 Huafang Zou1,2
Huafang Zou1,2 Qian Zhang3
Qian Zhang3 Jianxiang Liao1,2
Jianxiang Liao1,2 Dongfang Zou1
Dongfang Zou1 Zhanqi Hu1
Zhanqi Hu1 Bing Li1
Bing Li1 Li Chen1
Li Chen1 Jialun Wen1Xia Zhao1
Jialun Wen1Xia Zhao1 Victor Wei Zhang3
Victor Wei Zhang3 Dezhi Cao1,2*
Dezhi Cao1,2*- 1Department of Neurology, Shenzhen Children’s Hospital, Shenzhen, China
- 2Division of Epilepsy Surgery, Shenzhen Children’s Hospital, Shenzhen, China
- 3Department of Genomic Medicine, AmCare Genomics Lab, Guangzhou, China
Objective: Epilepsy, a prevalent neurological disorder, has multifaceted etiologies. Next-generation sequencing (NGS) has emerged as a robust diagnostic tool for this condition. This study aims to evaluate the detection efficiencies of different exome-based sequencing techniques.
Methods: Exome-based epilepsy panel tests, clinical exome sequencing (CES), and whole exome sequencing (WES) were conducted on 259 pediatric patients diagnosed with epilepsy. Single-nucleotide variants (SNVs) and copy number variants (CNVs) were interpreted based on each patient’s phenotypic presentation. Additionally, data concerning clinical symptoms, neuroimaging findings, treatment responses, and prognostic outcomes were collected and analyzed.
Results: The overall diagnostic yield was 32.8% (85/259), with a diagnostic yield of 40.0% for exome-based epilepsy panels, 30.1% for CES, and 27.8% for WES. We identified 82 cases with pathogenic or likely pathogenic SNVs and 4 cases with pathogenic CNVs, of which one case with both SNV and CNV. The most frequently detected gene was PRRT2, present in 10.0% (9/82) of cases. Epileptic syndromes were diagnosed in 66 patients, predominantly West Syndrome, Dravet Syndrome and Genetic Epilepsy with Febrile Seizures plus.
Conclusion: NGS is an effective method for uncovering the genetic foundations of pediatric epilepsy, with diagnostic yields varying based on the sequencing approach used. The growing preference for WES underscores its utility in complex cases, pointing to a trend towards more tailored diagnostic strategies.
1 Introduction
Epilepsy is a common neurological disorder in pediatrics characterized by a highly variable phenotype. Clinical manifestations range from self-limiting seizures to severe epileptic encephalopathy. The causes of epilepsy are diverse, including genetic abnormalities, structural changes, metabolic disorders, immune dysfunctions, and unknown etiologies (Scheffer et al., 2017). Genetic factors are responsible for 70%–80% of cases (Myers and Mefford, 2015a).
The genetic landscape of epilepsy is intricate, comprising CNVs, SNVs, small insertions or deletions (indels), and dynamic variations. There is significant phenotypic overlap among variants in various epilepsy-associated genes. The deployment of NGS has markedly enhanced the molecular diagnosis of epilepsy, facilitating rapid identification of causative genes, which supports prognosis assessment and the implementation of precision medicine. The clinical utility of NGS has been validated in conditions such as epileptic encephalopathies (Perucca et al., 2017b; Demos et al., 2019; Yang et al., 2019) as well as neurodevelopmental disorders where epilepsy is a secondary feature (Soden et al., 2014; Thevenon et al., 2016). Several diagnostic strategies, including gene panels, whole-exome sequencing (WES), and whole-genome sequencing (WGS), are currently employed to identify the genetic underpinnings of epilepsy. Generally, the diagnostic yield improves by analyzing a broader array of genes (Mercimek-Mahmutoglu et al., 2015; Mei et al., 2017).
In recent years, our department has adopted several diagnostic procedures for epilepsy, influenced by an evolving understanding of its genetic foundations and a reduction in the costs of high-throughput sequencing technologies. This study not only evaluates the efficacy and indications of various diagnostic approaches but also analyzes shifts in the trends of genetic testing strategies over time.
2 Materials and methods
2.1 Patients
A total of 259 Han Chinese pediatric patients from the Department of Neurology, Shenzhen Children’s Hospital were retrospectively studied. The inclusion criteria were: 1) clinically diagnosed with epilepsy by pediatric neurologists following the International League Against Epilepsy criteria (2017) (Fisher et al., 2017); 2) referred by pediatric neurologists for genetic tests between August 2018 and July 2021; 3) less than 18 years old at the time of epilepsy diagnosis; 4) patients whose epilepsy was not caused by head trauma or central nervous system infections. None of the evaluated individuals had consanguinity, and none were on medication for other neurological disorders during the evaluation. The patients’ family history, clinical presentation, electroencephalogram (EEG), brain magnetic resonance imaging (MRI), therapy options, and prognosis were collected and systematically reviewed. Aside from the balance between diagnostic yield and cost-effectiveness, the selection of different genetic testing methods was primarily based on the clinical presentation and the complexity of the case. Gene panel sequencing for epilepsy was recommended for patients with clear clinical indicators suggestive of specific epilepsy syndromes. For patients with more ambiguous or complex clinical features, Clinical exome sequencing (CES) was chosen to broaden the scope of genetic analysis beyond epilepsy-specific genes. WES was employed for patients with atypical or syndromic presentations, particularly when previous genetic testing did not yield a diagnosis or when the clinical features suggested a broader or more complex genetic basis. Mitochondrial genome sequencing was recommended if the patients presented suspicious mitochondrial disorder symptoms. The study was approved by the Shenzhen Children’s Hospital ethics committee. Written informed consent was obtained from all parents or their legal guardians.
2.2 Whole-exome sequencing
Peripheral blood from the patients and their parents (if available) was collected for DNA extraction using the Solpure Blood DNA Kit (Magen). DNA was fragmented to 300–500 bp using the Q800R Sonicator (Qsonica). Libraries were prepared with the Agilent Sureselect Human All Exon v5 kit (Agilent Technologies, Santa Clara, CA, USA) and were sequenced on the HiSeq 2000 (Illumina, San Diego, CA, USA). The average coverage depth was about 80–100×, with over 99% of the target regions. Fast software was used to process raw data to remove adapters and filter low-quality reads. Sequencing reads were mapped to the human reference genome (GRCh37/hg19) by NextGENe software (SoftGenetics, State College, PA, USA).
2.3 Clinical-exome sequencing and epilepsy gene panel
Custom-designed NimbleGen SeqCap probes (Roche NimbleGen, Madison, WI, United States) were used for in-solution hybridization. DNA samples were indexed and sequenced on the AmCareSeq-2000 (Amcare, Guangzhou, China). The average coverage depth was about 200×, with over 99% of the target regions covered by at least 20 reads. The processing methods of the raw data were consistent with those used for WES. The clinical-exome sequencing targeted coding exonic regions of approximately 5000 OMIM-recorded genes, known pathogenic variants from deep intronic or other non-coding regions. Similarly, epilepsy gene panel sequencing uses the same sequencing and data processing methods as CES. Still, it included 2,881 genes compiled through a comprehensive literature search (Pubmed, OMIM) and clinically available epilepsy panels.
2.4 Mitochondrial genome sequencing
Mitochondrial genome sequencing was conducted by long-range PCR (LR-PCR) and subsequent deep coverage sequencing (Zhang et al., 2012). In short, Mt-DNA was first amplified by LR-PCR and disrupted using ultrasonication. Mitochondrial libraries were established using the KAPA HTP Library Preparation Kit (Kapa Biosystems, USA). Sequencing was performed on HiSeq 2000 (Illumina). The average depth of sequencing was at least 5,000×. Variations were quantified by their percentage value. The lower limit of detection of variation heteroplasmy in the coding region is 2%.
2.5 Variant analysis and interpretation
Nucleotide variants identified in the aligned reads were extracted and analyzed using the NextGENe software (Version 2.4.2; SoftGenetics, State College, PA, United States). Variants were annotated using population databases (gnomAD, 1,000 Genomes, dbSNP) and variants databases (Clinvar, HGMD). In silico predictions were performed using the PolyPhen-2, SIFT, PROVEAN, MutationTaster, and GeneSplicer software tools. Sanger sequencing was performed to validate the variants identified by NGS. CNVs were interpreted based on the Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home), DECIPHER (https://decipher.sanger.ac.uk/), OMIM (https://www.omim.org/). The resolution was 100 kb with a bin size of 25 kb. Variants were classified as pathogenic (P), likely pthogenic (LP), variant of uncertain significance (VUS), likely benign (LB), or benign (B) according to the ACMG guideline (Richards et al., 2015b; Riggs et al., 2020).
2.6 Statistical analysis
Data were analyzed using SPSS software (version 23.0; IBM SPSS Statistics, Armonk, NY). Quantitative data were expressed as mean ± standard deviation (x ± s), while categorical data were presented as numbers and percentages (n%). The chi-squared test was used to compare categorical variables, including age at onset distribution, sex distribution, epilepsy classifications, syndromic diagnoses, the occurrence of developmental delay/intellectual disability (DD/ID), and findings on brain MRI. p-values less than 0.05 were considered statistically significant.
3 Results
3.1 Demographics and clinical characteristics
A total of 259 Han Chinese pediatric patients with epilepsy were included, and their demographic and clinical information are shown in Table 1. The male-to-female ratio was 1.49:1. Age at epilepsy onset ranged from 2 days to 14 years old (median 18 months). Only patients detected with pathogenic/likely pathogenic variants were considered to have causative variants, while patients detected with VUS or undetected variants were not included. Patients with causative variants were more likely to have onset from 1 month to the first year of life compared to those without causative variants (57.6% vs 28.7%, p < 0.001). The most frequent type of epilepsy was focal epilepsy (155/259, 59.8%). There were 66 patients diagnosed with epileptic syndromes, of which the most common were West syndrome (WS) (27/259, 10.4%), followed by Dravet syndrome (DS) (10/259, 3.9%), Genetic Epilepsy with Febrile Seizures plus (GEFS+) (10/259, 3.9%), and Lennox-Gastaut syndrome (LGS) (7/259, 2.7%). Other diagnosed epileptic syndromes included Ohtahara syndrome (OS), childhood absence epilepsy (CAE), benign familial infantile epilepsy (BFIE), Doose syndrome (epilepsy with myoclonic-astatic seizures, EMAS), Panayiotopoulos syndrome (PS), juvenile myoclonic epilepsy (JME), epilepsy and mental retardation limited to females (EFMR), electrical status epilepticus during sleep (ESES), and Benign childhood epilepsy with centrotemporal spikes (BECT). In addition, 139 of the 259 patients (53.7%) combined with DD or ID. Patients with causative variants were more likely to combine with DD/ID (70.6% vs45.4%, p = 0.001). Brain MRI found 143 (55.2%) patients showed neurologic abnormalities. The flowchart of the sequencing results was shown in Figure 1.
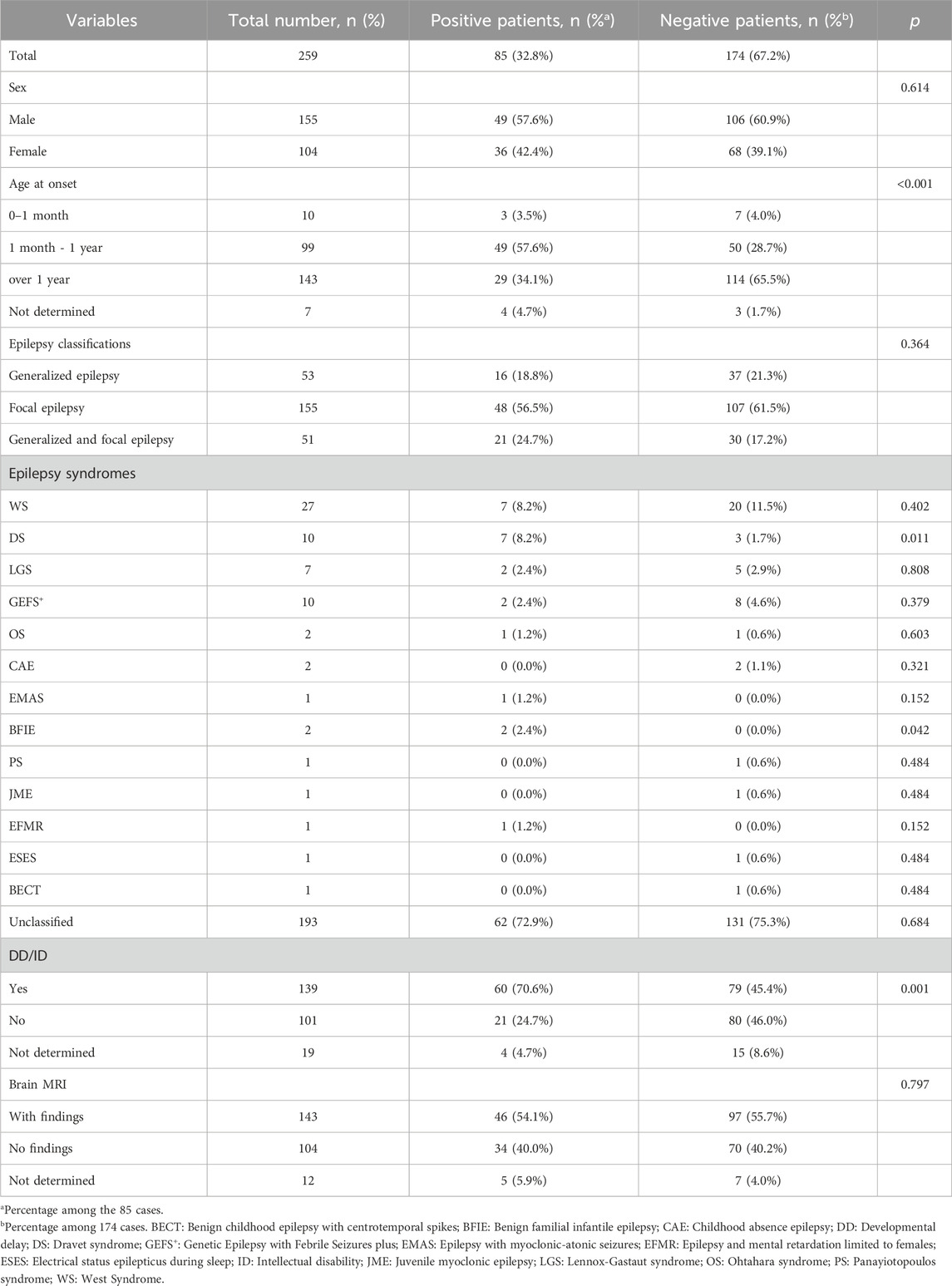
Table 1. Demographics and clinical information of the patients.
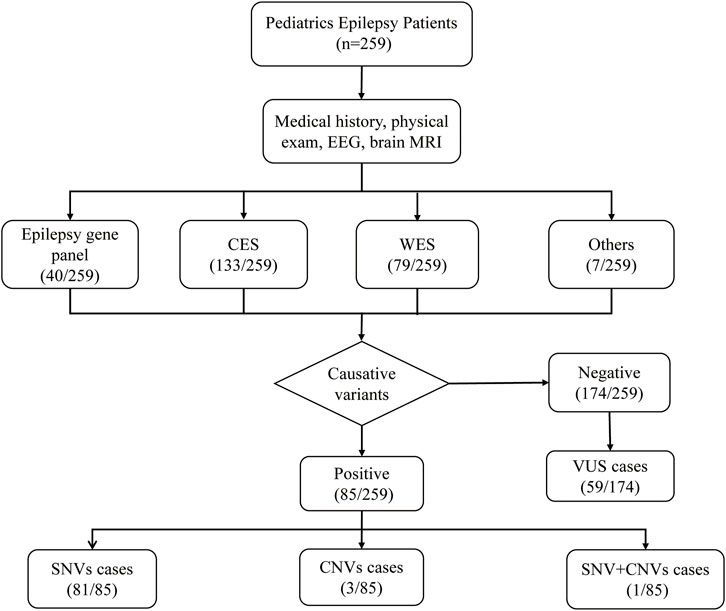
Figure 1. Schematic summarizing the 259 epilepsy cases.
3.2 Application of exome-based sequencing strategies
Overall, the majority of patients received CES (133/259, 51.4%), followed by WES (79/259, 30.5%) and epilepsy gene panel (40/259, 15.4%). However, the selection of sequencing methods had changed over the years (Figure 2A). CES (75.0%, 59.3%) constituted the majority in 2018–2019, while WES (0%, 6.2%) was the less common option. In 2020, CES retained its dominance (67.6%), but the preference for WES increased (16.2%), and the use of epilepsy gene panels decreased. In 2021–2022, WES became the preferred method (73.9%, 97.5%), surpassing CES (26.1%, 2.5%), with epilepsy gene panels being rarely chosen. Additionally, 62 of 259 cases conducted mitochondrial genome sequencing spontaneously (Figure 2B).
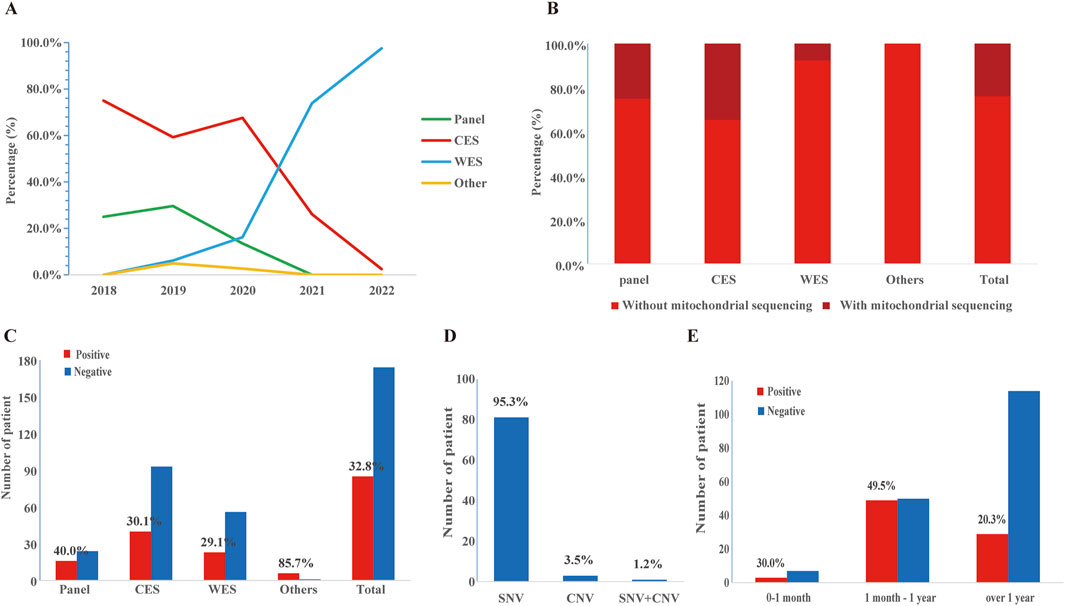
Figure 2. The application and diagnostic yield of different sequencing strategies. (A) The application of epilepsy gene panel, CES, and WES over time. (B) The application of mitochondrial genome sequencing. (C) Number of patients and diagnostic yield of epilepsy gene panel, CES, and WES. (D) Distribution of pathogenic/likely pathogenic (P/LP) single nucleotide variants (SNVs) and copy number variations (CNVs) in the study cohort. (E) Number of patients and detection rates at different ages of epilepsy onset. Detection rates were calculated as the ratio of the total number of positive cases in the age range of epilepsy onset. Additionally, please modify the sentence in Section 3.3 Diagnostic Yield (Line 377), changing “while CES (30.1%) and WES (27.8%)” to “while CES (30.1%) and WES (29.1%)”.
3.3 Diagnostic yield
A molecular diagnosis was established in 85 of 259 patients, resulting in a comprehensive diagnostic yield of 32.8% (Table 1). The diagnostic yield of each method was shown in Figure 2C. Surprisingly, the epilepsy gene panel had the highest diagnostic yield (40.0%), while CES (30.1%) and WES (27.8%) yielded similar results. As shown in Figure 2D, P/LP SNVs were found in 81 cases (95.3%) (including 3 cases with mitochondrial DNA (mt-DNA) variants), P/LP CNVs were found in 3 cases (3.5%), and concomitant likely pathogenic SNV and CNV were found in 1 case (1.2%). In addition, variants of uncertain significance possibly explaining the epilepsy of the patient were identified in 59 cases (59/259, 22.8%), which included 57 SNVs and 2 CNVs (Supplementary Table S1).
Patients who started epilepsy at 1 month to 1 year old had more P/LP variants (49.5%) than those onsets after 1 year old (20.3%) and before 1 month (30.0%) (Figure 2E). The molecular diagnostic rates in different clinical syndromes and their relevant genes were shown in Table 2. Among the 259 patients, West syndrome was clinically diagnosed in 27, seven of which were molecular diagnosed (detection rate: 25.9%). One SNV was found in NUS1, BRAF, CDKL5, IRF2BPL, SCN8A, COQ4, and KCNT1, respectively. 22q11.21 duplication was found in the same case that had a COQ4 variant. DS was clinically diagnosed in 10 cases, seven of which were molecular diagnosed (detection rate as 70.0%). Seven distinct SNVs were identified in SCN1A. Genetic Epilepsy with febrile seizures plus was clinically diagnosed in 10 cases, 2 of which were molecular diagnoses (detection rate as 20.0%). One of each SNV was found in SCN1A and KMT2D. Lennox-Gastaut syndrome was clinically diagnosed in 7 cases, two of which were molecularly diagnosed. (Detection rate as 28.6%). One of each SNV was found in ADSL and TSC2. Tuberous sclerosis was clinically diagnosed in 5 cases, and all of them were molecular diagnosed (detection rate as 100%). Other clinical syndromes and their related genes were listed in Table 2.
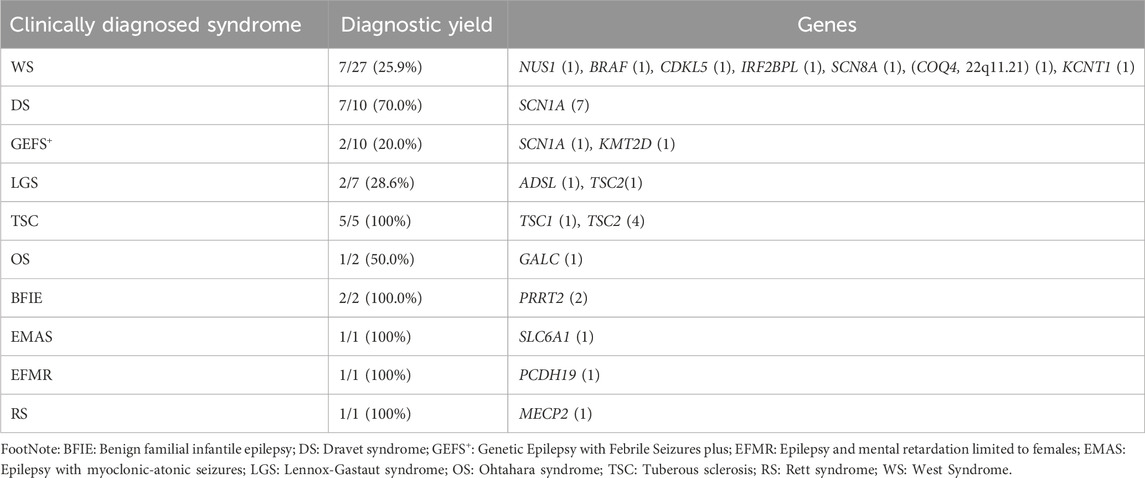
Table 2. Clinically diagnosed syndrome and associated genes.
3.4 Detection of single-nucleotide and copy-number variants
A total of 85 SNVs were identified in 82 cases, assigned to 44 nuclear genes and 2 mt-DNA genes (Supplementary Table S2). The most frequent nuclear gene was PRRT2 (9, 10.0%), followed by SCN1A (8, 9.8%) and TSC2 (7, 8.5%), SCN8A (4, 4.9%), TSC1 (3, 3.7%) (Figure 3). Two patients (2.4%) had variants in SLC6A1, PACS2, GALC, CSNK2B, CHD2, CDKL5, BRAF, RELN, and GJB2, respectively. Three SNVs in 2 mitochondrial genes, TRNL1 and ATP6, were discovered in 3 cases, respectively. According to the OMIM database, 63 variants (76.8%) were inherited in autosomal dominant mode, 9 variants (11.0%) were inherited in X-linked mode, seven variants (8.5%) were inherited in autosomal recessive mode, and 3 mitochondrial variants (3.7%) were maternally inherited. The origin of the variants was confirmed by trio-based sequencing or Sanger sequencing. Sixty of the SNVs were de novo (70.6%), and only 24 (28.2%) SNVs were inherited from the parents. (Figure 4).
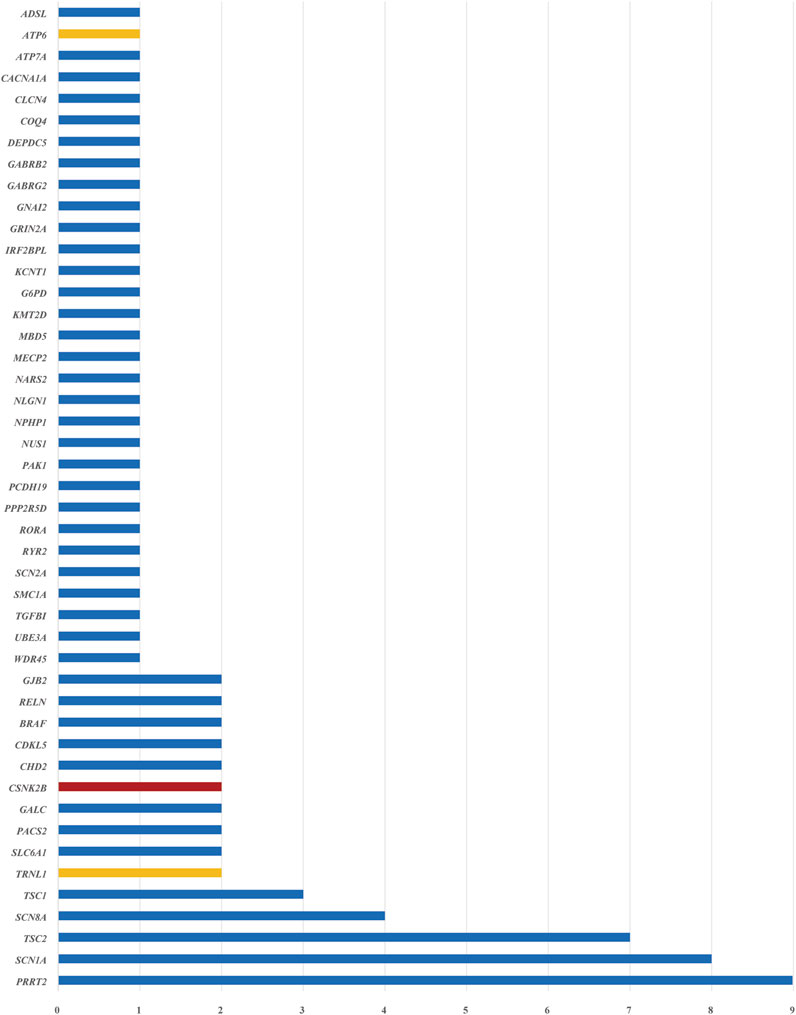
Figure 3. Disease-related genes were identified in 259 patients with epilepsy. The blue bars indicate nuclear genes, the orange bars indicate mitochondrial genes, and the red bar indicates a gene that was not included in the epilepsy gene panel. The horizontal axis represents the number of patients with pathogenic or likely pathogenic SNVs identified for each gene.
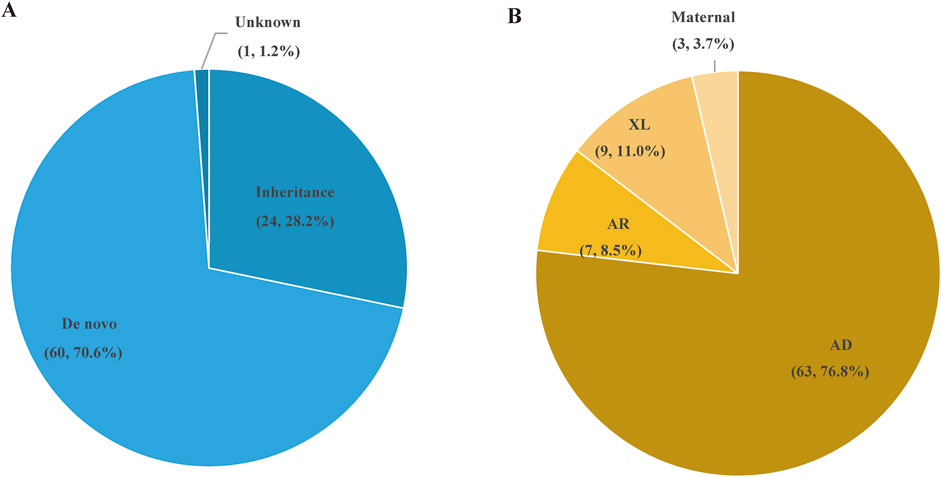
Figure 4. The types of variation and inheritance patterns in 84 P/LP patients with epilepsy. (A) Distribution of de novo and inherited genetic variants in the patient cohort. (B) Inheritance patterns of the genetic variants, including autosomal dominant (AD), autosomal recessive (AR), X-linked (XL), and maternal inheritance.
Four pathogenic/likely pathogenic CNVs were identified in 4 cases (4/259, 1.5%) (Supplementary Table S3). The length ranged from 398 kb to 5.56Mb, with 3 deletions and 1 duplication. The CNVs were located in chromosomes 1, 15, 16, and 22, respectively, and all were de novo. These CNVs contained or partially contained epilepsy-related genes, such as PRRT2 (16p11.2), ZBTB18 (1q43q44), and GABRB3 (15q11.2q13.1).
Interestingly, patient 133 presented ocular hypertelorism, brain hypoplasia, West syndrome, global developmental delay, and co-enzyme Q10 deficiency. CES detected likely pathogenic homozygous variants c.370G > A (p.G124S) in the COQ4 gene related to Leigh syndrome. Another likely pathogenic CNV was also found in the patient, 1.32 Mb duplication of 22q11.21, which contains 27 genes recorded in OMIM. The variants in COQ4 could explain epilepsy, brain hypoplasia, and DD. The duplicated region contained TBX1, which is related to DiGeorge syndrome or Velocardiofacial syndrome. The previously reported cases with 22q11.21 duplication syndrome showed DD, face anomaly, hearing damage, and congenital heart disease (Firth, 1993). Therefore, it is likely that both SNV and CNV influenced the patient’s phenotypes.
4 Discussion
In clinical practice, NGS has been demonstrated to be a useful diagnostic tool in diagnosing the cause of epilepsy. The identification of causative variants enables improvements in treatment options for patients. Our results revealed P/LP variants in 44 nuclear genes, with PRRT2, SCN1A, and TSC2 being the top three most frequently mutated, collectively accounting for 28.3% of all P/LP variants. The frequency of PRRT2 variants (10.0%) in this cohort, predominantly associated with BFIE, is notably higher than the 2%–6% reported in Western populations (Landolfi et al., 2021). PRRT2 plays a critical role in synaptic transmission by interacting with SNAP25 in the SNARE complex, a pathway essential for neurotransmitter release (Lee et al., 2012). Disruption of PRRT2 function may alter synaptic dynamics, contributing to seizure susceptibility. The strong familial inheritance pattern observed in PRRT2-related cases (77.8%) highlights the importance of integrating family history into genetic evaluations. The hotspot variation c.649dupC (p.R217Pfs*8), identified in six cases, further supports its role as a diagnostic marker for BFIE, with prior studies reporting this variant in 50%–70% of PRRT2-related epilepsy cases globally (Steinlein et al., 2012). SCN1A, present in 9.8% of cases, was primarily associated with DS, aligning with prior studies indicating frequencies ranging from 8%–15% in epilepsy cohorts (Zuberi et al., 2011; Olson et al., 2017; Yang et al., 2019). SCN1A encodes the Nav1.1 voltage-gated sodium channel ɑ-subunit, which is critical for maintaining inhibitory signaling in GABAergic neurons. Variations in SCN1A disrupt sodium channel function, impairing GABAergic inhibitory neurotransmission and leading to hyperexcitability, a hallmark of early-onset epilepsy (Catterall et al., 2010; Brunklaus et al., 2012). Notably, 87.5% of SCN1A variants in our cohort occurred de novo, consistent with global findings that 80%–90% of SCN1A variations arise de novo in DS patients (Depienne et al., 2009; Scheffer and Nabbout, 2019). Variability in seizure phenotypes among SCN1A-mutated patients suggests potential interactions with other ion channel genes, such as SCN2A and SCN8A, underscoring the polygenic complexity of SCN1A-related epilepsies (Lossin, 2009; Howell et al., 2015). TSC2 variations were identified in 8.5% of patients and were predominantly linked to earlier epilepsy onset and a higher prevalence of DD/ID, consistent with prior studies reporting TSC2 variations in 6%–10% of pediatric epilepsy cohorts (Crino, 2011).
Besides, TSC2, a critical component of the TSC1/TSC2 complex, regulates mTORC1 signaling, essential for synaptic plasticity, neural development, and cellular growth. Loss of TSC2 function leads to hyperactivation of mTOR, resulting in aberrant neural development and epileptogenesis. Patients with TSC2 variations may benefit from mTOR inhibitors like everolimus, which have demonstrated a >50% reduction in seizure frequency in TSC-related epilepsy patients (Krueger et al., 2013). Additionally, SCN1A-related network instability could exacerbate mTOR dysregulation, enhancing epileptogenesis (Howell et al., 2015). Similarly, PRRT2 disruptions, through their role in synaptic vesicle release, may indirectly impact mTOR signaling, highlighting a convergent mechanism linking ion channel dysfunction, synaptic regulation, and mTOR pathways. Variants in CSNK2B (OMIM: 115441) were identified in 2 patients through WES (Case 67 and 72), but this gene was not included in our epilepsy gene panel. CSNK2B causes Poirier-Bienvenu neurodevelopmental syndrome, which is characterized by early-onset epilepsy (median 5 months), clustered GTCS, myoclonic seizures, and DD (Bonanni et al., 2021). In addition, mt-DNA variants are also considered responsible for epilepsy and other neurological diseases (Rahman, 2012). We performed mitochondrial genome sequencing on 62 patients and detected 3 mt-DNA variants (3/259,1.2%). Each m.3243A > G variant was detected in case 78 and 96, respectively, which is a widespread variant in mitochondrial t-RNA and related to MELAS syndrome (Tanahashi et al., 2000). In case 136, another mt-DNA variant, m.8993T > G (p.L156R), was found in ATP6, which is related to Leigh syndrome (Ganetzky et al., 2019).
Additionally, 59 VUS were identified in this study, representing 22.8% of the cohort. Although VUS currently do not directly influence clinical decision-making, advances in functional studies, expanding genomic databases, and improved genotype-phenotype correlation frameworks could facilitated the reclassification of VUS over time. This reanalysis has the potential to improve diagnostic accuracy, support genetic counseling, and enable more precise treatment strategies (Richards et al., 2015a; Lindy et al., 2018). It is worth noting that there were five variations in CACNA1H. According to the ClinGen Epilepsy Gene Curation Expert Panel, CACNA1H is classified as a disputed gene due to insufficient evidence supporting its role as a monogenic cause of epilepsy (Helbig et al., 2018). While CACNA1H encodes a T-type calcium channel crucial for neuronal excitability, studies suggest its variants often act as susceptibility factors in polygenic or multifactorial contexts rather than as definitive monogenic causes (Weiss and Zamponi, 2020). Despite this, CACNA1H variants were included in this analysis to provide a comprehensive overview of all identified variants, given their potential role in seizure susceptibility and frequent detection in epilepsy cohorts. These findings should be interpreted cautiously in light of the gene’s uncertain clinical relevance as a monogenic cause. This also underscored the importance of refining gene classifications to clarify the precise role of CACNA1H in epilepsy through further functional studies and larger cohort analyses.
CNVs are a well-known genetic cause of epileptic syndromes and other neurological disorders. Our study established that the detection rate of CNVs was 1.5%, which was relatively lower than that of the previous studies (Jiang et al., 2021; Zou et al., 2021). The possible explanation is that CMA/CNV-seq is the first-tier diagnostic tool for CNV detection. If the patients detected P/LP CNVs that can explain their epilepsy through CMA/CNV-seq, they would not undergo additional genetic tests unless they were suspected of monogenic diseases.
Many prior cohort studies examined the effectiveness of various sequencing strategies in pediatric epileptic patients, with diagnostic yields ranging from 26.7% to 42% (Jang et al., 2019; Yang et al., 2019; Rochtus et al., 2020b; Chen et al., 2021). Gene selection principles have significant variability in different exome-based epilepsy gene panels, which range in size from 17 to 2,742 genes (Zhang et al., 2017; Yang et al., 2019). Mercimek-Mahmutoglu et al. (Mercimek-Mahmutoglu et al., 2015) reported an increasing diagnostic yield, from 10% to 48.5%, when the number of genes expanded from 35 to 265. However, the diagnostic yield did not always associate with a more significant number of genes. Kim et al. (Kim et al., 2021a) suggested an overall diagnostic yield to be 47.3% using an epilepsy gene panel containing 79–127 genes, while Chen et al. (Chen et al., 2021) achieved a diagnostic yield of 32.7% using WES. Similar results were seen in our study as well: We found that the epilepsy gene panel (40.0%) had a higher diagnostic yield than CES (30.1%) and WES (29.1%). A previous study in our department performed whole-genome sequencing (WGS) on 320 children with epilepsy. The overall diagnostic yield of WGS was 36.6% (Zou et al., 2021), comparable to our study (32.8%). The possible explanation could be that in earlier years, limited by the understanding of epilepsy genes and the cost considerations, genetic testing was restricted to patients with specific indications such as distinctive dysmorphic features, known family history, or indicative laboratory results. Subsequently, due to the increasing diagnostic needs and cost reduction, CES and WES were performed on more patients with non-specific phenotypes. Reducing diagnostic yield may be caused by the growing number of non-specific patients who received genetic tests. The preference for WES over epilepsy gene panels has grown over time.
Furthermore, our analysis revealed that the diagnostic yield varied significantly across patient subgroups, highlighting the interplay between epilepsy phenotypes and their genetic underpinnings. Patients diagnosed between 1 month and 1 year of age showed the highest diagnostic yield, which underscores the importance of early genetic testing in suspected genetic epilepsy syndromes (Kim et al., 2021a). This finding aligns with previous studies indicating that early-onset epilepsies are more likely to have a genetic basis (Rochtus et al., 2020b; Jiang et al., 2021). These aspects emphasize the need for a personalized approach in the genetic testing of pediatric epilepsy, taking into account the specific clinical presentation. Among the 139 cases (53.7%) with epilepsy accompanied by DD/ID, the diagnostic yield was significantly higher (70.6%) compared to patients without DD/ID (24.7%). These findings emphasize the distinct genetic basis of epilepsy with neurodevelopmental comorbidities, where pathogenic variants frequently contribute to both conditions. Genes like SCN1A and TSC2, identified in this study, are associated with syndromes like DS and tuberous sclerosis complex, both featuring epilepsy and developmental impairments. These results are consistent with prior research showing diagnostic yields of 60%–80% in similar cohorts, particularly when early-onset epilepsy and neurodevelopmental symptoms coexist (Lindy et al., 2018; Rochtus et al., 2020a). The co-occurrence of epilepsy with DD/ID provides phenotypic clarity, which aids in the interpretation of variants and increases the likelihood of identifying monogenic causes. Neurodevelopmental comorbidities often involve variations in pleiotropic genes, further underscoring the need for comprehensive genomic approaches (Myers and Mefford, 2015b). Focal epilepsies demonstrated a significantly higher diagnostic yield (56.5%) compared to generalized epilepsies (18.8%). This result, consistent with prior studies, reflects the relatively well-defined genetic basis of focal epilepsies, often involving single-gene variations in genes like SCN1A (Lindy et al., 2018; Kim et al., 2021b). In contrast, generalized epilepsies showed a more complex genetic architecture, including polygenic contributions and gene-environment interactions, which are less detectable with current sequencing platforms (Myers and Mefford, 2015b; Perucca et al., 2017a). Focal epilepsies also present with more specific clinical features, such as seizure semiology and focal imaging findings, enhancing the accuracy of variant interpretation. In comparison, the broader and overlapping phenotypes of generalized epilepsy complicate genetic analysis and reduce diagnostic yields. Moreover, the design of gene panels and exome approaches frequently prioritizes genes associated with focal epilepsy syndromes, potentially biasing diagnostic outcomes toward these phenotypes (Rochtus et al., 2020b). These findings collectively highlight the importance of integrating early and comprehensive genetic testing, tailored to the clinical presentation, to improve diagnostic outcomes across diverse epilepsy subtypes.
Our study elucidates the diagnostic utility of exome-based sequencing in pediatric epilepsy. While treatment and follow-up were conducted, the assessments during the follow-up did not systematically evaluate changes in the epilepsy syndromes. This limits our understanding of the dynamic and evolving nature of epilepsy and how it responds to treatment over time. Future research should include structured and periodic evaluations of epilepsy syndromes during follow-ups to capture potential changes in the epilepsy syndromes. Additionally, the sample size for this study was relatively small, which limits the generalizability of the findings. Efforts will be made in future studies to recruit a larger cohort, providing a deeper understanding of the utility of exome sequencing in pediatric epilepsy.
5 Conclusion
In summary, our study underscores the significant utility of exome-based sequencing in diagnosing pediatric epilepsy, revealing distinct advantages and evolving preferences among different sequencing methods. We observed a notable shift from targeted epilepsy gene panels, which provided the highest diagnostic yields, to whole-exome sequencing. This transition reflects WES’s broader diagnostic scope, capturing a more comprehensive array of genetic abnormalities beyond those traditionally associated with epilepsy. Notably, the diagnostic yield of patients from 1 month to 1 year old was the highest, emphasizing the critical role of early genetic testing in suspected genetic epilepsy syndromes. These findings advocate for a personalized approach to genetic testing in pediatric epilepsy, tailoring the sequencing method to specific clinical presentations and the age of onset. Future studies should continue to adapt and refine genetic testing strategies, accommodating the rapid evolution of sequencing technologies to maximize diagnostic yield and inform targeted treatment strategies.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: [https://pan.baidu.com/s/1f1shHg7ruJuQPlUnXLpYtQ?pwd=da49].
Ethics statement
The studies involving humans were approved by Shenzhen Children’s Hospital ethics committee. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participant’s legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
HZ: Formal Analysis, Writing–original draft. QZ: Formal Analysis, Writing–review and editing. JL: Data curation, Formal Analysis, Writing–review and editing. DZ: Data curation, Formal Analysis, Writing–review and editing. ZH: Data curation, Formal Analysis, Writing–review and editing. BL: Data curation, Formal Analysis, Writing–review and editing. LC: Data curation, Formal Analysis, Writing–review and editing. JW: Data curation, Formal Analysis, Writing–review and editing. XZ: Data curation, Formal Analysis, Writing–review and editing. VZ: Methodology, Software, Writing–review and editing. DC: Funding acquisition, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Guangdong High-level Hospital Construction Fund by Basic research of Shenzhen science and technology plan project (JCYJ20210324135211030) and Shenzhen Key Medical Discipline Construction Fund (No.SZXK033).
Acknowledgments
We thank the patients and their family members for taking part in this study. Thanks to Rowan Coates for his guidance on English grammar.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1496411/full#supplementary-material
References
Bonanni, P., Baggio, M., Duma, G. M., Negrin, S., Danieli, A., and Giorda, R. (2021). Developmental and epilepsy spectrum of Poirier-Bienvenu neurodevelopmental syndrome: description of a new case study and review of the available literature. Seizure 93, 133–139. doi:10.1016/j.seizure.2021.10.019
Brunklaus, A., Ellis, R., Reavey, E., Forbes, G., and Zuberi, S. (2012). Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 135 (8), 2329–2336. doi:10.1093/brain/aws151
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). NaV1. 1 channels and epilepsy. J. physiology 588 (11), 1849–1859. doi:10.1113/jphysiol.2010.187484
Chen, W., Qin, J., Shen, Y., Liang, J., Cui, Y., and Zhang, Y. (2021). Next generation sequencing in children with unexplained epilepsy: a retrospective cohort study. Brain Dev. 43 (10), 1004–1012. doi:10.1016/j.braindev.2021.05.014
Crino, P. B. (2011). mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 17 (12), 734–742. doi:10.1016/j.molmed.2011.07.008
Demos, M., Guella, I., DeGuzman, C., McKenzie, M. B., Buerki, S. E., Evans, D. M., et al. (2019). Diagnostic yield and treatment impact of targeted exome sequencing in early-onset epilepsy. Front. Neurol. 10, 434. doi:10.3389/fneur.2019.00434
Depienne, C., Trouillard, O., Saint-Martin, C., Gourfinkel-An, I., Bouteiller, D., Carpentier, W., et al. (2009). Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46 (3), 183–191. doi:10.1136/jmg.2008.062323
Firth, H. V. (1993). 22q11.2 duplication - retired chapter, for historical reference only. GeneReviews((R)).
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., et al. (2017). Operational classification of seizure types by the international League against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia 58 (4), 522–530. doi:10.1111/epi.13670
Ganetzky, R. D., Stendel, C., McCormick, E. M., Zolkipli-Cunningham, Z., Goldstein, A. C., Klopstock, T., et al. (2019). MT-ATP6 mitochondrial disease variants: phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum. Mutat. 40 (5), 499–515. doi:10.1002/humu.23723
Helbig, I., Riggs, E. R., Barry, C. A., Klein, K. M., Dyment, D., Thaxton, C., et al. (2018). The ClinGen Epilepsy Gene Curation Expert Panel—bridging the divide between clinical domain knowledge and formal gene curation criteria. Hum. Mutat. 39 (11), 1476–1484. doi:10.1002/humu.23632
Howell, K. B., McMahon, J. M., Carvill, G. L., Tambunan, D., Mackay, M. T., Rodriguez-Casero, V., et al. (2015). SCN2A encephalopathy: a major cause of epilepsy of infancy with migrating focal seizures. Neurology 85 (11), 958–966. doi:10.1212/WNL.0000000000001926
Jang, S. S., Kim, S. Y., Kim, H., Hwang, H., Chae, J. H., Kim, K. J., et al. (2019). Diagnostic yield of epilepsy panel testing in patients with seizure onset within the first year of life. Front. Neurol. 10, 988. doi:10.3389/fneur.2019.00988
Jiang, T., Gao, J., Jiang, L., Xu, L., Zhao, C., Su, X., et al. (2021). Application of trio-whole exome sequencing in genetic diagnosis and therapy in Chinese children with epilepsy. Front. Mol. Neurosci. 14, 699574. doi:10.3389/fnmol.2021.699574
Kim, S. Y., Jang, S. S., Kim, H., Hwang, H., Choi, J. E., Chae, J. H., et al. (2021a). Genetic diagnosis of infantile-onset epilepsy in the clinic: application of whole-exome sequencing following epilepsy gene panel testing. Clin. Genet. 99 (3), 418–424. doi:10.1111/cge.13903
Kim, S. Y., Jang, S. S., Kim, H., Hwang, H., Choi, J. E., Chae, J. H., et al. (2021b). Genetic diagnosis of infantile-onset epilepsy in the clinic: application of whole-exome sequencing following epilepsy gene panel testing. Clin. Genet. 99 (3), 418–424. doi:10.1111/cge.13903
Krueger, D. A., Wilfong, A. A., Holland-Bouley, K., Anderson, A. E., Agricola, K., Tudor, C., et al. (2013). Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann. neurology 74 (5), 679–687. doi:10.1002/ana.23960
Landolfi, A., Barone, P., and Erro, R. (2021). The spectrum of PRRT2-associated disorders: update on clinical features and pathophysiology. Front. Neurology 12, 629747. doi:10.3389/fneur.2021.629747
Lee, H.-Y., Huang, Y., Bruneau, N., Roll, P., Roberson, E. D., Hermann, M., et al. (2012). Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. 1 (1), 2–12. doi:10.1016/j.celrep.2011.11.001
Lindy, A. S., Stosser, M. B., Butler, E., Downtain-Pickersgill, C., Shanmugham, A., Retterer, K., et al. (2018). Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 59 (5), 1062–1071. doi:10.1111/epi.14074
Lossin, C. (2009). A catalog of SCN1A variants. Brain Dev. 31 (2), 114–130. doi:10.1016/j.braindev.2008.07.011
Mei, D., Parrini, E., Marini, C., and Guerrini, R. (2017). The impact of next-generation sequencing on the diagnosis and treatment of epilepsy in paediatric patients. Mol. Diagn Ther. 21 (4), 357–373. doi:10.1007/s40291-017-0257-0
Mercimek-Mahmutoglu, S., Patel, J., Cordeiro, D., Hewson, S., Callen, D., Donner, E. J., et al. (2015). Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 56 (5), 707–716. doi:10.1111/epi.12954
Myers, C. T., and Mefford, H. C. (2015a). Advancing epilepsy genetics in the genomic era. Genome Med. 7 (1), 91. doi:10.1186/s13073-015-0214-7
Myers, C. T., and Mefford, H. C. (2015b). Advancing epilepsy genetics in the genomic era. Genome Med. 7, 91–11. doi:10.1186/s13073-015-0214-7
Olson, H. E., Kelly, M., LaCoursiere, C. M., Pinsky, R., Tambunan, D., Shain, C., et al. (2017). Genetics and genotype–phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann. neurology 81 (3), 419–429. doi:10.1002/ana.24883
Perucca, P., Scheffer, I. E., Harvey, A. S., James, P. A., Lunke, S., Thorne, N., et al. (2017a). Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 131, 1–8. doi:10.1016/j.eplepsyres.2017.02.001
Perucca, P., Scheffer, I. E., Harvey, A. S., James, P. A., Lunke, S., Thorne, N., et al. (2017b). Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 131, 1–8. doi:10.1016/j.eplepsyres.2017.02.001
Rahman, S. (2012). Mitochondrial disease and epilepsy. Dev. Med. Child. Neurol. 54 (5), 397–406. doi:10.1111/j.1469-8749.2011.04214.x
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015a). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015b). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Rochtus, A., Olson, H. E., Smith, L., Keith, L. G., El Achkar, C., Taylor, A., et al. (2020a). Genetic diagnoses in epilepsy: the impact of dynamic exome analysis in a pediatric cohort. Epilepsia 61 (2), 249–258. doi:10.1111/epi.16427
Rochtus, A., Olson, H. E., Smith, L., Keith, L. G., El Achkar, C., Taylor, A., et al. (2020b). Genetic diagnoses in epilepsy: the impact of dynamic exome analysis in a pediatric cohort. Epilepsia 61 (2), 249–258. doi:10.1111/epi.16427
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 58 (4), 512–521. doi:10.1111/epi.13709
Scheffer, I. E., and Nabbout, R. (2019). SCN1A-related phenotypes: epilepsy and beyond. Epilepsia 60, S17–S24. doi:10.1111/epi.16386
Soden, S. E., Saunders, C. J., Willig, L. K., Farrow, E. G., Smith, L. D., Petrikin, J. E., et al. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6 (265), 265ra168. doi:10.1126/scitranslmed.3010076
Steinlein, O. K., Villain, M., and Korenke, C. (2012). The PRRT2 mutation c. 649dupC is the so far most frequent cause of benign familial infantile convulsions. Seizure 21 (9), 740–742. doi:10.1016/j.seizure.2012.07.006
Tanahashi, C., Nakayama, A., Yoshida, M., Ito, M., Mori, N., and Hashizume, Y. (2000). MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol. 99 (1), 31–38. doi:10.1007/pl00007403
Thevenon, J., Duffourd, Y., Masurel-Paulet, A., Lefebvre, M., Feillet, F., El Chehadeh-Djebbar, S., et al. (2016). Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin. Genet. 89 (6), 700–707. doi:10.1111/cge.12732
Weiss, N., and Zamponi, G. W. (2020). Genetic T-type calcium channelopathies. J. Med. Genet. 57 (1), 1–10. doi:10.1136/jmedgenet-2019-106163
Yang, L., Kong, Y., Dong, X., Hu, L., Lin, Y., Chen, X., et al. (2019). Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet. Med. 21 (3), 564–571. doi:10.1038/s41436-018-0091-8
Zhang, Q., Li, J., Zhao, Y., Bao, X., Wei, L., and Wang, J. (2017). Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin. Genet. 91 (5), 717–724. doi:10.1111/cge.12901
Zhang, W., Cui, H., and Wong, L. J. (2012). Comprehensive one-step molecular analyses of mitochondrial genome by massively parallel sequencing. Clin. Chem. 58 (9), 1322–1331. doi:10.1373/clinchem.2011.181438
Zou, D., Wang, L., Liao, J., Xiao, H., Duan, J., Zhang, T., et al. (2021). Genome sequencing of 320 Chinese children with epilepsy: a clinical and molecular study. Brain 144 (12), 3623–3634. doi:10.1093/brain/awab233
Keywords: epilepsy, seizure, next-generation sequencing, whole exome sequencing, genetic diagnosis
Citation: Zou H, Zhang Q, Liao J, Zou D, Hu Z, Li B, Chen L, Wen J, Zhao X, Zhang VW and Cao D (2025) Diagnostic efficiency of exome-based sequencing in pediatric patients with epilepsy. Front. Genet. 15:1496411. doi: 10.3389/fgene.2024.1496411
Received: 24 September 2024; Accepted: 11 December 2024;
Published: 21 January 2025.
Edited by:
Ayse Demirkan, University of Surrey, United KingdomReviewed by:
Emrah Yucesan, Istanbul University-Cerrahpasa, TürkiyeJeffrey Dennis Calhoun, Northwestern University, United States
Copyright © 2025 Zou, Zhang, Liao, Zou, Hu, Li, Chen, Wen, Zhao, Zhang and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dezhi Cao, Y2FvZGV6aGk4ODhAYWxpeXVuLmNvbQ==