 Munusamy Ajithkumar
Munusamy Ajithkumar Jonathan D’Ambrosio
Jonathan D’Ambrosio Marie-Agnès Travers
Marie-Agnès Travers Romain Morvezen
Romain Morvezen Lionel Degremont
Lionel Degremont- 1Ifremer, Ressources Biologiques et Environnement (RBE)-ASIM, La Tremblade, France
- 2SYSAAF, Station LPGP/INRAE, Campus de Beaulieu, Rennes, France
- 3IHPE, CNRS, Ifremer, Université de Montpellier, University Perpignan Via Domitia, Montpellier, France
Introduction: The blue mussel is one of the major aquaculture species worldwide. In France, this species faces a significant threat from infectious disease outbreaks in both mussel farms and the natural environment over the past decade. Diseases caused by various pathogens, particularly Vibrio spp., have posed a significant challenge to the mussel industry. Genetic improvement of disease resistance can be an effective approach to overcoming this issue.
Methods: In this work, we tested genomic selection in the blue mussel (Mytilus edulis) to understand the genetic basis of resistance to one pathogenic strain of Vibrio splendidus (strain 14/053 2T1) and to predict the accuracy of selection using both pedigree and genomic information. Additionally, we performed a genome-wide association study (GWAS) to identify putative QTLs underlying disease resistance. We conducted an experimental infection involving 2,280 mussels sampled from 24 half-sib families containing each two full-sib families which were injected with V. splendidus. Dead and survivor mussels were all sampled, and among them, 348 dead and 348 surviving mussels were genotyped using a recently published multi-species medium-density 60K SNP array.
Results: From potentially 23.5K SNPs for M. edulis present on the array, we identified 3,406 high-quality SNPs, out of which 2,204 SNPs were successfully mapped onto the recently published reference genome. Heritability for resistance to V. splendidus was moderate ranging from 0.22 to 0.31 for a pedigree-based model and from 0.28 to 0.36 for a genomic-based model.
Discussion: GWAS revealed the polygenic architecture of the resistance trait in the blue mussel. The genomic selection models studied showed overall better performance than the pedigree-based model in terms of accuracy of breeding values prediction. This work provides insights into the genetic basis of resistance to V. splendidus and exemplifies the potential of genomic selection in family-based breeding programs in M. edulis.
1 Introduction
Aquaculture is one of the fastest-growing food production industries, with an annual production of 130.9 million tons and a value of approximately 312.8 billion USD, while also maintaining a lower carbon footprint compared to terrestrial animals (Norman et al., 2019; FAO, 2024). Mussels are considered one of the major bivalve species cultured worldwide, with France ranking as the second largest European producer, producing 71,311 tons in 2022 (FAO, 2024). Two species as well as their hybrids are cultivated in France: the blue mussel Mytilus edulis, and the Mediterranean mussel Mytilus galloprovincialis. Production is distributed along the English Channel to the southwest coastline of France, and Mediterranean shores (Prou and Goulletquer, 2002). Mytilus species has been widely cultured due to its strong environmental adaptability, high nutritious value, and consumer preference (Prou and Goulletquer, 2002; Suplicy, 2020). French mussel production entirely relies on wild spat collection, mainly in Pays de Loire and in Nouvelle-Aquitaine regions (Prou and Goulletquer, 2002). Consequently, the French cultivated mussels are not genetically selected through selective breeding programs.
Massive mortality outbreaks have been reported in various bivalves species including mussels worldwide, significantly impacting production, causing economic losses, and negatively affecting ecosystems, with the cause often remaining unidentified (Degremont et al., 2007; Bódis et al., 2014; Burdon et al., 2014; Soon and Ransangan, 2019; Capelle et al., 2021; Lupo et al., 2021; Ericson et al., 2023). Since 2014, abnormal mussel mortality (AMM) outbreaks have been reported in French mussel farms, with mortality rates ranging from 10% to 99% depending on sites, seasons or years with peak mortality observed in the springs of 2014, 2016, and 2018 (Polsenaere et al., 2017; Degremont et al., 2019; Normand et al., 2022). Previous studies have identified several factors linked to AMM outbreaks, including seawater characteristics, pollution, mussel characteristics, culture practices, pathogens and climate change (review by Lupo et al., 2021). Various strains of Vibrio splendidus isolated from moribund mussels at sites affected by mass mortality have been linked to the AMM outbreaks (Bechemin et al., 2015; Ben Cheikh et al., 2016). Vibrio splendidus is a complex species comprising multiple strains, ranging from highly virulent to relatively innocuous (Ben Cheikh et al., 2016). Some virulent strains have been shown to be highly pathogenic to blue mussels, causing high mortality rates up to 90% within a week in experimental challenges (Ben Cheikh et al., 2016; Oden et al., 2016; Ben Cheikh et al., 2017). The virulence of these strains can vary based on several factors including mussel characteristics, environmental conditions, and seasons (Charles et al., 2020). While V. splendidus is not the direct cause of AMM, its consistent association with mortality outbreaks suggests it may play a contributory role under specific conditions. Recent studies on bivalve immune responses often lack a validated understanding of immune effectors or pathways, reflecting their reliance on innate rather than adaptive immunity, limiting the efficacy of vaccination strategies (Allam and Raftos, 2015; Rey-Campos et al., 2019). Selective breeding could be a useful approach to enhance the innate immune responses in bivalves (Degremont et al., 2015; Hollenbeck and Johnston, 2018). Understanding genetic basis of disease resistance is critical for its improvement through selective breeding.
Over the past decades, many commercially important bivalve species have demonstrated significant genetic improvement through mass selection, due to their high fecundity and short generation intervals (Gjedrem and Rye, 2018; Tan et al., 2020). Mass selection has been carried for growth traits in Chilean blue mussel Mytilus chilensis (Toro et al., 2004a; Toro et al., 2004b), and ploidy status for Mediterranean mussel M. galloprovincialis (Ajithkumar et al., 2025). Lately, a mass selection scheme implemented for resistance to AMM outbreaks in the blue mussel M. edulis resulted in a 34%–48% increase in survival after one generation of selection (Degremont et al., 2019). Although mass selection is effective, it may quickly lead to inbreeding if genetic diversity is not properly monitored (Hu et al., 2022). However, family based selective breeding programs have been initiated as an alternative strategy to mass selection, to estimate breeding values by combining phenotypic information and pedigree. These programs have targeted various traits across different mussel species, such as growth in M. edulis (Mallet et al., 1986), M. galloprovincialis (Nguyen et al., 2014; Pino-Querido et al., 2015; Díaz-Puente et al., 2020), M. chilensis (Alcapán et al., 2007; Guiñez et al., 2017), Hyriopsis cumingii (Jin et al., 2012; Bai et al., 2017), Perna calaniculus (Camara and Symonds, 2014); shell nacre color in H. cumingii (Bai et al., 2017); toxin accumulation and mantle color in M. galloprovincialis (Pino-Querido et al., 2015); and survival in M. edulis (Mallet et al., 1986).
Accurate estimations of breeding values are crucial for developing a successful breeding program and predicting the responses of traits of interest to selection. Advances in high-throughput genotyping technology have now made it possible to implement genomic selection effectively in aquaculture species (Boudry et al., 2021). Genomic selection is especially beneficial for traits that are costly or difficult to measure, such as flesh quality or disease resistance, and it can achieve similar or higher accuracies in estimated breeding values (EBVs) with less phenotypic data compared to traditional family-based selection (Regan et al., 2021; Yáñez et al., 2023). Furthermore, genomic selection enhances genetic gain by capturing genetic variation both within and between families (Boudry et al., 2021). Next-generation sequencing and genotyping-by-sequencing tools have been developed to detect genome-wide molecular markers for oyster, clam, abalone and scallop (Jiao et al., 2014; Ren et al., 2016; Wang et al., 2016; Nie et al., 2017; McCarty et al., 2022; Yang et al., 2022). However, these methods may not provide reliable markers for testing across different populations (e.g., between the training and breeding populations) because they sequence a large number of randomly distributed regions, and their effectiveness can vary with DNA quality, limiting their ability to produce consistent genomic analyses (Davey et al., 2011; Boudry et al., 2021). Alternatively, SNP arrays have been developed for several commercially important bivalve species, including silver-lipped pearl oyster (Pinctada maxima), with an Illumina ∼3k iSelect custom array (Jones et al., 2013a), the medium density bi-species (Pacific oyster Crassostrea gigas and European flat oyster Ostrea edulis) 57K SNP array (Gutierrez et al., 2017), the Pacific oyster (C. gigas), with a 190K SNP array (Qi et al., 2017), the Eastern Oyster (C. virginica), with a high density 566K and 66K SNP array (Guo et al., 2023), or medium density multi-species (M. edulis, M. galloprovincialis, M. trossulus, and M. chilensis) 60K SNP array (Nascimento-Schulze et al., 2023), all of which rely on commercial chip products with fixed sets of markers and specific genotyping platforms. SNP arrays have been applied to a range of aquaculture species for various purposes, such as uncovering the genetic architecture of traits, implementing genomic selection, characterizing genetic resources, pedigree monitoring, sex-determination, and inbreeding management. However, their use in bivalves has been relatively limited (Gutierrez et al., 2020; Jourdan et al., 2023).
In aquaculture, the salmon industry has been leading the way in genomic selection for several years (Odegård et al., 2014; Tsai et al., 2015; Correa et al., 2017; Robledo et al., 2018; Ajasa et al., 2024). To date, more frequently aquaculture species are following this trend such as rainbow trout Oncorhynchus mykiss, European sea bass Dicentrarchus labrax, Gilthead Sea Bream Sparus aurata, Nile tilapia Oreochromis niloticus, Channel catfish Ictalurus punctatus or whiteleg shrimp Litopenaeus vannamei [see for review (Houston et al., 2020; Boudry et al., 2021; Song et al., 2022; Yáñez et al., 2023)]. The recent advancements in genotyping tools have paved the way for exploring the potential of genomic selection in bivalves. Genomic selection has been investigated in the Portuguese oyster for morphometric traits, edibility traits and disease traits (Vu et al., 2021), in the American oyster for low salinity tolerance (McCarty et al., 2022),in the silver-lipped oyster for pearl quality traits (Zenger et al., 2019) and growth traits has been studied in the triangle sail mussel (Wang et al., 2022), European flat oyster (Penaloza et al., 2022) and Pacific oyster (Gutierrez et al., 2018; Jourdan et al., 2023). A few studies have been conducted in oysters showing the increase in accuracy of genomic selection over pedigree-based approaches for difficult to measure traits, such as disease resistance, meat content and color traits (Gutierrez et al., 2018; Gutierrez et al., 2020; Jourdan et al., 2023). To date, trials with low-density panels to reduce genomic evaluation costs have been conducted in several aquaculture species, indicating that developing cost-effective strategies for genomic selection will be pivotal in shaping modern aquaculture breeding programs (Kriaridou et al., 2020; Penaloza et al., 2022).
The primary aim of our study was to evaluate the potential of genomic selection for resistance to one pathogenic strain of V. splendidus in M. edulis. Using a multi-species Axiom Affymetrix 60K SNP array (Nascimento-Schulze et al., 2023), we first characterized the genetic structure and linkage disequilibrium of the blue mussel population. We then estimated genetic parameters for resistance to V. splendidus and performed GWAS to investigate its genetic architecture. Finally, we compared genomic and pedigree-based selection accuracies to optimize breeding programs.
2 Materials and methods
2.1 Family production
The 48 families of M. edulis used in this study are detailly described in (Ajithkumar et al., 2024b). Briefly, three wild mussel populations were sampled, two from the Atlantic coast (OLE-PON and YEU_001), and a third from the North of France (WIM), and transferred to the Ifremer hatchery in La Tremblade in the fall of 2016. Each mussel population was cleaned and placed in separate tanks containing unheated UV-treated, and filtered seawater (400 L per hour). To favor gametogenesis, mussels were fed a cultured phytoplankton diet (Isochrysis galbana, Tetraselmis suecica, and Skeletonema costatum). Two sets of crosses were performed in January 2017 (set 1) and in February 2017 (set 2). For each population, 100 mussels were individually placed in 400 mL beakers, and spawning was triggered by alternating cold (10°C) and warm seawater (20°C). Depending on the ripeness of the mussels and the sex ratio, four males for OLE-PON, and 11 males for YEU_001 were used in set 1, while 9 males were used for WIM in set 2. Within population, each male was mated with two females, producing in total 24 half-sib families, each containing two full-sib families. These 48 full-sib families consist of 8, 22 and 18 families from OLE-PON, YEU_001 and WIM population, respectively, all should be considered as outbred. Each full-sib family was grown separately in 30 L tanks filled with filtered and UV-treated seawater at 20°C until the pediveliger stage. Then, downwelling system were used until mussels reached 1 cm. At that size, they were transferred to our nursery in Bouin in April and May 2017 for set 1 and set 2, respectively. For each family, 1,000 spat were maintained in 15 L SEAPA© baskets, and all families were raised in a 20 m³ concrete raceway until the start of the experiment, which occurred in July 2018. More detailed on the larval and nursery culture are provided in Ajithkumar et al. (2024b).
2.2 Experimental infection and phenotyping
Detailed step-by-step protocol of the experimental infection is given in Ajithkumar et al. (2024b). Briefly, two experimental infections (EI_1 and EI_2) were conducted in July 2018. Among the 48 families (mean individual total weight of approximately 5 g), 24 families were randomly sampled and tested in El_1, and the others were tested in El_2. Additionally, a third experimental infection (EI_3) was performed, involving the same 24 families from El_1, to increase the genotype sample size due to some samples from El_1 were lost. Finally, three families tested in El_2 were also tested in El_1, leading to a total of 27 families tested in El_1, and one of those three was also tested in El_3 indicating that only one family was tested in each of the three experimental infections (see Supplementary Table S1 for details). For each experimental infection and family, a highly pathogenic strain of V. splendidus (strain 14/053 2T1) isolated during AMM outbreak in 2014 was injected in 30 mussels to investigate their resistance to this pathogen. This particular strain was selected for its high virulence, the highest among the 50 strains tested, and it belongs to a specific pathogenic group. The lethal dose-50 (LD50) was determined prior to the experimental infection by injecting varying concentrations of Vibrio splendidus strain into a large group of mussels. The selected dose was close to the LD50, ensuring adequate variance in the response for effective assessment of resistance. First, mussels were anesthetized using MgCl2 (50 g per L), and 50 μL of bacterial solution (109 bacteria/mL) was injected into the muscle. Then, ten injected mussels per family, for all the 24 families of one set were hold in one 120 L tank containing UV-filtered seawater. Three replicate tanks were used and, in each tank, water recirculation was maintained using a TECO®pump (Ravenna, Italy), which also maintained the seawater temperature at 17°C. Dead mussels were counted and sampled daily up to 72 h post-injection. The adductor muscle/gills of the dead mussels during the experiment and the surviving mussels at the end of the experiment were collected using scalpels disinfected with 70% ethanol and stored in 1.5 mL sterile tubes at room temperature.
2.3 Genotyping and quality control
Among the dead and alive animal sampled, around 14–15 mussels were genotyped per family (ranging from 13 to 16) (Table 1; Supplementary Table S1). The number of dead and alive per family was calculated in proportion to the mortality observed within each family in El_1 and in El_2. As the number of animals sampled in El_1 was not sufficient, 158 dead mussels and 35 live mussels were sampled from EI_3 (Supplementary Table S1). In total, 768 individuals were genotyped; 348 dead, 348 alive, and the remaining 72 were their parents (48 dams and 24 sires). DNA extraction and genotyping were performed by the Gentyane INRAE Platform (Clermont-Ferrand, France) using the multi species medium-density 60K SNP-array, Axiom_Myt_v1_r1 (Thermo Fisher Scientific, Waltham, Massachusetts, United States), which comprises 23,252 markers for M. edulis (Nascimento-Schulze et al., 2023). Quality controls on the 60K SNPs from the SNP array and genotyped individuals were performed as described in D'Ambrosio et al. (2019). Firstly, genotypes of all individuals were analyzed using the Axiom Analysis Suite software (AxAS; v.4.0.3.3) with the default best practice workflow suggested by the manufacturer, with few threshold modifications, which includes individual quality control and SNP quality control analysis (Dish Quality Control ≥0.20; Quality Control call rate ≥90; percent of passing samples ≥98; average call rate for passing samples ≥92%; call rate cutoff ≥95; Fisher’s Linear Discriminant ≥2.6). Consequently, 7,476 polymorphic SNPs were retained for further analysis. Subsequently, final quality control was performed using PLINK v1.9 software (Chang et al., 2015). Two individuals with an identity-by-descent value over 0.90 were considered as duplicated and both individuals were removed from the analysis. Only SNPs with a minor allele frequency (MAF) higher than 0.01 and those passing the Hardy-Weinberg equilibrium test (p-value <0.0000001) in the genotyped mussels were retained. After the quality control, data comprised of a total of 766 genotyped individuals for 3,406 SNPs.
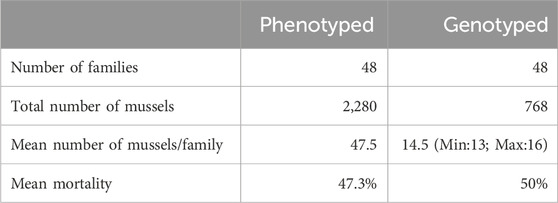
Table 1. Summary of the experimental infection using the pathogenic strain 14/053 2T1 of Vibrio splendidus in Mytilus edulis.
2.4 Parentage assignment
Parentage assignment was performed in the R package APIS (Griot et al., 2020) with a mismatch number set to 5%. The best 1,471 SNPs, selected with call rate greater than 90% and MAF value greater than 0.1 were used. Parentage assignment allowed the reconstruction of the pedigree of 647 offspring with assignment rates reaching 93.2% of the mussels having both parents assigned, while the remaining 47 mussels potentially from outside the cross-mating design, which were excluded from following analyses.
2.5 Genetic structure of the population
To evaluate potential genetic sub-structuring of populations and any associated biases, a principal component analysis (PCA) was performed using PLINK 1.9 (Chang et al., 2015) and the genetic structure was visualized using in RStudio (Team, 2024). Three individuals were identified as outliers beyond the population structure and were subsequently excluded from further analysis. Genetic differentiation between populations was measured through pairwise fixation index (FST) estimates using PLINK 1.9 (Chang et al., 2015).
2.6 SNP mapping, genome coverage and linkage disequilibrium estimation
All markers of the array along with their flanking regions were blasted using a BLASTn® procedure on the reference genome (Mytilus edulis genome assembly, xbMytEdul2, GenBank accession number: GCA_963676595.2). To map SNPs, considering the high polymorphism in the mussel genome, four mismatches were allowed over a length of around 71 base pairs. Only SNPs mapping to a unique position on the reference genome were retained for the subsequent stage of quality control as mentioned in previous section. Out of the 3,406 SNPs, only 2,204 matched our mapping criteria and were successfully positioned on the reference genome (Supplementary Table S2).
The pairwise linkage disequilibrium (LD) analysis was performed between all SNPs and adjacent markers for each linkage group and population to determine LD decay within the genome of M. edulis using Plink 1.9 (Chang et al., 2015).
2.7 Estimation of genetic parameters
2.7.1 PBLUP
Estimated breeding values, variance components, and heritability were calculated using the BLUPF90 software package (Misztal et al., 2014) through two different approaches: a linear mixed model with AIREMLF90 (Misztal et al., 2014) for assessing the trait on the observed scale, and a Gibbs analyses with THRGIBBS1F90 (Tsuruta and Misztal, 2006) for evaluating it on the underlying scale, based on pedigree-based relationship.
where
The EBV were estimated using BLUPF90 package and the variance components using AIREMLF90 and THRGIBBS1F90 programs. With the threshold model, the variance components were estimated using a Gibbs sampler with 100,000 iterations, 10,000 of burn-in and one sample was kept every 10 iterations for posterior analysis. Variance components were estimated using the average information restricted maximum likelihood algorithm (Gilmour et al., 1995). The h2 for linear model on observed scale transferred into underlying scale using the formulae from Dempster and Lerner (1950).
Heritability (
2.7.2 GBLUP
Estimated heritability was calculated using animal linear model with AIREMLF90 and animal threshold model with Gibbs sampling using THRGIBBS1F90. The GBLUP model uses the same approach as the PBLUP model, but
where
The population-specific analysis was performed using SNP markers to estimate heritability within population, and genetic correlation between populations. A high genetic correlation (0.99) was observed between populations (Supplementary Table S3).
2.7.3 ssGBLUP
Estimated heritability was calculated using animal linear model with AIREMLF90 and animal threshold model with Gibbs sampling using THRGIBBS1F90. The single-step GBLUP (ssGBLUP) model enhances the PBLUP and GBLUP model by fitting the H matrix, which integrates both genomic and pedigree data (Aguilar et al., 2010). The inverse of the H matrix was constructed as follows:
where G is as described above and A22 is the pedigree-based relationship matrix for genotyped animals.
2.8 Genome-wide association study
To identify SNPs associated with resistance to V. splendidus, a genome wide association study (GWAS) was performed using a mixed linear model association through ssGBLUP analysis. The postGSF90 module (Misztal et al., 2014) from the BLUPF90 package was used to estimate the effects of the SNPs (
where d is the vector of weights associated with the SNP effects and Z is the incidence matrix relating SNP effects to genomic breeding values.
Estimates of SNP effects (
with
2.9 Prediction accuracy
Prediction accuracy for the PBLUP, GBLUP, and ssGBLUP models was assessed using the ‘leave-one-out’ method, implemented with a linear model in BLUPF90 (Resende et al., 2012; Kristensen et al., 2018). In this approach, each observation is systematically excluded one at a time. The model is then trained on the remaining data, and the (G)EBV for the excluded individual is predicted by masking its phenotype.
The accuracy (r) of prediction was computed as the correlation between the (G) EBVs and the corrected phenotype (
The heritability value
2.9.1 Evaluation of the effect of SNP density on genomic predictions (GP)
SNP panels of varying densities were assessed by selecting subsets from the full QC-filtered SNP panel for each dataset. Panels of the following densities were tested: 500 SNPs, 1,000 SNPs, 1,500 SNPs, annotated SNPs (2,204), and all high-quality SNPs (3,406). SNPs for each panel were randomly sampled within each chromosome, with the number of SNPs chosen from each chromosome being proportional to the total number of high-quality SNPs per chromosome. To mitigate biases, we randomly generated five different SNP panels for each SNP density.
3 Results
3.1 Vibrio challenge
The cumulative mortality rate 72 h post-injection was 47%. At endpoint, mortality rates were 63% for EI_1, 41% for EI_2, and 37% for EI_3. Among mussel populations, the WIM population (54%) showed higher susceptibility to V. splendidus compared to the YEU_001 (45%) and OLE-PON (37%) populations (Supplementary Table S4). Mortality rates varied significantly among families upon exposure to V. splendidus, ranging from 17% to 83%. The mean mortality rates for all families are depicted in Figure 1.
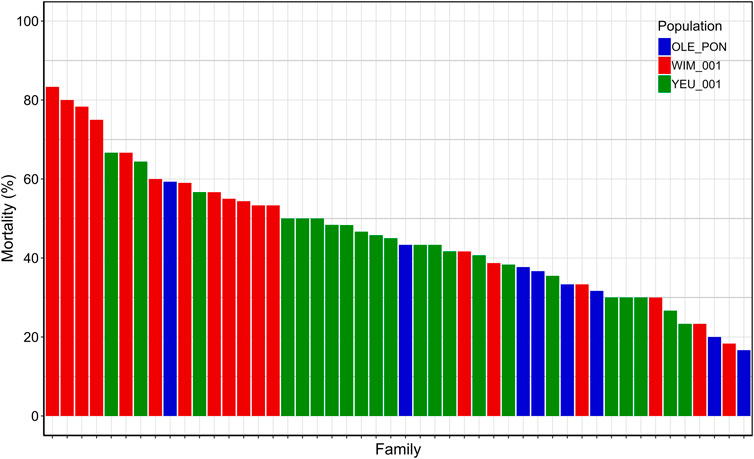
Figure 1. Final cumulative mortality 72 h post-injection for each family. Each bar represents a family and each color represent a population.
3.2 Population structure
Figure 2 illustrates the results of the principal component analysis (PCA), revealing the population structure of the mussel population. The first two PCA axes collectively account for over 15% of the total genetic variation. The YEU_001 and OLE-PON populations were generally homogeneous, although there were few stratifications within YEU_001. The WIM population have quite different genetic background from others, with two families whose offspring showed even greater isolation. Nevertheless, FST analysis revealed low genetic differentiation between populations. The mean genetic distances between populations are shown in Table 2, with FST values ranging from 0.02 to 0.03, suggesting genetic similarity across all three populations (Figure 3).
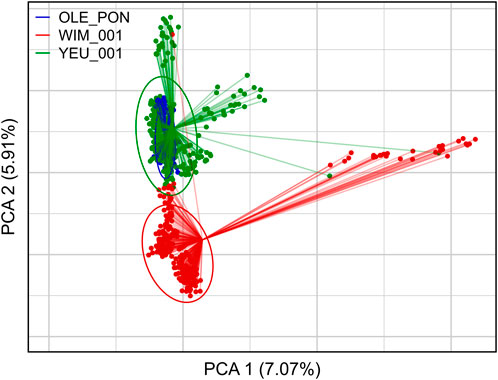
Figure 2. First two axes and associated variances of the principal component analysis (PCA) of the genetic diversity among the three populations of Mytilus edulis. The ellipses are constructed with axes defined as 1.5 times the standard deviation of the projections of individual coordinates on the axes. PCA was performed with 647 individuals and 3,406 SNPs.
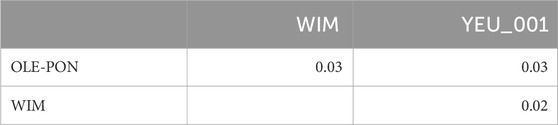
Table 2. Pairwise FST between populations of Mytilus edulis.
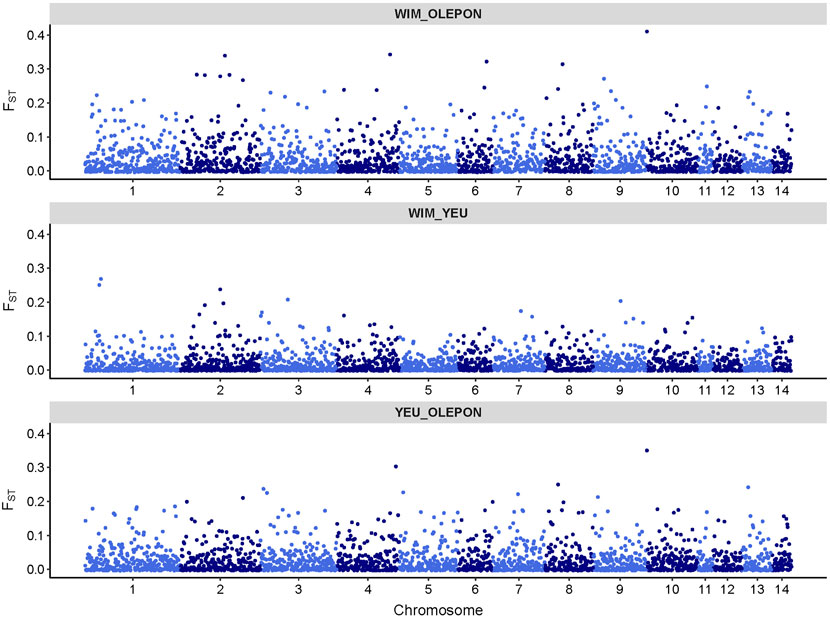
Figure 3. Genomic distribution of fixation index (FST) values as a function of chromosome position in the mussel genome for different studied population.
3.3 SNP mapping and genome coverage
2,204 SNPs were positioned on the reference genome, resulting a loss of 1,202 SNPs. The positions of markers on the chromosomes are illustrated in Supplementary Figure S1. The average SNP density per megabase (Mb) ranges from 0.57 to 2.37, varying among chromosomes and within chromosome (Supplementary Table S5). Approximately, only 9% of all 1 Mb segments contain more than 5 SNPs. SNP density exhibits non-uniformity throughout the genome, with each chromosome demonstrating varying densities. The lower marker density results in greater mean average distances between adjacent SNPs, ranged from 421 kb to 1739 kb depending on the chromosome.
3.4 Linkage disequilibrium analysis
Figure 4 illustrates that linkage disequilibrium (LD) decreases sharply as the distance between pairs of SNPs increases, with the most rapid decline occurring within the first 100 kb. Beyond this range, LD continues to decline and becomes more variable. The OLE-PON population consistently shows higher LD throughout the genome compared to other populations. On average, the LD values (r2) for SNPs less than 15 kb apart are 0.12 for OLE-PON, 0.10 for WIM, and 0.06 for YEU. Linkage disequilibrium values are generally low between adjacent SNPs for all the chromosomes, where distances between adjacent SNPs are larger.
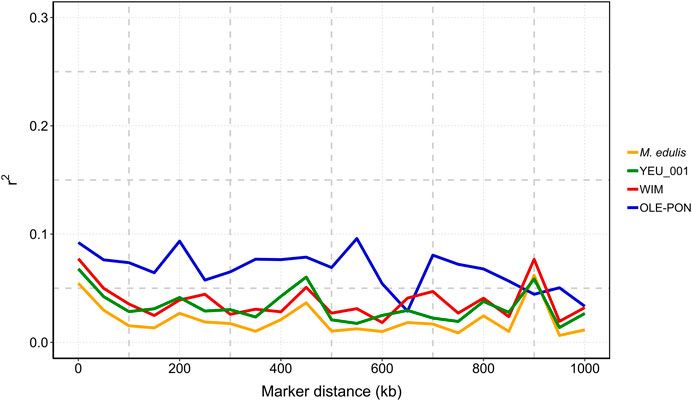
Figure 4. Linkage disequilibrium (r2) decay with physical distance between markers in each population and overall challenged to Vibrio splendidus. The X-axis is the physical location, and the Y-axis is the linkage disequilibrium value (r2).
3.5 Heritability
The estimates of heritability using the linear and Gibbs sampling models are summarized in Table 3. Pedigree-based heritability estimates for resistance to V. splendidus in M. edulis ranged from 0.22 to 0.31. Genomic heritability was slightly higher, varying between 0.33 and 0.36. The ssGBLUP based estimated heritability ranging from 0.28 to 0.33, which combines genomic and pedigree information.
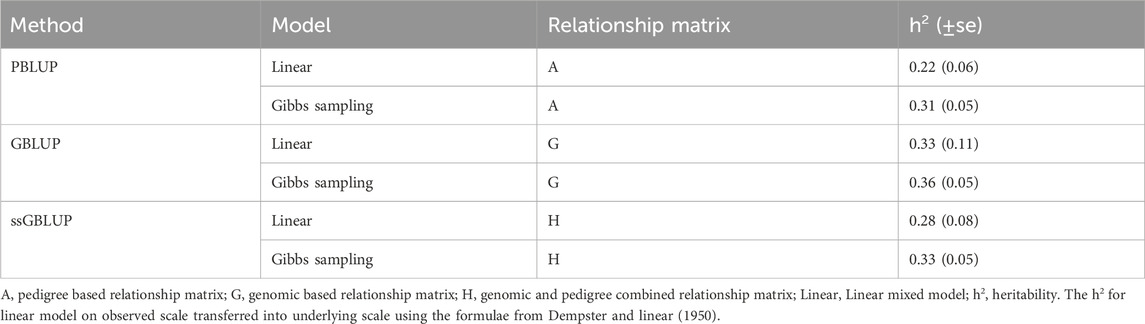
Table 3. Heritability for resistance to Vibrio splendidus in Mytilus edulis.
3.6 Genetic architecture
GWAS for resistance to V. splendidus suggest that this trait is likely to be impacted by multiple genomic regions. However, there were 20 SNPs that explained >0.5% of additive genetic variance on chr 1, chr 2, chr 3, chr 4, chr 5, chr 6, chr 8, chr 9, chr 13, and chr 14 (Figure 5). However, none of these markers explained more than 1.06% of the additive genetic variance (Figure 5; Table 4).
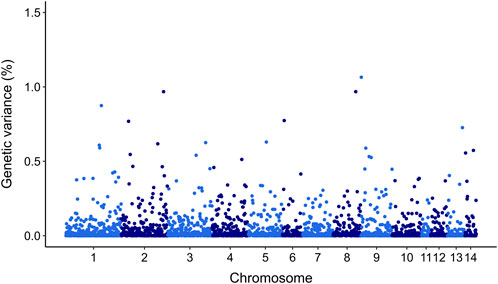
Figure 5. Manhattan plot of genetic variance explained by each SNP for resistance to Vibrio splendidus in Mytilus edulis using ssGBLUP approach. In X-axis SNP per chromosome and Y-axis percentage of genetic variance explained per each SNP.
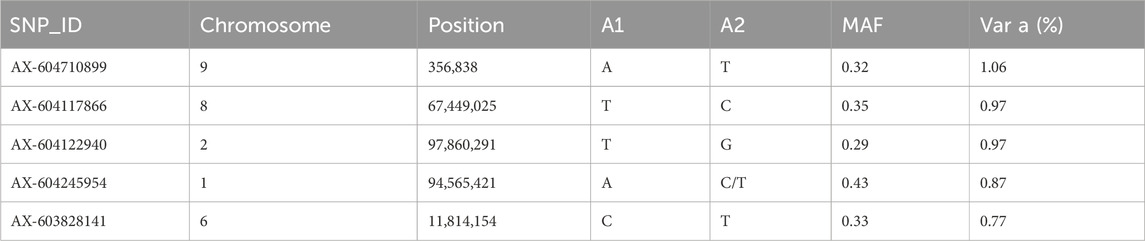
Table 4. Summary of top five SNPs associated with resistance to Vibrio splendidus in GWAS analysis (ssGBLUP) ranked with respect to genetic variance. Position = Physical position of SNP on the chromosome; A1 and A2 = Minor and major alleles, respectively; MAF = Minor allele frequency; var A = percentage of additive genetic variance explained by SNP.
3.7 Prediction accuracy
Accuracy with all data are 0.36, 0.43, 0.43 for PBLUP, GBLUP and ssGBLUP, respectively. Genomic selection (GBLUP and ssGBLUP) is better than PBLUP by 19%. Overall, prediction accuracy for genomic selection increased with the density of markers (Figure 6). Incorporating genomic information generally enhanced accuracy compared to pedigree-based estimation, except with 500 SNPs where PBLUP exhibited higher accuracy than GBLUP (Figure 6). With maximum training population and SNP subsets, genomic evaluation improved accuracy by 17%, 19%, 25%, and 19% for 1,000, 1,500, annotated (2,204), and all SNPs (3,406), respectively, compared to PBLUP. When comparing GBLUP and ssGBLUP models, the prediction accuracy was consistently favored the ssGBLUP model, except when using annotated SNPs in the GBLUP model (Figure 6).
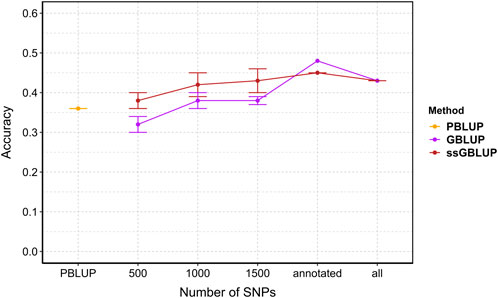
Figure 6. The estimated prediction accuracy of Vibrio splendidus resistance in Mytilus edulis using PBLUP, GBLUP and ssGBLUP across different marker densities. Each point is the average of 5 replicates. Error bars represent the standard error of the mean of 5 replicates. PBLUP - Pedigree based breeding values using all phenotyped animals, respectively. GBLUP - Genomic breeding values from only genotype animals, and ssGBLUP - Genomic breeding values from all genotyped and phenotyped animals obtained with a combined relationship matrix (H). Annotated: SNPs mapped onto the recently published reference genome (2,204); all: retained SNPs after quality control (3,406).
4 Discussion
In our study, we aimed to demonstrate the feasibility of genomic selection in a mussel breeding program in France. We used a recently developed multi species medium-density 60K SNP-array (Nascimento-Schulze et al., 2023) to perform genomic analysis. Combining multiple populations is a common approach for multi-population GP and combined GWAS analysis (VanRaden et al., 2009; Lund et al., 2011; Begum et al., 2012). However, there are three criteria for using this approach: first, the trait of interest is measured in the same way in different populations and genotyping is done using same SNP array; second, there is no G × E interaction in different populations; and finally, the genetic distance between the populations is minimal (Lund et al., 2011; Begum et al., 2012; Gebreyesus et al., 2019). As our study meets these requirements, combined population analyses were performed.
4.1 Genotyping quality and genome covering by selected SNPs
To the best of our knowledge, our study is the first to use the multi species medium-density 60K SNP-array (Nascimento-Schulze et al., 2023) to estimate genetic and genomic parameters in blue mussel (M. edulis). After all quality controls, we identified high-quality SNPs from 23,252 SNPs present on the array across 768 individuals. The necessity for stringent filtering of genotyping data is highlighted by the prevalence of poor-quality markers. One of the distinguishing features of the Mytilus species complex is its high degree of heterozygosity and abundance of mobile elements, which pose challenges in accurately identifying polymorphic SNPs (Gerdol et al., 2020; Sun et al., 2017). Compounding this difficulty, the current SNP array was developed using a whole-genome low coverage approach, which can lead to loss of polymorphic sites, especially across genetically diverse populations. A prior study validating this SNP array found 89.8% of the 23,252 SNPs to be polymorphic due to the close genetic match between the populations used for development and validation (Nascimento-Schulze et al., 2023). In contrast, after quality control in our study, only 14.6% of the SNPs were retained as polymorphic, likely due to genetic differences between our sampled population and the one used for array creation. This discrepancy highlights the influence of population structure on SNP retention and substantial genetic complexity and high polymorphism exhibited in mussel genome (Gerdol et al., 2020; Smietanka et al., 2014). This complexity stems from its evolutionary history of extensive gene flow among related species and a genome characterized by significant dispensable content. Indeed, about 30% of genes in Mytilus are dispensable, likely impacting SNP distribution and usability across populations (Gerdol et al., 2020). Out of 23,252 SNPs identified in M. edulis, without any other quality control, only 16,213 (70%) were annotated on the recently published reference genome of M. edulis. Assembly errors in the reference genome may rise from several factors, such as exceptionally high genetic polymorphism levels, non-Mendelian segregation of marker loci in paired crosses, and a significant occurrence of null alleles in genetic markers (Hedgecock et al., 2015). If we consider the other quality controls done, only 2,204 SNPs (64.8% of polymorphic, good quality SNPs, 9.4% of the total available SNPs) mapping successfully. This limited SNP coverage and sparse distribution across the linkage map could potentially lead to the omission of QTLs in specific regions, particularly in regions where markers are underrepresented. To improve SNP array quality and increase the number of informative SNPs, future work could include strategies such as pooled sequencing with a greater number of individuals, incorporating genetically diverse populations, or performing high-coverage whole-genome resequencing numbers of individuals from each population (Nascimento-Schulze et al., 2023). These adjustments would increase the SNP array’s robustness, thereby improving genome coverage, QTL detection power, and marker-trait association accuracy in M. edulis.
The bi-species Axiom Affymetrix 57K SNP array has been used in Pacific oysters, where applying the AxAS software’s best practice workflow led to a notable reduction in the number of informative SNPs. Specifically, Gutierrez et al. (2018) reported 23,000 informative SNPs from 820 individuals, Vendrami et al. (2019) identified 21,499 SNPs from 232 individuals, and Jourdan et al. (2023) obtained 14,500 SNPs from 2,420 individuals. This reduction is largely attributed to the complex genetic structure of molluscs, stemming from the highly polymorphic nature of their genomes (Gerdol et al., 2020; Jiao et al., 2021; Song et al., 2021), and is further influenced by the genetic relationship between the training population used for array design and the breeding candidates in selective breeding program (Houston et al., 2020). However, recent studies on bivalves have demonstrated that a moderate number of high-quality markers (1,000–3,000) could suffice for accurate predictions (Gutierrez et al., 2018; Kriaridou et al., 2020; Penaloza et al., 2022).
4.2 Linkage disequilibrium
Linkage disequilibrium (LD) at the genome level plays a crucial role in the efficacy of breeding programs, influencing genetic variance and the accuracy of association analyses (Goddard and Hayes, 2009; Siol et al., 2017). In our study, values for r2 ranged between 0.07 and 0.09 for SNPs within a distance of 10 kb and from 0.03 to 0.08 within 50 kb in the populations studied, suggesting limited short-range LD at current SNP marker densities. LD levels decreased to less than 0.05 at 100 kb in two populations. The high LD in OLE-PON is probably an overestimate due to a smaller effective population size (Ne) than in the other populations. Overall, LD between adjacent markers within each population was predominantly less than 0.1 within 2 kb, indicating a rapid decline in LD within the blue mussel genome. Therefore, these LD estimates should be treated with caution. This swift decay suggests a historically large effective population size and high recombination rate, reflecting substantial genetic diversity within the population (Ellegren and Galtier, 2016). Moreover, LD values are population-specific, and influenced by evolutionary factors such as natural selection, mutation, genetic drift, origin and migration, as well as molecular forces including historical recombination events, and breeding history such as historical effective population sizes, intensity and direction of artificial selection, population admixture, and mating patterns (Du et al., 2007). Our findings confirm the low LD in M. edulis populations, consistent with previous studies on bivalves (Jones et al., 2013b; Vera et al., 2022; Jourdan et al., 2023). However, the sparse SNP distribution and generally low LD values (r2 < 0.1) suggest that increasing marker density is essential to improve prediction accuracy and QTL detection.
4.3 Population structure
FST is widely applied to evaluate genetic differentiation between/among populations (Hu et al., 2022). The low FST values (FST < 0.03) observed in our study suggest minimal genetic differentiation among mussel populations, indicating a lack of significant genetic structure. This phenomenon may be attributed to similar selection pressure and limited gene flow among the mussel populations, irrespective of geographic location. Similar to our findings, wild populations of M. edulis sampled from the Atlantic coast of France and from the Northern France to Germany had low Fst value (<0.003) using ancestry-informative SNPs (Simon et al., 2020). In addition, similar findings have been reported in other studies, such as pairwise FST (<0.02) among wild edible cockle using SNPs information (Vera et al., 2022) and among wild populations of Pacific oyster using allozymes and microsatellites markers (Appleyard and Ward, 2006). The PCA result showed that the WIM breeders formed a substructure with allele frequencies that differed from those of the other populations. This divergence may reduce the accuracy of EBV prediction in WIM populations and may be a cause of masking of the genetic architecture, as observed in a study of resistance to nodavirosis in sea bass (Griot et al., 2021). However, this clustering is inconsistent with the low mean FST values (<0.03), suggesting limited genetic differentiation and a degree of genetic relatedness among the populations. The two families whose offspring showed greater isolation from others in the WIM population may be due to the peculiar characteristics of the parents, which drive the first axis of the PCA.
4.4 Heritability
Our study presents the first report of heritability estimates for resistance to V. splendidus experimental infection in M. edulis based on genome-wide SNPs. We observed moderate heritability for V. splendidus resistance (0.22–0.36), which are higher compared to our previous study using the same population. This increase may be attributed to the inclusion of a third experimental infection in this study, despite the overall lower mortality rate (Ajithkumar et al., 2024b). Disease resistance to pathogens in bivalves seems to be a heritable trait, with moderate to high heritability in oysters, clams, and abalone, ranging from 0.21 to 0.63 (Degremont et al., 2015; Brokordt et al., 2017; Smits et al., 2020). Studies on oysters have shown varying levels of heritability (h2: 0.09–0.54) against different Vibrio spp. pathogens at different life stages (Azema et al., 2017; Nordio et al., 2021; Zhai et al., 2021; Dietrich et al., 2022). When comparing heritability estimates across different methods, both GBLUP and ssGBLUP consistently showed higher heritability estimates than pedigree-based methods. This difference is likely due to the genomic relationship matrix constructed based on genome-wide SNPs information can capture both within and between-family genetic variance, whereas traditional pedigree-based selection only captures genetic variance between families (Boudry et al., 2021). The higher heritability was observed with GBLUP, which may be due to the selective genotyping approach (348 dead and 348 alive individuals genotyped), potentially biasing heritability estimates and limiting the capture of family genetic variation. However, the ssGBLUP approach, which includes both genotyped and non-genotyped individuals, provides a more precise heritability estimate by capturing family variation from both genotyped and phenotyped individuals. To date, numerous studies across aquaculture species have similarly demonstrated that GBLUP methods provide higher estimated heritability and greater accuracy compared to PBLUP (Tsai et al., 2015; Gutierrez et al., 2018). These results underscore the presence of genetic variation for resistance to V. splendidus in our mussel populations, and highlight significant opportunities for enhancing disease resistance through selective breeding programs, whether using pedigree-based or genomic selection strategies.
4.5 Genome-wide association study
QTL detection in our populations posed challenges due to limited number of markers and individuals. Given the data in the current study do suggest a polygenic nature of resistance to V. splendidus, exploiting all markers to calculate genomic breeding values for resistance may be the most effective approach. Our association analyses suggest that resistance against V. splendidus exhibits a polygenic architecture without major QTLs. Similar findings have been reported for bacterial disease resistance in various aquaculture species including, Atlantic salmon Salmo salar (Correa et al., 2015), Coho salmon Oncorhynchus kisutch (Barría et al., 2018), Gilthead Sea Bream (Palaiokostas et al., 2016), European seabass (Oikonomou et al., 2022), and Pacific Oyster (Yang et al., 2022). For instance, a study on catfish identified four QTLs associated with columnaris resistance using a high-density SNP array (Geng et al., 2015), highlighting the importance of high-density SNP array for GWAS studies. Furthermore, multiple studies on rainbow trout concluded that a panel of approximately 5,000 evenly distributed SNPs across the genome was effective in identifying multiple QTLs for resistance to bacterial cold water disease due to the strong long-range LD in breeding populations (Vallejo et al., 2017; Vallejo et al., 2018; Vallejo et al., 2022). The SNP array used in this study had a limited number and quality of SNPs, and the mussel genome showed a very low level of LD. Our study used 2,204 SNPs, which may not provide sufficient coverage given the rapid LD decay and unevenly distributed SNPs across the genome, potentially leading to the omission of important QTLs. Future studies would benefit from incorporating high-quality SNP markers (>30K SNPs) and a larger number of phenotyped individuals (>1,000), which could significantly enhance the power and precision of QTL detection for V. splendidus resistance in blue mussels (Jones et al., 2013b; Barría et al., 2018). Additionally, developing an F2 generation by crossing parental lines with clearly contrasting phenotypic traits (resistant and susceptible) would provide an ideal basis for QTL mapping (Gu et al., 2011). In F2 populations, genotype segregation combined with divergent phenotypic differences provides strong conditions for QTL identification (Geng et al., 2015). Family-based mapping in these populations further strengthens QTL detection power by minimizing recombination between QTLs and linked markers, thereby improving the accuracy of marker-trait associations (Geng et al., 2015; Gonen et al., 2015). Moreover, family-based association mapping is particularly effective for detecting rare alleles associated with QTLs, which may be challenging to identify in other population structures (Geng et al., 2015).
4.6 Prediction accuracy
The accuracy of genomic selection is affected by several factors, including the relationship between training and validation animals, sample size in the reference population, marker density, effective population size, LD structure, underlying trait architecture and heritability of the trait (Yáñez et al., 2023). Therefore, the lower prediction accuracies observed here may reflect the marker density or underlying genetic architecture of the trait. The choice of genomic selection model for breeding programs requires a prior understanding of the genetic architecture of the selected trait(s). In the current study on M. edulis populations, the genetic contribution to the observed variation in resistance to V. splendidus was largely polygenic in nature. For the improvement of polygenic traits, GBLUP is the most reliable model and typically provides the highest prediction accuracy for highly polygenic traits, while the Bayesian models are preferable for traits controlled by few large effect loci in genomic selection (Legarra et al., 2015; Yáñez et al., 2023).
In our study, genomic selection improves accuracy of up to 19% compared to pedigree selection. A key consideration for the commercial implementation of genomic selection in shellfish aquaculture is the high cost of genotyping. Reference population size and marker density are two key factors for effectively reducing the cost of genomic selection (Song et al., 2022). One strategy to enhance the economic viability of genomic selection is to use a low-density SNP panel, which can reduce costs while achieving prediction accuracy similar to that of medium-density panels (Kriaridou et al., 2020). The prediction accuracies for genomic models in our study ranged from 0.32 to 0.48 for resistance to V. splendidus (with SNP densities ranging from 500 to ∼3,400), whereas the accuracy of PBLUP was 0.36. This result is slightly lower than the ranges reported for disease-related traits in other bivalve species. For instance, genomic selection prediction accuracies from GBLUP models for resistance to Ostreid herpesvirus (OsHV-1-lvar) ranged from 0.68 to 0.76 in the Pacific oyster (Gutierrez et al., 2020). Prediction accuracies for growth-related traits using the GBLUP model in other bivalves are relatively similar, e.g., 0.52–0.73 in the Pacific oyster (Gutierrez et al., 2018; Jourdan et al., 2023), 0.67–0.79 in the Portuguese oyster (Vu et al., 2021), 0.25–0.48 in the Zhikong scallop (Wang et al., 2016) and >0.83 in European flat oyster (Penaloza et al., 2022). Other reports on genomic prediction accuracies for disease-related traits in finfish aquaculture species show the prediction accuracies as low as 0.21, reviewed in Houston et al. (2020). However, this result highlight that genomic selection is a useful approach to increase resistance to V. splendidus in our blue mussel populations.
Overall, our results showed that genomic methods predict better accuracy (25%–33%) for resistance to V. splendidus using ∼2000 SNPs in a family-based design compared to pedigree-based estimation. This indicates that substantial improvements in the rate of genetic gain can be achieved through genomics-based selection techniques. It also increases the feasibility of using a low-density SNP panel for genomic selection in mussel breeding for Vibrio resistance, as low-density genotyping is significantly more cost-effective than high-density SNP arrays. Furthermore, studies on disease resistance in the Pacific oyster, growth traits in the European flat oyster, and heat tolerance in the Pacific abalone have shown that low-density SNP panels of around 1,000–2000 SNPs can achieve EBV accuracies similar to those obtained with medium-density arrays (Gutierrez et al., 2020; Kriaridou et al., 2020; Liu et al., 2022; Penaloza et al., 2022). Similar findings in multiple aquatic species have shown that low-density panels can achieve higher accuracies than the pedigree-based approach, making them a viable alternative for identifying candidates with the highest genetic merit for complex traits such as growth and disease resistance (Kriaridou et al., 2020).
Although the mussel genome is 1.4 Gb in size, our study suggests that a relatively low number of genetic markers can still achieve high prediction accuracy, with a rapid LD decay observed across all populations. In this study, we highlight the possibility of reducing the genotyping costs associated with genomic prediction approaches, caution should be exercised regarding the smallest marker density. Our study found that using only 500 SNPs in the GBLUP model resulted in an estimated decrease in the accuracy of genomic breeding values (GEBVs) for resistance to V. splendidus by 11% compared to PBLUP. It is important to note that when both pedigree information and genotypes are available, using ssGBLUP is preferable, as it demonstrates superior accuracy compared to PBLUP. Furthermore, our findings emphasize that annotated SNPs on the M. edulis genome provided more information about the studied population and led to higher prediction accuracy than using all SNPs in either the GBLUP or ssGBLUP model. This difference could be due to even distribution of phenotypes among genotyped individuals (50% mortality), or unannotated markers may introduce noise, thereby affecting the accuracy of GEBV estimation. Further investigations using more SNPs (>10K SNPs), and larger reference population (>1,000 individuals) hold potential for genomic selection to further increase the prediction accuracy for host resistance to V. splendidus in farmed mussel populations.
5 Conclusion
Our study estimated moderate heritability for resistance to V. splendidus in blue mussel populations using both pedigree and genomic data from a challenge experiment. GWAS analysis indicates that resistance to V. splendidus appears to be polygenic, suggesting that genomic selection is likely to be more effective than marker-assisted selection. We found that genomic selection can improve accuracy by up to 19% compared to pedigree-based selection. Additionally, our results highlight the potential for reducing the number of markers, which could make genomic selection more cost-effective. However, better genomic tools are essential, as the existing SNP arrays lack number of high-quality SNPs, leading to inadequate genome coverage and density. Increasing the number of genotyped individuals is crucial for boosting GWAS power and prediction accuracy. Overall, selective breeding emerges as a promising approach for improving resistance to V. splendidus in blue mussels, with genomic selection offering substantial potential for increasing genetic gains.
Data availability statement
The data presented in the study are deposited in the Sextant (Ifremer) repository as independent phenotype and genotype datasets accessible at https://doi.org/10.12770/e4707865-a8e3-4c9f-af99-e37bcd2f507d.
Author contributions
MA: Data curation, Formal Analysis, Software, Visualization, Writing–original draft, Writing–review and editing. JD: Software, Writing–review and editing. M-AT: Methodology, Writing–review and editing. RM: Software, Supervision, Writing–review and editing. LD: Conceptualization, Funding acquisition, Methodology, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by DPAM of the French Ministries of Ecology and Agriculture through the research program “MORBLEU” (Grant No. 18/2216164F) and the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 956697.
Acknowledgments
We would like to thank René Robert from LERPC-Ifremer and his colleagues for sampling mussel populations, Hatchery and nursery teams for their help to grow and protect the mussels inside the secured facilities, Delphine Tourbiez for her assistance to experimental infection. We express our sincere thanks to Vincent Pailler for helping to annotate the SNPs on the reference genome.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1487807/full#supplementary-material
References
Aguilar, I., Misztal, I., Johnson, D. L., Legarra, A., Tsuruta, S., and Lawlor, T. J. (2010). Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 93 (2), 743–752. doi:10.3168/jds.2009-2730
Ajasa, A. A., Boison, S. A., Gjoen, H. M., and Lillehammer, M. (2024). Accuracy of genomic prediction using multiple Atlantic salmon populations. Genet. Sel. Evol. 56 (1), 38. doi:10.1186/s12711-024-00907-5
Ajithkumar, M., Dégremont, L., Garcia, C., Ledu, C., and Benabdelmouna, A. (2025). Response to selection for cytogenetic status and their relationship with mortality in Mytilus edulis and Mytilus galloprovincialis in France. Aquaculture 597, 741912. doi:10.1016/j.aquaculture.2024.741912
Ajithkumar, M., Lillehammer, M., Travers, M.-A., Maurouard, E., Aslam, M. L., and Dégremont, L. (2024b). Genetic parameters for resistance to field mortality outbreaks and resistance to a pathogenic strain of Vibrio splendidus in Mytilus edulis, Mytilus galloprovincialis and natural hybrid. Aquaculture 590 (0044-8486), 741034. doi:10.1016/j.aquaculture.2024.741034
Alcapán, A. C., Nespolo, R. F., and Toro, J. E. (2007). Heritability of body size in the Chilean blue mussel (Mytilus chilensisHupé 1854): effects of environment and ageing: heritability of body size: effects of environment and aging. Aquac. Res. 38 (3), 313–320. doi:10.1111/j.1365-2109.2007.01678.x
Allam, B., and Raftos, D. (2015). Immune responses to infectious diseases in bivalves. J. Invertebr. Pathol. 131, 121–136. doi:10.1016/j.jip.2015.05.005
Appleyard, S. A., and Ward, R. D. (2006). Genetic diversity and effective population size in mass selection lines of Pacific oyster (Crassostrea gigas). Aquaculture 254 (1-4), 148–159. doi:10.1016/j.aquaculture.2005.10.017
Azema, P., Lamy, J. B., Boudry, P., Renault, T., Travers, M. A., and Degremont, L. (2017). Genetic parameters of resistance to Vibrio aestuarianus, and OsHV-1 infections in the Pacific oyster, Crassostrea gigas, at three different life stages. Genet. Sel. Evol. 49, 23. doi:10.1186/s12711-017-0297-2
Bai, Z. Y., Li, Q. Q., Han, X. K., and Li, J. L. (2017). Estimates of genetic parameters and genotype by environment interactions for shell nacre color and growth traits in the purple freshwater pearl mussel Hyriopsis cumingii. Aquacult Int. 25 (6), 2079–2090. doi:10.1007/s10499-017-0170-x
Barría, A., Christensen, K. A., Yoshida, G. M., Correa, K., Jedlicki, A., Lhorente, J. P., et al. (2018). Genomic predictions and genome-wide association study of resistance against piscirickettsia salmonis in Coho salmon (Oncorhynchus kisutch) using ddRAD sequencing. G3-Genes Genom Genet. 8 (4), 1183–1194. doi:10.1534/g3.118.200053
Bechemin, C., Soletchnik, P., Polsenaere, P., Le Moine, O., Pernet, F., Protat, M., et al. (2015). Episodes de mortalité massive de moules bleues observés en 2014 dans les Pertuis charentais. Bull. Epidémiologie, Santé animale alimentaion (67), 6–9.
Begum, F., Ghosh, D., Tseng, G. C., and Feingold, E. (2012). Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 40 (9), 3777–3784. doi:10.1093/nar/gkr1255
Ben Cheikh, Y., Travers, M. A., and Le Foll, F. (2017). Infection dynamics of a V. splendidus strain pathogenic to Mytilus edulis: in vivo and in vitro interactions with hemocytes. Fish. Shellfish Immun. 70, 515–523. doi:10.1016/j.fsi.2017.09.047
Ben Cheikh, Y., Travers, M. A., Morga, B., Godfrin, Y., and Le Foll, F. (2016). First evidence for a Vibrio strain pathogenic to Mytilus edulis altering hemocyte immune capacities. Fish. Shellfish Immun. 53, 91. doi:10.1016/j.fsi.2016.03.140
Bódis, E., Tóth, B., and Sousa, R. (2014). Massive mortality of invasive bivalves as a potential resource subsidy for the adjacent terrestrial food web. Hydrobiologia 735 (1), 253–262. doi:10.1007/s10750-013-1445-5
Boudry, P., Allal, F., Aslam, M. L., Bargelloni, L., Bean, T. P., Brard-Fudulea, S., et al. (2021). Current status and potential of genomic selection to improve selective breeding in the main aquaculture species of International Council for the Exploration of the Sea (ICES) member countries. Aquac. Rep. 20, 100700. doi:10.1016/j.aqrep.2021.100700
Brokordt, K., González, R., Farías, W., Winkler, F. E., and Lohrmann, K. B. (2017). First insight into the heritable variation of the resistance to infection with the bacteria causing the withering syndrome disease in Haliotis rufescens abalone. J. Invertebr. Pathol. 150, 15–20. doi:10.1016/j.jip.2017.08.014
Burdon, D., Callaway, R., Elliott, M., Smith, T., and Wither, A. (2014). Mass mortalities in bivalve populations: a review of the edible cockle Cerastoderma edule (L.). Estuar. Coast Shelf S 150, 271–280. doi:10.1016/j.ecss.2014.04.011
Camara, M. D., and Symonds, J. E. (2014). Genetic improvement of New Zealand aquaculture species: programmes, progress and prospects. New Zeal J. Mar. Fresh 48 (3), 466–491. doi:10.1080/00288330.2014.932291
Capelle, J. J., Garcia, A. B., Kamermans, P., Engelsma, M. Y., and Jansen, H. M. (2021). Observations on recent mass mortality events of marine mussels in the Oosterschelde, The Netherlands. Aquacult Int. 29 (4), 1737–1751. doi:10.1007/s10499-021-00713-6
Chang, C. C., Chow, C. C., Tellier, L. C. A. M., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
Charles, M., Trancart, S., Oden, E., and Houssin, M. (2020). Experimental infection of Mytilus edulis by two Vibrio splendidus-related strains: determination of pathogenicity level of strains and influence of the origin and annual cycle of mussels on their sensitivity. J. Fish. Dis. 43 (1), 9–21. doi:10.1111/jfd.13094
Correa, K., Bangera, R., Figueroa, R., Lhorente, J. P., and Yáñez, J. M. (2017). The use of genomic information increases the accuracy of breeding value predictions for sea louse (Caligus rogercresseyi) resistance in Atlantic salmon (Salmo salar). Genet. Sel. Evol. 49, 15. doi:10.1186/s12711-017-0291-8
Correa, K., Lhorente, J. P., López, M. E., Bassini, L., Naswa, S., Deeb, N., et al. (2015). Genome-wide association analysis reveals loci associated with resistance against Piscirickettsia salmonis in two Atlantic salmon (Salmo salar L.) chromosomes. Bmc Genomics 16, 854. doi:10.1186/s12864-015-2038-7
D'Ambrosio, J., Phocas, F., Haffray, P., Bestin, A., Brard-Fudulea, S., Poncet, C., et al. (2019). Genome-wide estimates of genetic diversity, inbreeding and effective size of experimental and commercial rainbow trout lines undergoing selective breeding. Genet. Sel. Evol. 51, 26. doi:10.1186/s12711-019-0468-4
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M., and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12 (7), 499–510. doi:10.1038/nrg3012
Degremont, L., Ernande, B., Bedier, E., and Boudry, P. (2007). Summer mortality of hatchery-produced Pacific oyster spat (Crassostrea gigas). I. Estimation of genetic parameters for survival and growth. Aquaculture 262 (1), 41–53. doi:10.1016/j.aquaculture.2006.10.025
Degremont, L., Garcia, C., and Allen, S. K. (2015). Genetic improvement for disease resistance in oysters: a review. J. Invertebr. Pathol. 131, 226–241. doi:10.1016/j.jip.2015.05.010
Degremont, L., Maurouard, E., Rabiller, M., and Glize, P. (2019). Response to selection for increasing resistance to the spring mortality outbreaks in Mytilus edulis occurring in France since 2014. Aquaculture 511, 734269. doi:10.1016/j.aquaculture.2019.734269
Dempster, R.E., and Lerner, I.M. (1950). Heritability of threshold characters. Genetics 35 (2), 212-236. doi:10.1093/genetics/35.2.212
Díaz-Puente, B., Guiñez, R., Pita, A., Miñambres, M., and Presa, P. (2020). Genotype by environment interaction for shell length in Mytilus galloprovincialis. J. Exp. Mar. Biol. Ecol. 522, 151252. doi:10.1016/j.jembe.2019.151252
Dietrich, J. P., Hicks, M. B. R., Hard, J. J., Nichols, K. M., Langdon, C. J., Divilov, K., et al. (2022). Heritability estimates of disease resistance to Vibrio coralliiyticus in Pacific oyster (Crassostrea gigas) larvae from a selective broodstock program. Aquaculture 560, 738492. doi:10.1016/j.aquaculture.2022.738492
Du, F. X., Clutter, A. C., and Lohuis, M. M. (2007). Characterizing linkage disequilibrium in pig populations. Int. J. Biol. Sci. 3 (3), 166–178. doi:10.7150/ijbs.3.166
Ellegren, H., and Galtier, N. (2016). Determinants of genetic diversity. Nat. Rev. Genet. 17 (7), 422–433. doi:10.1038/nrg.2016.58
Ericson, J. A., Venter, L., Copedo, J. S., Nguyen, V. T., Alfaro, A. C., and Ragg, N. L. C. (2023). Chronic heat stress as a predisposing factor in summer mortality of mussels, Perna canaliculus. Aquaculture 564, 738986. doi:10.1016/j.aquaculture.2022.738986
FAO (2024). The state of world fisheries and aquaculture 2024 – blue transformation in action. Rome.
Gebreyesus, G., Buitenhuis, A. J., Poulsen, N. A., Visker, M. H. P. W., Zhang, Q., van Valenberg, H. J. F., et al. (2019). Combining multi-population datasets for joint genome-wide association and meta-analyses: the case of bovine milk fat composition traits. J. Dairy Sci. 102 (12), 11124–11141. doi:10.3168/jds.2019-16676
Geng, X., Sha, J., Liu, S. K., Bao, L. S., Zhang, J. R., Wang, R. J., et al. (2015). A genome-wide association study in catfish reveals the presence of functional hubs of related genes within QTLs for columnaris disease resistance. Bmc Genomics 16, 196. doi:10.1186/s12864-015-1409-4
Gerdol, M., Moreira, R., Cruz, F., Gomez-Garrido, J., Vlasova, A., Rosani, U., et al. (2020). Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 21 (1), 275. doi:10.1186/s13059-020-02180-3
Gilmour, A. R., Thompson, R., and Cullis, B. R. (1995). Average information REML: an efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51 (4), 1440–1450. doi:10.2307/2533274
Gjedrem, T., and Rye, M. (2018). Selection response in fish and shellfish: a review. Rev. Aquacult 10 (1), 168–179. doi:10.1111/raq.12154
Goddard, M. E., and Hayes, B. J. (2009). Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10 (6), 381–391. doi:10.1038/nrg2575
Gonen, S., Baranski, M., Thorland, I., Norris, A., Grove, H., Arnesen, P., et al. (2015). Mapping and validation of a major QTL affecting resistance to pancreas disease (salmonid alphavirus) in Atlantic salmon (Salmo salar). Heredity 115 (5), 405–414. doi:10.1038/hdy.2015.37
Griot, R., Allal, F., Brard-Fudulea, S., Morvezen, R., Haffray, P., Phocas, F., et al. (2020). APIS: an auto-adaptive parentage inference software that tolerates missing parents. Mol. Ecol. Resour. 20 (2), 579–590. doi:10.1111/1755-0998.13103
Griot, R., Allal, F., Phocas, F., Brard-Fudulea, S., Morvezen, R., Bestin, A., et al. (2021). Genome-wide association studies for resistance to viral nervous necrosis in three populations of European sea bass (Dicentrarchus labrax) using a novel 57k SNP array DlabChip. Aquaculture 530, 735930. doi:10.1016/j.aquaculture.2020.735930
Gu, X., Feng, C., Ma, L., Song, C., Wang, Y., Da, Y., et al. (2011). Genome-wide association study of body weight in chicken F2 resource population. Plos One 6 (7), e21872. doi:10.1371/journal.pone.0021872
Guiñez, R., Toro, J. E., Krapivka, S., Alcapán, A. C., and Oyarzún, P. A. (2017). Heritabilities and genetic correlation of shell thickness and shell length growth in a mussel, Mytilus chilensis (Bivalvia:Mytilidae). Aquac. Res. 48 (4), 1450–1457. doi:10.1111/are.12981
Guo, X. M., Puritz, J. B., Wang, Z. W., Proestou, D., Allen, S., Small, J., et al. (2023). Development and evaluation of high-density SNP arrays for the eastern oyster Crassostrea virginica. Mar. Biotechnol. 25 (1), 174–191. doi:10.1007/s10126-022-10191-3
Gutierrez, A. P., Matika, O., Bean, T. P., and Houston, R. D. (2018). Genomic selection for growth traits in pacific oyster (Crassostrea gigas): potential of low-density marker panels for breeding value prediction. Front. Genet. 9, 391. doi:10.3389/fgene.2018.00391
Gutierrez, A. P., Symonds, J., King, N., Steiner, K., Bean, T. P., and Houston, R. D. (2020). Potential of genomic selection for improvement of resistance to ostreid herpesvirus in Pacific oyster (Crassostrea gigas). Anim. Genet. 51 (2), 249–257. doi:10.1111/age.12909
Gutierrez, A. P., Turner, F., Gharbi, K., Talbot, R., Lowe, N. R., Peñaloza, C., et al. (2017). Development of a medium density combined-species SNP array for pacific and European oysters (Crassostrea gigas and Ostrea edulis). G3-Genes Genom Genet. 7 (7), 2209–2218. doi:10.1534/g3.117.041780
Hedgecock, D., Shin, G., Gracey, A. Y., Van Den Berg, D., and Samanta, M. P. (2015). Second-Generation Linkage Maps for the Pacific Oyster Crassostrea gigas Reveal Errors in Assembly of Genome Scaffolds. G3-Genes Genomes Genetics 5, 2007–2019. doi:10.1534/g3.115.019570
Hollenbeck, C. M., and Johnston, I. A. (2018). Genomic tools and selective breeding in molluscs. Front. Genet. 9, 253. doi:10.3389/fgene.2018.00253
Houston, R. D., Bean, T. P., Macqueen, D. J., Gundappa, M. K., Jin, Y. H., Jenkins, T. L., et al. (2020). Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 21 (7), 389–409. doi:10.1038/s41576-020-0227-y
Hu, Y. M., Li, Q., Xu, C. X., Liu, S. K., Kong, L. F., and Yu, H. (2022). Genetic variability of mass-selected and wild populations of Iwagaki oyster (Crassostrea nippona) revealed by microsatellites and mitochondrial COI sequences. Aquaculture 561, 738737. doi:10.1016/j.aquaculture.2022.738737
Jiao, W. Q., Fu, X. T., Dou, J. Z., Li, H. D., Su, H. L., Mao, J. X., et al. (2014). High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: building up an integrative genomic framework for a bivalve mollusc. DNA Res. 21 (1), 85–101. doi:10.1093/dnares/dst043
Jiao, Z. X., Tian, Y., Hu, B. Y., Li, Q., and Liu, S. K. (2021). Genome structural variation landscape and its selection signatures in the fast-growing strains of the pacific oyster, Crassostrea gigas. Mar. Biotechnol. 23 (5), 736–748. doi:10.1007/s10126-021-10060-5
Jin, W., Bai, Z. Y., Fu, L. L., Zhang, G. F., and Li, J. L. (2012). Genetic analysis of early growth traits of the triangle shell mussel, Hyriopsis Cumingii, as an insight for potential genetic improvement to pearl quality and yield. Aquacult Int. 20 (5), 927–933. doi:10.1007/s10499-012-9518-4
Jones, D. B., Jerry, D. R., Forêt, S., Konovalov, D. A., and Zenger, K. R. (2013a). Genome-wide SNP validation and mantle tissue transcriptome analysis in the silver-lipped pearl oyster, pinctada maxima. Mar. Biotechnol. 15 (6), 647–658. doi:10.1007/s10126-013-9514-3
Jones, D. B., Jerry, D. R., Khatkar, M. S., Raadsma, H. W., and Zenger, K. R. (2013b). A high-density SNP genetic linkage map for the silver-lipped pearl oyster, Pinctada maxima: a valuable resource for gene localisation and marker-assisted selection. Bmc Genomics 14, 810. doi:10.1186/1471-2164-14-810
Jourdan, A., Morvezen, R., Enez, F., Haffray, P., Lange, A., Vetois, E., et al. (2023). Potential of genomic selection for growth, meat content and colour traits in mixed-family breeding designs for the Pacific oyster Crassostrea gigas. Aquaculture 576, 739878. doi:10.1016/j.aquaculture.2023.739878
Kriaridou, C., Tsairidou, S., Houston, R. D., and Robledo, D. (2020). Genomic prediction using low density marker panels in aquaculture: performance across species, traits, and genotyping platforms. Front. Genet. 11, 124. doi:10.3389/fgene.2020.00124
Kristensen, P. S., Jahoor, A., Andersen, J. R., Cericola, F., Orabi, J., Janss, L. L., et al. (2018). Genome-wide association studies and comparison of models and cross-validation strategies for genomic prediction of quality traits in advanced winter wheat breeding lines. Front. Plant Sci. 9, 69. doi:10.3389/fpls.2018.00069
Legarra, A., Croiseau, P., Sanchez, M. P., Teyssèdre, S., Sallé, G., Allais, S., et al. (2015). A comparison of methods for whole-genome QTL mapping using dense markers in four livestock species. Genet. Sel. Evol. 47, 6. doi:10.1186/s12711-015-0087-7
Liu, J. Y., Peng, W. Z., Yu, F., Shen, Y. W., Yu, W. C., Lu, Y. S., et al. (2022). Genomic selection applications can improve the environmental performance of aquatics: a case study on the heat tolerance of abalone. Evol. Appl. 15 (6), 992–1001. doi:10.1111/eva.13388
Lund, M. S., de Roos, A. P. W., de Vries, A. G., Druet, T., Ducrocq, V., Fritz, S., et al. (2011). A common reference population from four European Holstein populations increases reliability of genomic predictions. Genet. Sel. Evol. 43 (1), 43. doi:10.1186/1297-9686-43-43
Lupo, C., Bougeard, S., Le Bihan, V., Blin, J. L., Allain, G., Azema, P., et al. (2021). Mortality of marine mussels Mytilus edulis and M. galloprovincialis: systematic literature review of risk factors and recommendations for future research. Rev. Aquacult 13 (1), 504–536. doi:10.1111/raq.12484
Mallet, A. L., Freeman, K. R., and Dickie, L. M. (1986). The genetics of production characters in the blue mussel Mytilus edulis. I. A preliminary analysis. Aquaculture 57 (1-4), 133–140. doi:10.1016/0044-8486(86)90190-0
McCarty, A. J., Allen, S. K., and Plough, L. V. (2022). Genome-wide analysis of acute low salinity tolerance in the eastern oyster Crassostrea virginica and potential of genomic selection for trait improvement. G3-Genes Genom Genet. 12 (1), jkab368. doi:10.1093/g3journal/jkab368
Misztal, I., Tsuruta, S., Lourenco, D. A. L., Aguilar, I., Legarra, A., and Vitezica, Z. (2014). BLUPF90 and related programs (BGF90).
Nascimento-Schulze, J. C., Bean, T. P., Penaloza, C., Paris, J. R., Whiting, J. R., Simon, A., et al. (2023). SNP discovery and genetic structure in blue mussel species using low coverage sequencing and a medium density 60 K SNP-array. Evol. Appl. 16 (5), 1044–1060. doi:10.1111/eva.13552
Nguyen, T. T. T., Hayes, B. J., and Ingram, B. A. (2014). Genetic parameters and response to selection in blue mussel (Mytilus galloprovincialis) using a SNP-based pedigree. Aquaculture 420, 295–301. doi:10.1016/j.aquaculture.2013.11.021
Nie, H. T., Yan, X. W., Huo, Z. M., Jiang, L. W., Chen, P., Liu, H., et al. (2017). Construction of a high-density genetic map and quantitative trait locus mapping in the manila clam ruditapes philippinarum. Sci. Rep-Uk 7, 229. doi:10.1038/s41598-017-00246-0
Nordio, D., Khtikian, N., Andrews, S., Bertotto, D., Leask, K., and Green, T. (2021). Adaption potential of Crassostrea gigas to ocean acidification and disease caused by Vibrio harveyi. Ices J. Mar. Sci. 78 (1), 360–367. doi:10.1093/icesjms/fsaa080
Norman, R. A., Crumlish, M., and Stetkiewicz, S. (2019). The importance of fisheries and aquaculture production for nutrition and food security. Rev. Sci. Tech. Oie 38 (2), 395–407. doi:10.20506/rst.38.2.2994
Normand, J., Benabdelmouna, A., Louis, W., and Grizon, J. (2022). MYTILOBS Campagne 2020-2021. Réseau d'observation des moules d'élevage sur la côte Atlantique et dans la Manche. Edition 2022.
Odegård, J., Moen, T., Santi, N., Korsvoll, S. A., Kjoglum, S., and Meuwissen, T. H. E. (2014). Genomic prediction in an admixed population of Atlantic salmon (Salmo salar). Front. Genet. 5, 402. doi:10.3389/fgene.2014.00402
Oden, E., Burioli, E. A. V., Trancart, S., Pitel, P. H., and Houssin, M. (2016). Multilocus sequence analysis of Vibrio splendidus related-strains isolated from blue mussel Mytilus sp during mortality events. Aquaculture 464, 420–427. doi:10.1016/j.aquaculture.2016.07.024
Oikonomou, S., Samaras, A., Tekeoglou, M., Loukovitis, D., Dimitroglou, A., Kottaras, L., et al. (2022). Genomic selection and genome-wide association analysis for stress response, disease resistance and body weight in European seabass. Animals 12 (3), 277. doi:10.3390/ani12030277
Palaiokostas, C., Ferraresso, S., Franch, R., Houston, R. D., and Bargelloni, L. (2016). Genomic prediction of resistance to pasteurellosis in Gilthead Sea Bream (Sparus aurata) using 2b-RAD sequencing. G3-Genes Genom Genet. 6 (11), 3693–3700. doi:10.1534/g3.116.035220
Penaloza, C., Barria, A., Papadopoulou, A., Hooper, C., Preston, J., Green, M., et al. (2022). Genome-wide association and genomic prediction of growth traits in the European flat oyster (Ostrea edulis). Front. Genet. 13, 926638. doi:10.3389/fgene.2022.926638
Pino-Querido, A., Alvarez-Castro, J. M., Guerra-Varela, J., Toro, M. A., Vera, M., Pardo, B. G., et al. (2015). Heritability estimation for okadaic acid algal toxin accumulation, mantle color and growth traits in Mediterranean mussel (Mytilus galloprovincialis). Aquaculture 440, 32–39. doi:10.1016/j.aquaculture.2015.01.032
Polsenaere, P., Soletchnik, P., Le Moine, O., Gohin, F., Robert, S., Pepin, J. F., et al. (2017). Potential environmental drivers of a regional blue mussel mass mortality event (winter of 2014, Breton Sound, France). J. Sea Res. 123, 39–50. doi:10.1016/j.seares.2017.03.005
Prou, J., and Goulletquer, P. (2002). The French mussel industry: present status and perspectives. Bull. Aquac. Assoc. Can. 102 (3), 17–23.
Qi, H. G., Song, K., Li, C. Y., Wang, W., Li, B. S., Li, L., et al. (2017). Construction and evaluation of a high-density SNP array for the Pacific oyster (Crassostrea gigas). Plos One 12 (3), e0174007. doi:10.1371/journal.pone.0174007
Regan, T., Bean, T. P., Ellis, T., Davie, A., Carboni, S., Migaud, H., et al. (2021). Genetic improvement technologies to support the sustainable growth of UK aquaculture. Rev. Aquacult 13 (4), 1958–1985. doi:10.1111/raq.12553
Ren, P., Peng, W. Z., You, W. W., Huang, Z. K., Guo, Q., Chen, N., et al. (2016). Genetic mapping and quantitative trait loci analysis of growth-related traits in the small abalone Haliotis diversicolor using restriction-site-associated DNA sequencing. Aquaculture 454, 163–170. doi:10.1016/j.aquaculture.2015.12.026
Resende, M. F. R., Muñoz, P., Resende, M. D. V., Garrick, D. J., Fernando, R. L., Davis, J. M., et al. (2012). Accuracy of genomic selection methods in a standard data set of loblolly pine (pinus taeda L.). Genetics 190 (4), 1503–1510. doi:10.1534/genetics.111.137026
Rey-Campos, M., Moreira, R., Gerdol, M., Pallavicini, A., Novoa, B., and Figueras, A. (2019). Immune tolerance in Mytilus galloprovincialis hemocytes after repeated contact with Vibrio splendidus. Front. Immunol. 10, 1894. doi:10.3389/fimmu.2019.01894
Robledo, D., Matika, O., Hamilton, A., and Houston, R. D. (2018). Genome-wide association and genomic selection for resistance to amoebic gill disease in atlantic salmon. G3-Genes Genom Genet. 8 (4), 1195–1203. doi:10.1534/g3.118.200075
Simon, A., Arbiol, C., Nielsen, E. E., Couteau, J., Sussarellu, R., Burgeot, T., et al. (2020). Replicated anthropogenic hybridisations reveal parallel patterns of admixture in marine mussels. Evol. Appl. 13 (3), 575–599. doi:10.1111/eva.12879
Siol, M., Jacquin, F., Chabert-Martinello, M., Smykal, P., Le Paslier, M. C., Aubert, G., et al. (2017). Patterns of genetic structure and linkage disequilibrium in a large collection of pea germplasm. G3-Genes Genom Genet. 7 (8), 2461–2471. doi:10.1534/g3.117.043471
Smietanka, B., Burzynski, A., Hummel, H., and Wenne, R. (2014). Glacial history of the European marine mussels Mytilus, inferred from distribution of mitochondrial DNA lineages. Heredity 113 (3), 250–258. doi:10.1038/hdy.2014.23
Smits, M., Enez, F., Ferraresso, S., Dalla Rovere, G., Vetois, E., Auvray, J. F., et al. (2020). Potential for genetic improvement of resistance to perkinsus olseni in the manila clam, ruditapes philippinarum, using DNA parentage assignment and mass spawning. Front. Vet. Sci. 7, 579840. doi:10.3389/fvets.2020.579840
Song, H., Guo, X. M., Sun, L. N., Wang, Q. H., Han, F. M., Wang, H. Y., et al. (2021). The hard clam genome reveals massive expansion and diversification of inhibitors of apoptosis in Bivalvia. Bmc Biol. 19 (1), 15. doi:10.1186/s12915-020-00943-9
Song, H. L., Dong, T., Yan, X. Y., Wang, W., Tian, Z. H., Sun, A., et al. (2022). Genomic selection and its research progress in aquaculture breeding. Rev. Aquacult 15, 274–291. doi:10.1111/raq.12716
Soon, T. K., and Ransangan, J. (2019). Extrinsic factors and marine bivalve mass mortalities: an overview. J. Shellfish Res. 38 (2), 223–232. doi:10.2983/035.038.0202
Sun, J., Zhang, Y., Xu, T., Zhang, Y., Mu, H., Zhang, Y., et al. (2017). Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1 (5), 0121. doi:10.1038/s41559-017-0121
Suplicy, F. M. (2020). A review of the multiple benefits of mussel farming. Rev. Aquacult 12 (1), 204–223. doi:10.1111/raq.12313
Tan, K., Zhang, H. K., and Zheng, H. P. (2020). Selective breeding of edible bivalves and its implication of global climate change. Rev. Aquacult 12 (4), 2559–2572. doi:10.1111/raq.12458
Team, P. (2024). RStudio: integrated development environment for R. Posit software. Boston, MA: PBC. URL.
Toro, J. E., Alcapán, A. C., Ojeda, J. A., and Vergara, A. M. (2004a). Selection response for growth rate (shell height and live weight) in the Chilean blue mussel (Mytilus chilensis Hupe 1854). J. Shellfish Res. 23 (3), 753–757.
Toro, J. E., Alcapán, A. C., Vergara, A. M., and Ojeda, J. A. (2004b). Heritability estimates of larval and spat shell height inthe Chilean blue mussel (Mytilus chilensisHupe 1854)produced under controlled laboratory conditions. Aquac. Res. 35 (1), 56–61. doi:10.1111/j.1365-2109.2004.00985.x
Tsai, H. Y., Hamilton, A., Tinch, A. E., Guy, D. R., Gharbi, K., Stear, M. J., et al. (2015). Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. Bmc Genomics 16, 969. doi:10.1186/s12864-015-2117-9
Tsuruta, S., and Misztal, I. (2006). THRGIBBS1F90 for estimation of variance components with threshold and linear models. J. Anim. Sci. 84, 15.
Vallejo, R. L., Evenhuis, J. P., Cheng, H., Fragomeni, B. O., Gao, G., Liu, S., et al. (2022). Genome-wide mapping of quantitative trait loci that can be used in marker-assisted selection for resistance to bacterial cold water disease in two commercial rainbow trout breeding populations. Aquaculture 560, 738574. doi:10.1016/j.aquaculture.2022.738574
Vallejo, R. L., Liu, S., Gao, G., Fragomeni, B. O., Hernandez, A. G., Leeds, T. D., et al. (2017). Similar genetic architecture with shared and unique quantitative trait loci for bacterial cold water disease resistance in two rainbow trout breeding populations. Front. Genet. 8, 156. doi:10.3389/fgene.2017.00156
Vallejo, R. L., Silva, R. M. O., Evenhuis, J. P., Gao, G., Liu, S., Parsons, J. E., et al. (2018). Accurate genomic predictions for BCWD resistance in rainbow trout are achieved using low-density SNP panels: evidence that long-range LD is a major contributing factor. J. Anim. Breed. Genet. 135, 263–274. doi:10.1111/jbg.12335
VanRaden, P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91 (11), 4414–4423. doi:10.3168/jds.2007-0980
VanRaden, P. M., Van Tassell, C. P., Wiggans, G. R., Sonstegard, T. S., Schnabel, R. D., Taylor, J. F., et al. (2009). Invited review: reliability of genomic predictions for North American holstein bulls. J. Dairy Sci. 92 (1), 16–24. doi:10.3168/jds.2008-1514
Vendrami, D. L. J., Houston, R. D., Gharbi, K., Telesca, L., Gutierrez, A. P., Gurney-Smith, H., et al. (2019). Detailed insights into pan-European population structure and inbreeding in wild and hatchery Pacific oysters (Crassostrea gigas) revealed by genome-wide SNP data. Evol. Appl. 12 (3), 519–534. doi:10.1111/eva.12736
Vera, M., Maroso, F., Wilmes, S. B., Hermida, M., Blanco, A., Fernández, C., et al. (2022). Genomic survey of edible cockle (Cerastoderma edule) in the Northeast Atlantic: a baseline for sustainable management of its wild resources. Evol. Appl. 15 (2), 262–285. doi:10.1111/eva.13340
Vu, S. V., Gondro, C., Nguyen, N. T. H., Gilmour, A. R., Tearle, R., Knibb, W., et al. (2021). Prediction accuracies of genomic selection for nine commercially important traits in the Portuguese oyster (Crassostrea angulata) using DArT-seq technology. Genes 12 (2), 210. doi:10.3390/genes12020210
Wang, J. P., Li, L., and Zhang, G. F. (2016). A high-density SNP genetic linkage map and QTL analysis of growth-related traits in a hybrid family of oysters (Crassostrea gigas × Crassostrea angulata) using genotyping-by-sequencing. G3-Genes Genom Genet. 6 (5), 1417–1426. doi:10.1534/g3.116.026971
Wang, Z. Y., Hu, H. H., Sun, T. Y., Li, X., Lv, G. L., Bai, Z. Y., et al. (2022). Genomic selection for improvement of growth traits in triangle sail mussel (Hyriopsis cumingii). Aquaculture 561, 738692. doi:10.1016/j.aquaculture.2022.738692
Yáñez, J. M., Barría, A., López, M. E., Moen, T., Garcia, B. F., Yoshida, G. M., et al. (2023). Genome-wide association and genomic selection in aquaculture. Rev. Aquacult 15 (2), 645–675. doi:10.1111/raq.12750
Yang, B., Zhai, S. Y., Zhang, F. Q., Wang, H. B., Ren, L. T., Li, Y. J., et al. (2022). Genome-wide association study toward efficient selection breeding of resistance to Vibrio alginolyticus in Pacific oyster, Crassostrea gigas. Aquaculture 548, 737592. doi:10.1016/j.aquaculture.2021.737592
Zenger, K. R., Khatkar, M. S., Jones, D. B., Khalilisamani, N., Jerry, D. R., and Raadsma, H. W. (2019). Genomic selection in aquaculture: application, limitations and opportunities with special reference to marine shrimp and pearl oysters. Front. Genet. 9, 693. doi:10.3389/fgene.2018.00693
Keywords: mussels, mortality, breeding program, prediction accuracy, GWAS, linkage disequilibrium, Vibrio
Citation: Ajithkumar M, D’Ambrosio J, Travers M-A, Morvezen R and Degremont L (2025) Genomic selection for resistance to one pathogenic strain of Vibrio splendidus in blue mussel Mytilus edulis. Front. Genet. 15:1487807. doi: 10.3389/fgene.2024.1487807
Received: 28 August 2024; Accepted: 27 November 2024;
Published: 03 January 2025.
Edited by:
Roger L. Vallejo, Agricultural Research Service (USDA), United StatesReviewed by:
Agustin Barria, Benchmark Genetics, NorwayRama Bangera, Akvaforsk Genetics Center, Norway
Copyright © 2025 Ajithkumar, D’Ambrosio, Travers, Morvezen and Degremont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Munusamy Ajithkumar, YWppdGhrdW1hci5tdW51c2FteUBpZnJlbWVyLmZy
†These authors have contributed equally to this work and share last authorship