentation are not commonly observed in patients with DSH. Furthermore, the generalized distribution of lesions differed from DSH in the present study which is a localized pigmentary dermatosis. Although the hypopigmentation is caused by the missing melanocytes in the two cases, the current case study showed no degenerative melanocyte morphology in hypopigmented lesions, thereby indicating that different mechanisms might underlie the formation of hypopigmentation in DSH and the SASH1-related dyschromatosis (Kondo et al., 2008).
Clinical signs of FPHH consist of progressive diffuse hyperpigmented lesions, multiple café-au-lait macules, intermingled with scattered hypopigmented maculae, and lentigines (Amyere et al., 2011; Zanardo et al., 2004). KITLG gene is identified to be its pathogenic gene (Amyere et al., 2011). FPHH resembles the present cases in the generalized lentigo-like lesions on the body. However, the dyschromatosis also presented with symmetrical hypopigmented macules or patches and lacked other classical manifestations of FPHH, such as diffuse hyperpigmentation and café-au-lait macules. Histologically, FPHH has been reported to be characterized by an unchanged number of melanocytes and a Caucasian-like distribution pattern of melanosomes within keratinocytes in all lesions (Amyere et al., 2011; Zanardo et al., 2004). Alteration in melanocytes’ number participated in the SASH1-related dyschromatosis and the distribution pattern of melanosomes within keratinocytes is consistent with the previously reported distribution of melanosomes in keratinocytes from different skin types. These features distinguished our case from FPHH.
Other than the peculiar type of dyschromatosis (Zhou et al., 2013; Zhong et al., 2019; Wu et al., 2020; Liu et al., 2021; Cao et al., 2021; Yang et al., 2023), the SASH1 mutations are also associated with another phenotype of skin pigmentation disorder: multiple lentigines (Shellman et al., 2015; Zhang et al., 2016; Wang et al., 2017; Araki et al., 2021; Kim et al., 2023). To date, a total of 22 SASH1 mutations have been found associated with skin pigmentation changes (S587N was reported in the Chinese Journal of Dermatology and Chinese Journal of Dermatovenereology, S507F and c.49_54dupCCCCAGfanfa, c.1284 + 4A>G were reported in Chinese Journal of Dermatovenereology) (Table 1). Among them, mutations S519N, S516I, and S587N are associated with both multiple lentigines and the peculiar dyschromatosis phenotype (Table 1). The difference between these two phenotypes of pigmentary disorder with SASH1 mutations relates to the presence or absence of hypopigmentation. Additionally, the scattered and mild hypopigmented spots and macules are also observed in SASH1-related multiple lentigines as reported by Shellman et al. (2015) and Zhang et al. (2016), respectively, thereby indicating that multiple lentigines might develop into the dyschromatosis under certain circumstance. In addition, both conditions were found related to the changes in the number of melanocytes in the epidermis: the number of melanocytes in normally and hyper-pigmented skin were similar but approximately twice than the normal controls in both multiple lentigines (Shellman et al., 2015) and dyschromatosis related with SASH1 mutations. Hence, we conclude that these two conditions related to SASH1 might be the same disease spectrum because of greater resemblance in their clinical phenotype, pathologic characteristics, and molecular genetics.
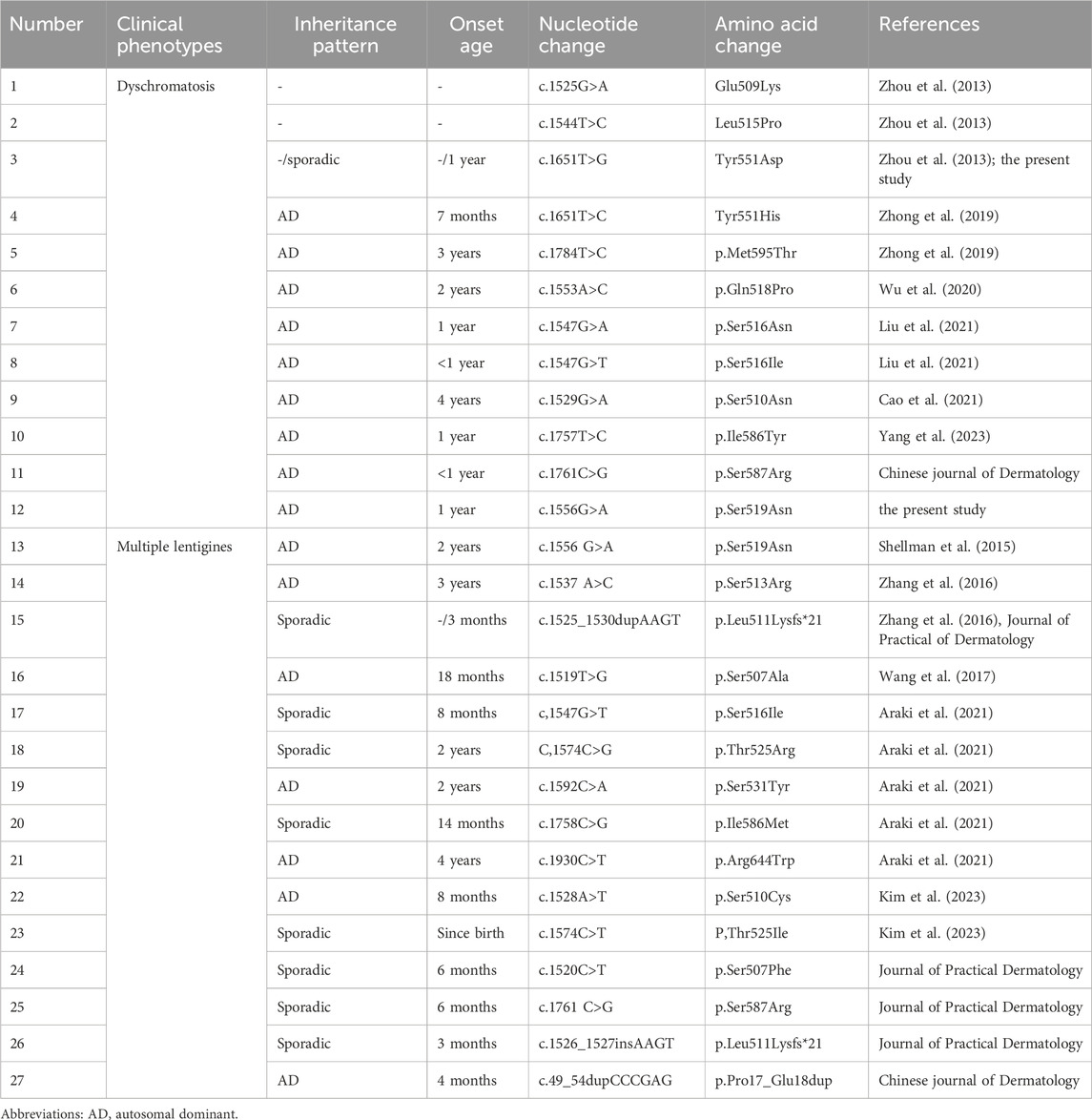
Table 1. SASH1 mutations associated with pigmentation disorders.
Overall, we reported a phenotype of dyschromatosis with SASH1 mutations in a five-generation Chinese family and a sporadic case and examined the clinicopathologic features of this peculiar dyschromatosis. The clinical phenotypic, pathologic, and genetic differences between SASH1-related dyschromatosis and other dyschromatosis of autosomal dominant inheritance including DUH, DSH, or FPHH suggest that the SASH1-related dyschromatosis might be a distinct entity. Additionally, the high consistency in clinical manifestation, pathological expression, and genetic basis between the SASH1-related dyschromatosis and SASH1-related multiple lentigines suggests that these two conditions might be different phenotypes of a disease spectrum. Our findings not only help enhance a more comprehensive understanding of SASH1-related dyschromatosis but also provide a useful hint for the diagnosis of pigmentary dermatosis.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
TW: formal analysis, methodology, writing–original draft, and writing–review and editing. DL: methodology and writing–review and editing. YD: conceptualization, funding acquisition, and writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (No. 81472864).
We are deeply grateful to all of the members of this family for their enthusiastic participation and willingness to donate their time and information.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1414129/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Premature canities of individual IV 9. Individual IV 9 was 30-year-old, and his hair had turned almost completely white. His hair was dyed, and the color of the proximal hair was white.
SUPPLEMENTARY FIGURE S2 | Clinical features of an additional individual with similar manifestations. (A) Hypopigmented patches with hyperpigmented macules on his elbows. (B) Hypopigmented patches with hyperpigmented macules on the dorsa of his hand. (C) Diffuse hypopigmentation on his buttocks, and scattered macules of hyper- and hypopigmentation on his trunk. (D) Diffuse hypopigmentation with interspersed spots of hyperpigmentation on his lower limbs and some small islands of normally pigmented skin within the hypopigmentation.
Amyere, M., Vogt, T., Hoo, J., Brandrup, F., Bygum, A., Boon, L., et al. (2011). KITLG mutations cause familial progressive hyper- and hypopigmentation. J. Invest Dermatol 131, 1234–1239. doi:10.1038/jid.2011.29
Araki, Y., Okamura, K., Saito, T., Matsumoto, K., Natsuga, K., Nishimoto, J., et al. (2021). Five novel mutations in SASH1 contribute to lentiginous phenotypes in Japanese families. Pigment. Cell Melanoma Res. 34, 174–178. doi:10.1111/pcmr.12930
Cao, L., Zhang, R., Yong, L., Chen, S., Zhang, H., Chen, W., et al. (2021). Novel missense mutation of SASH1 in a Chinese family with dyschromatosis universalis hereditaria. BMC Med. Genomics 14, 168. doi:10.1186/s12920-021-01014-w
Cui, Y. X., Xia, X. Y., Zhou, Y., Gao, L., Shang, X. J., Ni, T., et al. (2013). Novel mutations of ABCB6 associated with autosomal dominant dyschromatosis universalis hereditaria. PLoS One 8, e79808. doi:10.1371/journal.pone.0079808
Gupta, A., Sharma, Y., Dash, K. N., Verma, S., Natarajan, V. T., and Singh, A. (2015). Ultrastructural investigations in an autosomal recessively inherited case of dyschromatosis universalis hereditaria. Acta Derm. Venereol. 95, 738–740. doi:10.2340/00015555-2030
Hurbain, I., Romao, M., Sextius, P., Bourreau, E., Marchal, C., Bernerd, F., et al. (2018). Melanosome distribution in keratinocytes in different skin types: melanosome clusters are not degradative organelles. J. Invest Dermatol. 138, 647–656. doi:10.1016/j.jid.2017.09.039
Kim, J. Y., Kwon, I. J., and Lee, S. E. (2023). Two novel mutations in SASH1 identified in a familial and a sporadic generalized lentiginosis phenotype in Koreans. Clin. Exp. Dermatol 48, 1171–1173. doi:10.1093/ced/llad199
Kondo, T., Suzuki, T., Mitsuhashi, Y., Ito, S., Kono, M., Komine, M., et al. (2008). Six novel mutations of the ADAR1 gene in patients with dyschromatosis symmetrica hereditaria: histological observation and comparison of genotypes and clinical phenotypes. J. Dermatol 35, 395–406. doi:10.1111/j.1346-8138.2008.00493.x
Liu, H., Li, Y., Hung, K. K., Wang, N., Wang, C., Chen, X., et al. (2014). Genome-wide linkage, exome sequencing and functional analyses identify ABCB6 as the pathogenic gene of dyschromatosis universalis hereditaria. PLoS One 9, e87250. doi:10.1371/journal.pone.0087250
Liu, J. W., Habulieti, X., Wang, R. R., Ma, D. L., and Zhang, X. (2021). Two novel SASH1 mutations in Chinese families with dyschromatosis universalis hereditaria. J. Clin. Lab. Anal. 35, e23803. doi:10.1002/jcla.23803
Miyamura, Y., Suzuki, T., Kono, M., Inagaki, K., Ito, S., Suzuki, N., et al. (2003). Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am. J. Hum. Genet. 73, 693–699. doi:10.1086/378209
Shellman, Y. G., Lambert, K. A., Brauweiler, A., Fain, P., Spritz, R. A., Martini, M., et al. (2015). SASH1 is involved in an autosomal dominant lentiginous phenotype. J. Invest Dermatol 135, 3192–3194. doi:10.1038/jid.2015.292
Wang, J., Zhang, J., Li, X., Wang, Z., Lei, D., Wang, G., et al. (2017). A novel denovo mutation of the SASH1 gene in a Chinese family with multiple lentigines. Acta Derm. Venereol. 97, 530–531. doi:10.2340/00015555-2575
Wu, N., Tang, L., Li, X., Dai, Y., Zheng, X., Gao, M., et al. (2020). Identification of a novel mutation in SASH1 gene in a Chinese family with dyschromatosis universalis hereditaria and genotype-phenotype correlation analysis. Front. Genet. 11, 841. doi:10.3389/fgene.2020.00841
Yang, Y., Jiang, N., Mai, J. Q., Yang, S., Xiao, Y., and Liu, S. (2023). Uncovering a new SASH1 mutation associated with dyschromatosis universalis hereditaria using whole-exome-sequencing: a case report. Med. Baltim. 4, e34448. doi:10.1097/MD.0000000000034448
Zanardo, L., Stolz, W., Schmitz, G., Kaminski, W., Vikkula, M., Landthaler, M., et al. (2004). Progressive hyperpigmentation and generalized lentiginosis without associated systemic symptoms: a rare hereditary pigmentation disorder in south-east Germany. Acta Derm. Venereol. 84, 57–60. doi:10.1080/00015550310005780
Zhang, C., Li, D., Zhang, J., Chen, X., Huang, M., Archacki, S., et al. (2013). Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J. Invest Dermatol 133, 2221–2228. doi:10.1038/jid.2013.145
Zhang, J., Cheng, R., Liang, J., Ni, C., Li, M., and Yao, Z. (2016). Lentiginous phenotypes caused by diverse pathogenic genes (SASH1 and PTPN11): clinical and molecular discrimination. Clin. Genet. 90, 372–377. doi:10.1111/cge.12728
Zhong, W. L., Wang, H. J., Lin, Z. M., and Yang, Y. (2019). Novel mutations in SASH1 associated with dyschromatosis universalis hereditaria. Indian J. Dermatol Venereol. Leprol. 85, 440. doi:10.4103/ijdvl.IJDVL_360_17
Keywords: SASH1, dyschromatosis, dyschromatosis universalis hereditaria, multiple lentigines, ABCB6
Citation: Wang T, Li D and Deng Y (2025) Case report: Clinicopathological characteristics of SASH1 mutation-related dyschromatosis: a rethinking of the classification of dyschromatosis. Front. Genet. 16:1414129. doi: 10.3389/fgene.2025.1414129
Received: 08 April 2024; Accepted: 10 February 2025;
Published: 06 March 2025.
Edited by:
Peiguang Wang, Anhui Medical University, ChinaReviewed by:
Yanhua Liang, Southern Medical University, ChinaCopyright © 2025 Wang, Li and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunhua Deng, eWhkZW5nQG1haWxzLnRqbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.