
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet., 21 October 2024
Sec. Livestock Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1466382
 Tolegen Assanbayev1Rakhmetolla Akilzhanov1Tlekbol Sharapatov1
Tolegen Assanbayev1Rakhmetolla Akilzhanov1Tlekbol Sharapatov1 Rakhimbek Bektayev2Diana Samatkyzy3
Rakhimbek Bektayev2Diana Samatkyzy3 Daniyar Karabayev2
Daniyar Karabayev2 Aidana Gabdulkayum3
Aidana Gabdulkayum3 Asset Daniyarov2,4
Asset Daniyarov2,4 Saule Rakhimova3
Saule Rakhimova3 Ulan Kozhamkulov3Dos Sarbassov5
Ulan Kozhamkulov3Dos Sarbassov5 Ainur Akilzhanova3*
Ainur Akilzhanova3* Ulykbek Kairov2*
Ulykbek Kairov2*The horse (Equus caballus) is a domesticated animal with great significance in human civilization and history, having played a crucial role in transportation, agriculture, and warfare. Over millennia, intentional breeding has resulted in the creation of approximately 500 distinct horse breeds, each selected for specific performance qualities, appearance, and behavior (Petersen et al., 2013). The earliest evidence of horse domestication dates back to the Eneolithic Botai culture (3500 BCE) in prehistoric Northern Kazakhstan, where horses continue to hold cultural significance (Outram et al., 2009; Levine, 1999; Sarbassova, 2015). Although domestication in Botai occurred independently of the main domestication path, horses have been an essential aspect of steppe pastoralism in the region of modern Kazakhstan since the Bronze Age (Kyselý and Peške, 2022; Frachetti and Benecke, 2009; Outram et al., 2012). As a result, traditional selection over hundreds and thousands of years has shaped the Kazakh horse breed (Kabylbekova et al., 2024).
Zhabe is an intrabreed type of Kazakh horse that originated in Western Kazakhstan and is currently used throughout the country (Figure 1A). This type is known for its strong, slightly rough constitution and high endurance. Horses of this type are characterized by a coarse head, a short fleshy neck, a wide and deep body, a broad back, a muscular croup, and strong, bony legs. They also have a thick, long mane and tail, short fetlocks on the legs, and dense skin. Their colors are typically bay or dark red, but can also be mousey, gray, or black (Dmitriev and Ėrnst, 1989). In state farm conditions, Kazakh horses, including Zhabe, have been selectively bred for increased size and weight. They are well-adapted to traditional Kazakh methods of seasonal pasturing and are bred in herds, even inharsh winter climatic conditions, to produce working horses, meat, and milk (Omarov et al., 2019).
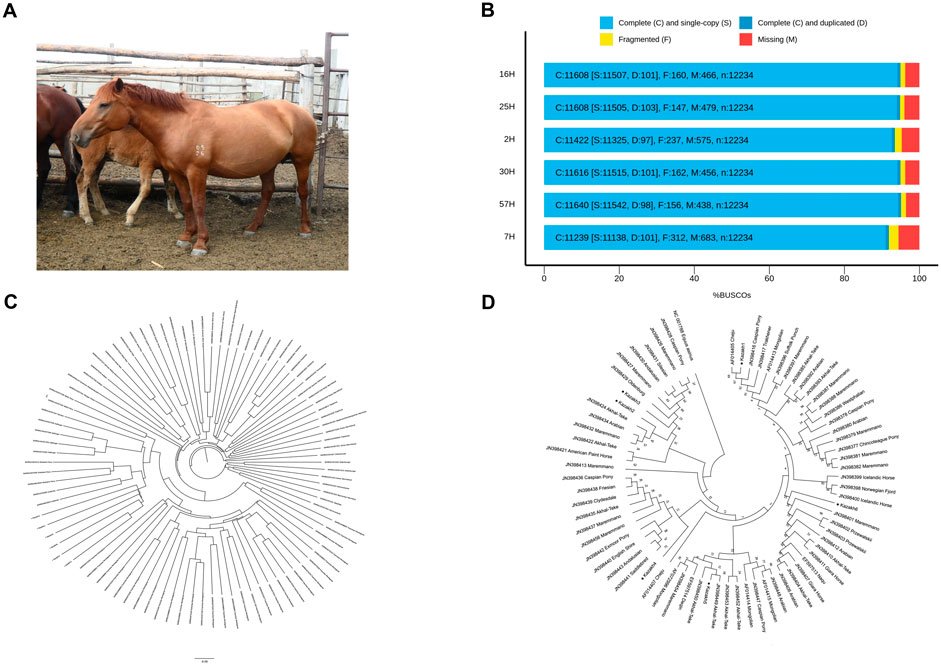
Figure 1. De novo genome assembly of Kazakh horse of Zhabe type. (A) Representative image of Kazakh horse of Zhabe traditional type. (B) BUSCO assessment results: each column represents the percentage of identified BUSCO genes in genome samples. (C) Genomic relationships between the Kazakh horse and other horse breeds shown by a neighbor-joining phylogeny tree (samples 2–57H are respectively presented as Kazakh1-6). (D) Neighbor-joining bootstrap consensus tree reconstructed from the mtDNA control region sequences of Kazakh horse and other horse breeds (samples 2–57H are respectively presented as Kazakh1-6).
Previous studies have characterized Kazakh horses using array-based genotyping, RNA-seq, and WGBS-seq (Pozharskiy et al., 2023; Liu et al., 2018; Yu et al., 2021; Liu et al., 2023). This study presents the first high-quality genome assemblies for six Kazakh horses of the Zhabe type, providing a valuable resource for genetic research and comparative genomics. Conserving genetic diversity is vital for the present and future maintenance of the valuable traits of the breed (Bruford et al., 2015). It is also widely acknowledged that comprehensive molecular genetic data characterizing inter- and intraspecies diversity is important for the efficient management of genetic resources economically important animal varieties (Ruane, 2000; Simianer, 2005; Toro et al., 2009). Here, we present six new de novo genome assemblies, generated using Oxford Nanopore Technology, for Kazakh horses of the Zhabe traditional type.
Peripheral blood samples from six horses (2H, 7H, 16H, 25H, 30H, and 57H) were collected in 1 mL volumes at “Akzhar Ondiris” horse farm (51°32′07.4″N 77°27′16.9″E) in Pavlodar region of Kazakhstan (Figure 1A). All samples were anticoagulated with EDTA and refrigerated at 4°C. The phenotypic characteristics of these horses are detailed in Supplementary Table S1. Genomic DNA was extracted from the samples using Illustra Blood Kit (Cytiva, United State) and Gentra Puregene Blood Kit (Qiagen, Germany) following the manufacturers’ protocols. The concentration and quality of the extracted DNA were checked using a Qubit fluorometer (Invitrogen, United State), a Nanodrop 2000 spectrophotometer (Thermo Scientific, United State), and 1% agarose gel electrophoresis. This high-molecular-weight DNA was then used for library construction and subsequent Nanopore sequencing.
To generate Oxford Nanopore long reads, 3 µg of genomic DNA was randomly sheared to obtain a target size of 20 kbp using g-TUBE (Covaris, United State) and processed according to the Ligation Sequencing Kit (SQK-LSK110) protocol (Oxford Nanopore Technologies, United Kingdom). For genome sequencing, at least 1 µg of sheared DNA from each sample was utilized for library construction. DNA fragments were repaired using NEBNext FFPE Repair Mix (New England Biolabs, United State). End repair and A-tailing were performed using the NEBNext End Repair/dA-Tailing Module kit (New England Biolabs, United State), followed by ligation of Oxford Nanopore sequencing adapters with the NEBNext Quick Ligation Module (E6056) (New England Biolabs, United State). The constructed libraries were sequenced on R9.4.1 flow cells of PromethION sequencer (Oxford Nanopore Technologies, United Kingdom) for 72 h. Basecalling of the raw signal data was performed using Guppy v.5.1.13, which also trimmed adapters and removed low-quality sequencing reads with a Q-score below 9.0. All DNA samples were sequenced with an average coverage of 26X. A summary of the sequenced reads is provided in Supplementary Table S2.
Draft assemblies were produced using one round of Flye v.2.9.2 (Kolmogorov et al., 2019), followed by a polishing round with Oxford Nanopore Technologies (ONT) reads using Medaka v.1.11.1 (https://github.com/nanoporetech/medaka). To evaluate the quality of the final assemblies, we aligned the ONT contigs to EquCab3.0 reference genome assembly (NCBI Accession No. GCF_002863925.1) and assessed them with QUAST v.5.2.0 (Gurevich et al., 2013). Considering the advanced sequencing ability of ONT, the longest contig among the assembled genomes was 92.32 Mb, and the largest contig N50 was 28.26 Mb. The completeness of the genome assemblies was further assessed using BUSCO v.5.4.6 (Simão et al., 2015), which compared the genome against the laurasiatheria_odb10 database containing 12,234 orthologous genes. BUSCO assessment scores ranged from 93% to 95% (Figure 1B; Table 1), indicating high completeness for the obtained assemblies.
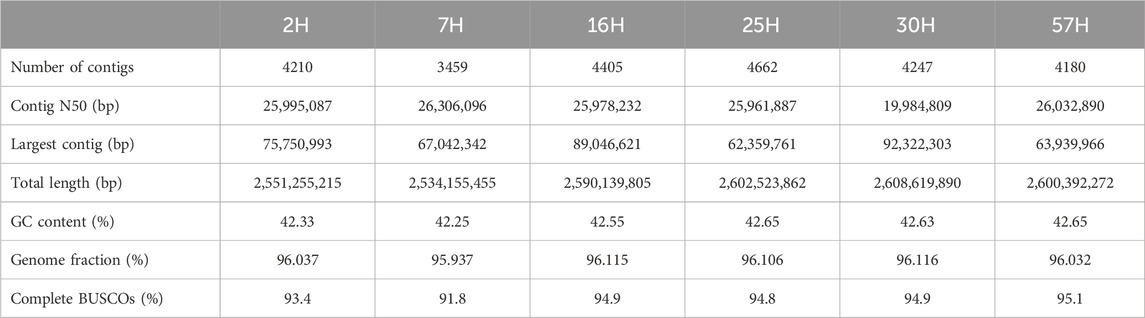
Table 1. QUAST metrics and BUSCO assessment results of the sequencing data.
To identify SNVs and indels, the wf-human-variation Epi2Me Labs pipeline from ONT (https://github.com/epi2me-labs/wf-human-variation) was used. Samples were analyzed using Clair3 v.1.0.4, which identified small variants in ONT reads (Supplementary Table S3). The number of identified SNVs ranged from 6,336,129 to 7,101,556, while the number of identified indels ranged between 549,718 and 820,662 across samples.
Phylogenetic analysis and tree construction were performed using VCF-kit v.0.2.6 (Cook and Andersen, 2017) and MEGA software v.11.0.13 (Tamura et al., 2021). The neighbor-joining tree was constructed using 1,331,674 mutation points from a merged VCF file (Figure 1C) containing data from Kazakh horses and 88 additional horse samples (Jagannathan et al., 2019) deposited in the European Nucleotide Archive (ENA) database (https://www.ebi.ac.uk/ena/). At the autosomal genetic level, Kazakh Zhabe horses formed a distinct cluster and a separate group compared to other horse breeds. Additionally, a multiple sequence alignment of all mitochondrial D-loop sequences was performed in MEGA using the built-in MUSCLE (Edgar, 2004) alignment option to construct a consensus tree. The analysis included 71 samples of the control region and mtDNA from our assemblies, as well as 25 different horse breeds deposited in the National Center for Biotechnology Information (NCBI) GenBank database (http://www.ncbi.nlm.nih.gov/). All sequences were processed using blastn v.2.12.0+ (Camacho et al., 2009) to extract an early part of the control region (400 bp in the position between 15,469 and 15,868). The consensus tree (Figure 1D) was built using the Neighbor-Joining method (Saitou and Nei, 1987) with 1,000 bootstrap iterations. The D-loop region sequence of the donkey (Equus asinus, NCBI Accession No. NC001788) was used as an outgroup. While phylogeny reconstruction showed Kazakh horse mtDNA sequences are widespread and distributed across many different clusters in the tree, two samples (Kazakh1 and Kazakh5) from the assembled genomes formed a distinct clade with Cheju and Akhal-Teke horses. These results are consistent with previous studies (Gemingguli et al., 2016) reporting tightly linked mtDNA genetic relationships between these breeds. It can be suggested that the Kazakh horse breed has a mixed origin in the maternal lineage, likely due to the use of horse populations in trade and military campaigns, which moved them to distant locations, where they interbred with indigenous populations. The observed lack of Kazakh horse samples clustering in the phylogenetic tree constructed from mtDNA control region sequences may indicate high levels of variability, which, in turn, means that the Kazakh breed may serve as an important reservoir of genetic biodiversity. It is of particular significance for horses, as a species, because its wild ancestors are now extinct and sources of biodiversity that could be used to maintain their functions in certain environments are limited.
All sequence data presented in this study are deposited in the NCBI Sequence Read Archive (SRA) repository and are publicly available under accession numbers SRX18227458-18227464. The obtained genome assemblies were submitted and registered under the following NCBI GenBank accession numbers: GCA_029814115.1, GCA_029814095.1, GCA_029784105.1, GCA_029814075.1, GCA_029784085.1, GCA_029814055.1.
The animal study was reviewed and approved by the Ethics Committee of National Laboratory Astana, Nazarbayev University. The study was conducted in accordance with the local legislation and institutional requirements.
TA: Conceptualization, Project administration, Resources, Writing–review and editing. RA: Conceptualization, Project administration, Resources, Writing–review and editing. TS: Conceptualization, Project administration, Resources, Writing–review and editing. RB: Formal Analysis, Investigation, Software, Visualization, Writing–original draft. DiS: Methodology, Validation, Writing–review and editing. DK: Formal Analysis, Investigation, Software, Visualization, Writing - original draft. AG: Methodology, Validation, Writing–review and editing. AD: Data curation, Software, Writing–review and editing. SR: Investigation, Methodology, Validation, Writing–review and editing. UK: Investigation, Methodology, Validation, Writing–review and editing. DoS: Conceptualization, Funding acquisition, Supervision, Writing–review and editing. AA: Conceptualization, Funding acquisition, Investigation, Supervision, Writing–review and editing. UK: Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Visualization, Writing–original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research has been funded by the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan program targeted funding #AP14869903 and #BR18574184.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1466382/full#supplementary-material
Bruford, M. W., Ginja, C., Hoffmann, I., Joost, S., Orozco-terWengel, P., Alberto, F. J., et al. (2015). Prospects and challenges for the conservation of farm animal genomic resources, 2015-2025. Front. Genet. 6, 314. doi:10.3389/fgene.2015.00314
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinforma. 10, 421–429. doi:10.1186/1471-2105-10-421
Cook, D. E., and Andersen, E. C. (2017). VCF-kit: assorted utilities for the variant call format. Bioinformatics 33, 1581–1582. doi:10.1093/bioinformatics/btx011
Dmitriev, N. G., and Ėrnst, L. K. (1989). Animal genetic resources of the USSR (Rome: Food and Agriculture Organization of the United Nations).
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids Res. 32, 1792–1797. doi:10.1093/nar/gkh340
Frachetti, M., and Benecke, N. (2009). From sheep to (some) horses: 4500 years of herd structure at the pastoralist settlement of Begash (south-eastern Kazakhstan). Antiquity 83, 1023–1037. doi:10.1017/S0003598X00099324
Gemingguli, M., Iskhan, K. R., Li, Y., Qi, A., Wunirifu, W., Ding, L. Y., et al. (2016). Genetic diversity and population structure of Kazakh horses (Equus caballus) inferred from mtDNA sequences. Genet. Mol. Res. 15. doi:10.4238/gmr.15048618
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi:10.1093/bioinformatics/btt086
Jagannathan, V., Gerber, V., Rieder, S., Tetens, J., Thaller, G., Drögemüller, C., et al. (2019). Comprehensive characterization of horse genome variation by whole-genome sequencing of 88 horses. Anim. Genet. 50, 74–77. doi:10.1111/age.12753
Kabylbekova, D., Assanbayev, T. S., Kassymbekova, S., and Kantanen, J. (2024). Genetic studies and breed diversity of Kazakh native horses: a comprehensive review. Adv. Life Sci. 11, 18–27.
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi:10.1038/s41587-019-0072-8
Kyselý, R., and Peške, L. (2022). New discoveries change existing views on the domestication of the horse and specify its role in human prehistory and history–a review. Archeol. Rozhl. 74, 299–345. doi:10.35686/AR.2022.15
Levine, M. A. (1999). Botai and the origins of horse domestication. J. Anthropol. Archaeol. 18, 29–78. doi:10.1006/jaar.1998.0332
Liu, L., Zhang, Y., Ma, H., Cao, H., and Liu, W. (2023). Integrating genome-wide methylation and transcriptome-wide analyses to reveal the genetic mechanism of milk traits in Kazakh horses. Gene 856, 147143. doi:10.1016/j.gene.2022.147143
Liu, L. L., Fang, C., and Liu, W. J. (2018). Identification on novel locus of dairy traits of Kazakh horse in Xinjiang. Gene 677, 105–110. doi:10.1016/j.gene.2018.07.009
Omarov, M., Akimbekov, A., Assanbayev, T., Temirzhanova, A., Ussenova, L., Uahitov, Z., et al. (2019). Meat and dairy productivity of Jabe Kazakh horses of different factory lines. Ad alta-Journal Interdiscip. Res. 9, 81–89.
Outram, A. K., Kasparov, A., Stear, N. A., Varfolomeev, V., Usmanova, E., and Evershed, R. P. (2012). Patterns of pastoralism in later Bronze Age Kazakhstan: new evidence from faunal and lipid residue analyses. J. Archaeol. Sci. 39, 2424–2435. doi:10.1016/j.jas.2012.02.009
Outram, A. K., Stear, N. A., Bendrey, R., Olsen, S., Kasparov, A., Zaibert, V., et al. (2009). The earliest horse harnessing and milking. Science 323, 1332–1335. doi:10.1126/science.1168594
Petersen, J. L., Mickelson, J. R., Cothran, E. G., Andersson, L. S., Axelsson, J., Bailey, E., et al. (2013). Genetic diversity in the modern horse illustrated from genome-wide SNP data. PloS one 8, e54997. doi:10.1371/journal.pone.0054997
Pozharskiy, A., Abdrakhmanova, A., Beishova, I., Shamshidin, A., Nametov, A., Ulyanova, T., et al. (2023). Genetic structure and genome-wide association study of the traditional Kazakh horses. animal 17, 100926. doi:10.1016/j.animal.2023.100926
Ruane, J. (2000). A framework for prioritizing domestic animal breeds for conservation purposes at the national level: a Norwegian case study. Conserv. Biol. 14, 1385–1393. doi:10.1046/j.1523-1739.2000.99276.x
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi:10.1093/oxfordjournals.molbev.a040454
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi:10.1093/bioinformatics/btv351
Simianer, H. (2005). Decision making in livestock conservation. Ecol. Econ. 53, 559–572. doi:10.1016/j.ecolecon.2004.11.016
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi:10.1093/molbev/msab120
Toro, M. A., Fernández, J., and Caballero, A. (2009). Molecular characterization of breeds and its use in conservation. Livest. Sci. 120, 174–195. doi:10.1016/j.livsci.2008.07.003
Keywords: Kazakh horse, oxford nanopore technologies (ONT), de novo assembly, Kazakhstan, whole genome sequencing (WGS)
Citation: Assanbayev T, Akilzhanov R, Sharapatov T, Bektayev R, Samatkyzy D, Karabayev D, Gabdulkayum A, Daniyarov A, Rakhimova S, Kozhamkulov U, Sarbassov D, Akilzhanova A and Kairov U (2024) Whole genome sequencing and de novo genome assembly of the Kazakh native horse Zhabe. Front. Genet. 15:1466382. doi: 10.3389/fgene.2024.1466382
Received: 17 July 2024; Accepted: 07 October 2024;
Published: 21 October 2024.
Edited by:
Filippo Biscarini, National Research Council (CNR), ItalyReviewed by:
Giulia Moscatelli, National Research Council (CNR), ItalyCopyright © 2024 Assanbayev, Akilzhanov, Sharapatov, Bektayev, Samatkyzy, Karabayev, Gabdulkayum, Daniyarov, Rakhimova, Kozhamkulov, Sarbassov, Akilzhanova and Kairov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ainur Akilzhanova, YWtpbHpoYW5vdmFAbnUuZWR1Lmt6; Ulykbek Kairov, dWx5a2Jlay5rYWlyb3ZAbnUuZWR1Lmt6
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.