 Ciro Gianmaria Amoroso
Ciro Gianmaria Amoroso Giuseppe Andolfo
Giuseppe Andolfo1 Introduction
1.1 Hazelnut cultivation and challenges
Hazelnuts are trees belonging to the Betulaceae family and Corylus genus (Wani et al., 2020). Due to their delicious flavor profile, nutrient composition, and antioxidant properties, hazelnuts are widely used as whole nuts or as processed foods. Due to their highly appreciated properties, popular Corylus species are cultivated across the globe, including Corylus avellana, widely cultivated in Europe; C. americana, predominantly found in North America; and C. heterophylla and C. mandshurica, extensively utilized in Asia (Botta et al., 2019). Thus far, several pathogens such as Xanthomonas sp., Pseudomonas sp., Botrytis cinerea, Alternaria sp., Cytospora sp. Phytophthora sp. and various pests compromise nut production (Guerrero et al., 2014; Battilani et al., 2018; Sun et al., 2023), which also constantly faces environmental stress (Allegrini et al., 2022).
1.2 Limitations of classical breeding and the potential of CRISPR-Cas9
Plants are constantly engaged in a struggle for survival and adaptation, conventional breeding techniques have allowed the development of hazelnut cultivars with improved characteristics especially related to cold resistance and yield (Wang G. X. et al., 2018; Botta et al., 2019; Mehlenbacher and Molnar, 2021). However, the efficiency of classical breeding approaches depends on the availability of genomic resources and may be limited in commercial varieties due to the introduction of undesired genetic traits during breeding steps. Furthermore, the classical breeding approach has been recognized as a time-consuming process that requires multiple generations and years to introduce and fix desirable traits (Tester and Langridge, 2010). Significant support for hazelnut genetic research and improvement may come from new genome editing techniques that are revolutionizing plant breeding programs and functional studies. In particular, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 technology has been successfully applied in various fruit trees and nuts, becoming a conventional technique for enhancing biotic and abiotic stress tolerance in plants (Wang X. et al., 2018; Chang et al., 2022). The CRISPR-Cas9 system employs the Cas9 nuclease, able to induce DNA double-strand breaks (DSB) (Puchta, 2017). Once a DSB is created, the cell’s natural repair mechanisms come into play, leading to non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ often leads to insertions or deletions (indels) at the break site, which can result in gene knockout, while HDR can be utilized for precise edits when a donor template is provided (Khan et al., 2018). The guidance for these modifications is provided by guide RNAs (gRNAs) that are designed to effectively guide the Cas9 nuclease on the intended target sites (Hsu et al., 2013). The success of these modifications heavily relies on the design of highly specific gRNAs (Filippova et al., 2019). For example (Evangelista et al., 2024), suggested that the design of gRNAs targeting specific domains of hazelnut allergenic genes could reduce unintended effects caused by complete gene silencing. This approach would enhance the hypo-allergenicity of plants without compromising gene fitness (Tran et al., 2021). Indeed, this strategy has been employed in previous studies where the Cas9 enzyme was directed toward specific domains associated with plant stress susceptibility (Tran et al., 2021). Thereby, the CRISPR-Cas9 technology has been defined as a simple, highly efficient, specific, and cost-effective method that can facilitate functional genetic studies and the generation of transgene-free edited plants in a shorter period compared to classical breeding.
1.3 gRNA design for CRISPR-Cas9 in hazelnut
Numerous web-based tools have been developed to facilitate the design of gRNAs across a variety of plant species. Available online software can be used for selecting optimal sgRNA targets based on user-defined parameters (Uniyal et al., 2019; Kornel et al., 2019; Haeussler et al., 2016; Liu et al., 2017; Bae et al., 2014a). However, to date, user-friendly software integrating the genomes of Corylus species are not yet available, which presents a significant gap for researchers in this area. Crucial support for the application of CRISPR-Cas9 in hazelnut came from recent studies that released genome assemblies of different Corylus species, providing insights into genetic diversity and evolutionary gene relationships (Li et al., 2021; Lucas et al., 2021; Zhao et al., 2021; Brainard et al., 2024). High-quality genome sequences and curated gene prediction are essential for identifying suitable targets and gRNA design (Mohr et al., 2016). However, several factors influence the gRNA effectiveness, efficiency, and uniqueness of target genes, such as the sequences matching on the target gene, the position of the Protospacer Adjacent Motif (PAM) sequence, the accessibility of target sites within the chromatin structure (Jensen et al., 2017), and the formation of secondary structures (Riesenberg et al., 2022). Indeed, it has been shown that self-folding free energy strongly influences cleavage efficiency (Wang et al., 2019). Therefore, gRNA activity is predicted by specific methods providing on and off-target scores for evaluating the potential cutting efficiency of gRNAs on target genes and on potential unintended genomic loci (Mohr et al., 2016). However, currently available tools for gRNA design do not allow for determining their secondary structure (Hassan et al., 2021). Guides are predicted by assessing their activity through various tools that have been developed (Bae et al., 2014b; Montague et al., 2014; Moreno-Mateos et al., 2015; Chuai et al., 2018; Concordet and Haeussler, 2018; Cui et al., 2018). Algorithms such as Rule Set one and Rule Set 2 have been developed for on-target activity prediction (Gagnon et al., 2014; Heigwer et al., 2014; Wang et al., 2014; Xu et al., 2015). These algorithms take into account features like nucleotide composition, GC content, and positional characteristics to forecast gRNA efficacy with the objective of enhancing gRNA design by maximizing on-target activity. Conversely, for predicting off-target effects, algorithms like CFD (Cutting Frequency Determination), Mismatch count, and MIT specificity have been developed (Cong et al., 2013; Hsu et al., 2013; Mali et al., 2013; Doench et al., 2014; Doench et al., 2016). These algorithms employ scoring systems based on mismatches and sequence features to anticipate potential off-target activity of gRNAs. Recent studies pointed out the reliability and accuracy of the CFD score compared to the MIT score and Mismatch Count method in predicting off-target effects during gRNA design for CRISPR-Cas9 applications in plants (Liu et al., 2020; Naeem et al., 2020). The development of dedicated databases (DB) is a real support for molecular biologists in genome editing programs. Available user-friendly tools lacked Corylus reference genomes, and bioinformatics software for custom analysis requires advanced command-line skills. This limitation made it difficult for researchers to access simple and intuitive interfaces for designing gRNAs. Additionally, gene editing studies require the identification of duplicated target genes (paralogs). Plant genomes frequently host gene groups that have evolved from a common ancestor retaining overlapping or redundant functions. This poses a challenge to functional genetics research and makes gRNA design a crucial step (Bhuyan et al., 2023). Therefore, an atlas could support the selection of gRNAs for the simultaneous silencing of duplicated genes, or for utilizing of Homologs Direct Repair approaches (Aksoy et al., 2022). In this view, the development of a comprehensive DB containing all this information represents a significant advantage for one of the most critical steps in CRISPR-Cas9 application.
To this end, we released the single and multi-target Cas9 guide database (SiMul-db) including gRNAs libraries, guide self-folding free energy, paralog gene lists, and protein domain annotations for C. americana, C. avellana, C. heterophylla, and C. mandshurica. Moreover, we included Arabidopsis thaliana in the orthology analysis for comparative proposes. Finally, we reported two examples of guide identification for singular and multiple editing of B. cinerea susceptible genes in C. avellana.
2 Value of the data
• SiMul-db represents a valuable genomic resource for scientists involved in hazelnut breeding programs.
• Paralog identification will facilitate the selection of gRNA for multi-copy gene targets.
• Orthology inference will permit the transfer of gene function from model species to Corylus genes.
3 Materials and methods
3.1 Data sources
To develop a comprehensive and user-friendly database of Cas9 guide sequences for hazelnut plants, we used the European hazelnut (C. avellana) ‘Tombul’ genome (v2.4) and its gene model annotation as reported by Lucas et al. (2021). The C. avellana genome sequence (GCA_901000735.2_CavTom2PMs-1.0_genomic.fna), gene model annotation (GCA_901000735.2_CavTom2PMs-1.0_genomic.gbff) and relative protein sequences were downloaded from the GenBank site (https://ftp.ncbi.nih.gov/genomes/genbank/plant/). We also used a genome assembly (Camericanavar_rush_835_v1.0.fa) of the American hazelnut (C. americana) accession ‘Rush’ (Brainard et al., 2024) and the genome assemblies (Chr_genome_assembly_changed.fa and Cma.genome.chr.fa) of two wild Asian varieties (C. heterophylla Fisch. and C. mandshurica Maxim.) (Zhao et al., 2021; Li et al., 2021), as well as A. thaliana genome assembly (Araport11) for comparative purposes (https://www.arabidopsis.org/).
3.2 CRISPR-Cas9 guide RNA design
To obtain the Corylus whole-genome gRNA libraries we used the reference gRNAs database (RD)-build model implemented in CRISPR-Local software using -U 15 -D 3 settings (Sun et al., 2019). The reference genomes (.fa) and corresponding gene annotations (.gff) of C. avellana, C. americana, C. heterophylla, and C. mandshurica were used as input files. The screening of all possible on-target gRNAs and their scoring were based on the Rule Set 2 algorithm (Doench et al., 2016). While the prediction of the effects of each off-target site with the highest cutting frequency determination (CFD) score for each gRNA, was realized by the SeqMap program (Jiang and Wong, 2008). All target and off-target data determined across the entire genome are exported into RD format (Supplementary Tables S1–S4), which includes information about guide sequence, physical position, the relative position against transcription start site, on-target score, and potential off-target sites with the highest CFD score for each gRNA for every locus. Database (DB)-search model was used to obtain sorted results from all annotated Cor a genes. Paralogs (PL)-search model was used to extract gRNAs matching multi-gene targets.
3.3 Orthology relationships, paralog genes identification and protein domains annotation
To provide a deep understanding of the evolution and diversification of genes in Corylus plants, we used OrthoFinder v2.5.1 package tools (Emms and Kelly, 2019). Simultaneously, C. americana, C. avellana C. heterophylla, C. mandshurica, and A. thaliana proteomes were analyzed, with default settings. In this package, the BLAST tool was used for fast sequence similarity searches among protein sequences. The clustering of genes was inferred using the MCL clustering algorithm; an unrooted gene tree was inferred for each orthogroup using DendroBLAST (Kelly and Maini, 2013). The protein domain architecture was annotated using Pfam database implemented in InterProScan v5.69–101.0 software (Jones et al., 2014) with default setting.
3.4 Prediction of RNAs secondary structure
The RNA secondary structure prediction and comparison were calculated with RNAfold software implemented in the ViennaRNA package (version 2.6.4) (Lorenz et al., 2011). Specifically, the propensity to form secondary structures was determined by calculating the self-folding free energy (ΔG expressed in kcal/mol) of the guide sequence using the -d2 option as the default dangling-end model, allowing a single nucleotide to contribute with all its possible favorable interactions.
4 Results and discussion
4.1 Cas9 gRNA sequences and orthogroups identification
Over thirteen million gRNAs were predicted in the four Corylus genome assemblies available to date (Table 1). Future updates to SiMul-db will incorporate newly sequenced Corylus genome assemblies, further expanding the database and increasing the number of available species for gRNA design. The guide on-target values range from 0 to 1, and gRNAs with higher on-target scores are considered to perform better (Bae et al., 2014a). Considering the high number of obtained gRNAs, we selected gRNAs with on-target score higher than 0.66, obtaining a subset of 1,025,628 gRNAs that were considered top rank (Doench et al., 2016; Haeussler et al., 2016). Interestingly, 71,262 gRNAs were classified as multi-target gRNAs (Table 1). On average, non-functional guide sequences had significantly higher potential for self-folding than functional ones (Wong et al., 2015). To hone gRNA evaluation, we estimated the self-folding free energy (ΔG) to determine guide propensity to form secondary structures. (Wong et al., 2015). Generally, gRNA will fold within itself when the ΔG value is more negative, which hinders pairing with the on-target (KesavanNair, 2023). According to Jensen et al. (2017), the ability of Cas9 endonuclease to efficiently cleave the target is greater for ΔG values comprised between −2 and 0 kcal/mol. In our database, ∼80% of best gRNAs showed a ΔG > −2 kcal/mol. Furthermore, our dataset was implemented with an orthology analysis between Corylus proteomes, and including A. thaliana as an outgroup genome. A. thaliana was chosen as a reference due to its widespread use as a model species and the extensive knowledge about its genes (Cao et al., 2011). Orthology analysis allowed the identification of 21,237 orthogroups (Supplementary Tables S5, S6; Supplementary Figure S1). The identification of orthogroups between Corylus spp. and A. thaliana can speed up the discovery of target genes and potential paralogs for future genome editing studies (Mota et al., 2020). Moreover, SiMul-db was implemented with gene domain predictions that could allow the selection of specific gRNAs tailored on domain of interests (Supplementary Tables S7–S10). Finally, we selected the best gRNAs with higher on-target scores and lower CFD scores for each gene model predicted in the four Corylus genomes (Table 1).

Table 1. Genome editing gRNA libraries for four hazelnut species.
4.2 Framework of SiMul database
SiMul-db is a user-friendly research tool for the selection of the best Cas9 guides in hazelnut species. Corylus genomic data, including genome sequences, gene models, and protein sequences were processed to generate SiMul-db (Figure 1). Protein domain information have been obtainted consulting the Protein family database (Pfam) implemented in InterProScan software (Jones et al., 2014). Additionally, the proteome of reference model A. thaliana was included in orthology analysis for comparative purposes. Guide prediction and comparative analysis allowed to provide Cas9-gRNAs libraries and to identify homolog groups, respectively (Figure 1). While ΔG estimation provided additional information for a more accurate selection of guides. Therefore, through SiMul-db workflow users can identify single or multi-target genes (Armario Najera et al., 2019). Users can choose the best gRNAs considering the efficiency (on target, CFD, and ΔG) scores, or specific target region of the coding sequence, such as specific predicted domains (Supplementary Tables S7–S10). While for duplicated genes SiMul-db suggests common gRNA sequences for multi-editing (Figure 1). This streamlined approach allows for efficient and accurate guide selection for biotechnology assisted breeding in the Corylus genus.
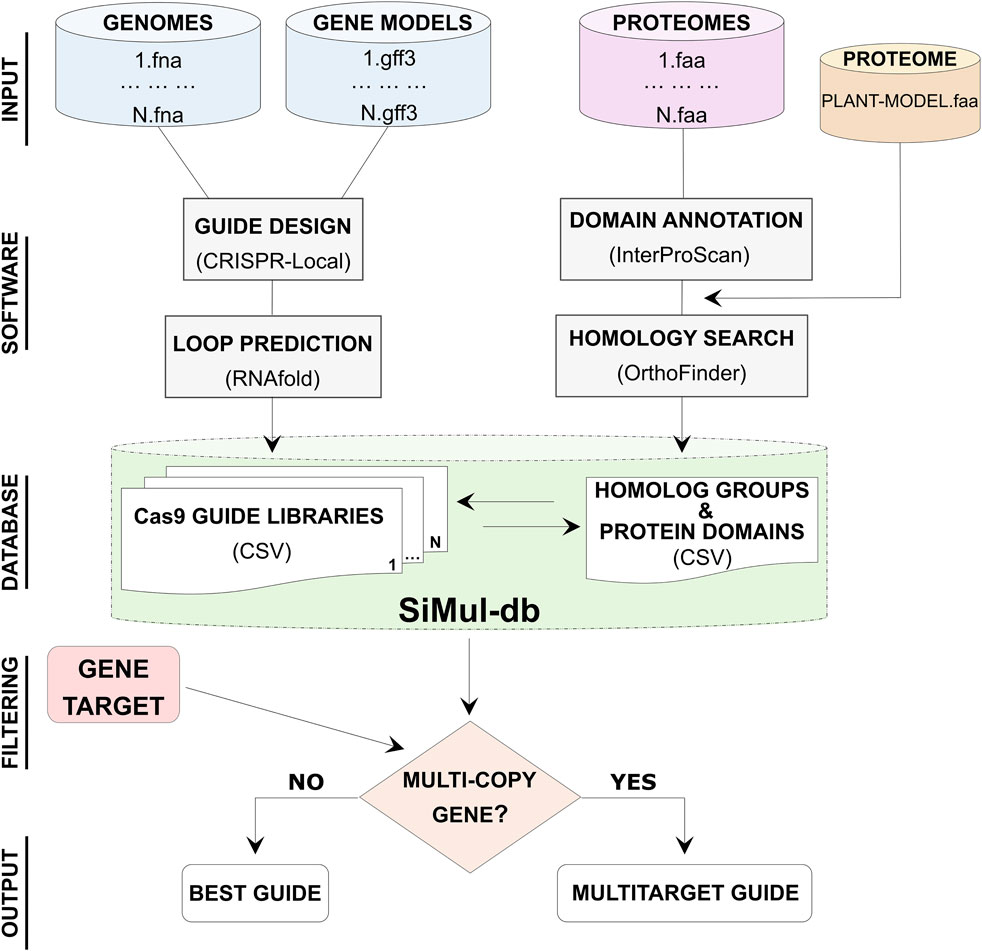
Figure 1. Diagram showing the workflow steps (data elaboration and primary curation) for the development of single and multi-target Cas9 gRNA database (SiMul-db). Best guide refers to gRNA with I) the top on-target, CFD and ΔG scores, or II) more suitable protein-coding region.
4.3 Filtering of single and multi-targeting gRNAs
Specific hazelnut genes related to agricultural traits, metabolic pathways, or responses to biotic and abiotic stresses could be selected using SiMul-db. For example, nuts are vulnerable to B. cinerea, commonly known as “gray mold”, a fungal pathogen affecting various plant species (Romanazzi and Feliziani, 2014). This pathogen can infect multiple parts of hazelnut, including fruits, inducing significant yield losses and quality deterioration (Guerrero et al., 2014). SiMul-db can assist in identifying potential genes and provide valuable insights for the success of genome editing strategies. Below we provide two strategies for the selection of single and multiple gRNAs for targeting genes involved in B. cinerea interaction. Previous studies allowed the identification of two genes, AtDND1 (AT5G15410) and AtPUB17 (AT1G29340), potentially involved in plant-pathogen susceptibility (Sun et al., 2017; Ramirez Gaona et al., 2023). In particular, the silencing of AtDND1 and AtPUB17 has been shown to reduce susceptibility to B. cinerea (Supplementary Table S7). Therefore, exploring SiMul-db, researchers can easily reveal the Corylus orthologs (OG0011955: CamerRush.05G196000.1, Cav05g20890.1, EVM0018229.1, and CmaG0015144.1) to AtDND1 (Supplementary Table S5), and find the best guide for each identified orthologous gene (Supplementary Table S11). Furthermore, three paralogs to AtPUB17 were found in C. avellana (Cav02g18830.1, Cav02g18860.1, Cav02g18960.1). By querying SiMul-db, it was possible to identify a single gRNA that could be used for silencing all three paralogs simultaneously (Supplementary Table S12).
5 Conclusion
SiMul-db emerges as an innovative tool for accelerating gRNA selection for genome editing in hazelnuts. It provides lists of gRNAs with high on-target efficiency, low off-target effects, and relative self-folding free energy of the guide sequences. For the first time, the evolutionary relationships of Corylus spp. are consolidated into a unique database, which reduces the risk of undesired off-target effects and enhances the accuracy of CRISPR-Cas9. Even in the absence of efficient agrobacterium-mediated transformation protocols, SiMul-db can be consulted with alternative transformation methods, such as transient CRISPR-Cas9 modifications (Son and Park, 2022). Furthermore, future implementations of SiMul-db will include other plant species, making genome editing more accessible to researchers. This will facilitate plant genome editing programs and functional studies, ultimately boosting agricultural productivity and plant resilience.
6 Direct link to deposited data and information to users
The CRISPR-Cas9 gRNA dataset of four Corylus species can be accessed at FIGSHARE with the following link https://figshare.com/s/3ac61758f15226572aef. The candidate gRNAs identified from CRISPR-Local could be exported in GFF format and imported into the IGV genome browser (Thorvaldsdóttir et al., 2013) for comparison and visual inspection (Supplementary Figure S1). The Supplementary Material (Supplementary Tables S1–S8) for this article can be found online at: https://figshare.com/s/3ac61758f15226572aef. Users can download and use the data freely for research purpose only with acknowledgment to us and quoting this paper as a reference to the data.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://figshare.com/s/3ac61758f15226572aef.
Author contributions
CGA: Data curation, Formal Analysis, Investigation, Resources, Writing–original draft. GA: Conceptualization, Data curation, Formal Analysis, Investigation, Project administration, Supervision, Visualization, Writing–review and editing.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1467316/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | A typical window in the Integrative Genomics Viewer (IGV) software. Displayed are the gene model Cav01g18280.1 located on chromosome 1 of the C. avellana genome and the gRNA predicted by CRISPR-Local.
References
Aksoy, E., Yildirim, K., Kavas, M., Kayihan, C., Yerlikaya, B. A., Çalik, I., et al. (2022). General guidelines for CRISPR/Cas-based genome editing in plants. Mol. Biol. Rep. 49, 12151–12164. doi:10.1007/s11033-022-07773-8
Allegrini, A., Salvaneschi, P., Schirone, B., Cianfaglione, K., and Di Michele, A. (2022). Multipurpose plant species and circular economy: Corylus avellana L. as a study case. Front. Biosci. 27, 11. doi:10.31083/j.fbl2701011
Armario Najera, V., Twyman, R. M., Christou, P., and Zhu, C. (2019). Applications of multiplex genome editing in higher plants. Curr. Opin. Biotechnol. 59, 93–102. doi:10.1016/j.copbio.2019.02.015
Bae, S., Kweon, J., Kim, H. S., and Kim, J.-S. (2014a). Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 11, 705–706. doi:10.1038/nmeth.3015
Bae, S., Park, J., and Kim, J.-S. (2014b). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. doi:10.1093/bioinformatics/btu048
Battilani, P., Chiusa, G., Arciuolo, R., Somenzi, M., Fontana, M., Castello, G., et al. (2018). Diaporthe as the main cause of hazelnut defects in the Caucasus region. Phytopathol. Mediterr. 57. doi:10.14601/Phytopathol_Mediterr-22872
Bhuyan, S. J., Kumar, M., Ramrao Devde, P., Rai, A. C., Mishra, A. K., Singh, P. K., et al. (2023). Progress in gene editing tools, implications and success in plants: a review. Front. Genome 5, 1272678. doi:10.3389/fgeed.2023.1272678
Botta, R., Molnar, T. J., Erdogan, V., Valentini, N., Torello Marinoni, D., and Mehlenbacher, S. A. (2019). “Hazelnut (Corylus spp.) breeding,” in Advances in plant breeding strategies: nut and beverage crops. Editors J. M. Al-Khayri, S. M. Jain, and D. V. Johnson (Cham: Springer International Publishing), 157–219. doi:10.1007/978-3-030-23112-5_6
Brainard, S. H., Sanders, D. M., Bruna, T., Shu, S., and Dawson, J. C. (2024). The first two chromosome-scale genome assemblies of American hazelnut enable comparative genomic analysis of the genus Corylus. Plant Biotechnol. J. 22, 472–483. doi:10.1111/pbi.14199
Cao, J., Schneeberger, K., Ossowski, S., Günther, T., Bender, S., Fitz, J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. doi:10.1038/ng.911
Chang, Y., Song, X., Zhang, Q., Zhang, P., Lei, X., and Pei, D. (2022). Robust CRISPR/Cas9 mediated gene editing of JrWOX11 manipulated adventitious rooting and vegetative growth in a nut tree species of walnut. Sci. Hortic. 303, 111199. doi:10.1016/j.scienta.2022.111199
Chuai, G., Ma, H., Yan, J., Chen, M., Hong, N., Xue, D., et al. (2018). DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 19, 80. doi:10.1186/s13059-018-1459-4
Concordet, J.-P., and Haeussler, M. (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245. doi:10.1093/nar/gky354
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/cas systems. Science 339, 819–823. doi:10.1126/science.1231143
Cui, Y., Xu, J., Cheng, M., Liao, X., and Peng, S. (2018). Review of CRISPR/Cas9 sgRNA design tools. Interdiscip. Sci. Comput. Life Sci. 10, 455–465. doi:10.1007/s12539-018-0298-z
Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. doi:10.1038/nbt.3437
Doench, J. G., Hartenian, E., Graham, D. B., Tothova, Z., Hegde, M., Smith, I., et al. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. doi:10.1038/nbt.3026
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi:10.1186/s13059-019-1832-y
Evangelista, A. R., Amoroso, C. G., Nitride, C., and Andolfo, G. (2024). Seed storage allergens tackled via next-generation research assistant. Front. Food. Sci. Technol. 4, 1372770. doi:10.3389/frfst.2024.1372770
Filippova, J., Matveeva, A., Zhuravlev, E., and Stepanov, G. (2019). Guide RNA modification as a way to improve CRISPR/Cas9-based genome-editing systems. Biochimie 167, 49–60. doi:10.1016/j.biochi.2019.09.003
Gagnon, J. A., Valen, E., Thyme, S. B., Huang, P., Ahkmetova, L., Pauli, A., et al. (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 9, e98186. doi:10.1371/journal.pone.0098186
Guerrero, J. C., Pérez, S. F., Ferrada, E. Q., Cona, L. Q., and Bensch, E. T. (2014). Phytopathogens of hazelnut (Corylus avellana L.) in southern Chile. Acta Hortic., 269–273. doi:10.17660/ActaHortic.2014.1052.36
Haeussler, M., Schönig, K., Eckert, H., Eschstruth, A., Mianné, J., Renaud, J.-B., et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148. doi:10.1186/s13059-016-1012-2
Hassan, M. M., Chowdhury, A. K., and Islam, T. (2021). “In silico analysis of gRNA secondary structure to predict its efficacy for plant genome editing,” in CRISPR-cas methods. Springer protocols handbooks. Editors M. T. Islam,, and K. A. Molla (New York, NY: Humana). doi:10.1007/978-1-0716-1657-4_2
Heigwer, F., Kerr, G., and Boutros, M. (2014). E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123. doi:10.1038/nmeth.2812
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi:10.1038/nbt.2647
Jensen, K. T., Fløe, L., Petersen, T. S., Huang, J., Xu, F., Bolund, L., et al. (2017). Chromatin accessibility and guide sequence secondary structure affect CRISPR -Cas9 gene editing efficiency. FEBS Lett. 591, 1892–1901. doi:10.1002/1873-3468.12707
Jiang, H., and Wong, W. H. (2008). SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics 24, 2395–2396. doi:10.1093/bioinformatics/btn429
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi:10.1093/bioinformatics/btu031
Kelly, S., and Maini, P. K. (2013). DendroBLAST: approximate phylogenetic trees in the absence of multiple sequence alignments. PLoS ONE 8 (3), e58537. doi:10.1371/journal.pone.0058537
KesavanNair, L. R. (2023). Computational design of guide RNAs and vector to knockout LasR gene of Pseudomonas aeruginosa. Gene Genome, 100028. doi:10.1016/j.ggedit.2023.100028
Khan, S., Mahmood, M. S., Rahman, S., Zafar, H., Habibullah, S., Khan, Z., et al. (2018). CRISPR/Cas9: the Jedi against the dark empire of diseases. J. Biomed. Sci. 25, 29. doi:10.1186/s12929-018-0425-5
Kornel, L., Tessa, G. M., Maximilian, K., Yamila, N. T. C., Håkon, T., and Eivind, V. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47 (W1), W171–W174. doi:10.1093/nar/gkz365
Li, Y., Sun, P., Lu, Z., Chen, J., Wang, Z., Du, X., et al. (2021). The Corylus mandshurica genome provides insights into the evolution of Betulaceae genomes and hazelnut breeding. Hortic. Res. 8, 54. doi:10.1038/s41438-021-00495-1
Liu, G., Zhang, Y., and Zhang, T. (2020). Computational approaches for effective CRISPR guide RNA design and evaluation. Comput. Struct. Biotechnol. J. 18, 35–44. doi:10.1016/j.csbj.2019.11.006
Liu, H., Ding, Y., Zhou, Y., Jin, W., Xie, K., and Chen, L.-L. (2017). CRISPR-P 2.0: an improved CRISPR-cas9 tool for genome editing in plants. Mol. Plant 10, 530–532. doi:10.1016/j.molp.2017.01.003
Lorenz, R., Bernhart, S. H., Höner zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F., et al. (2011). ViennaRNA package 2.0. Algorithms Mol. Biol. 6, 26. doi:10.1186/1748-7188-6-26
Lucas, S. J., Kahraman, K., Avşar, B., Buggs, R. J. A., and Bilge, I. (2021). A chromosome-scale genome assembly of European hazel (Corylus avellana L.) reveals targets for crop improvement. Plant J. 105, 1413–1430. doi:10.1111/tpj.15099
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi:10.1126/science.1232033
Mehlenbacher, S. A., and Molnar, T. J. (2021). “Hazelnut breeding,” in Plant breeding reviews. Editor I. Goldman (Wiley), 9–141. doi:10.1002/9781119828235.ch2
Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., and Perrimon, N. (2016). CRISPR guide RNA design for research applications. FEBS J. 283, 3232–3238. doi:10.1111/febs.13777
Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., and Valen, E. (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407. doi:10.1093/nar/gku410
Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J.-D., Fernandez, J. P., Mis, E. K., Khokha, M. K., et al. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988. doi:10.1038/nmeth.3543
Mota, A. P. Z., Fernandez, D., Arraes, F. B. M., Petitot, A.-S., De Melo, B. P., De Sa, M. E. L., et al. (2020). Evolutionarily conserved plant genes responsive to root-knot nematodes identified by comparative genomics. Mol. Genet. Genomics 295, 1063–1078. doi:10.1007/s00438-020-01677-7
Naeem, M., Majeed, S., Hoque, M. Z., and Ahmad, I. (2020). Latest developed strategies to minimize the off-target effects in CRISPR-cas-mediated genome editing. Cells 9, 1608. doi:10.3390/cells9071608
Puchta, H. (2017). Applying CRISPR/Cas for genome engineering in plants: the best is yet to come. Curr. Opin. Plant Biol. 36, 1–8. doi:10.1016/j.pbi.2016.11.011
Ramirez Gaona, M., Van Tuinen, A., Schipper, D., Kano, A., Wolters, P. J., Visser, R. G. F., et al. (2023). Mutation of PUB17 in tomato leads to reduced susceptibility to necrotrophic fungi. Plant Biotechnol. J. 21, 2157–2159. doi:10.1111/pbi.14127
Riesenberg, S., Helmbrecht, N., Kanis, P., Maricic, T., and Pääbo, S. (2022). Improved gRNA secondary structures allow editing of target sites resistant to CRISPR-Cas9 cleavage. Nat. Commun. 13, 489. doi:10.1038/s41467-022-28137-7
Romanazzi, G., and Feliziani, E. (2014). “Botrytis cinerea (gray mold),” in Postharvest decay (Elsevier), 131–146. doi:10.1016/B978-0-12-411552-1.00004-1
Son, S., and Park, S. R. (2022). Challenges facing CRISPR/Cas9-Based genome editing in plants. Front. Plant Sci. 13, 902413. doi:10.3389/fpls.2022.902413
Sun, J., Liu, H., Liu, J., Cheng, S., Peng, Y., Zhang, Q., et al. (2019). CRISPR-Local: a local single-guide RNA (sgRNA) design tool for non-reference plant genomes. Bioinformatics 35, 2501–2503. doi:10.1093/bioinformatics/bty970
Sun, J., Zhang, X., Zheng, J., Liu, G., and Chen, L. (2023). Importance of cell wall permeability and cell wall degrading enzymes during infection of Botrytis cinerea in hazelnut. Forests 14, 565. doi:10.3390/f14030565
Sun, K., Van Tuinen, A., Van Kan, J. A. L., Wolters, A.-M. A., Jacobsen, E., Visser, R. G. F., et al. (2017). Silencing of DND1 in potato and tomato impedes conidial germination, attachment and hyphal growth of Botrytis cinerea. BMC Plant Biol. 17, 235. doi:10.1186/s12870-017-1184-2
Tester, M., and Langridge, P. (2010). Breeding technologies to increase crop production in a changing world. Science 327, 818–822. doi:10.1126/science.1183700
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinforma. 14 (2), 178–192. doi:10.1093/bib/bbs017
Tran, M. T., Doan, D. T. H., Kim, J., Song, Y. J., Sung, Y. W., Das, S., et al. (2021). CRISPR/Cas9-based precise excision of SlHyPRP1 domain(s) to obtain salt stress-tolerant tomato. Plant Cell Rep. 40, 999–1011. doi:10.1007/s00299-020-02622-z
Uniyal, A. P., Mansotra, K., Yadav, S. K., and Kumar, V. (2019). An overview of designing and selection of sgRNAs for precise genome editing by the CRISPR-Cas9 system in plants. Biotech 9, 223. doi:10.1007/s13205-019-1760-2
Wang, D., Zhang, C., Wang, B., Li, B., Wang, Q., Liu, D., et al. (2019). Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 10, 4284. doi:10.1038/s41467-019-12281-8
Wang, G. X., Ma, Q. H., Zhao, T. T., and Liang, L. S. (2018a). Resources and production of hazelnut in China. Acta Hortic., 59–64. doi:10.17660/ActaHortic.2018.1226.8
Wang, T., Wei, J. J., Sabatini, D. M., and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-cas9 system. Science 343, 80–84. doi:10.1126/science.1246981
Wang, X., Tu, M., Li, Z., Wang, Y., and Wang, X. (2018b). Current progress and future prospects for the clustered regularly interspaced Short palindromic Repeats (CRISPR) genome editing technology in fruit tree breeding. Crit. Rev. Plant Sci. 37, 233–258. doi:10.1080/07352689.2018.1517457
Wani, I. A., Ayoub, A., Bhat, N. A., Dar, A. H., and Gull, A. (2020). “Hazelnut,” in Antioxidants in vegetables and nuts - properties and health benefits. Editors G. A. Nayik,, and A. Gull (Singapore: Springer Singapore), 559–572. doi:10.1007/978-981-15-7470-2_29
Wong, N., Liu, W., and Wang, X. (2015). Wu-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 16, 218. doi:10.1186/s13059-015-0784-0
Xu, H., Xiao, T., Chen, C.-H., Li, W., Meyer, C. A., Wu, Q., et al. (2015). Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25, 1147–1157. doi:10.1101/gr.191452.115
Keywords: gRNA design, Corylus sp., paralogs, orthology analysis, gene editing
Citation: Amoroso CG and Andolfo G (2024) SiMul-db: a database of single and multi-target Cas9 guides for hazelnut editing. Front. Genet. 15:1467316. doi: 10.3389/fgene.2024.1467316
Received: 19 July 2024; Accepted: 22 November 2024;
Published: 16 December 2024.
Edited by:
Maurice H. T. Ling, University of Newcastle (Singapore), SingaporeReviewed by:
Adison Choonkit Wong, Singapore Institute of Technology, SingaporeDibyajyoti Pramanik, Iowa State University, United States
Copyright © 2024 Amoroso and Andolfo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Andolfo, Z2l1c2VwcGUuYW5kb2xmb0B1bmluYS5pdA==