
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 29 November 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1477291
 Jorge Alfredo Bevilacqua1*†
Jorge Alfredo Bevilacqua1*† Abdullah Mohammed Al-Salti2Abubaker Al Madani3Armando Alves da Fonseca4Hacer Durmus5Josiah Chai6Ali Alshehri7Márcia Gonçalves Ribeiro8
Abdullah Mohammed Al-Salti2Abubaker Al Madani3Armando Alves da Fonseca4Hacer Durmus5Josiah Chai6Ali Alshehri7Márcia Gonçalves Ribeiro8 Paulo Sgobbi9
Paulo Sgobbi9 Sergey S. Nikitin10
Sergey S. Nikitin10 Steven Vargas11Adriana Furtado12
Steven Vargas11Adriana Furtado12 Nathan Thibault13Roberto Araujo14
Nathan Thibault13Roberto Araujo14 Nadia Daba13
Nadia Daba13Introduction: Hereditary myopathies arise due to numerous pathogenic variants occurring in distinct genes, which amount to several hundred. Limb–girdle muscular dystrophies (LGMDs) constitute a heterogeneous group of neuromuscular disorders involving more than 30 genes. Clinically, LGMD is characterized by limb–girdle muscular weakness (LGMW). Late-onset Pompe disease is an important disorder with a differential diagnosis for LGMD, where next-generation sequencing (NGS) plays a crucial role in accurate and prompt diagnosis. The sensitivity and specificity of a 10-gene NGS panel have been previously evaluated for the prevalent forms of recessive LGMD (LGMD-R) and Pompe disease in Latin American patients with LGMW of unknown cause. This project aims to identify the regional relative prevalence of frequent LGMD-R subtypes and Pompe disease in a larger geographic area and to diagnose patients with LGMW by identifying genetic variants of LGMD-R and Pompe disease.
Methods and Results: This 21-country multicentric analysis enrolled 2,372 patients with LGMW from 2017 to 2018. Sequencing analysis was performed using the Illumina NextSeq 500 system, and variant interpretation was performed according to the American College of Medical Genetics and Genomics guidelines. Pathogenic or likely pathogenic variants were seen in 11% of patients (n = 261). Among the positive cases, NGS effectively diagnosed 86.2% and 13.8% of patients with LGMD and Pompe disease, respectively. The most prevalent pathogenic acid α-glucosidase (GAA) variant identified was c.-32-13T > G.
Conclusion: The study adds to the knowledge of the relative occurrence of various subtypes of LGMD worldwide. The inclusion of GAA in the NGS panel to investigate patients with LGMW is a powerful diagnostic approach to screen for late-onset Pompe disease.
Hereditary myopathies encompass a broad spectrum of genetically determined muscular disorders caused by several hundred pathogenic variants in distinct genes (Benarroch et al., 2024). Among these, limb–girdle muscular dystrophies (LGMDs) comprise a diverse group of hereditary diseases distinguished by progressive limb–girdle muscular weakness (LGMW) and dystrophic features in the muscle biopsy (Nigro and Savarese, 2014; Khadilkar et al., 2018; Straub et al., 2018; Wicklund, 2019). Currently, there are 32 subtypes of LGMD. Depending on inheritance pattern, LGMD is classified into two main categories: autosomal dominant (LGMD-D) and autosomal recessive (LGMD-R). LGMD-D represents 10% of the LGMD, encompassing five subtypes (LGMD-D1 to D5), whereas 90% are LGMD-R, comprising 27 forms (LGMD-R1 to R27), each of which is caused by different pathogenic gene variants, with considerable phenotypical overlap (Straub et al., 2018; Wicklund, 2019; National Organization for Rare Disorders, 2021; Benarroch et al., 2024).
The diagnosis of a specific LGMD and obtaining appropriate prevalence estimates for each LGMD subtype are challenged by the low reported incidence rates of 1 in 14,500 to 1 in 123,000 (Liu et al., 2019; National Organization for Rare Disorders, 2021). Additionally, there is a huge phenotype overlap between LGMD and several other forms of LGMW (Nigro and Savarese, 2014; Khadilkar et al., 2018; Liu et al., 2019). Among these, Pompe disease is a rare, metabolic, autosomal recessive disease associated with the GAA (acid α-glucosidase) gene whose clinical signs and symptoms overlap with those of LGMD-R (Werneck et al., 2013; Lévesque et al., 2016; Lorenzoni et al., 2018). The estimated incidence of Pompe disease is 1 in 40,000 births in the United States (National Organization for Rare Disorders, 2021). Proximal muscle weakness of the shoulder and pelvic girdles (LGMW) and impairment of respiratory function are characteristic features of late-onset Pompe disease (LOPD) (Schüller et al., 2012; Lévesque et al., 2016).
The screening diagnostic method in patients suspected to have Pompe disease is an enzymatic analysis from dried blood spot (DBS). Molecular analysis confirms the biochemical diagnosis, and is especially useful for the early diagnosis of individuals with LGMW, including LGMD-R subtypes and Pompe disease (Reddy et al., 2017; Tsai et al., 2017; Bevilacqua et al., 2020). Next-generation sequencing (NGS), has induced a paradigm shift in the effective diagnosis of diseases with diverse phenotypes such as LGMD or LGMW (Málaga et al., 2019). As such, this study aimed to investigate the relative prevalence of LGMD-R and Pompe disease in a large cohort of individuals with genetically unclassified LGMW from 21 countries using a 10-gene NGS panel that includes nine genes for LGMD-R (CAPN3, DYSF, SGCA, SGCB, SGCD, SGCG, FKRP, ANO5, TCAP) and the gene for Pompe disease (GAA). The secondary objective of this study was to identify the region-wise relative prevalence of LGMD-R forms and Pompe disease to create awareness about the overlapping phenotype of Pompe disease with LGMD.
This multicenter investigation included a large collection of samples from 21 countries, encompassing Brazil, Mexico, Colombia, Russia, Malaysia, Turkey, Saudi Arabia, Israel, Oman (Gulf), Panama, Peru, Chile, Hong Kong, Kazakhstan, South Africa, Singapore, Dominican Republic, Costa Rica, Ecuador, Guatemala, and El Salvador. Global epidemiology, countrywide and region-wide prevalence, and local technical logistic capacity were considered for the selection of the countries. The project was designed and conducted as per the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines. IPPMG at the Universidade Federal do Rio de Janeiro approved the research with number 3.642.024.
The inclusion criteria for patients were as follows: male and female patients aged 5 years and above; with progressive LGMW without definitive diagnosis per molecular and/or immunohistochemical analysis; with or without elevated serum creatine kinase (CK) activity; and not previously tested for Pompe disease (enzyme activity or GAA genetic testing). Patients with Pompe disease with a previous positive test (enzyme activity or GAA genetic testing) were excluded from the study.
The gene panel was prescribed by a neuromuscular specialist in accordance with the clinical inclusion criteria, but specific clinical data or phenotype descriptions of the patients were not collected as part of this study. Genomic DNA was extracted from peripheral dried blood spots of each patient and collected on filter paper for molecular testing. The samples (obtained by convenience sampling) were received between 2017 and 2018. The 10-gene NGS panel was developed at the Diagnósticos Laboratoriais Especializados (DLE), Sao Paulo, Brazil. Further NGS testing was processed at the DLE as described previously (Bevilacqua et al., 2020). The NGS panel was chosen based on worldwide prevalence, national and regional epidemiology, and local technical capacity. It comprised nine recessive genes for LGMD (CAPN3, DYSF, SGCA, SGCB, SGCD, SGCG, FKRP, ANO5, TCAP) and one gene for Pompe disease (GAA) (Supplementary Table S1) (Straub et al., 2018; Wicklund, 2019; Benarroch et al., 2020; Bevilacqua et al., 2020; Schiava et al., 2022).
Variants were classified based on the American College of Medical Genetics and Genomics as “pathogenic,” “likely pathogenic,” “variant of uncertain significance,” “likely benign,” or “benign” with respect to a disease and its inheritance pattern (Ellard et al., 2020). Targeted NGS was performed with more than 98% coverage of target regions at 20X or greater for each gene in the panel using Agilent SureSelectQXT target enrichment. Specific deep intronic variants were targeted, and sequencing of flanking intron/exon regions up to 25 base pairs was performed. A custom SureSelectQXT kit (Agilent Technology) was used for the enrichment of these coded and flanked intronic regions. The Illumina NextSeq 500 system was used for sequencing these enriched regions as previously described (Bevilacqua et al., 2020).
Following DNA sequencing, the sequence alignment was conducted using the Burrows–Wheeler Aligner software. This process involved analyzing the sequencing data by comparing it to the standard genome GRCh37 (hg19). The aligned data were used for variant calling (single-nucleotide variants and indels) with the SAMtools software, and annotation was performed using Ensembl Variant Effect Predictor (Bevilacqua et al., 2020).
To ensure accurate sample identification and obtain high-quality DNA, quality control (QC) procedures were conducted alongside the sequencing analysis. Essential steps of NGS, such as library preparation, target capturing, and data sequencing, underwent thorough quality checks. Additionally, mapping, and variant QC metrics were calculated for the sequencing output, allowing the exclusion and reevaluation of any failed samples. QualiMap software was used to check the quality of the sequencing analysis and call of variants. The data are expressed in percentages (Bevilacqua et al., 2020).
This study enrolled 2,372 patients across 21 countries between 2017 and 2018. The countries that participated in the study are shown in Supplementary Figure S1. Most of the patients were adults (≥18 years [57%]). The country-wise distribution of the enrolled patients and the number of patients who were positive for the studied LGMD subtypes and Pompe disease are shown in Supplementary Table S2.
Of the 2,372 patients, 11% (n = 261) tested positive for LGMD-R or Pompe disease. Among the 261 positive cases, 86.2% (n = 225) of the patients had LGMD-R, and 13.8% (n = 36) were diagnosed with Pompe disease. Figure 1 shows the frequency of patients with LGMD-R subtypes and Pompe disease among the 261 confirmed cases. Of the enrolled population, the majority had LGMD-R2 (dysferlinopathy; 27%, n = 69), whereas LGMD-R6 (δ-sarcoglycanopathy; <1%, n = 1) (from Brazil) was the least commonly reported subtype.
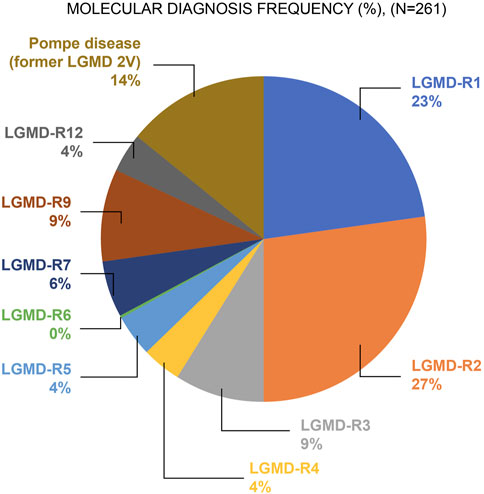
Figure 1. Frequency of individuals with confirmed LGMD-R subtype and Pompe disease (N = 261) Abbreviations: LGMD: Limb–girdle muscular dystrophy; LGMD-R: Recessive limb–girdle muscular dystrophy.
The regional relative prevalence of LGMD-R subtypes across the 21 countries that participated in the study is represented in Supplementary Figure S2. Of the 69 patients diagnosed with LGMD-R2, the highest number of cases (n = 26) was reported from Brazil, followed by Mexico (n = 11), Turkey (n = 9), and Saudi Arabia (n = 8).
The frequencies of the most prevalent LGMD-R subtypes in 14 participating countries are presented in Supplementary Figure S3. LGMD-R2 was observed in the highest number of patients (n = 26 of 83 patients; the total screened population was 697) in Brazil.
Pompe disease was the third most frequent cause of LGMW in the cohort, reported in 36 (13.8%) patients. Table 1 displays the age groups of the patients with Pompe disease. Although only male and female patients aged ≥5 years were considered based on the study inclusion criteria, these younger patients (n = 3) were included and screened as an exception because of special requests from healthcare professionals. Pompe disease was reported in 11 countries, with the highest number of cases from Brazil (n = 11). However, no patients with Pompe disease were reported from the remaining 10 countries (Colombia, Malaysia, Oman [Gulf], Panama, Chile, Hong Kong, Kazakhstan, Dominican Republic, Costa Rica, and Ecuador).

Table 1. Percentage of patients with Pompe disease in different age groups.
The commonly encountered pathogenic GAA variant identified in the population was c.-32-13T > G, which was detected in a heterozygous state at a frequency of 20%. The other GAA variants detected included c.2238G > C and c.2417C > T at a frequency of 5% each. The c.119G > A variant was identified in 4% of the cases, whereas c.1343G > C and c.1552-3C > G were identified in 3% of the cases.
LGMD is a diverse group of neuromuscular diseases with substantial phenotypical overlap with other forms of myopathy, including Pompe disease, manifesting as LGMW (National Organization for Rare Disorders, 2021). Although more than 30 LGMD-associated genes are known, many affected patients remain undiagnosed because of the current challenges associated with gene detection methods and clinical overlap with other forms of myopathy-producing LGMW (Wicklund, 2019; National Organization for Rare Disorders, 2021). This diagnostic NGS project provides insights into the relative prevalence of the most common LGMD-R subtypes and Pompe disease across 21 countries, in some of which, there is no access to genetic diagnosis. In this study covering 2,372 patients, 225 patients were diagnosed with LGMD and 36 with Pompe disease through NGS. The findings of this study underscore the significance of genetic testing in the diagnosis of various disorders that present similar clinical symptoms.
The benefit of the gene panel in identifying genetic variants was reported with the inclusion of the GAA gene because 5% of the total population (121/2,372 individuals) were found to harbor GAA variants. Molecular diagnosis would have been missed without the inclusion of GAA in the gene panel. The most common GAA variant identified in this study was c.-32-13T>G (Supplementary Tables S3, S4).
Thirty-six (13.8%) patients from 11 countries, including Brazil, Mexico, Russia, Turkey, Saudi Arabia, Israel, Peru, South Africa, Singapore, Guatemala, and El Salvador, were diagnosed with Pompe disease. Interestingly, even among the countries that enrolled a small number of patients, such as Peru, South Africa, Singapore, Guatemala, and El Salvador, patients with Pompe disease were identified among those with LGMW.
The LOPD prevalence in a large group of patients in the United States, analyzed using a 35-gene NGS panel, was 0.8% (38/4,656 cases) (Nallamilli et al., 2018), whereas the LOPD prevalence in our previous Latin American study was 0.4% (9/2,103 cases) (Bevilacqua et al., 2020). The relative prevalence of 1.5% (36/2,372 individuals) was higher than the reported prevalence percentages. Therefore, promoting genetic testing for LGMD and Pompe disease among a representative global population is urgently needed for early disease management and awareness creation regarding the overlapping phenotypes of Pompe with LGMD and other forms of LGMW.
Pathogenic variants of the CAPN3 gene cause calpainopathy (LGMD-R1), which is typically observed in patients aged 2–40 years (Nallamilli et al., 2018). In this study, a greater number of pathogenic variants were observed in the DYSF and CAPN3 genes. This result indicated that allelic heterogeneity was higher in these genes than in the other genes in the panel. This study shows that dysferlinopathy and calpainopathy are frequently occurring LGMD-R subtypes in the enrolled population (DYSF-associated LGMD-R2: 69/261 [26.44%] and CAPN3-associated LGMD-R1:61/261 [23.37%]). These findings are consistent with those of Nallamilli et al. (2018), who conducted a study on 4,656 patients with clinically suspected LGMD across the United States (LGMD-R1: 214/1259 LGMD-positive cases [17%]; LGMD-R2: 201/1259 LGMD-positive cases [16%]; N = 4,656) (Nallamilli et al., 2018) and the observations in our previous Latin American study (LGMD-R1: 90/335 LGMD-positive cases [26.87%]; LGMD-R2: 127/335 LGMD-positive cases [37.91%]; N = 2,103) (Bevilacqua et al., 2020). It is worth mentioning that the actual prevalence of LGMD-R2 may be much higher in some countries included and may not be represented in the present study as some patients with LGMD-R2 were included in the Latin American pilot study (37.91%) (Bevilacqua et al., 2020; Cerino et al., 2022) and ongoing studies.
To our knowledge, this is the first attempt at region-wise LGMD mapping across the 21 participating countries some of which did not have access to NGS testing. The results of this study are consistent with those of NGS projects conducted across different geographic regions. Nallamilli et al. (2018) and Töpf et al. (2020) conducted NGS testing among 4,656 North American patients and 1,001 European and Middle Eastern patients using a 35-gene panel and a 170-gene panel, respectively (Nallamilli et al., 2018; Töpf et al., 2020). Both studies included patients with undiagnosed LGMW and/or hyperCKemia. Moreover, 8 of the 12 genes in the study by Nallamilli et al. (2018)—ANO5, SGCA, CAPN3, FKRP, SGCB, SGCB, DYSF, and GAA (Nallamilli et al., 2018)—were part of the 10-gene panel used in the present study.
The first limitation of this study is the low diagnostic yield, which may be due to a lack of proper understanding of the gene panel and the inclusion of a limited number of genes in the panel. Future research using gene panels with a greater number of genes can contribute to expanding the knowledge in this area. The limited patient population, particularly in certain countries, could be considered as the second limitation of this study. Additionally, the ethnicity of the patients could not be considered because of the unavailability of relevant information. The third limitation of this study was that disease severity in relation to genetic variants was not included. Further research on the validation and optimization of NGS panels, consideration of confounding factors, and a more comprehensive representation of diverse populations, including ethnicity and specific clinical findings, is necessary. Although NGS testing offers improved diagnostic capabilities, the long turnaround time must be considered. The period from the date that a laboratory received the testing kit from different countries to the final NGS result was 20 days.
The findings from this study reveal the benefits of the NGS approach in the genetic testing of LGMD and Pompe in a patient population of 2,372 from 21 countries. A confirmed diagnosis of LGMD-R or Pompe disease was made in 11% of the patients through NGS. Pompe disease was identified in 13.8% of the total diagnosed population, including patients from 11 participating countries. The most commonly identified GAA variant was c.-32-13T>G. NGS is a reliable test for the identification of patients with muscular weakness given that access to NGS technology and expertise is not a challenge. Hence, we propose that NGS be employed as a frontline diagnostic tool in individuals with suspected muscular diseases of genetic origin.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://d19rvjna3xg7q0.cloudfront.net/data/consolidado.tsv, FL00001.
The studies involving humans were approved by IPPMG at the Universidade Federal do Rio de Janeiro with number 3.642.024. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
JB: Conceptualization, Formal Analysis, Writing–review and editing. AA-S: Data curation, Writing–review and editing. AbA: Formal Analysis, Writing–review and editing. ArA: Data curation, Writing–review and editing. HD: Data curation, Writing–review and editing. JC: Data curation, Writing–review and editing. AA: Data curation, Writing–review and editing. MR: Data curation, Writing–review and editing. PS: Conceptualization, Data curation, Formal Analysis, Writing–review and editing. SN: Conceptualization, Data curation, Formal Analysis, Writing–review and editing. SV: Conceptualization, Data curation, Formal Analysis, Writing–review and editing. AF: Conceptualization, Data curation, Formal Analysis, Writing–review and editing. NT: Formal Analysis, Writing–review and editing. RA: Conceptualization, Data curation, Formal Analysis, Writing–review and editing. ND: Conceptualization, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Sanofi funded this project. Sanofi provided funding for logistic support and editorial assistance for the development of this manuscript.
We thank the patients, clinicians, and center nursing staff who participated in this NGS diagnostic study funded by Sanofi. Sanofi provided funding for logistic support for the development of this manuscript. BioQuest Solutions provided editorial support for the preparation of this manuscript, and Sanofi funded the support.
JAB, AMA, AAM, AAF, HD, JC, AA, PS, SSN, SV, and RA have received advisory board honoraria from Sanofi. JAB has received travel support and lecture and speaker honoraria from Sanofi. MGR has nothing to disclose. AF, RA, NT, and ND are employees of Sanofi and hold shares and stock options. The funder had the following involvement in the study: logistic support and editorial assistance for the development of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1477291/full#supplementary-material
CK, Creatine kinase; D, Dominant; DLE, Diagnósticos Laboratoriais Especializados; GAA, Acid α-glucosidase; LGMD, Limb–girdle muscular dystrophy; LGMW, Limb–girdle muscular weakness; LOPD, Late-onset Pompe disease; NGS, Next-generation sequence; QC, Quality control; R, Recessive.
Benarroch, L., Bonne, G., Rivier, F., and Hamroun, D. (2020). The 2021 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 30, 1008–1048. doi:10.1016/j.nmd.2020.11.009
Benarroch, L., Bonne, G., Rivier, F., and Hamroun, D. (2024). The 2024 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 34, 126–170. doi:10.1016/j.nmd.2023.12.007
Bevilacqua, J. A., Guecaimburu Ehuletche, M. D. R., Perna, A., Dubrovsky, A., Franca, M. C., Vargas, S., et al. (2020). The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J. Rare Dis. 15, 11. doi:10.1186/s13023-019-1291-2
Cerino, M., González-Hormazábal, P., Abaji, M., Courrier, S., Puppo, F., Mathieu, Y., et al. (2022). Genetic profile of patients with limb-girdle muscle weakness in the Chilean population. Genes 13, 1076. doi:10.3390/genes13061076
Ellard, S., Baple, E. L., Callaway, A., Berry, I., Forrester, N., Turnbull, C., et al. (2020). ACGS best Practice Guidelines for variant classification in rare disease 2020. Available at: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf (Accessed June 21, 2021).
Khadilkar, S. V., Patel, B. A., and Lalkaka, J. A. (2018). Making sense of the clinical spectrum of limb girdle muscular dystrophies. Pract. Neurol. 18, 201–210. doi:10.1136/practneurol-2017-001799
Lévesque, S., Auray-Blais, C., Gravel, E., Boutin, M., Dempsey-Nunez, L., Jacques, P.-E., et al. (2016). Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing. Orphanet J. Rare Dis. 11, 8. doi:10.1186/s13023-016-0390-6
Liu, W., Pajusalu, S., Lake, N. J., Zhou, G., Ioannidis, N., Mittal, P., et al. (2019). Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet. Med. 21, 2512–2520. doi:10.1038/s41436-019-0544-8
Lorenzoni, P. J., Kay, C. S. K., Higashi, N. S., D’Almeida, V., Werneck, L. C., and Scola, R. H. (2018). Late-onset Pompe disease: what is the prevalence of limb-girdle muscular weakness presentation? Arq. Neuropsiquiatr. 76, 247–251. doi:10.1590/0004-282x20180018
Málaga, D. R., Brusius-Facchin, A. C., Siebert, M., Pasqualim, G., Saraiva-Pereira, M. L., Souza, C. F. M. D., et al. (2019). Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 42, 197–206. doi:10.1590/1678-4685-gmb-2018-0092
Nallamilli, B. R. R., Chakravorty, S., Kesari, A., Tanner, A., Ankala, A., Schneider, T., et al. (2018). Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transl. Neurol. 5, 1574–1587. doi:10.1002/acn3.649
National Organization for Rare Disorders (2021). Limb-girdle muscular dystrophies. Available at: https://rarediseases.org/rare-diseases/limb-girdle-muscular-dystrophies/#:∼:text=The%20prevalence%20of%20LGMD%20is,and%202J%20are%20extremely%20rare (Accessed April 28, 2021).
Nigro, V., and Savarese, M. (2014). Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta. Myol. 33, 1–12.
Reddy, H. M., Cho, K.-A., Lek, M., Estrella, E., Valkanas, E., Jones, M. D., et al. (2017). The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J. Hum. Genet. 62, 243–252. doi:10.1038/jhg.2016.116
Schiava, M., Marchesoni, C., García De Rosa, M. L., Estrada, N., Cejas, L. L., Pardal, A., et al. (2022). Genetic characterization of limb girdle muscular dystrophies and Pompe disease in a large Argentine cohort. Neurol. Perspect. 2, 123–133. doi:10.1016/j.neurop.2022.03.003
Schüller, A., Wenninger, S., Strigl-Pill, N., and Schoser, B. (2012). Toward deconstructing the phenotype of late-onset Pompe disease. Am. J. Med. Genet. C Semin. Med. Genet. 160C, 80–88. doi:10.1002/ajmg.c.31322
Straub, V., Murphy, A., Udd, B., Corrado, A., Aymé, S., Bönneman, C., et al. (2018). 229th ENMC international workshop: limb girdle muscular dystrophies – nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 28, 702–710. doi:10.1016/j.nmd.2018.05.007
Töpf, A., Johnson, K., Bates, A., Phillips, L., Chao, K. R., England, E. M., et al. (2020). Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. 22, 1478–1488. doi:10.1038/s41436-020-0840-3
Tsai, A. C., Hung, Y., Harding, C., Koeller, D. M., Wang, J., and Wong, L. C. (2017). Next generation deep sequencing corrects diagnostic pitfalls of traditional molecular approach in a patient with prenatal onset of Pompe disease. Am. J. Med. Genet. A 173, 2500–2504. doi:10.1002/ajmg.a.38333
Werneck, L. C., Lorenzoni, P. J., Kay, C. S. K., and Scola, R. H. (2013). Muscle biopsy in Pompe disease. Arq. Neuropsiquiatr. 71, 284–289. doi:10.1590/0004-282X20130022
Keywords: next-generation sequencing, late-onset Pompe disease, limb-girdle muscular dystrophies, limb-girdle muscular weakness, acid α-glucosidase
Citation: Bevilacqua JA, Al-Salti AM, Al Madani A, Alves da Fonseca A, Durmus H, Chai J, Alshehri A, Ribeiro MG, Sgobbi P, Nikitin SS, Vargas S, Furtado A, Thibault N, Araujo R and Daba N (2024) Detection of gene variants associated with recessive limb–girdle muscular weakness and Pompe disease in a global cohort of patients through the application of next-generation sequencing analysis. Front. Genet. 15:1477291. doi: 10.3389/fgene.2024.1477291
Received: 07 August 2024; Accepted: 12 November 2024;
Published: 29 November 2024.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Patryk Lipiński, Maria Sklodowska-Curie Medical Academy, PolandCopyright © 2024 Bevilacqua, Al-Salti, Al Madani, Alves da Fonseca, Durmus, Chai, Alshehri, Ribeiro, Sgobbi, Nikitin, Vargas, Furtado, Thibault, Araujo and Daba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge Alfredo Bevilacqua, amJldmlsYWNAbWVkLnVjaGlsZS5jbA==
†ORCID: Jorge Alfredo Bevilacqua, orcid.org/0000-0002-0525-9308
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.