 Miaomiao Xin1
Miaomiao Xin1 Xin Guan
Xin Guan Yi Li
Yi Li Hongsheng Sun
Hongsheng Sun Min Fu
Min Fu- 1Department of Rheumatology and Immunology, Shandong Provincial Hospital Affiliated to Shandong First Medical University (Shandong Provincial Hospital), Jinan, Shandong, China
- 2Department of Rheumatology and Immunology, Xing’an League People’s Hospital, Ulan Hot, China
- 3Department of Radiology, Shandong Provincial Hospital Affiliated to Shandong First Medical University (Shandong Provincial Hospital), Jinan, Shandong, China
- 4Department of Joint Surgery and Sports Medicine, Shandong Provincial Hospital Affiliated to Shandong First Medical University (Shandong Provincial Hospital), Jinan, Shandong, China
Background: Developmental dysplasia of the hip (DDH) is one of the most common developmental disorders worldwide, caused by a combination of genetic and environmental factors.
Methods: To investigate the genetic etiology of DDH in a proband (a 27-year-old male), we reviewed the patient’s clinical data and collected peripheral blood samples from the proband and his parents. Genomic DNA was extracted, and polymerase chain reaction (PCR) amplification was performed. Clinical whole-exome sequencing (WES) using next-generation sequencing (NGS) was conducted to identify potential mutation sites, which were then validated through Sanger sequencing. Bioinformatics analysis was performed to assess the pathogenicity of the identified variant, and 3D protein modeling was conducted to predict its impact on protein structure.
Results: The proband presented with pain in bilateral hips, and based on clinical symptoms, laboratory findings and imaging studies, the final diagnosis was considered to be acetabular dysplasia with overlapping secondary synovial chondromatosis. Family history revealed similar symptoms in the proband’s father, while the grandparents and other family members were unaffected. The patient underwent bilateral total hip arthroplasty and synovectomy. NGS and Sanger sequencing identified a heterozygous missense mutation in the COL2A1 gene (ex13, c.823C > T; p.Arg275Cys) in both the proband and his father, while this mutation was absent in the mother. Bioinformatic analysis indicated that the c.823C > T (p.Arg275Cys) variant is pathogenic, and structural modeling demonstrated that the substitution of arginine with cysteine at residue 275 altered the protein structure.
Conclusion: Our findings highlight the diagnostic utility of NGS in identifying precise genetic causes of DDH. The identification of the COL2A1 gene mutation in this present case represents a novel clinical phenotype, expanding the spectrum of disorders associated with COL2A1 mutations.
1 Introduction
Developmental dysplasia of the hip (DDH) is a complex musculoskeletal disorder with a broad spectrum of clinical manifestations, ranging from asymptomatic cases with mild radiological abnormalities to more severe forms, including minor joint instability, acetabular dysplasia, subluxation, and complete dislocation of the hip joint (Basit et al., 2016). While mild dysplasia may resolve spontaneously, more severe hip deformities, if misdiagnosed or improperly treated during early stages, tend to worsen progressively, resulting in chronic hip pain, restricted movement, abnormal gait, and eventually degenerative arthritis in adulthood (Zhao et al., 2023).
DDH arises from an interplay of multiple genetic and environmental factors (Zhao et al., 2023). The inheritance pattern of DDH is generally thought to follow an autosomal incomplete dominant model (Zhao et al., 2023). However, the significant genetic heterogeneity observed among DDH cases means that the condition does not adhere to classical Mendelian inheritance (Basit et al., 2017). Current genetic studies on DDH have focused primarily on specific chromosomal regions, particularly chromosomes 1, 3, 12, 17, and 20 (Kenanidis et al., 2020). Several genes have been implicated in the pathogenesis of DDH, including C-X3-C motif chemokine receptor 1 (CX3CR1), growth differentiation factor 5 (GDF5), collagen type I alpha 1 chain (COL1A1), Asporin (ASPN), and vitamin D receptor (VDR) (Zamborsky et al., 2019). Additionally, interleukin 6, homeobox genes (HOXD9 and HOXB9), pregnancy-associated plasma protein A2 (PAPPA2), transforming growth factor-beta 1 (TGF-β1), T-box transcription factor (TBX4), ubiquinol-cytochrome C reductase complex chaperone (UQCC), ribosome biogenesis factor (BMS1), and fibroblast growth factor 2 (FGF2) have also been linked to the development of DDH (Zamborsky et al., 2019).
Environmental risk factors associated with DDH include breech position, female gender, family history of DDH, first-born, physical limitations within the uterus (macrosomia, oligohydramnios, multiple pregnancies), and incorrect swaddling position (swaddling infants in the adducted and extended position) (Nandhagopal et al., 2024; Ortiz-Neira et al., 2012).
The COL2A1 gene, located on chromosome 12q13.11, contains 54 exons and encodes the α1 chain of type II procollagen (Barat-Houari et al., 2016). Three pro-α1 chains intertwine to form the procollagen molecule, which undergoes further processing into fibrils that are then cross-linked to form mature type II collagen (Col-II) fibers, which are important components of cartilage structure and function (Barat-Houari et al., 2016).
Mutations in the COL2A1 gene give rise to a diverse group of disorders collectively termed type II collagenopathies. Zhang et al. summarized the relationship between COL2A1 gene mutations and type II collagenopathy phenotypes, finding that N-propeptide regional mutations (especially exon 2) lead to mild phenotypes, while C-propeptide regional mutations cause severe and lethal phenotype (Zhang et al., 2020). Non-substitutions mutations (such as deletions, duplications, and insertions) are more likely to lead to mild phenotypes, except for small deletion mutations, which, unless causing frameshifts, typically result in severe or lethal phenotypes (Zhang et al., 2020).
Research on the relationship between COL2A1 and acetabular dysplasia, including case reports and genetic linkage analyses related to hip joint involvement in type II collagenopathies, has shown that COL2A1 is a key player. Namaqualand Hip Dysplasia (NHD) represents a milder form of spondyloepiphyseal dysplasia (SED), characterized primarily by progressive arthropathy of the hip joint (Agenbag et al., 2020). A linkage analysis spanning four generations within a South American family indicated an association between NHD and COL2A1 gene (Sher et al., 1991). Further supporting this connection, Agenda et al. discovered a missense pathogenic variant, c.2014G > T, in the COL2A1 gene among 23 individuals from a five-generation South African family affected by NHD (Agenbag et al., 2020). This finding underscores that mutations in the COL2A1 gene are directly linked to the development of the NHD phenotype. Additionally, Granch and colleagues have demonstrated that polymorphisms in the COL2A1 gene are also associated with osteoarthritis (OA) secondary to DDH (Granchi et al., 2002).
In the study of the pathogenesis of DDH, COL2A1 is also used as an indicator to evaluate cartilage metabolic capacity. Feng et al. investigated the relationship between DDH progression and cartilage metabolism disorders by analyzing cartilage tissues from patients with DDH, OA, and femoral neck fractures using histochemical staining, immunohistochemistry, and Western blot methods. Their findings revealed that the levels of aggrecan and Col-II in the DDH group were significantly lower than those in the control group and even lower than those in the OA group. Additionally, collagenous fibrils in the DDH group appeared highly rare and obscure (Feng et al., 2017). Normal articular cartilage is characterized by abundant aggrecan and Col-II, which may provide osmotic function and mechanical support, enabling the cartilage to resist compression (Feng et al., 2017). The reduction and disorganization of aggrecan and Col-II in DDH patients can lead to further joint cartilage degeneration and structural lesions, which would probably result in an articular cartilage metabolic disorder and hip joint dysfunction (Feng et al., 2017).
Here, we report a case of acetabular dysplasia accompanied by secondary synovial chondromatosis (SSC), whose father also suffers from the same conditions. To investigate the underlying etiology of these conditions, whole-exome sequencing (WES) was performed in the proband, revealing a COL2A1 mutation. This report also reviews recent progress in the genetic understanding of these disorders.
2 Materials and methods
2.1 Compliance with ethical standards
This study was approved by the Medical Ethics Committee of Shandong Provincial Hospital (SWYX:2022-449). Clinical and laboratory procedures were conducted after obtaining written informed consent from all participants. All procedures adhered to the principles outlined in the Helsinki Declaration.
2.2 Next-generation sequencing (NGS) and variant calling
Peripheral blood DNA extraction: Peripheral blood samples were collected from the proband and his parents using EDTA-anticoagulated tubes. Genomic DNA was extracted from the samples using the QIAamp DNA Blood Midi KIT (Qiagen, Germany) in accordance with the manufacturer’s instructions and quantified using both a NanoDrop spectrophotometer and a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, United States).
NGS analysis: WES was performed on the proband’s genomic DNA, which was sent to Hangzhou Dean Medical Laboratory Center for bioinformatics analysis. The analysis targeted coding regions of 5,075 genes associated with hereditary diseases, with a specific focus on genes linked to inherited bone diseases and chondrosarcoma. High-quality variants were identified through filtering and screening processes, followed by annotation using major genetic databases, including NCBI, dbSNP, OMIM, ESP, ExAC, ClinVar, and Thousand Genomes. The pathogenicity of identified variants was further evaluated using predictive bioinformatics tools such as SIFT, PolyPhen-2, and MutationTaster.
2.3 Sanger sequencing to verify the variant
Polymerase chain reaction (PCR) amplification and Sanger sequencing were conducted to confirm the candidate mutation. Primers were designed to target the identified mutation site, and PCR amplification was performed on the genomic region containing the suspected mutation. The PCR products were stored appropriately before being subjected to Sanger sequencing using an ABI 3130xl genetic analyzer. Sequencing results were aligned and compared with NCBI reference sequences using Chromas software to confirm the mutation.
2.4 Bioinformatic analysis
Conservation across species was assessed using the T-Coffee tool (https://www.ebi.ac.uk/jdispatcher/msa/tcoffee/). For the protein modeling, wild-type protein models were generated using the SWISS-MODEL tool (https://swissmodel.expasy.org/). The spatial structure and altered residue of the mutant protein were visualized using PyMOL software to assess the structural impact of the identified mutation.
3 Results
3.1 Clinical characteristics, diagnosis and treatment process
3.1.1 The proband
The proband is a 27-year-old male who presented with bilateral hip pain lasting 6 months, which had progressively worsened over the past 2 months. Initially, the pain began following physical exertion and worsened with activity but did not resolve with rest. The patient reported no significant worsening at night. Associated symptoms included mild low back pain, but there was no heel pain, joint swelling, or deformity of small joints in the limbs. The patient sought treatment at a local hospital, where magnetic resonance imaging (MRI) of the lumbar spine revealed Schmorl’s nodes from T11 to L5 vertebrae. Based on these findings, the patient was diagnosed with seronegative spondyloarthropathy and prescribed oral non-steroidal anti-inflammatory drugs (NSAIDs), which provided partial symptom relief. However, 2 months prior to admission to our hospital, the bilateral hip pain worsened and he was admitted to our hospital.
Since the onset of the disease, the patient has reported no history of fever, cough, muscle soreness, headache, urinary urgency, frequent urination, abdominal pain, or ophthalmia. His body weight remained stable, and he denied smoking or alcohol consumption. Additionally, the patient had no history of chronic diseases, such as hypertension, diabetes, coronary heart disease, or cerebrovascular disease. He also denied any history of infectious diseases (e.g., hepatitis or tuberculosis), exposure to infectious individuals, psoriasis, or chronic diarrhea. There was no personal or family history of other significant medical conditions. The patient had received all routine childhood vaccinations. His parents were not consanguineous, but his father had experienced similar symptoms.
On physical examination, the patient’s vital signs were within normal ranges: body temperature, 36.9°C; heart rate, 87 beats per minute; respiratory rate, 21 breaths per minute; blood pressure, 140/82 mmHg; and oxygen saturation, 99% on ambient air. The patient appeared well-developed and well-nourished, although his gait was impaired. No abnormalities were observed in his conjunctiva or vision, and mucosal surfaces were dry without evidence of oral, vulvar, or perianal ulcers. Skin examination revealed no rashes or lymphadenopathy. Cardiopulmonary and abdominal examinations were unremarkable. Upon musculoskeletal examination, the skin overlying the hips showed no redness, swelling, or ulceration, and the local temperature was normal. Moderate tenderness was detected in the anterior space of the left hip joint. Patrick’s test was positive bilaterally, suggesting hip joint pathology. Joint mobility was restricted on both sides, with ranges of motion measured at 100°flexion, 0° extension, 20° pronation, and 20° supination. Sensory function, muscle strength, and peripheral circulation were intact in both lower limbs, with palpable and adequate pulses in the bilateral dorsalis pedis arteries.
Comprehensive laboratory and imaging examinations were performed. Liver function, lactate dehydrogenase, creatine kinase, and biochemical test results were normal, except for alkaline phosphatase, which was elevated at 139 U/L (reference range: 45–125 U/L). Routine blood, urine and stool tests were unremarkable. Erythrocyte sedimentation rate, C-reactive protein, anti-streptolysin, and Mycobacterium tuberculosis T-cell detection tests showed no abnormalities. Complement 3, complement 4, and immunoglobulin levels were within normal limits. Bone metabolism evaluation revealed elevated total procollagen type I amino-terminal peptide at 128.40 ng/mL (reference range: 9.06–76.24 ng/mL) and a 25-dihydroxyvitamin D level of 6.62 ng/mL (reference range: 20–100 ng/mL). Testing for human leukocyte antigen (HLA)-B27 was negative. Autoimmune markers, including rheumatoid factor, anti-cyclic citrullinated peptide antibody, anti-RA33 antibody, antinuclear antibody, and anti-SA antibody, were all negative. Radiographic imaging identified several abnormalities. A plain X-ray of the pelvis showed dysplastic osteoarthrosis of the acetabulum. Full-length anteroposterior radiographs of both lower limbs demonstrated OA and SSC in the knees, with no abnormalities observed in the ankles. Computed tomography (CT) with three-dimensional reconstruction of the hip joints (Figure 1) confirmed bilateral dysplastic osteoarthrosis of the acetabulum and SC. MRI of the cervical spine indicated degenerative changes and intervertebral disc herniation at the C3/4 and C5/6 levels. MRI of the sacroiliac joints (Figure 2) initially suggested “bilateral sacroiliac arthritis.” However, following a review by two additional radiologists, the sacroiliac joint surfaces were considered smooth, and the edema signal did not support the diagnosis of sacroiliac arthritis. Based on the clinical presentation and imaging findings, the diagnosis of acetabular dysplasia with overlapping SSC was established.
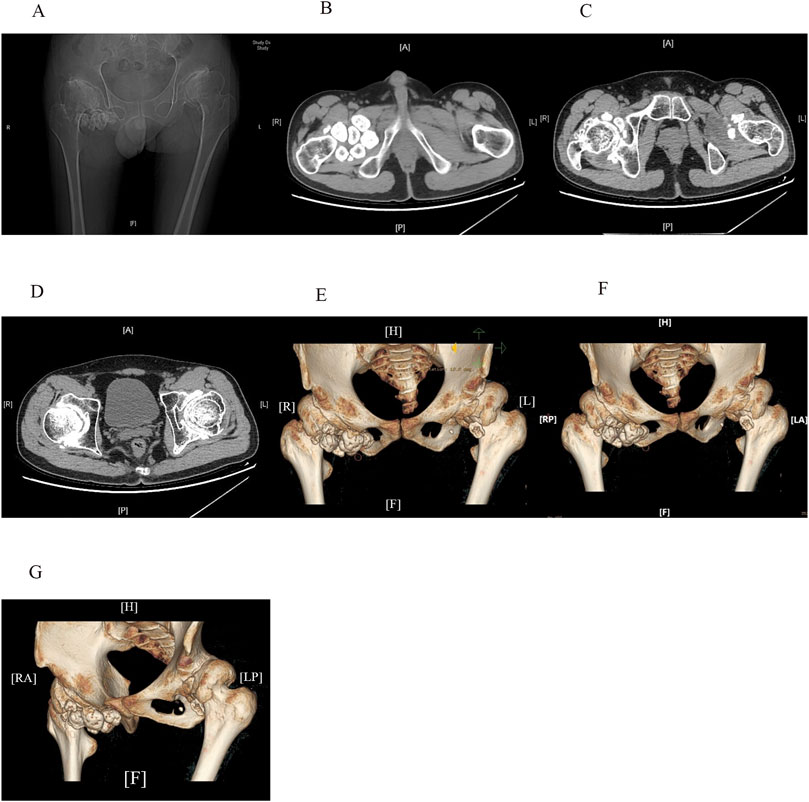
Figure 1. CT scans (A–D) and three-dimensional reconstruction images (E–G) of the hip joints reveal shallow acetabula with increased acetabular indices and bone hyperplasia along the joint margins bilaterally. The femoral heads appear hypertrophic and flattened, with anterolateral displacement and thick, short femoral necks. The neck-stem angle of the femur is increased, and the hip joint spaces are nonuniformly narrowed. Articular surface sclerosis and subarticular cystic changes are present on both sides. Enlargement of the bilateral hip joint capsules is evident, with multiple pomegranate seed-like high-density foci, particularly on the right side. Soft tissue swelling can also be observed around the joint.
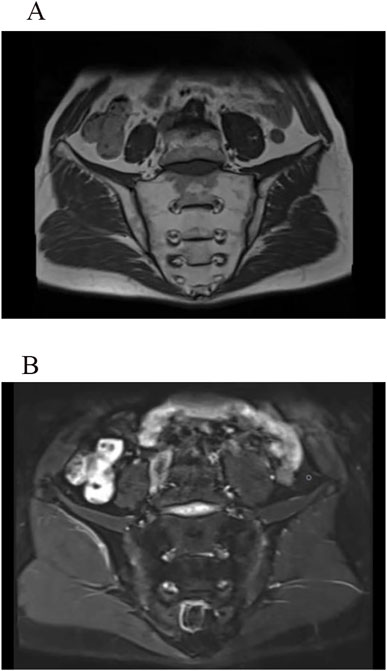
Figure 2. MRI images of the sacroiliac joints show long T1 (A) and T2 (B) signals on both the sacral and iliac surfaces. The articular surfaces appear smooth on T1WI, without signs of bone erosion, new bone formation, or abnormal fat deposition. The joint spaces are normal, with no evidence of pathological narrowing.
After evaluation by osteoarticular surgeons, the patient was diagnosed with acetabular dysplasia accompanied by SSC. The patient’s general condition was assessed to be good, and no significant contraindications to surgery were identified based on the completed examinations. To alleviate pain, restore hip function and improve quality of life, bilateral total hip arthroplasty and synovectomy were performed under general anesthesia with endotracheal intubation. During the surgery, multiple smooth, loose bodies of varying sizes, with white or milky-white appearance, were identified, along with a large, rounded mass (Figure 3). Postoperatively, the patient received symptomatic and supportive care. Instructions were provided to maintain clean dressings, follow a nutrient-rich diet to support recovery, and perform exercises to enhance hip flexion. The patient was advised to practice dorsiflexion to prevent thrombosis and scheduled for follow-up in the Outpatient Department of Bone and Joint Surgery 1 month post-operation.
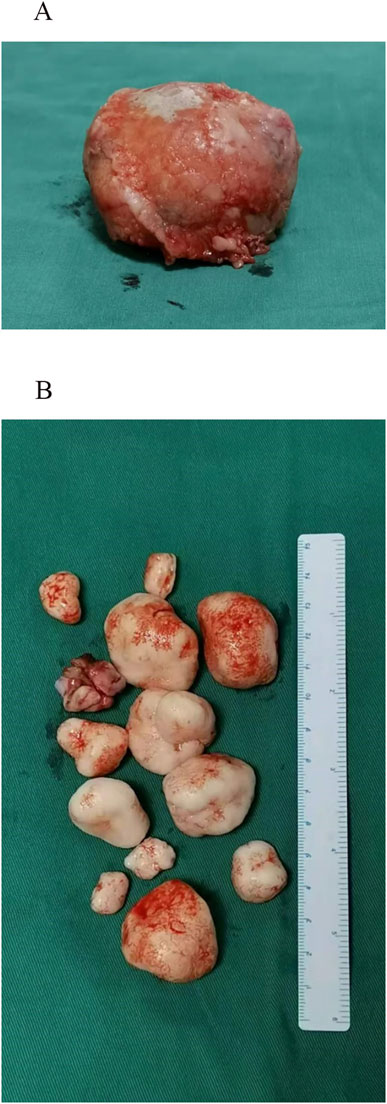
Figure 3. Intraoperative visualization. (A) A large, round-like tumor body observed during the procedure. (B) Multiple smooth, white or milky-white loose bodies of varying sizes, ranging from several millimeters to several centimeters in diameter, were identified during surgery.
3.1.2 The Proband’s father
The proband’s father also experienced similar symptoms of bilateral hip pain, but without restricted hip movement or a limp, indicating milder symptoms. He had never received a formal diagnosis or treatment. After the diagnosis of his son, the father underwent X-ray imaging of the pelvis and bilateral knees (Figure 4). The pelvic X-ray revealed dysplastic osteoarthrosis of the acetabulum, bilateral SSC, and dislocation of the left hip joint. Anteroposterior and lateral radiographs of both knees demonstrated OA, bilateral SC, and the presence of osteochondroma.
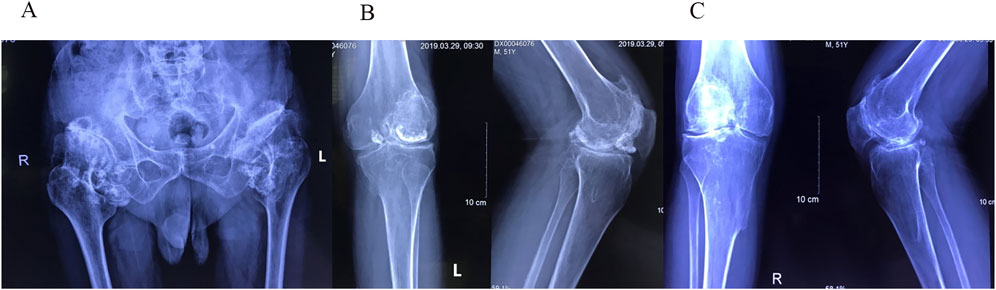
Figure 4. (A) Pelvic radiograph showing shallow acetabula with increased acetabular indices and bone hyperplasia along the joint margins bilaterally. The femoral heads appear hypertrophic and flattened with anterolateral displacement. The left hip joint demonstrates dislocation, indicated by overlapping of the femoral head and acetabular shadow, along with superior displacement of the femur. Both femoral necks are thick and short, and the neck-stem angles are increased bilaterally. The right hip joint space is unevenly narrowed, with sclerosis of the articular surface and degeneration of the subarticular capsule in the weight-bearing area. Enlargement of the bilateral hip joint capsules is observed, with multiple pomegranate seed-like high-density foci, particularly on the right side. Soft tissue swelling is present around the joint. (B) Anteroposterior radiograph of the left knee joint and (C) anteroposterior radiograph of the right knee joint show narrowing of the joint spaces, low patellar position (patella baja), and slight outward displacement of the patella on both sides. Bone hyperplasia and sclerosis of the joint surfaces are visible along the joint margins. Multiple pomegranate seed-like high-density foci of varying sizes are evident within the joint spaces, especially in the left knee joint, where a larger focus is located anteriorly. Bony protrusions are observed on the medial sides of the upper tibiae bilaterally. These protrusions are continuous with the tibial cortex and connected to the medullary cavity, consistent with osteochondromas.
3.2 Genetic analysis
Through WES technology, we can identify all variants (including point mutations and small indels) in the exons of 5,075 genes and their adjacent ±20 bp intronic regions in the patient. The detection is performed using a standardized target region capture sequencing platform, with quality control metrics as follows: an average coverage of 98.99% and an average sequencing depth of 124.86. Subsequently, the identified variants are annotated and compared with disease-related databases such as Online Mendelian Inheritance in Man (OMIM)and Human Phenotype Ontology (HPO) to determine if they match known clinical phenotypes. This process helps us understand whether the discovered genetic variants are associated with specific diseases or symptoms.
NGS revealed that the proband carried a pathogenic heterozygous missense mutation in the COL2A1 gene, specifically in exon 13 at c.823C > T (p.Arg275Cys) (Table 1). No other pathogenic mutations, suspected pathogenic variants, or variants of unknown clinical significance were identified in genes related to chondrosarcoma. Sanger sequencing confirmed the presence of the same COL2A1 mutation in both the proband and his father. The heterozygous missense mutation (c.823C > T) in exon 13 of the COL2A1 gene resulted in the substitution of arginine with cysteine at the p.Arg275 position (p.Arg275Cys). The proband’s mother tested negative for this mutation (Table 2; Figures 5, 6).

Table 1. The results of clinical whole-exome high-throughput sequencing of the proband.

Table 2. Fixed-site Sanger sequencing results for the proband and his parents for the COL2A1 gene.
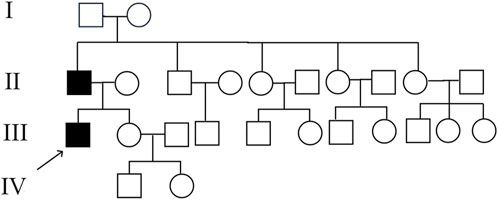
Figure 5. Pedigree of the proband’s family. Both the proband and his father were affected, while his grandparents and other family members did not exhibit similar symptoms.
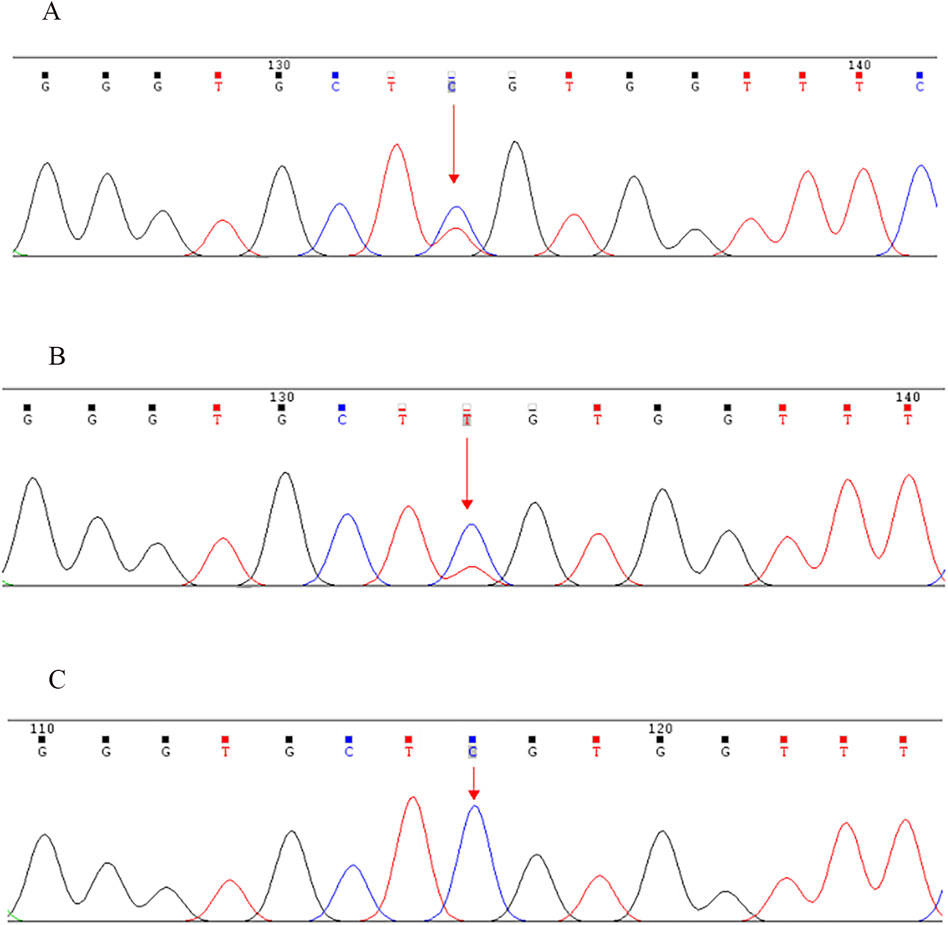
Figure 6. Sanger sequencing validation of the COL2A1 exon 13 c.823C > T (p.Arg275Cys) mutation in the proband and his parents. (A) Proband (mutation detected), (B) Father (mutation detected), (C) Mother (mutation not detected). Note: NCBI reference sequence: GCAGGGTGCTCGTGGTTTCCC.
3.3 Bioinformatic analysis
Conservation analysis indicated that the arginine residue at position p.Arg275 is highly conserved across multiple species, underscoring its potential functional importance (Table 3). Structural modeling revealed that the substitution of arginine with cysteine at residue 275 led to alterations in the protein structure (Figure 7). The c.823C > T variant’s pathogenicity was assessed via bioinformatic tools, including SIFT, PolyPhen-2, and MutationTaster. Referring to the HumanDiv database, Polyphen-2 scores ≥0.957 suggest the amino acid substitution is probably damaging; scores between 0.453 and 0.956 indicate the substitution is possibly damaging; and scores ≤0.452 imply the substitution is benign. SIFT scores span from 0 to 1, with amino acid substitution scores < 0.05 indicating a prediction of deleteriousness, and scores ≥0.05 suggesting the substitution is tolerated. The c.823C > T (p. Arg275Cys) in the COL2A1 gene was predicted to be deleterious: MutationTaster (0.999, disease-causing), SIFT (0.010, damaging), and PolyPhen-2 (1.000, probably damaging) (Figure 8).
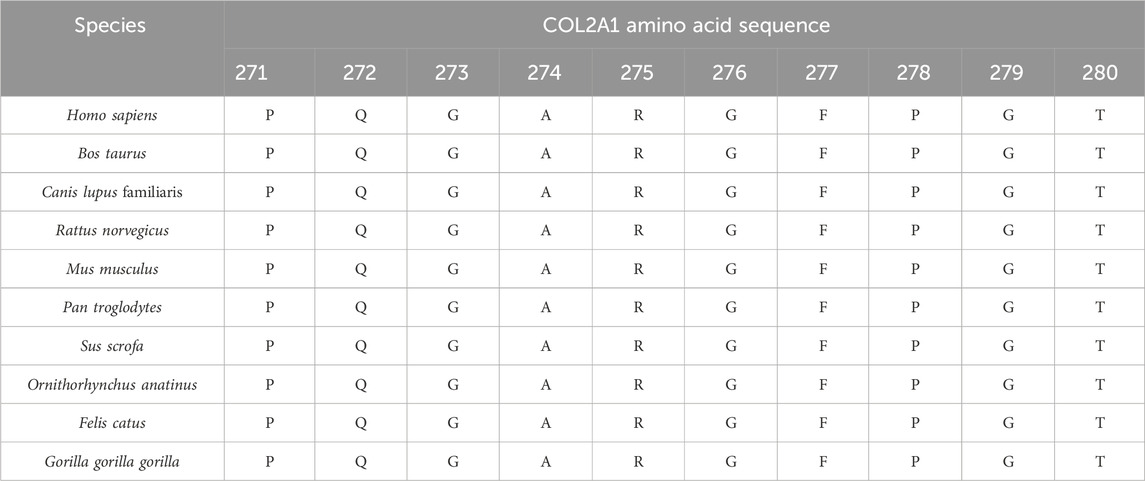
Table 3. Conservation analysis of the arginine residue at site p.Arg275 in COL2A1.
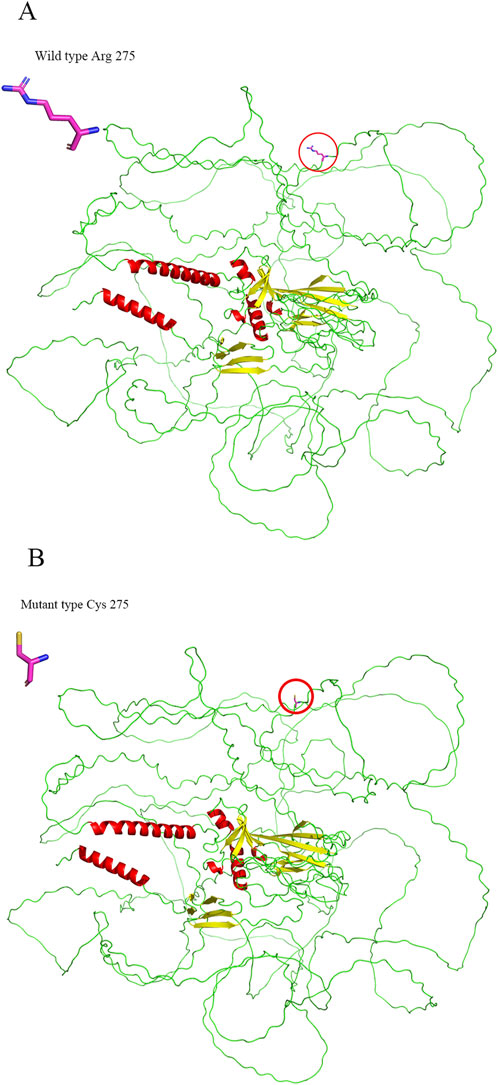
Figure 7. 3D structural comparison of the wild-type (A) and mutant (B) p.Arg275Cys forms of COL2A1.
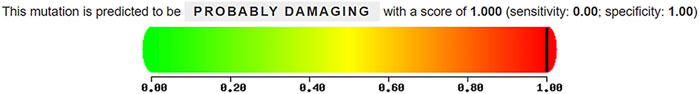
Figure 8. Pathogenicity prediction of the c.823C > T (p. Arg275Cys) missense mutation in COL2A1 using PolyPhen-2.
4 Discussion
Dysplastic osteoarthrosis of the acetabulum is a hip joint disorder that develops in adults due to long-term biomechanical abnormalities resulting from congenital acetabular dysplasia (Tian et al., 2003). Key pathological changes associated with this condition include secondary OA, subchondral pseudocysts, and chronic joint dislocations (Tian et al., 2003). Acetabular dysplasia is diagnosed based on specific radiographic criteria, including a shortened acetabular roof, a center-edge angle of less than 30°, with or without increased acetabular inclination (acetabular index > 45°), and femoral head exposure exceeding 25% on anteroposterior pelvic radiographs (Tian et al., 2003). Radiographic features include the presence of OA alongside a shallow or steep acetabulum (Tian et al., 2003). Early radiographic signs include thickening of the white line of the acetabular cap, resembling an “eyebrow,” with pointed or tilted outer margins (Tian et al., 2003). As the condition progresses, sclerosis of the acetabular roof enlarges, subchondral cystic changes develop, and joint space narrows within the weight-bearing region (Tian et al., 2003).
SC is characterized by the formation of multiple cartilaginous or osteochondral loose bodies within the synovium (Adelani et al., 2008). On X-ray and CT imaging, SC is typically identified by multiple high-density calcified foci within the joint (Alexander et al., 1987).
The radiographic findings in our patients were consistent with acetabular dysplasia and SC. The X-ray images revealed classic features of acetabular dysplasia, such as shallow and flattened acetabula, excessive tilt, and incomplete femoral head coverage. CT imaging demonstrated multiple pomegranate seed-like high-density foci in both hip joints, consistent with calcified cartilage nodules. Overall, these imaging findings provided sufficient evidence to establish a definitive diagnosis of acetabular dysplasia with overlapping SC.
Given the similarity of symptoms between the proband and his father, the possibility of a genetic etiology was investigated. Genetic testing identified a COL2A1 gene mutation in both the proband and his father. Specifically, a heterozygous missense mutation (c.823C > T) was detected in exon 13 of the COL2A1 gene, resulting in an amino acid substitution from arginine to cysteine at position 275 (p. Arg275Cys). The underlying pathogenesis may involve the mutation leading to congenital acetabular dysplasia, which subsequently initiates a cascade of pathological changes affecting both bone and soft tissues. These abnormalities alter the morphology and biomechanics of the hip joint, contributing to the development of SC and secondary arthritis, which in turn exacerbate joint damage and structural deterioration.
The clinical phenotype associated with the c.823C > T (p.Arg275Cys) mutation identified in this study differs from previously reported cases in the literature (Hildebrand, 2015; Matsui et al., 2009). The presence of acetabular dysplasia in these patients expands the phenotypic spectrum of COL2A1 mutations. The p.Arg275Cys mutation in the COL2A1 gene has been previously reported to manifest as spondyloepiphyseal dysplasia congenita (SEDC), spondyloarthritis, early-onset osteoarthritis (EO-OA), and Czech dysplasia (Hoornaert et al., 2006). The current focus is to distinguish these cases from other type II collagenopathies that present with overlapping clinical features. The following section discusses type II collagenopathies with clinical presentations and mutation sites similar to those observed in these patients.
4.1 Discussion on type II collagenopathies
4.1.1 SED
SED refers to a group of rare genetic skeletal disorders primarily affecting the spine and epiphyses, with the predominant clinical features including disproportionate short stature, thoracocyllosis, and progressive degeneration of multiple joints (Liu, 2014). Radiographic findings often reveal platyspondyly, epiphyseal dysplasia, and narrowed joint spaces (Liu, 2014). Inheritance patterns of SED include autosomal dominant, autosomal recessive, and X-linked recessive modes (Liu, 2014). According to the Nosology and Classification of Genetic Skeletal Disorders (2010 revision), SED is categorized into nine clinical types, which include SEDC and Czech dysplasia, among others (Warman et al., 2011).
4.1.1.1 SEDC
SEDC predominantly affects the vertebrae and the proximal epiphyses of long bones (Liu, 2014). Clinical manifestations in patients with SEDC are diverse and include short stature and skeletal deformities (Li et al., 2015). Common spinal abnormalities include scoliosis and platyspondyly (flattened vertebral bodies) (Li et al., 2015). Additionally, atlantoaxial instability, often resulting from odontoid hypoplasia, may lead to cervical cord compression (Al Kaissi et al., 2019; Tofield and Mackinnon, 2003). Hip deformities, such as avascular necrosis-like changes in the acetabulum and bilateral femoral epiphyses, are frequent and may cause hip pain and reduced walking endurance (Li et al., 2015). Patients with SEDC may also present with hearing loss, cleft palate, and ocular complications, including myopia and retinal detachment (Dahiya et al., 2000). Radiographic features of SEDC typically include flattened and oval-shaped vertebral bodies, scoliosis, absence of ossification at the epiphyseal ends of the femoral heads, and irregular dysplasia of the femoral neck (Anderson et al., 1990). Other common findings include a flattened acetabular crest, deformities of the hip and knee joints, and disruptions in the ossification of long bones (Anderson et al., 1990).
SEDC is commonly associated with mutations in the COL2A1 gene (Anderson et al., 1990). In their study, Zhang et al. reported that among 87 identified cases of SEDC, the majority of mutations were missense mutations (80 cases), with 57 of these occurring at the Gly site of the Gly-X-Y repeat sequence of the protein (Zhang et al., 2020). Additionally, the p.Arg989Cys site was identified as a potential mutation hotspot, indicating its significance in the pathogenesis of the disorder (Silveira et al., 2015).
4.1.1.2 Czech dysplasia
Czech dysplasia is an autosomal dominant skeletal disorder characterized by early-onset progressive OA, normal stature, and brachydactyly, most commonly affecting the third and fourth toes (Burrage et al., 2013). Other features include mild platyspondyly with atypical vertebral endplates, progressive hearing loss, and the absence of cleft palate or ophthalmologic abnormalities (Burrage et al., 2013; Hoornaert et al., 2007). Radiographic findings often reveal short metacarpals and metatarsals, reduced intervertebral spaces, and the presence of osteochondromatosis (Burrage et al., 2013) or synovial osteochondromatosis (Moreira et al., 2023).
In cases where typical clinical features are absent but a family history of early-onset arthritis is present, WES can provide valuable diagnostic insights. Czech dysplasia is distinguishable from other type II collagenopathies by a single missense mutation in the COL2A1 gene (R275C, c.823C > T) (Matsui et al., 2009). A previous report described a 3.5-year-old female patient with a family history of early-onset arthritis who presented only with prominent knees (Burrage et al., 2013). WES identified a c. 823C > T (p.R275C) mutation in the COL2A1 gene, confirming the diagnosis of Czech dysplasia (Burrage et al., 2013). This case illustrates how WES enables early diagnosis in the presymptomatic stage, facilitating anticipatory guidance and genetic counseling (Burrage et al., 2013).
The two patients described in this study presented with bilateral hip pain, and imaging revealed acetabular dysplasia. However, no typical spinal abnormalities associated with SED, such as flattened vertebrae or odontoid hypoplasia, were observed. Additionally, there were no signs of extra spinal involvement characteristic of SED, such as short tubular bones, absent epiphyseal ossification, cleft palate, ocular complications, or sensorineural hearing loss. Furthermore, neither patient exhibited the hallmark features of Czech dysplasia, such as shortened third and fourth toes. Based on these findings, SED can be excluded as a diagnosis for the two patients.
4.1.2 EO-OA
EO-OA, also referred to as premature OA, is typically secondary to inflammatory conditions or biomechanical abnormalities, such as osteochondrodysplasia, and often presents as part of a syndrome (Aury-Landas et al., 2016). Idiopathic EO-OA, which results from congenital or hereditary factors rather than trauma or infection, is rare, particularly in non-syndromic families without associated dysplasia or other underlying pathologies (Aury-Landas et al., 2016). The clinical features of EO-OA include the development of distal interphalangeal osteophytes, known as Heberden’s nodes, and progressive cartilage degeneration in the knees, hips, and other joints, and affected individuals frequently experience intermittent joint pain and swelling (Ala-Kokko et al., 1990). In previous studies, the average age of onset for EO-OA was reported to be 19 years (Rukavina et al., 2014).
Mutations in the COL2A1 gene have been confirmed as a cause of EO-OA (Husar-Memmer et al., 2013). In 1995, one of 45 patients with familial EO-OA was identified as carrying a COL2A1 mutation (Ritvaniemi et al., 1995). Additionally, four EO-OA probands were found to carry the same mutation (p. Arg275Cys) in the COL2A1 gene (Carlson et al., 2006; Hoornaert et al., 2006; Lopponen et al., 2004; Williams et al., 1993).
According to established diagnostic criteria for EO-OA, which include radiographic evidence of OA, a body mass index (BMI) ≤30, an age of onset ≤50 years, and involvement of at least one joint site, the proband in this study meets the diagnostic criteria for EO-OA (Aury-Landas et al., 2016). However, the EO-OA observed in the proband is likely secondary to acetabular dysplasia and SC, rather than idiopathic EO-OA, given the underlying biomechanical abnormalities in the hip joints.
4.2 Discussion on differential diagnoses
Adult DDH must be differentiated from other conditions that cause hip pain, such as avascular necrosis of the femoral head (ANFH), hip OA, and ankylosing spondylitis (AS). When imaging reveals multiple loose bodies, it is essential to distinguish SC-associated findings from pigmented villonodular synovitis (PVNS).
When AD is accompanied by cystic degeneration of the femoral head, it should be differentiated from ANFH (Deng, 2014). Cystic degeneration in AD develops secondary to the underlying biomechanical abnormalities and often involves both the femoral head and the acetabulum, particularly in the weight-bearing areas (Deng, 2014). These cysts are typically adjacent to the joint surface, with smooth, well-defined margins and sclerotic edges. In some cases, a vacuum phenomenon may be observed (Deng, 2014). The morphology of the femoral head in AD remains largely preserved, though joint space narrowing and joint subluxation are common (Deng, 2014). In contrast, ANFH primarily affects the femoral head, with well-developed hip sockets and cystic degeneration restricted to the femoral head (Liu et al., 2010). Acetabular involvement is uncommon in ANFH, and the cystic lesions often exhibit mixed densities, poorly defined boundaries, and variable sizes, sometimes appearing as crescent-shaped or fissured areas (Liu et al., 2010). In advanced stages, the femoral head may fracture, collapse, and deform, with joint space narrowing occurring only in later stages (Deng, 2014). Joint subluxation is rare in ANFH, and patients often have a history of corticosteroid use, alcohol abuse, or trauma (Deng, 2014).
In late stages, AD may lead to secondary OA, which requires differentiation from primary degenerative hip OA. While both conditions share similar pathological mechanisms, the presence of AD serves as a precursor in secondary OA (Deng, 2014). AD-related OA tends to present at a younger age and is more common in females (Deng, 2014). In such cases, osteogenesis, sclerosis, and cystic degeneration predominantly affect the acetabulum, with larger cystic lesions and common joint subluxation (Deng, 2014). Conversely, primary degenerative OA is associated with normal acetabular development, occurs more frequently in males, and is characterized by bone hyperplasia and smaller cystic changes primarily involving the femoral head. In primary OA, the cysts are often chiseled and small, and joint subluxation is uncommon (Deng, 2014).
AS is more common in adolescents and mainly affects axial joints, particularly the sacroiliac joints and spine, often causing structural changes and strongly associated with the HLA-B27 (Pedersen and Maksymowych, 2019). The proband sought medical attention due to bilateral hip pain, and an initial MRI of the sacroiliac joints revealed findings suggestive of sacroiliitis, which may have led the attending physician to consider AS as a possible diagnosis. However, it is essential to carefully evaluate the patient’s symptoms, laboratory findings, and imaging data to arrive at an accurate diagnosis. From a symptomatic perspective, low back pain is a hallmark feature of AS, often presenting early in the disease. However, in the proband’s case, low back pain was not a predominant symptom, and he did not report severe nocturnal pain. His symptoms worsened with activity and did not improve with rest, which is inconsistent with inflammatory back pain typically seen in AS. Additionally, the patient tested negative for HLA-B27, further reducing the likelihood of an AS diagnosis. The imaging findings also did not support AS. Although MRI initially indicated “sacroiliitis,” further analysis revealed that the sacroiliac joint surfaces were relatively smooth, and the observed inflammatory edema signal was inconsistent with sacroiliac arthritis. The sacroiliitis-like changes might instead be attributed to mechanical stress on the sacroiliac joints, potentially caused by prolonged use of crutches and poor posture, such as leaning forward. Therefore, a diagnosis of AS or spondyloarthritis could not be confirmed.
PVNS, also known as tenosynovial giant cell tumor (TGCT), is a benign lesion characterized by synovial villous nodular hyperplasia and hemosiderin deposition (Li et al., 2021). Malignant transformation and metastasis of PVNS are rare (Li et al., 2021). The clinical presentations of PVNS and SC are similar, with both conditions initially manifesting as hip pain and reduced range of motion (Startzman et al., 2016). Conservative treatment often fails to relieve symptoms in either disease (Xie et al., 2015). However, imaging studies can help differentiate the two conditions. PVNS is typically characterized by a slightly high-density soft tissue mass around the joint on CT, with varying degrees of adjacent bone destruction. The soft tissue mass in PVNS is often separated by fibrous septa (Han et al., 2010). In contrast, the CT scan of the proband’s hip joint revealed multiple high-density foci consistent with calcified nodules, without any evidence of bone destruction. These findings effectively ruled out PVNS as a diagnosis in this case.
4.3 Discussion on research progress regarding the genetics of DDH and its role in early diagnosis and treatment
Although extensive research has explored the genetic underpinnings of DDH, no definitive susceptibility genes have been identified (Wen et al., 2023). In a systematic review, Wen et al. pointed out that the genes most closely associated with DDH are CX3CR1, ASPN, COL1A1, HOX, and GDF5, with GDF5 receiving the most attention in the literature (Wen et al., 2023). In addition to these well-studied susceptibility genes, Wen et al. also summarized that animal models have implicated other genes, including TENM3, UFSP2, and WISP2 in the development of DDH (Wen et al., 2023). Futhermore, epigenetic changes also contribute to the pathogenesis of DDH. According to Wen et al., these include abnormal methylation of gene promoters, the altered expression of microRNAs such as miR-1-3p, miR-129-5p, and miR-140, and dysregulation of the long non-coding RNA (lncRNA) H19 (Wen et al., 2023).
Researchers have proposed that two distinct genetic pathways may underlie the development of DDH. The first involves genes responsible for the formation and development of acetabular cartilage and bone, while the second regulates genes involved in the development of the hip joint capsule and surrounding soft tissues (Wynne-Davies, 1970). Current research primarily focuses on these two pathways to better understand the molecular regulatory mechanisms involved in DDH (Zhao et al., 2023). Thus, identifying these genetic patterns and susceptibility genes is essential for clarifying the complex pathogenesis of DDH and holds potential for improving early detection, diagnosis, and treatment (Zhao et al., 2023). Advances in genetic research have highlighted the importance of identifying candidate and susceptibility genes for DDH (Zhao et al., 2023). In parallel, the development of genetic screening tools, including intrauterine diagnostic techniques, offers promising avenues for early detection and prenatal diagnosis (Zhao et al., 2023). Early genetic diagnosis could facilitate timely interventions, potentially improving patient outcomes (Zhao et al., 2023).
For children with a high risk of DDH but without obvious symptoms, the risk of developing DDH can be assessed by detecting these specific gene mutations (Agenbag et al., 2020). The detection of the COL2A1 gene can be done through direct cycle sequencing and NGS methods (Agenbag et al., 2020). Although the COL2A1 gene contains 54 exons, direct cycle sequencing is still a viable method for variant detection due to its relatively small overall size (Agenbag et al., 2020). However, NGS offers a better choice in terms of efficiency and coverage. NGS can sequence one or more target genes or even the entire exon group at the same time, thereby accurately identifying gene mutations that lead to hereditary diseases (Agenbag et al., 2020).
The earlier DDH is detected, the better the treatment outcome (Wang et al., 2023). Studies have shown that the effectiveness of treating DDH at the age of 8 is not superior to not treating it (Thomas, 2015). Therefore, early detection and diagnosis are key to treatment. Clinical screening methods for DDH in newborns and infants include physical examination, ultrasonography at 6 weeks, and X-ray screening at 4–6 months (Ji et al., 2022). These clinical screening methods have a low detection rate for DDH patients who later need hip joint replacement surgery, and there is a risk of over-treatment (Paton, 2017). Therefore, genetic screening should be supplemented for high-risk infants (Ji et al., 2022). For children without related gene mutations, regular testing and physical correction should be adopted as much as possible to avoid excessive treatment; for children with related gene mutations, long-term follow-up monitoring should be carried out simultaneously with treatment to determine the abnormal development of the hip joint (Ji et al., 2022).
The main purpose of early treatment of DDH is to restore the concentric relationship between the acetabulum and the femoral head, usually based on non-surgical treatment (Wang et al., 2023). When the age increases or the closed reduction correction is not possible, surgical treatment is needed to improve gait, alleviate pain, and prolong the lifespan of the hip joint (Wang et al., 2023). Most untreated adult DDH has a poor prognosis, while early diagnosis and treatment can improve disease outcomes, reduce the probability of needing surgical treatment later, and alleviate the burden of the disease on patients, families, and society (Wang et al., 2023).
In the present case, secondary SC was associated with congenital acetabular dysplasia, affecting both the hip and knee joints. This report provides a concise overview of research advances in the genetics of SC.
4.4 Discussion on research progress in the genetics of SC
Extensive research has been conducted to understand the pathogenesis of SC. Traditionally, SC has been considered to result from chondroid metaplasia of the joint synovium (Brucher et al., 2014). However, several studies have highlighted a possible association between SC and chromosomal abnormalities or gene mutations. Sciot et al. reported a case of SC involving clonal chromosomal changes and proposed that SC could be classified as a true neoplastic disorder (Sciot et al., 1998). Similarly, Mertens et al. analyzed the cytogenetics of four SC cases and identified the loss of chromosomal band 10q26 and rearrangements in 1p13 and 12q13, further supporting the theory of clonal proliferation in SC (Mertens et al., 1996). In a study by Totoki et al., an FN1-ACVR2A in-frame gene fusion was identified in one case of SC during a genomic investigation of benign and malignant cartilaginous neoplasms (Totoki et al., 2014). When analyzing seven additional cases of SC, the same gene fusion was detected in one other case. Subsequent studies by Amary et al. (2019) and Agaram et al. (2020) confirmed the presence of FN1 and/or ACVR2A gene rearrangements in SC using fluorescence in situ hybridization (FISH) and RNA sequencing. Additionally, Fukutani et al. (2022) identified a homozygous mutation (C/C genotype) in the Gli1 gene (rs2228226 G > C) in two cases of SC involving the temporomandibular joint. These findings suggest a genetic component in the development of SC.
This study has some limitations. Firstly, clinical whole exome high-throughput sequencing cannot fully cover certain genes or gene regions that are highly repetitive, low complexity, or pseudogene regions. The detection scope does not include genomic structural variations (such as large heterozygous deletions, duplications, and inversion rearrangements), large heterozygous insertion mutations, or mutations located in regulatory regions or deep intronic regions. Secondly, although these imaging findings provided sufficient evidence to establish a definitive diagnosis of acetabular dysplasia with overlapping SC, unfortunately, surgical specimens were not collected for pathological confirmation.
5 Conclusion
SC is a rare disease characterized by nonspecific symptoms that complicate diagnosis, often resulting in delayed detection and prolonged patient suffering. Consequently, when assessing young and middle-aged men presenting with hip pain, rheumatologists should consider conditions beyond AS and conduct a thorough inquiry into the patient’s family history to facilitate accurate differential diagnosis. To date, COL2A1 gene mutations have not been reported in cases of acetabular dysplasia with SC. This study provides new evidence linking COL2A1 gene mutations to acetabular dysplasia with SC, demonstrating that NGS can enhance diagnostic precision. These findings expand the clinical spectrum of COL2A1 mutations, introducing new phenotypes associated with this gene.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: The data that support the findings of this study are openly available in ClinVar databases at https://www.ncbi.nlm.nih.gov/clinvar/RCV003228897.3/, Variation record RCV003228897.3; Submission Accession SCV002754582.
Ethics statement
The studies involving humans were approved by the Medical Ethics Committee of the Shandong Provincial Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MX: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–original draft. XG: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–original draft. JY: Investigation, Methodology, Resources, Supervision, Writing–review and editing. YL: Investigation, Methodology, Resources, Supervision, Writing–review and editing. ZM: Investigation, Methodology, Resources, Supervision, Writing–review and editing. HS: Conceptualization, Formal Analysis, Methodology, Project administration, Resources, Supervision, Writing–review and editing. MF: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 82001744) and the Shandong Provincial Natural Science Foundation (No. ZR2021MH264).
Acknowledgments
We thank the patient and his family for their contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adelani, M. A., Wupperman, R. M., and Holt, G. E. (2008). Benign synovial disorders. J. Am. Acad. Orthop. Surg. 16 (5), 268–275. doi:10.5435/00124635-200805000-00005
Agaram, N. P., Zhang, L., Dickson, B. C., Swanson, D., Sung, Y. S., Panicek, D. M., et al. (2020). A molecular study of synovial chondromatosis. Genes Chromosom. Cancer 59 (3), 144–151. doi:10.1002/gcc.22812
Agenbag, G., Vorster, A., Julius, S., Ramesar, R., and Beighton, P. (2020). Namaqualand hip dysplasia in South Africa: the molecular determinant elucidated. S Afr. Med. J. 111 (1), 57–60. doi:10.7196/SAMJ.2020.v111i1.14561
Ala-Kokko, L., Baldwin, C. T., Moskowitz, R. W., and Prockop, D. J. (1990). Single base mutation in the type II procollagen gene (COL2A1) as a cause of primary osteoarthritis associated with a mild chondrodysplasia. Proc. Natl. Acad. Sci. U. S. A. 87 (17), 6565–6568. doi:10.1073/pnas.87.17.6565
Alexander, J. E., Holder, J. C., Mcconnell, J. R., and Fontenot, E. (1987). Synovial osteochondromatosis. Am. Fam. Physician 35 (2), 157–161.
Al Kaissi, A., Ryabykh, S., Pavlova, O. M., Ochirova, P., Kenis, V., Chehida, F. B., et al. (2019). The Managment of cervical spine abnormalities in children with spondyloepiphyseal dysplasia congenita: observational study. Med. (Baltimore) 98 (1), e13780. doi:10.1097/MD.0000000000013780
Amary, F., Perez-Casanova, L., Ye, H., Cottone, L., Strobl, A. C., Cool, P., et al. (2019). Synovial chondromatosis and soft tissue chondroma: extraosseous cartilaginous tumor defined by FN1 gene rearrangement. Mod. Pathol. 32 (12), 1762–1771. doi:10.1038/s41379-019-0315-8
Anderson, I. J., Goldberg, R. B., Marion, R. W., Upholt, W. B., and Tsipouras, P. (1990). Spondyloepiphyseal dysplasia congenita: genetic linkage to type II collagen (COL2AI). Am. J. Hum. Genet. 46 (5), 896–901.
Aury-Landas, J., Marcelli, C., Leclercq, S., Boumediene, K., and Bauge, C. (2016). Genetic determinism of primary early-onset osteoarthritis. Trends Mol. Med. 22 (1), 38–52. doi:10.1016/j.molmed.2015.11.006
Barat-Houari, M., Sarrabay, G., Gatinois, V., Fabre, A., Dumont, B., Genevieve, D., et al. (2016). Mutation update for COL2A1 gene variants associated with type II collagenopathies. Hum. Mutat. 37 (1), 7–15. doi:10.1002/humu.22915
Basit, S., Albalawi, A. M., Alharby, E., and Khoshhal, K. I. (2017). Exome sequencing identified rare variants in genes HSPG2 and ATP2B4 in a family segregating developmental dysplasia of the hip. BMC Med. Genet. 18 (1), 34. doi:10.1186/s12881-017-0393-8
Basit, S., Hannan, M. A., and Khoshhal, K. I. (2016). Developmental dysplasia of the hip: usefulness of next generation genomic tools for characterizing the underlying genes - a mini review. Clin. Genet. 90 (1), 16–20. doi:10.1111/cge.12755
Brucher, N., Faruch-Bilfeld, M., Molinier, F., Brouchet-Gomez, A., Lapegue, F., and Sans, N. (2014). Primary synovial osteochondromatosis of the first interphalangeal joint of the foot: a case report. Diagn Interv. Imaging 95 (4), 451–453. doi:10.1016/j.diii.2013.10.003
Burrage, L. C., Lu, J. T., Liu, D. S., Moss, T. J., Gibbs, R., Schlesinger, A. E., et al. (2013). Early childhood presentation of Czech dysplasia. Clin. Dysmorphol. 22 (2), 76–80. doi:10.1097/MCD.0b013e32835fff39
Carlson, K. M., Yamaga, K. M., Reinker, K. A., Hsia, Y. E., Carpenter, C., Abe, L. M., et al. (2006). Precocious osteoarthritis in a family with recurrent COL2A1 mutation. J. Rheumatol. 33 (6), 1133–1136.
Dahiya, R., Cleveland, S., and Megerian, C. A. (2000). Spondyloepiphyseal dysplasia congenita associated with conductive hearing loss. Ear Nose Throat J. 79 (3), 178–182. doi:10.1177/014556130007900312
Deng, J. L. (2014). X-ray diagnosis and differential diagnosis of adult acetabular dysplasia. Orthop. J. China 22 (11), 971–975. doi:10.3977/j.issn.1005-8478.2014.11.03
Feng, W. J., Wang, H., Shen, C., Zhu, J. F., and Chen, X. D. (2017). Severe cartilage degeneration in patients with developmental dysplasia of the hip. IUBMB Life 69 (3), 179–187. doi:10.1002/iub.1606
Fukutani, T., Toratani, S., Kanda, T., Matsui, K., Yamasaki, S., Sumi, K., et al. (2022). Two cases of temporomandibular synovial chondromatosis associated with Gli1 gene mutation. Int. J. Environ. Res. Public Health 19 (8), 4702. doi:10.3390/ijerph19084702
Granchi, D., Stea, S., Sudanese, A., Toni, A., Baldini, N., and Giunti, A. (2002). Association of two gene polymorphisms with osteoarthritis secondary to hip dysplasia. Clin. Orthop. Relat. Res. 403, 108–117. doi:10.1097/00003086-200210000-00018
Han, H., Wang, Y., Yin, M., and Xu, H. (2010). Imaging manifestations of pigmented villonodular synovitis. Huazhong Univ. Sci. Technolog. Med. Sci. 39 (2), 258–260+271. doi:10.3870/j.issn.1672-0741.2010.02.028
Hildebrand, B. (2015). Mutation in the type II collagen gene (COL2A1) as a cause of early osteoarthritis and juvenile idiopathic arthritis-A pseudorheumatoid arthritis variant. Semin. Arthritis Rheum. 44 (6), e22. doi:10.1016/j.semarthrit.2015.01.001
Hoornaert, K. P., Dewinter, C., Vereecke, I., Beemer, F. A., Courtens, W., Fryer, A., et al. (2006). The phenotypic spectrum in patients with arginine to cysteine mutations in the COL2A1 gene. J. Med. Genet. 43 (5), 406–413. doi:10.1136/jmg.2005.035717
Hoornaert, K. P., Marik, I., Kozlowski, K., Cole, T., Le Merrer, M., Leroy, J. G., et al. (2007). Czech dysplasia metatarsal type: another type II collagen disorder. Eur. J. Hum. Genet. 15 (12), 1269–1275. doi:10.1038/sj.ejhg.5201913
Husar-Memmer, E., Ekici, A., Al Kaissi, A., Sticht, H., Manger, B., Schett, G., et al. (2013). Premature osteoarthritis as presenting sign of type II collagenopathy: a case report and literature review. Semin. Arthritis Rheum. 42 (4), 355–360. doi:10.1016/j.semarthrit.2012.05.002
Ji, C., Li, C. W., and Deng, L. F. (2022). Research progress in the prevention and treatment of developmental dysplasia of the hip. Chin. J. Orthop. 42 (01), 54–64. doi:10.3760/cma.j.cn121113-20210914-00555
Kenanidis, E., Gkekas, N. K., Karasmani, A., Anagnostis, P., Christofilopoulos, P., and Tsiridis, E. (2020). Genetic predisposition to developmental dysplasia of the hip. J. Arthroplasty 35 (1), 291–300.e1. doi:10.1016/j.arth.2019.08.031
Li, H., Ma, L., Wang, B., Cui, Y., and Xiao, T. (2015). Identification of a novel mutation of the COL2A1 gene in a Chinese family with spondyloepiphyseal dysplasia congenita. Eur. Spine J. 24 (8), 1813–1819. doi:10.1007/s00586-015-3999-6
Li, W., Hu, P., and Bi, S. (2021). Research progress of pigmented villonodular synovitis. Int. J. Orthop. Sci. 42 (01), 40–44. doi:10.3969/j.issn.1673-7083.2021.01.010
Liu, J. P., Zhang, S. Q., and Chen, W. H. (2010). Radiographic imaging feature and differential diagnosis of early femoral head necrosis. China J. Orthop. Traumatology 23 (5), 344–348. doi:10.3969/j.issn.1003-0034.2010.05.008
Liu, L. (2014). “The clinical diagnosis and gene mutation analysis of spinal epiphyseal dysplasia,”. Master’s Thesis (Beijing: Peking Union Medical College).
Lopponen, T., Korkko, J., Lundan, T., Seppanen, U., Ignatius, J., and Kaariainen, H. (2004). Childhood-onset osteoarthritis, tall stature, and sensorineural hearing loss associated with Arg75-Cys mutation in procollagen type II gene (COL2A1). Arthritis Rheum. 51 (6), 925–932. doi:10.1002/art.20817
Matsui, Y., Michigami, T., Tachikawa, K., Yamazaki, M., Kawabata, H., and Nishimura, G. (2009). Czech dysplasia occurring in a Japanese family. Am. J. Med. Genet. A 149A (10), 2285–2289. doi:10.1002/ajmg.a.33010
Mertens, F., Jonsson, K., Willen, H., Rydholm, A., Kreicbergs, A., Eriksson, L., et al. (1996). Chromosome rearrangements in synovial chondromatous lesions. Br. J. Cancer 74 (2), 251–254. doi:10.1038/bjc.1996.346
Moreira, L. A., Carvalho, D. R., Santos, S. C. L., Silva, C. C. E., Ferreira, B. S. A., Cunha, B. M. D., et al. (2023). Czech dysplasia mimicking rheumatoid arthritis: case series and literature review. Mod. Rheumatol. 34 (4), 705–710. doi:10.1093/mr/road070
Nandhagopal, T., Tiwari, V., and De Cicco, F. L. (2024). Developmental dysplasia of the hip. Treasure Island (FL): StatPearls Publishing.
Ortiz-Neira, C. L., Paolucci, E. O., and Donnon, T. (2012). A meta-analysis of common risk factors associated with the diagnosis of developmental dysplasia of the hip in newborns. Eur. J. Radiol. 81 (3), e344–e351. doi:10.1016/j.ejrad.2011.11.003
Paton, R. W. (2017). Screening in developmental dysplasia of the hip (DDH). Surgeon 15 (5), 290–296. doi:10.1016/j.surge.2017.05.002
Pedersen, S. J., and Maksymowych, W. P. (2019). The pathogenesis of ankylosing spondylitis: an update. Curr. Rheumatol. Rep. 21 (10), 58. doi:10.1007/s11926-019-0856-3
Ritvaniemi, P., Korkko, J., Bonaventure, J., Vikkula, M., Hyland, J., Paassilta, P., et al. (1995). Identification of COL2A1 gene mutations in patients with chondrodysplasias and familial osteoarthritis. Arthritis Rheum. 38 (7), 999–1004. doi:10.1002/art.1780380717
Rukavina, I., Mortier, G., Van Laer, L., Frkovic, M., Dapic, T., and Jelusic, M. (2014). Mutation in the type II collagen gene (COL2AI) as a cause of primary osteoarthritis associated with mild spondyloepiphyseal involvement. Semin. Arthritis Rheum. 44 (1), 101–104. doi:10.1016/j.semarthrit.2014.03.003
Sciot, R., Dal Cin, P., Bellemans, J., Samson, I., Van Den Berghe, H., and Van Damme, B. (1998). Synovial chondromatosis: clonal chromosome changes provide further evidence for a neoplastic disorder. Virchows Arch. 433 (2), 189–191. doi:10.1007/s004280050235
Sher, C., Ramesar, R., Martell, R., Learmonth, I., Tsipouras, P., and Beighton, P. (1991). Mild spondyloepiphyseal dysplasia (Namaqualand type): genetic linkage to the type II collagen gene COL2A1. Am. J. Hum. Genet. 48 (3), 518–524.
Silveira, K. C., Bonadia, L. C., Superti-Furga, A., Bertola, D. R., Jorge, A. A., and Cavalcanti, D. P. (2015). Six additional cases of SEDC due to the same and recurrent R989C mutation in the COL2A1 gene–the clinical and radiological follow-up. Am. J. Med. Genet. A 167 (4), 894–901. doi:10.1002/ajmg.a.36954
Startzman, A., Collins, D., and Carreira, D. (2016). A systematic literature review of synovial chondromatosis and pigmented villonodular synovitis of the hip. Phys. Sportsmed. 44 (4), 425–431. doi:10.1080/00913847.2016.1216238
Thomas, S. R. (2015). A review of long-term outcomes for late presenting developmental hip dysplasia. Bone Jt. J. 97 (6), 729–733. doi:10.1302/0301-620X.97B6.35395
Tian, J., Bi, W., and Meng, F. (2003). Imaging diagnosis of acetabular dysplastic coxarthrosis in adult. Chin. J. Radiol. 37 (02), 135–139.
Tofield, C. E., and Mackinnon, C. A. (2003). Cleft palate repair in spondyloepiphyseal dysplasia congenita: minimizing the risk of cervical cord compression. Cleft Palate Craniofac J. 40 (6), 629–631. doi:10.1597/02-159
Totoki, Y., Yoshida, A., Hosoda, F., Nakamura, H., Hama, N., Ogura, K., et al. (2014). Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res. 24 (9), 1411–1420. doi:10.1101/gr.160598.113
Wang, K. Z., Zhou, J. S., Shi, Z. J., Hu, Y. C., Zhang, H., Chen, X. D., et al. (2023). Guideline for the diagnosis and treatment of developmental dysplasia of the hip in China (2023 edition). Chin. J. Anat. Clin. 28 (8), 493–511. doi:10.3760/cma.j.cn101202-20230612-00145
Warman, M. L., Cormier-Daire, V., Hall, C., Krakow, D., Lachman, R., Lemerrer, M., et al. (2011). Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A 155A (5), 943–968. doi:10.1002/ajmg.a.33909
Wen, J., Ping, H., Kong, X., and Chai, W. (2023). Developmental dysplasia of the hip: a systematic review of susceptibility genes and epigenetics. Gene 853, 147067. doi:10.1016/j.gene.2022.147067
Williams, C. J., Considine, E. L., Knowlton, R. G., Reginato, A., Neumann, G., Harrison, D., et al. (1993). Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg75-->Cys mutation in the procollagen type II gene (COL2A1). Hum. Genet. 92 (5), 499–505. doi:10.1007/BF00216458
Wynne-Davies, R. (1970). Acetabular dysplasia and familial joint laxity: two etiological factors in congenital dislocation of the hip. A review of 589 patients and their families. J. Bone Jt. Surg. Br. 52 (4), 704–716. doi:10.1302/0301-620x.52b4.704
Xie, G. P., Jiang, N., Liang, C. X., Zeng, J. C., Chen, Z. Y., Xu, Q., et al. (2015). Pigmented villonodular synovitis: a retrospective multicenter study of 237 cases. PLoS One 10 (3), e0121451. doi:10.1371/journal.pone.0121451
Zamborsky, R., Kokavec, M., Harsanyi, S., Attia, D., and Danisovic, L. (2019). Developmental dysplasia of hip: perspectives in genetic screening. Med. Sci. Basel 7 (4), 59. doi:10.3390/medsci7040059
Zhang, B., Zhang, Y., Wu, N., Li, J., Liu, H., and Wang, J. (2020). Integrated analysis of COL2A1 variant data and classification of type II collagenopathies. Clin. Genet. 97 (3), 383–395. doi:10.1111/cge.13680
Keywords: COL2A1, mutation, synovial chondromatosis, developmental dysplasia of the hip, the hip joints
Citation: Xin M, Guan X, Yang J, Li Y, Man Z, Sun H and Fu M (2025) Mutation in the COL2A1 gene is associated with acetabular dysplasia. Front. Genet. 15:1521412. doi: 10.3389/fgene.2024.1521412
Received: 01 November 2024; Accepted: 20 December 2024;
Published: 20 January 2025.
Edited by:
Jordi Pérez-Tur, Spanish National Research Council (CSIC), SpainReviewed by:
Jose-Noel Ibrahim, Lebanese American University, LebanonJulia Metzger, University of Veterinary Medicine Hannover, Germany
Copyright © 2025 Xin, Guan, Yang, Li, Man, Sun and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongsheng Sun, MTM4NjkxOTI1MDlAMTI2LmNvbQ==; Min Fu, ZnVtbXNkQDE2My5jb20=
†These authors have contributed equally to this work and share last authorship