 Yuanyuan Jiang
Yuanyuan Jiang Ping Jia
Ping Jia Xiaoying Feng
Xiaoying Feng Dingding Zhang
Dingding Zhang- 1Marfan Research Group, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
- 2Department of Neurosurgery Nursing, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
- 3College of Medical Technology, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 4Sichuan Provincial Key Laboratory for Genetic Disease, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
Marfan syndrome (MFS) is an inherited disorder that affects the connective tissues and mainly presents in the bones, eyes, and cardiovascular system, etc. Aortic pathology is the leading cause of death in patients with Marfan syndrome. The fibrillin-1 gene (FBN1) is a major gene involved in the pathogenesis of MFS. It has been shown that the aortic pathogenesis of MFS is associated with the imbalances of the transforming growth factor-beta (TGF-β) signaling pathway. However, the exact molecular mechanism of MFS is unclear. Animal models may partially mimic MFS and are vital to the study of MFS. Several species of animals have been used for MFS studies, including chicks, cattle, mice, pigs, zebrafishes, Caenorhabditis elegans, and rabbits. These models were developed spontaneously or in combination with genetic engineering techniques. This review is to describe the TGF-β signaling pathway in MFS and the potential application of animal models to provide new therapeutic strategies for patients with MFS.
1 Introduction
Marfan syndrome (MFS, OMIM: #154700) is a complex systemic connective tissue disorder that is generally inherited in an autosomal dominant manner. Aortic pathology is the primary factor causing death in patients with MFS (Lazea et al., 2021; Spencer, 2024). Multiple bone defects were first reported in 1896 by French pediatrician Antoine-Bernard and officially named MFS in 1931 (Lloyd, 1934). In McKusick. (1955) proposed that MFS is a genetic disease of connective tissue from a clinical point of view. It was not until 1986 that Sakai et al. (1986) isolated a new connective tissue protein called fibrillin-1 from human fibroblast culture medium. Fibrillin was found abundant in tissues affected in MFS patients, particularly in the aortic root, acute aortic coarctation, disproportionate growth of long bones and lens ectasia (Lazea et al., 2021). The prevalence of MFS is 20/100,000, but there isn’t any clinically confirmed MFS prevalence rates on the basis of Ghent-I or Ghent-II nosology (Groth et al., 2015). First survey of MFS prevalence in the Danish Unified Healthcare System was based on Ghent-II nosology. Groth et al. (2015) presented a prevalence of MFS of 6.5/100,000 in Denmark in 2014, and the prevalence of MFS in Denmark in 2014 is 41% higher than that of the Danish prevalence rate published almost 20 years ago. (4.6/100,000) (Fuchs, 1997). In the 1970s, the average life expectancy for MFS patients was 32 years. The introduction of aortic root replacement therapy led to a rise in the average life expectancy of MFS patients to 41 years in 1995. Today, most MFS patients can live to around 72 years with proper management (Coelho and Almeida, 2020).
This disease can frequently lead to fatal cardiovascular disease in the neonatal period or as progressively more severe cardiovascular disease in adolescence and adulthood. Veiga-Fernandez et al. (2020) demonstrated that the mortality rate within the initial 15 months of prenatal suspected cases of early-onset MFS (EOMS) was 73.68%, and the proportion of deaths in the first 5 years of postnatal confirmed patients was 61.1%. Because cardiovascular complications may not occur in early stages of MFS (Wozniak-Mielczarek et al., 2019; van Elsacker et al., 2022), early diagnosis of MFS is necessary though difficult.
Thoracic aortic disease in most patients with MFS begins as an asymptomatic enlargement of the aortic root, which gradually increases in size over time to form an aneurysm. The enlargement of the aortic aneurysm may eventually lead to acute ascending aortic dissection (called type A dissection) (Kraehenbuehl et al., 2012). Type A dissection is a life-threatening complication of MFS that can lead to a shortened life expectancy in MFS (Wang et al., 2021; Asano et al., 2022; Farag et al., 2023). Remarkably, a minority of MFS patients have type B aortic dissection. Typically, type B aortic dissection occurs without significant enlargement of the descending aorta and enlargement of the aortic root. Type B aortic dissection is less acutely fatal than in MFS patients with type A aortic dissection (Kraehenbuehl et al., 2012; Yildiz et al., 2023). Type A aortic dissection correlates with high morbidity and mortality, with the majority of people with MFS dying of complications of aortic dissection or rupture until the advent of aortic surgery, mostly before the age of 45 years ago (Silverman et al., 1994; Sharma, 2009). Presently, proper diagnosis and treatment of thoracic aortic aneurysms in patients with MFS can prevent most acute type A aortic dissection. With the development of aortic root replacement therapy since the 1970s, the life expectancy of patients with MFS approaches that of the general population (Kuzmanovic et al., 2004; Song et al., 2012; Asano et al., 2022).
In addition to thoracic aortic disease, MFS impacts other organs and tissues of the patients. Symptoms include: tall stature, disproportionately long limbs, abnormally curved spine, protruding thorax, depressed sternum, atrophied lungs, abnormally depressed acetabulum, enlarged spinal canal of the lumbar vertebral segments, and tattooing of the skin (Loeys et al., 2010; Pollock et al., 2021). Certain presentations of MFS exhibit similarities with other conditions, such as Loeys-Dietz syndrome (LDS) and Shprintzen-Goldberg syndrome (SGS) (Verstraeten et al., 2016). The clinical diagnosis of MFS requires the identification of features present throughout the system, which can be assisted by genetic testing. The MFS Ghent nomenclature was revised in 2010 to emphasize the importance of FBN1 testing (Loeys et al., 2010).
Approximately 3,077 mutations in FBN1 have been reported to date, with 1815 missense mutations (http://www.umd.be/FBN1/). More than 1,700 FBN1 mutations have been identified to be potentially contributing to the development of MFS (Groth et al., 2017). The NCBI database identifies 505 organisms that are homologous to human FBN1 (https://www.ncbi.nlm.nih.gov/gene/2200/ortholog/?scope=7776&term=FBN1), such as mouse, rat, pig and zebrafish. Using these animals with specific pathogenic variants as models is considered suitable for studying the early-onset and severe symptoms of MFS(Tae et al., 2016; de Souza et al., 2021). It is therefore critical to understand how alterations in FBN1 lead to this multi-effect pathophysiology to determine appropriate therapies. Basic research in animal models of MFS and clinical trials of molecularly-targeted drugs have provided new therapeutic strategies for patients with MFS.
2 MFS related genes
In Sakai et al. (1986) proposed the diagnostic criteria for MFS. The initial set of criteria was formulated in Berlin in 1986. These criteria were established primarily to help clinicians to determine which patients should be categorized as suffering from this disease. In 1996, new diagnostic criteria called Ghent-I nosology (Ghent I) was established. FBN1 gene was identified as a susceptibility gene for MFS (DePaepe et al., 1996). The Ghent-II nosology were modified in 2010. Loeys et al. (2010) underlined the importance of thoracic aortic disease. The revised Ghent-II nosology specifically focus on the significant overlap between Sphrintzene-Goldberg syndrome (SGS), Loeyse-Dietz syndrome (LDS), and MFS, including potentially similar involvement of bones, aortic roots, skin, and dura mater. Occasionally, SGS and vascular Ehlerse-Danlos syndrome (vEDS) overlap with that of MFS in the vascular system, dura mater, skin, and bone. The difference between MFS and related diseases is shown in Table 1. These diseases, along with non-syndromic aneurysmal syndromes, are linked to abnormal TGF-β signaling (Verstraeten et al., 2016).
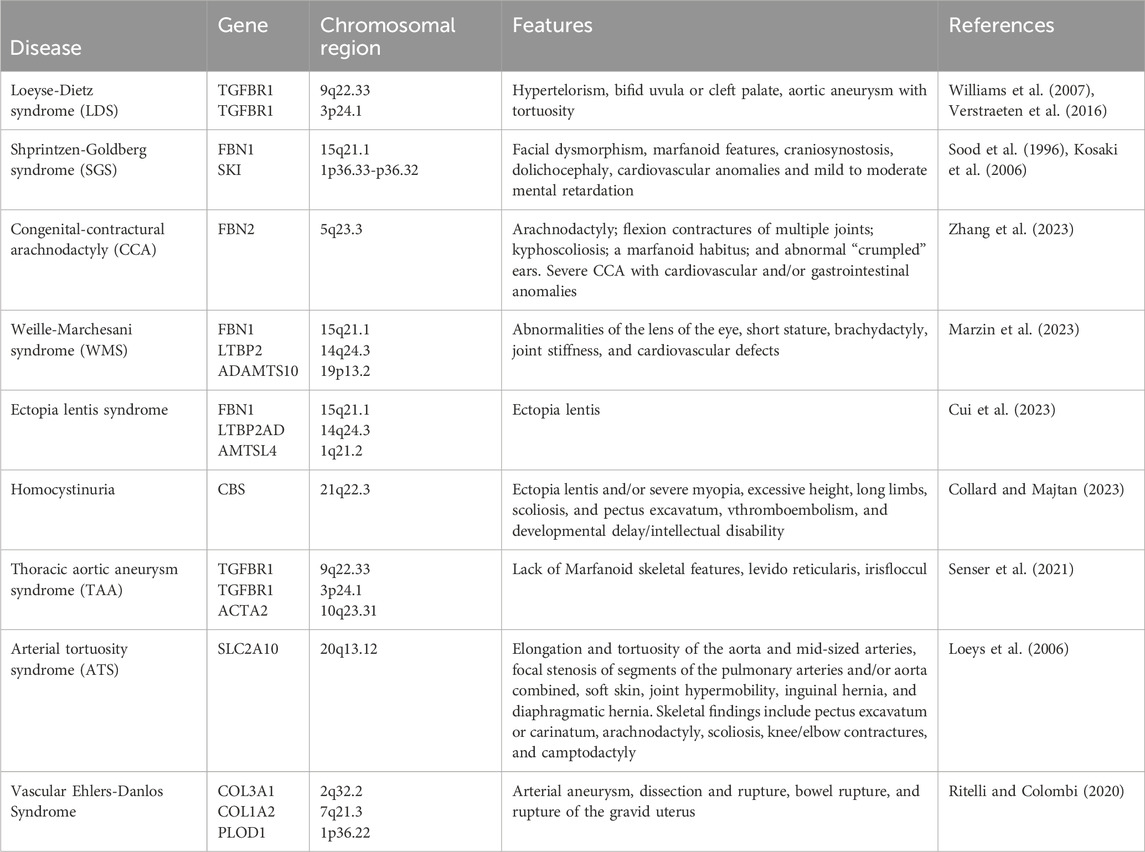
Table 1. Differences between MFS and related diseases.
2.1 FBN1 gene in MFS
The FBN1 gene is located at 15q21.1, the cDNA is about 200 kb long and contains 65 exons with GC-rich sequences upstream of the exons (Gong et al., 2019; Lin et al., 2021). The FBN1 precursor consists of 2,871 amino acids and contains a total of 6 structural regions FBN1 mutations have been observed in more than 90% of the cases of MFS (Manuel Becerra-Munoz et al., 2018). Mutations in FBN1 occur throughout most the gene. FBN1 missense mutations account for 53%-56.1%, truncation variants 33%-36.8%, intronic variants 7.1%-13%, and total genomic rearrangements 1.8%-2.9% (Yang et al., 2018; Arnaud et al., 2021). Identification of FBN1 genotypes for specific MFS phenotypes is complicated by the interfamilial and intrafamilial variability in the clinical features of MFS. Missense mutations in exons 24 to 32 are associated with severe EMOS (Milewicz and Duvic, 1994; Faivre et al., 2009).
2.2 FBN2 gene in MFS
The fibrillin-2 gene (FBN2, formerly known as Fib5) on chromosome 5q23-q31 is inextricably linked to fibrillin-1. The two proteins have the exact same structural domain structure as well as the same number and order of sequence motifs. At the amino acid level, structural domains B and D of fibrillin-1 and fibrillin-2 are 80% identical (Zhang et al., 1995; Dietz et al., 2005; Beene et al., 2013). Studies have demonstrated that the developmental expression of FBN2 precedes that of FBN1 (Robinson and Godfrey, 2000). FBN2 is involved in the development of elastic fiber formations, while FBN1 primarily preserves the functionality of elastic structures. FBN2 is universally expressed in elastic tissues, but FBN1 is primarily located in tissues that are subjected to stress and weight-bearing. Gupta et al. found the FBN2 gene mutation sites in a premature infant who met the diagnostic criteria for MFS and her brother as well. Gupta et al. (2002), Gupta et al. (2004) presented MFS due to FBN2 gene mutation in three children under 6 years in Mexico, with persistent dilatation by aortic echocardiography for more than 5 years.
2.3 TGFBR genes in MFS
Stheneur et al. (2008) showed that in 457 patients with MFS or related disorders the detection rates of TGFBR1/2 mutated genes were 6.2% and 4.8% in classical MFS and 6.2% and 4.6% in incomplete classical MFS. De Cario et al. (2018) found in 75 patients with MFS that TGFBR2 had a total of 10 polymorphisms in TGFBR2 and 6 polymorphisms in TGFBR1. TGFBR1 is located on chromosome 9q22.33 and consists of nine exons encoding the TGFBR1 protein. Somers et al. (2016) proposed that the 6Ala allele of the TGFBR1 could be regarded as a low-frequency variant in MFS patients. TGFBR2 is located on chromosome 3p24.1 (Mizuguchi et al., 2004). Mutations in the TGFBR2 are tied to MFS in individuals who do not exhibit significant ocular symptoms (Bitarafan et al., 2020). In nematode models, mutations in TGFBR2 associated with MFS or MFS-like syndromes might cause structural perturbations in TGFBR2, leading to the exposure of surface structural domains, changes in subcellular localization patterns, and effect the transport of TGFBR1 indirectly (Singh et al., 2006; Lin et al., 2019).
2.4 LTBP genes in MFS
The underlying TGF-β binding protein (LTBP) is a protein that targets TGF-β to the ECM by interacting with fibrillin-1 (Todorovic et al., 2005). Fibrillin is structurally related to the LTBP gene family out of which four have been identified (LTBP1, LTBP2, LTBP3, and LTBP4). All of these genes contain multiple tandem copies of the cb-EGF motif with two distinct 8-cysteine repeats in fibronectin and LTBP. One of these is the LTBP motif, which contains 8 cysteine residues that cluster internally in Marfan syndrome and associated microfibrillar diseases (Rifkin et al., 2018).
Sticchi et al. (2018) identified a patient with MFS in aortic symptoms of mutations in the FBN1, NOTCH1, LTBP1, and TGFBR3 genes. Ramona et al. showed that c.1642C > T (p.Arg548*) of LTBP2 may be associated with ocular manifestations of MFS, MVP and funnel chest. In 2010, Desir et al. observed that MFS might be related to mutations in the LTBP2 gene (Desir et al., 2010). In 2019, Morlino et al. noted that two Romani individuals exhibited a phenotype resembling MFS due to the presence of the homozygous p.R299X variant in the LTBP2 (Morlino et al., 2019). Bertoli-Avella et al. indicated that patients with LTBP3 gene mutations exhibited elastic fiber breakage, as well as increased accumulation of collagen and proteoglycans within the aortic wall tissue (Bertoli-Avella et al., 2015). They found that individuals with mutations in this gene exhibited severe cardiovascular symptoms that closely resembled those in MFS patients. Korneva et al. (2019) detected that the absence of LTBP3 attenuated elastic fiber breakage and focal dilatation in a mouse model of MFS lacking fibrillin-1. However, MFS mouse models with spinal deformities persisted in the absence of LTBP3 (Zilberberg et al., 2015). MFS mice lacking LTBP3 had increased survival and suppressed Smad2/3 and Erk1/2 activation in the aorta. Aortic aneurysms disappeared in MFS mice (Bertoli-Avella et al., 2015; Zilberberg et al., 2015). These data suggested that the latent TGF-β complex composed of LTBP3/TGF-β may contribute to the progression of aortic disease in MFS.
3 TGF-β pathway abnormalities in MFS
TGF-β family play critical roles in embryonic development, adult tissue homeostasis and repair. Genetic studies in animals demonstrated that the TGF-β signaling pathway was correlated with MFS (Goumans and ten Dijke, 2018; Gensicke et al., 2020). An integral role of the TGF-β signaling pathway in the pathogenesis of MFS thoracic aortic aneurysms was identified by knocking out the FBN1 gene in MFS model mice (Gensicke et al., 2020). FBN1 protein modulates the levels of activated TGF-β in the extracellular matrix through its interaction with a complex comprising LAP, LTBP, and TGF-β. Extracellular activation of this complex is an essential condition for the biological activity of TGF-β regulation. Upon activation of this complex, TGF-β transmits signals to two membrane-bound TGF-β receptors via costimulatory binding. Furthermore, signals are transmitted from the cell membrane to the nucleus via Smad-dependent or Smad-independent pathways (Derynck and Zhang, 2003; Shi and Massagué, 2003; Tang et al., 2018). FBN1 protein mutations causing abnormal TGF-β pathway signalling in MFS are displayed in Figure 1.
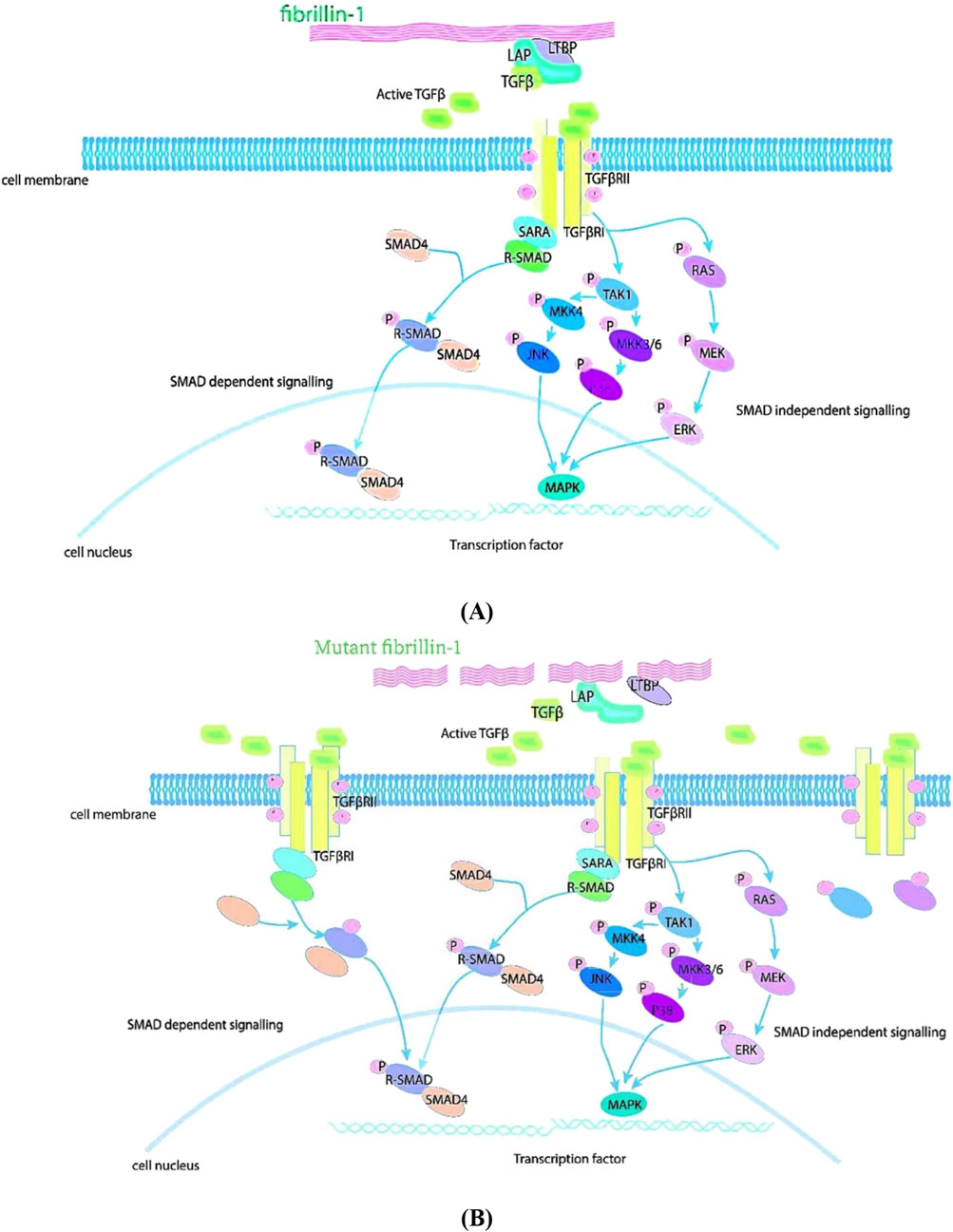
Figure 1. TGF-β pathway signaling in MFS. FBN1 has a role in regulating TGF-β. (A) FBN1 binds to a large latent complex composed of LAP, LTBP and TGF-β and regulates the concentration of activated TGF-β in the matrix. Upon activation of the complex, TGF-β binds TGF-β receptors and signals. This signal can be transmitted from the cell membrane to the nucleus via Smad-dependent or Smad-independent pathways. (B) Mutations in FBN1 cause the release of large amounts of active TGF-β1 from the extracellular matrix, leading to over-activation of the TGF-β signaling pathway and accelerating the destruction of the extracellular matrix.
3.1 SMAD dependent signaling
The inactive TGF-β precursor consists of 390–442 amino acids, and the TGF-β precursor has three specialized regions consisting of an N-terminal hydrophobic signal peptide region, a 249-amino-acid latently relevant peptide region, and a C-terminal region (Shi and Massagué, 2003; Goumans and ten Dijke, 2018; Tang et al., 2018). Inactive TGF-β precursors are endopeptidase cleaved in the Golgi apparatus, and thereby forms a small latent complex. The mature TGF-β homodimer contained therein is formed by non-covalent bonding with two latent peptides and is normally secreted by the cell as a large latent complex together with LTBP. LTBP anchors inactive TGF-β to the extracellular matrix. Inactive TGF-β releases biologically active TGF-β from the latent complex through the interaction of latent-associated peptides with a variety of proteins. Upon binding of activated TGF-β to cell-surface receptors, TGF-β ligands stimulate the assembly of serine/threonine kinase complexes, which pass through the cytoplasm and protein phosphorylation initiates signal transduction (Shi and Massagué, 2003; Akhurst and Hata, 2012; Goumans and ten Dijke, 2018).
Activated TGF-βR1 specifically recognizes and phosphorylates R-Smad (receptor-regulated Smads). The R-Smad substrate activates the TGF-β receptor complex via the SMAD receptor activation anchor (SARA). Phosphorylation of R-Smad reduces its affinity for SARA and leads to the formation of a heterodimer with Smad4. Activated R-Smad forms a heterodimeric complex with Smad4. The heterodimeric complex then rapidly translocates to the nucleus. Upon entry into the nucleus, this complex interacts with transcription factors containing sequence-specific DNA-binding affinities at promoter sites to regulate gene expression. Smad4 is translocated to the nucleus only when bound to R-Smad, which can autonomously move from the cytoplasm to the nucleus in the absence of Smad4. However, when Smad4 is blocked or absent, R-Smad can translocate but lacks the ability to completely signal the nucleus through gene expression. This implies that the primary role of Smad4 is to regulate transcription rather than to transmit TGF-β signaling from the cytoplasm to the nucleus (Johnsen et al., 2002; Ten Dijke et al., 2002; Derynck and Zhang, 2003; Shi and Massagué, 2003).
3.2 SMAD independent signaling
In addition to Smad-independent transcription, TGF-β activates Smad independent signaling cascades. Smad independent signaling pathway includes the Erk, JNK, and p38 MAPK kinase pathways, and the mechanisms by which TGF-β activates Erk, JNK or p38 MAPK and their biological consequences remain to be elucidated. JNK and p38 MAPK signaling is activated by various MAPK kinases (MAPKKKs) in response to many stimuli. Rapid activation of Ras by TGF-β is associated with the participation of Ras in TGF-β induced Erk MAPK signaling (Derynck and Zhang, 2003; Holm et al., 2011; Mu et al., 2012). Smad independent signaling such as NO, angiotensin, WNT, NOTCH and PI3K/AKT are also related to MFS progression (Nataatmadja et al., 2013; Dong et al., 2023).
Gensicke et al. (2020) analyzed the expression levels of the atypical regulators ERK and p38 in cardiac tissues of the FBN1C1039G/+ MFS mouse model by Western blotting. Rouf et al. (2017) measured mouse aneurysms by thoracic echocardiography and determined levels of phosphorylated Erk1/2 (p-Erk1/2) and pSmad2 in aortic tissue. These results indicated that aneurysms in MFS model mice are linked to Erk1/2 Smad and p38 signaling. Sato et al. (2018) showed that the downstream protein of Ras, pRaf1, and pERK1/2 were significantly elevated by Western blotting. Inhibition of the Ras-induced Erk signaling pathway reduced aneurysm growth in mice. These results showed that activation of Ras/Erk MAPK signaling could induce TGF-β expression, being expected to result in aortic aneurysm growth. Chung et al. (2007) were the first ones to demonstrate that endothelial dysfunction in the thoracic aorta of patients with MFS was probably due to downregulation of NOS-induced NO production by Akt or endothelial cells. de la Fuente-Alonso et al. (2021) reported that NO overactivation of sGC-PRKG signaling induced MFS thoracic aortic disease. These pointed to the importance of NO in the progression of thoracic aortic lesions. Angiotensin II (Ang-II) directly induced aortic dilatation in a mouse model of MFS, affecting TGF-β synthesis and receptor expression. Ang-II interacts with TGF-β signaling (Yu and Jeremy, 2018). Jespersen et al. (2022) detected Notch 3 expression in aortic tissue from the FBN1mgR/mgR MFS mouse model and human MFS. Notch3 levels were elevated in both the MFS mouse model and human MFS. Thus, aortic abnormalities in MFS are combined with increased Notch3 activation. Granata et al. (2017) modeled the vasculature of human induced pluripotent stem cells (MFS-hipscs). Smooth muscle cells (SMCs) of MFS-hipscs origin recapitulated the pathology of Marfan’s aorta with high levels of KLF4 and P-p38, which was validated in patient samples. These results suggested that krppel-like factor 4 (KLF4) controls the p38 pathway to regulate SMC apoptosis. Activation of the mechanistic target of rapamycin (mTOR) pathway is also thought to play a role in aortic aneurysm formation in patients with MFS. The mTOR signaling pathway was shown to be significantly activated during aneurysm development, and inhibition of mTOR signaling reduced the development of aortic coarctation in animal models (Sarbassov et al., 2005; Laplante and Sabatini, 2012; Saxton and Sabatini, 2017).
4 Treatment of MFS
Pharmacological treatments for MFS aim to limit the rate of aortic dilatation and slow the progression of cardiovascular disease. However, MFS cannot be completely cured, and research into drug therapy remains a hot topic (Chiu, 2022).
β-blockers have been recommended as first-line therapeutic agents for MFS(Bowman et al., 2019; Deleeuw et al., 2021). β-blockers are classic drugs that selectively bind to β-adrenergic receptors, thereby antagonizing the agonistic effects of neurotransmitters and catecholamines on β receptors (Koo et al., 2017). These drugs reduce heart rate and myocardial contractility by blocking β1 and β2 receptors distributed in the heart and blood vessels, thereby reducing myocardial oxygen consumption (Fici et al., 2023). Furthermore, β-blockers inhibit the release of renin and decrease the production of angiotensin II, thus reducing systemic vascular resistance (Paolillo et al., 2021). This mechanism of action not only helps to reduce cardiac afterload and lower blood pressure, but also reduces dilatory pressure in the aortic root (Messerli et al., 2023). In patients with Marfan syndrome, dilatation of the aortic root is a serious problem that can lead to fatal aortic coarctation or rupture. β-blockers mitigate the likelihood of these complications by decelerating the rate of aortic root dilation through the above mechanisms. Several clinical trials have confirmed that β-blockers effectively reduce the rate of aortic root dilation in MFS patients (Koo et al., 2017; Jondeau et al., 2023). Although β-blockers have demonstrated efficacy in the management of Marfan syndrome, they may be associated with some side effects. Common cardiovascular adverse effects include hypotension and bradycardia, which may lead to exacerbation of heart failure in severe cases, especially in elderly patients or at high doses (Richards and Tobe, 2014; Vargas et al., 2023). It may induce or exacerbate bronchial asthma due to the blocking of β2 receptors on bronchial smooth muscle. Hypogonadism, anaphylactic rash, and gastrointestinal distress are also common side effects (Tatu et al., 2019). Therefore, the challenge of using β-blockers as a primary therapy for the prevention of aortic complications in patients with MFS is enormous.
In mice model of MFS, angiotensin receptor enkephalin inhibitors delay ascending aortic dilatation more than ARB alone (Yu and Jeremy, 2018). Thus, angiotensin receptor enkephalin inhibitor therapy may be a novel approach for the treatment of MFS (Brooke et al., 2008; Habashi et al., 2011). Angiotensin-converting enzyme inhibitors (ACEIs) reduce arterial pressure and delay atherosclerosis. Mutations in the FBN1 gene enhance TGF-β signaling, whereas ACEIs reduce TGF-β signaling (Cavanaugh et al., 2017a). Therefore, ACEIs can be recommended for the treatment of patients with MFS. The study of animal models of MFS such as zebrafish, mice, pigs and in vitro models of human induced pluripotent stem cells would also help to determine the efficacy and safety of novel drugs for MFS.
“Bentall-de Bono” aortic root replacement has enhanced the lifespan of MFS patients (Ornelas-Casillas and Garcia-Arias, 2022). This surgery should be considered as early as possible when patients with MFS are labelled as high risk for ascending aortic risk factors (Dallan et al., 2021; Hlavicka et al., 2022; Ornelas-Casillas and Garcia-Arias, 2022). Lens ectasia is a common manifestation of ocular involvement in patients with MFS. Eye complications such as glaucoma and long-term blindness can be prevented by early surgical intervention. Sahay et al. described a microscope-guided lens aspiration technique that can be applied to treat anterior lens dislocation in children (Sahay et al., 2019). About 60% of patients with MFS have scoliosis. When the condition is severe, it can cause significant skeletal deformities, pain, and restrictive ventilatory dysfunction. It is recommended that patients with MFS may be treated surgically with symptoms (Lumban Tobing and Akbar, 2020; Rava et al., 2020). In 2017, Wang et al. helped an MFS couple birth a healthy newborn by using preimplantation genetic diagnosis (PGD).The PGD approach can yield healthy offspring for families at high risk for genetic disorders (Wang et al., 2017).
5 Animal models of MFS
At present, animal models for MFS are categorized into two groups: spontaneous animal models and experimental animal models. Animal models of MFS primarily mimic the aortic coarctation phenotype of human MFS. Although many studies have been conducted to investigate the molecular mechanisms of TGF-β and other signaling pathways in MFS with the knockout FBN1 mouse model, there are fewer reports on other homologous animal models of MFS. The advantages and disadvantages of the MFS animal model are highlighted in Table 2.
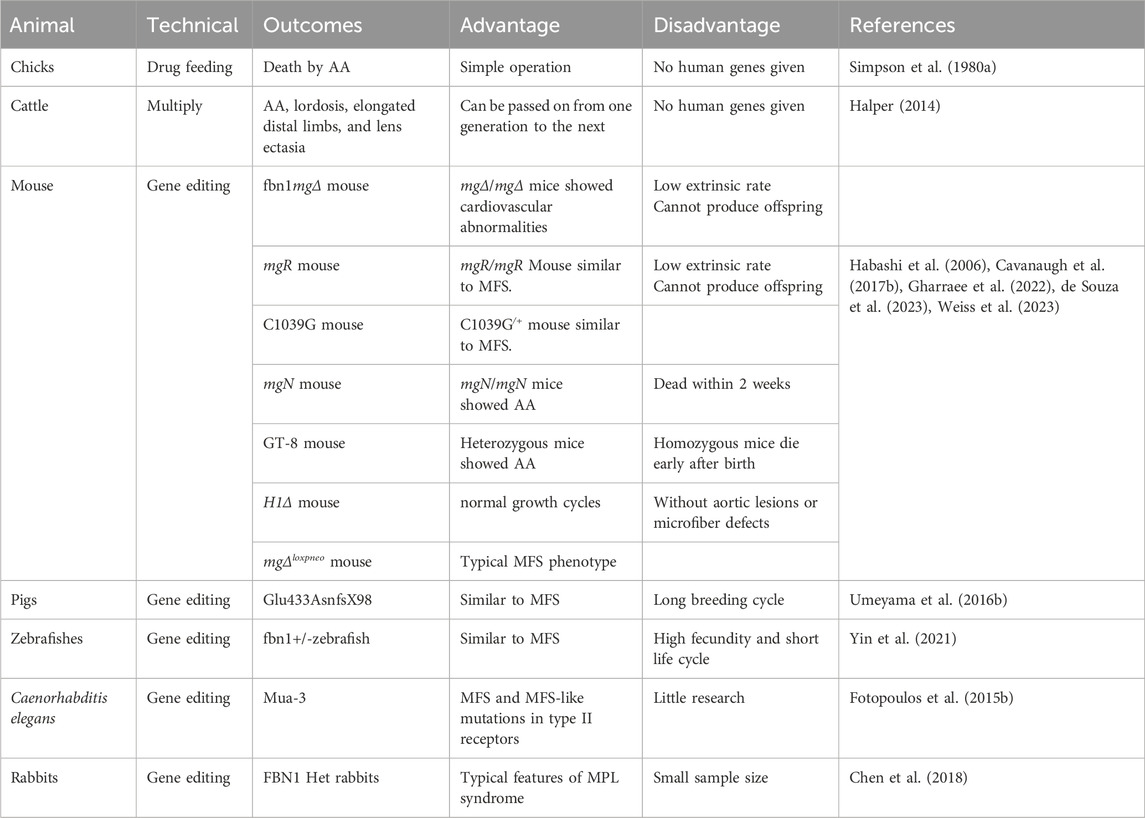
Table 2. Advantages and disadvantages of animal models of MFS.
5.1 Spontaneous animal models
5.1.1 Cattle model of MFS
Bovine MFS is an inherited disease with many of the clinical and pathologic manifestations of human MFS. The main manifestations are lens ectasia, aortic dilatation, aneurysms and ruptures. In 1990, Besser et al. described a phenotypically normal purebred bull that produced 72 calves from 41 females. Seven of these calves were infected. Clinical examination of siblings of infected calves revealed no skeletal, ocular or cardiovascular lesions. Neonatal calves affected are known as bovine MFS(BMFS), which suffered from scoliosis, distal limb overgrowth, and lens ectasia. These symptoms were phenotypically similar to human MFS (Besser et al., 1990). In 1994, Potter, K A and T E Besser tracked 10 BMFS cattle. Autopsy results showed that six BMFS cattle died of severe cardiovascular lesions (Potter and Besser, 1994). Morphological and ultrastructural revealed severe degenerative elastic fiber disease in the aortic tissue of BMFS. BMFS cattle with vvg-staining of sections displayed an increased ultrastructural density of elastic fibers in the skin, lungs and neck, and an increase in peripheral microprogenic fibers.
Cardiovascular lesions in BMFS are close to human MFS visually and under microscope. Therefore, BMFS can be a valuable model for exploring the molecular pathogenesis and therapeutic approaches to human MFS. However, BMFS are costly and have long breeding cycles. BMFS is rarely studied with MFS at present.
5.2 Experimental animal models
5.2.1 Chicks model of MFS
In 1980, Simpson et al. (1980b) observed aneurysms in patients with MFS, chicks fed a copper-deficient diet and turkeys fed b-aminopropionitrile (BAPN) by light and electron microscopy. Broken elastic fibers and atrophied smooth muscle cells in the abdominal aorta were detected by Orcein-Van Gieson staining of sections and electron microscopy. The results indicated similar aneurysm phenotypes in chicks and humans. Nevertheless, this MFS-like Chicks model has not been widely used ever since.
5.2.2 Mouse model of MFS
Most animal models of MFS have been created by gene editing mice. The mice models include mgΔ, mgR, C1039G, mgN, H1Δ, and mgΔloxpneo mice.
5.2.2.1 MgΔ mice
In Pereira et al. (1997) established the FBN1mgΔ mice model. In a C57BL/6J mice background, exons 19–24 of the 6 kb long FBN1 were replaced with a neomycin-resistant expression cassette (neoR) using gene editing techniques. Expression analysis showed that the transcript level of the mgΔ allele was 90% lower than that of the normal FBN1 allele. Heterozygous mgΔ/+ mice had normal life expectancy, but all mgΔ/mgΔ mice died around 3 weeks after birth. FBN1mgΔ/+ animals were histologically indistinguishable from wild-type mice. FBN1mgΔ/mgΔ mice had normal bones, but all suffered from cardiovascular complications, such as AA/AD. The transcript level of the mgΔ allele was 90% lower than that of the normal FBN1 allele.
The neoR box sequence possibly interferes with the expression of the mutant allele, thus limits the dominant-negative effect of the mutation. Targeting experiments in mice suggest that the FBN1 protein is primarily involved in tissue homeostasis.
5.2.2.2 MgR mice
In Pereira et al. (1999) used gene editing in a C57BL/6 mouse background to clone the target mgR allele from the PGKneo cassette into the region of exons 18 and 19 and replaced exons 19 to 24 with the neomycin (neo) gene. Both mice with heterozygous and pure mutants of mgR were born in the anticipated proportions and did not exhibit any abnormal characteristics at birth. The mgR/mgR mice exhibited clinically significant scoliosis and increased thoracodorsal-ventral diameters and died of aortic coarctation in early adulthood compared to mgR/+ mice (Pereira et al., 1999). Similar Gene-targeted mutant mgR mice with low expression of FBN1 resulted in MFS-like manifestations. Histopathological analysis of aortic specimens demonstrated the presence of medial calcification, inflammatory fibroproliferative response, and inflammation-mediated lysis of the entrapped aneurysm.
5.2.2.3 C1039G mice
In Judge et al. (2004) overexpressed an FBN1 (C1663R) mutant in a normal mouse background using yeast artificial chromosome transgenesis. The C1039G mutation was localized to exon 25 of the cb-EGF-like structural domain encoded by the mouse FBN1 gene (Judge et al., 2004). The FBN1C1039G/C1039G mice often died perinatally from vascular anomalies. The FBN1C1039G/+ mice were under a normal life cycle with abnormalities of the aorta and typical skeletal abnormalities (Judge et al., 2004). Heterozygous mice with characteristics similar to MFS in humans exhibit impaired microfibre deposition, skeletal malformations and progressive deterioration of aortic wall structure.
5.2.2.4 MgN mice
In Carta et al. (2006) created the FBN1 gene deletion (mgN) mouse model. An exon of the FBN1 gene containing an ATG codon and signal peptide coding sequence of approximately 700 bp was replaced with a phosphoglycerol kinase neo cassette and an alkaline phosphatase gene containing an internal ribosomal entry site. mgN/+ mice had a normal life cycle and phenotype. However, mgN/mgN mice died within 2 weeks of birth from ruptured aortic aneurysms, impaired lung function, and/or diaphragmatic collapse. Deletion of one or both FBN1 alleles in the context of FBN1 deletion resulted in embryonic death. This suggests that FBN1 is critical for substrate assembly and the initiation of embryonic development.
5.2.2.5 H1Δ mice
In 2010, two novel knock-in mice were born. Charbonneau et al. performed an experiment involving C57Bl/6 mice with Cre recombinase inserted into the Rosa 26 locus, which was mated with FBN1 mice tagged with an enhanced green fluorescent protein (eGFP). The specific lox locus was integrated into the targeting vector and the lox66 and lox77 motifs were reversed by cre-mediated recombination. eGFP coding sequence was inverted into the frame after exon 32. cbEGF region was truncated. FBN1 was tagged with eGFP. A mice strain named GT-8 was successfully established. GT-8 homozygous mice died early after birth. Heterozygous mice had abnormal skin and dilated aorta but normal growth cycles (Charbonneau et al., 2010). Another mice model called H1Δ (heterozygous 1 structural domain deletion) was created through cre-mediated removal of FBN1 exon 7 (bordered by loxP sites) in a second generation of mice on a C57Bl/6 background. H1Δ mice had a normal growth cycle and no aortopathy or microfibrillar defects.
5.2.2.6 MgΔloxpneo mice
In Lima et al. (2010) modified the neoR (mgΔloxpneo) of the mgΔ mouse model with lox-P sequences within the context of the C57BL/6 and 129/Sv animal models by gene editing. mgΔloxpneo from the C57BL/6 and 129/Sv backgrounds were mated with CD1 females to produce the F1 generation. Heterozygous mgΔloxpneo of the F1 generation displayed no significant phenotype. Subsequently, the heterozygotes of the F1 generation were hybridized. The heterozygous mgΔloxpneo mice all exhibited typical MFS phenotypes, including aortic, skeletal (primarily retroconvex), and respiratory (emphysema) phenotypes. Both crosses had normal lifespan and reproduction (Lima et al., 2010). The phenotype of the MFS model mice established by the gene editing techniques described above was dominated by aortic lesions. Clinical variants in the mice indicated that epigenetic factors were associated with disease severity.The overall expression level of FBN1 was strongly negatively correlated with the severity of the phenotype, and thisconfirmed the relevant role of mutated FBN1 in the pathogenesis of MFS.
5.2.3 Pigs model of MFS
The advancement of reproductive biology led to genetically engineered pigs as models for human genetic diseases. Although several models of monogenic diseases exist, replication of human diseases caused by haploinsufficiency remains a major challenge. Investigating the gene regulatory mechanisms associated with haploinsufficiency can be of great practical value in establishing reliable pig models of MFS.
In Umeyama et al. (2016a) created an FBN1 mutant (fbn1mut) clone pig (+/Glu433AsnfsX98) by using genome editing and somatic cell nuclear transplantation. This model exhibited phenotypes similar to human MFS symptoms, as scoliosis, funnel chest, and aortic anomalies. Second generation (G2) pure mutant pigs exhibited typical MFS symptoms and survived for up to 28 days (Umeyama et al., 2016a). In 2022, Jack et al. established the fbn1mut/+ (Glu433AsnfsX98/WT) porcine model by somatic cell nuclear transfer (SCNT) technique. The first method involved transferring blastocyst-stage SCNT embryos into recipient reserve sows after 5 days of long-term culture. The symptoms of MFS related to scoliosis, concave chest, delayed epiphyseal mineralization and aortic wall elastic fibre abnormalities were demonstrated in 4 of 8 fbn1mut/+ pigs. Many of these symptoms were observed in the neonatal period. The second method of cloning pigs were SCNT embryos transplanted at the early cleavage stage without prolonged culture. There were two pigs that had no abnormalities in the neonatal period and developed symptoms at maturity. In vitro manipulation of embryos (including culture) can epigenetically alter gene expression. In the fourth generation of cloned fbn1mut/+ male pigs, MFS symptoms were observed in G1 ∼ G4 stage pigs at 662 days postnatal age. Cardiovascular lesions were the predominant symptom in individuals in the G2 ∼ G4 stages. The FBN1Glu433AsnfsX98/WT genotype were characterized by a later onset of MFS. The phenotypic diversity and neonatal morbidity observed in G0 cloned animals appeared to decrease after G1 (Jack et al., 2022b). The fbn1mut/+ profile recapitulated the variable and delayed pathogenesis of MFS. However, predicting and controlling the kinetics of symptom onset in porcine models of MFS still remains a major challenge for future investigations.
5.2.4 Zebrafishes model of MFS
Zebrafish models have the advantage of clear embryonic optics, high fecundity, low rearing costs, short life cycles, ease of experimental manipulation and the fact that approximately 70% of human genes have functional homologues in zebrafish (Fontana et al., 2018).
In Chen et al. (2006) produced knockout embryos by injecting 2 independent splice-site morpholino (MO) targeting the zebrafish FBN1 gene into Tg (fli1/EGFP) embryos. Tg embryos exhibited dilated caudal vessels, head and ocular vessels at 30–31 h after fertilization (Chen et al., 2006). In 2021, the first fbn1+/− zebrafish model was built by using CRISPR/Cas9 gene editing technology. This model was designed to mimic the genetic defects in human FBN1. The fbn1+/− zebrafish exhibited morphological and cardiovascular abnormalities in the juvenile stage, with marked pigmentation, increased body length, and body thinning (Yin et al., 2021). Genomic DNA was extracted from F1 embryos (hpf) 24 h after fertilization. F0 with genetic mutations were selected for mating to produce F1 progeny. f1 fbn1+/− adults were mated with Tg wild fish to produce F2 transgenic fish. f2 fbn1+/− heterozygous fish carried the mutants, which indicated that the fbn1+/− zebrafish transgenic line was successfully constructed. The first fbn1+/− zebrafish model was successfully constructed using CRISPR/Cas9 gene editing tools. This model manifested obvious morphological and cardiovascular abnormalities similar to those of MFS.
5.2.5 Caenorhabditis elegans model of MFS
Mua-3 is the mammalian homologue of fibrillin-1 in Caenorhabditis elegans, the cause of MFS. Previous studies have suggested that there were possible conserved interactions between mua-3 and the TGF-β pathway in Crypto-bacterium nematodes. Crypto-bacterium cryptic rod nematodes can be used to further mimic MFS in worms (Fotopoulos et al., 2015a). In 2019, Lin et al. identified MFS and MFS-like mutations in the type II (LTA motifs including leucine, threonine, and alanine) receptor leading to mistransportation of the receptor through the cryptic rod nematode model of Hidradenitis elegans (Lin et al., 2019).
In MFS-like patients, three heterozygous missense mutations W521R, R528H and R537P in the TGF-β type II receptor correspond to W580R, R587H and R596P, respectively, in the cryptic nematode type II receptor DAF-4. The three-dimensional structure of the structural pairs of the c-terminal domains of the type II TGF-β receptor was investigated. The LTA motif of the kinase structural domain was exposed externally. It can interact with other proteins. The structure and function of the human type II TGF-β receptor were modelled using Pymol software. The modeling results indicate that mutations in MFS-like symptoms interfere with the structural domain located on the exposed surface of the type II TGF-β receptor. This modifies its engagement with cytoplasmic transport and/or regulatory proteins, or the activity of the receptor. This model clearly demonstrates that the function of the type II receptor carrying the MFS-like mutation is determined by somatic assays. In a wild-type background, the increase in somatotype is similar to the predominant nature of these mutations found in MFS-like patients. Cryptobacterium cryptic nematode MFS-like syndrome model provides a new paradigm for MFS-like syndrome receptor transporter-disease linkage.
5.2.6 Rabbits model of MFS
Rabbits are a classic animal model species for cardiovascular diseases such as atherosclerosis, and are frequently used animal model for eye disease study.
Chen et al. (2018) truncated the C-terminal end of the FBN1 gene by trans cytoplasmic microinjection of Cas9 mRNA and single-stranded rna (sgRNA) into fertilized embryos of rabbits using CRISPR/Cas9 technology. The heterozygous rabbit model showed muscular dystrophy, ocular syndrome, aortic dilatation and lipodystrophy in the MPL syndrome clinically (Chen et al., 2018). FBN1 rabbits had a high mortality rate, were prone to lung infection, pneumothorax and dilated ascending aorta found in the dead rabbits. The cause of death was probably aortic root dilatation and MPL lung phenotype. The ascending aorta of FBN1 Het rabbits was less elastic and flattened compared to the normal ascending aorta of the WT controls. In addition, the ascending and abdominal aorta were dilated in the FBN1 Het rabbits compared to the normal ascending aorta in the WT control group. FBN1 Het rabbits displayed skin atrophy, skin laxity and slow hair growth compared to WT rabbits. Gastrocnemius and quadriceps muscles were significantly reduced in FBN1 Het rabbits. H&E staining and statistical analysis illustrated significant thinning of muscle fibers in FBN1 Het rabbits. FBN1 Het rabbit muscle atrophy, X-ray examination of the FBN1 Het rabbit and the WT rabbit indicated that the length and diameter of the femur and tibia of the FBN1 Het rabbit were abnormal. In addition, H&E staining demonstrated a decrease in bone marrow cells and bone marrow adipocytes in FBN1 Het rabbits. Toluidine blue staining showed that osteoblasts were significantly reduced in the bone deposition area of FBN1 Het rabbits. These observations indicated that FBN1 mutations induced the typical phenotype of MPL syndrome in FBN1 Het rabbits.
The current animal models used to study MFS are mainly based on mice. Mice reproduce quickly and in short cycles, and this model is readily available and relatively low cost (Summers, 2024). Although mouse models of MFS exhibit certain similarities to the condition in patients, they do not precisely replicate the human disease state, particularly in terms of long-term aortic dilatation and the development of coarctation (Summers, 2024). Pigs and humans share similarities in several anatomical and physiological characteristics and have body sizes suitable for a variety of surgical procedures and clinical evaluations (Jack et al., 2022a; Salinas et al., 2022). The pig model with higher maintenance costs and more resources and space are its drawbacks. Zebrafish share a highly homologous genome with humans and have a short reproductive cycle that takes up little space (Abrial et al., 2022). It makes zebrafish an ideal model for studying the molecular mechanisms and gene function of Marfan syndrome. Nevertheless, the zebrafish cardiovascular system is anatomically different from humans, which may affect research on certain cardiovascular disorders (Wang et al., 2024).
6 Prospectives
MFS is an inherited disorder that is rare but pathogenic. Fibrillin 1 mutations overactivate TGF-β, which leads to an imbalance in the TGF-β signaling pathway and a signaling cascade. In addition to the TGF-β signaling pathway, NO, angiotensin, WNT, NOTCH, PI3K/AKT, and other signaling pathways are disregulated in MFS. PI3K/AKT and mTOR have been associated with aortic lesion formation in MFS. However, the specific molecular mechanism of MFS still remains unclear. Treatment of MFS includes surgery and drug therapy, which is necessary for patients with high risk factors in the aorta or other organs, especially for neonatal MFS. Pharmacological treatment is a current research hotspot, and the search for new targeted drugs is predicated on the need to pass experiments with MFS model animals as well as clinical trials, and the most used model is the MFS mouse model at present. In addition to the mouse model, the rabbit model can be modeled to study the ocular phenotype of MFS, and the zebrafish model has cardiovascular characteristics to study the cardiovascular phenotype of MFS.
However, there are challenges in the research for MFS treatment. First, certain animal models exhibit unusual clinical presentations, such as no or mild AA/AC, disproportionate bone growth, lens ectasia and other clinical symptoms. Second, only part of animal genomes is available. Third, the applicability of findings from MFS animal models to the human population is constrained. Although animal models of MFS are diverse, only the rabbit model presented the symptoms of lens ectasia. In addition, researchers should consider factors such as minimal interference, reproducibility, and cost-effectiveness while conducting animal experiments. Hence, a high-quality MFS animal model should possess the following attributes: (i) obvious cardiovascular abnormalities (e.g., aortic dilatation/clamping); (ii) similarity to human MFS in skeletal malformations (e.g., curvature of spine, disproportionate growth of bones); (iii) lens ectasia similar to human MFS; and (iv) animal have homologs of human MFS-causing genes; (v) the animal model results specifically and plausibly reflect the pathologic process of human MFS.
Animal models with diverse characteristics are critical for exploring the pathophysiology and new therapeutic approaches for MFS patients. Each animal model used to simulate human MFS has pros and cons. Gene-edited animal models focus on the study of clinical manifestations, pathologic alterations, and therapeutic approaches for MFS, offering significant insights into the outward appearance and management strategies for MFS. In addition to FBN1 gene-edited animal models, other relevant gene-edited animal models are necessary to study the molecular mechanisms and gene therapies of human MFS, conjunct with novel molecular methodologies aimed at modifying the regulation of distinct genes in specific tissues in a targeted manner. Genetic models of MFS have been examined from the causative genes to the cellular and tissue levels, providing insights into the progression of MFS pathology from embryonic stages to adulthood.
The establishment of animal models of MFS can be used to probe the pathogenesis, molecular mechanisms, and pharmacological treatments of human MFS. The role of animal models in MFS is essential due to variances in anatomy, physiology, and genetics between humans and animals. These models are employed to replicate particular disease characteristics and offer significant insights to investigators. Hence, continuous endeavors have concentrated on developing a more optimized animal model that replicates the etiology and therapeutic effects associated with MFS in humans. The future trend of animal models may provide engaging insights into the comprehending of pathological and genetic characteristics, as well as novel therapeutic approaches for MFS.
Author contributions
YJ: Writing–original draft. PJ: Data curation, Formal Analysis, Writing–original draft. XF: Methodology, Writing–original draft. DZ: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from Chengdu Municipal Health Commission (2024025), the Key Research and Development Project of the Sichuan Provincial Science and Technology Department (2023YFS0311), Science and Technology Bureau of Chengdu (YF06-00070-SN), and the University of Electronic Science and Technology of China (ZYGX2021YGLH217).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrial, M., Basu, S., Huang, M., Butty, V., Schwertner, A., Jeffrey, S., et al. (2022). Latent TGFβ-binding proteins 1 and 3 protect the larval zebrafish outflow tract from aneurysmal dilatation. Dis. Model Mech. 15 (3), dmm046979. doi:10.1242/dmm.046979
Akhurst, R. J., and Hata, A. (2012). Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11 (10), 790–811. doi:10.1038/nrd3810
Arnaud, P., Milleron, O., Hanna, N., Ropers, J., Ould Ouali, N., Affoune, A., et al. (2021). Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 23 (7), 1296–1304. doi:10.1038/s41436-021-01132-x
Asano, K., Cantalupo, A., Sedes, L., and Ramirez, F. (2022). Pathophysiology and therapeutics of thoracic aortic aneurysm in marfan syndrome. Biomolecules 12 (1), 128. doi:10.3390/biom12010128
Beene, L. C., Wang, L. W., Hubmacher, D., Keene, D. R., Reinhardt, D. P., Annis, D. S., et al. (2013). Nonselective assembly of fibrillin 1 and fibrillin 2 in the rodent ocular zonule and in cultured cells: implications for marfan syndrome. Investigative Ophthalmol. and Vis. Sci. 54 (13), 8337–8344. doi:10.1167/iovs.13-13121
Bertoli-Avella, A. M., Gillis, E., Morisaki, H., Verhagen, J. M. A., de Graaf, B. M., van de Beek, G., et al. (2015). Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 65 (13), 1324–1336. doi:10.1016/j.jacc.2015.01.040
Besser, T. E., Potter, K. A., Bryan, G. M., and Knowlen, G. G. (1990). An animal model of the Marfan syndrome. Am. J. Med. Genet. 37 (1), 159–165. doi:10.1002/ajmg.1320370137
Bitarafan, F., Razmara, E., Khodaeian, M., Keramatipour, M., Kalhor, A., Jafarinia, E., et al. (2020). Three Novel Variants identified in FBN1 and TGFBR2 in seven Iranian families with suspected Marfan syndrome. Mol. Genet. and Genomic Med. 8 (8), e1274. doi:10.1002/mgg3.1274
Bowman, M. A. H., Eagle, K. A., and Milewicz, D. M. (2019). Update on clinical trials of losartan with and without β-blockers to block aneurysm growth in patients with marfan syndrome: a review. Jama Cardiol. 4 (7), 702–707. doi:10.1001/jamacardio.2019.1176
Brooke, B. S., Habashi, J. P., Judge, D. P., Patel, N., Loeys, B., and Dietz, H. C. (2008). Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 358 (26), 2787–2795. doi:10.1056/NEJMoa0706585
Carta, L., Pereira, L., Arteaga-Solis, E., Lee-Arteaga, S. Y., Lenart, B., Starcher, B., et al. (2006). Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 281 (12), 8016–8023. doi:10.1074/jbc.M511599200
Cavanaugh, N. B., Qian, L., Westergaard, N. M., Kutschke, W. J., Born, E. J., and Turek, J. W. (2017a). A novel murine model of marfan syndrome accelerates aortopathy and cardiomyopathy. Ann. Thorac. Surg. 104 (2), 657–665. doi:10.1016/j.athoracsur.2016.10.077
Cavanaugh, N. B., Qian, L., Westergaard, N. M., Kutschke, W. J., Born, E. J., and Turek, J. W. (2017b). A novel murine model of marfan syndrome accelerates aortopathy and cardiomyopathy. Ann. Thorac. Surg. 104 (2), 657–665. doi:10.1016/j.athoracsur.2016.10.077
Charbonneau, N. L., Carlson, E. J., Tufa, S., Sengle, G., Manalo, E. C., Carlberg, V. M., et al. (2010). In vivo studies of mutant fibrillin-1 microfibrils. J. Biol. Chem. 285 (32), 24943–24955. doi:10.1074/jbc.M110.130021
Chen, E., Larson, J. D., and Ekker, S. C. (2006). Functional analysis of zebrafish microfibril-associated glycoprotein-1 (Magp 1) in vivo reveals roles for microfibrils in vascular development and function. Blood 107 (11), 4364–4374. doi:10.1182/blood-2005-02-0789
Chen, M., Yao, B., Yang, Q., Deng, J., Song, Y., Sui, T., et al. (2018). Truncated C-terminus of fibrillin-1 induces Marfanoid-progeroid-lipodystrophy (MPL) syndrome in rabbit. Dis. Models and Mech. 11 (4), dmm031542. doi:10.1242/dmm.031542
Chiu, H.-H. (2022). An update of medical care in Marfan syndrome. Tzu chi Med. J. 34 (1), 44–48. doi:10.4103/tcmj.tcmj_95_20
Chung, A. W. Y., Yeung, K. A., Cortes, S. F., Sandor, G. G. S., Judge, D. P., Dietz, H. C., et al. (2007). Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br. J. Pharmacol. 150 (8), 1075–1083. doi:10.1038/sj.bjp.0707181
Coelho, S. G., and Almeida, A. G. (2020). Marfan syndrome revisited: from genetics to the clinic. Rev. Port. De. Cardiol. 39 (4), 215–226. doi:10.1016/j.repc.2019.09.008
Collard, R., and Majtan, T. (2023). Genetic and pharmacological modulation of cellular proteostasis leads to partial functional rescue of homocystinuria-causing cystathionine-beta synthase variants. Mol. Cell. Biol. 43, 664–674. doi:10.1080/10985549.2023.2284147
Cui, R. Z., Hodge, D. O., and Mohney, B. G. (2023). Incidence and de novo mutation rate of Marfan syndrome and risk of ectopia lentis. J. Aapos 27 (5), 273.e1–273.e4. doi:10.1016/j.jaapos.2023.07.006
Dallan, L. R. P., Dallan, L. A. O., Duncan Santiago, J. A., Ribeiro Dias, R., Manuel de Almeida Brandao, C., and Jatene, F. B. (2021). Bentall-de Bono procedure for acute aortic dissection. Multimedia Man. Cardiothorac. Surg. 10. doi:10.1510/mmcts.2021.014
De Cario, R., Sticchi, E., Lucarini, L., Attanasio, M., Nistri, S., Marcucci, R., et al. (2018). Role of TGFBR1 and TGFBR2 genetic variants in Marfan syndrome. J. Vasc. Surg. 68 (1), 225–233. doi:10.1016/j.jvs.2017.04.071
de la Fuente-Alonso, A., Toral, M., Alfayate, A., Jesus Ruiz-Rodriguez, M., Bonzon-Kulichenko, E., Teixido-Tura, G., et al. (2021). Aortic disease in Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO. Nat. Commun. 12 (1), 262. doi:10.1038/s41467-021-22933-3
Deleeuw, V., De Clercq, A., De Backer, J., and Sips, P. (2021). An overview of investigational and experimental drug treatment strategies for marfan syndrome. J. Exp. Pharmacol. 13, 755–779. doi:10.2147/jep.S265271
DePaepe, A., Devereux, R. B., Dietz, H. C., Hennekam, R. C. M., and Pyeritz, R. E. (1996). Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet. 62 (4), 417–426. doi:10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425 (6958), 577–584. doi:10.1038/nature02006
Desir, J., Sznajer, Y., Depasse, F., Roulez, F., Schrooyen, M., Meire, F., et al. (2010). LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur. J. Hum. Genet. 18 (7), 761–767. doi:10.1038/ejhg.2010.11
de Souza, R. B., Gyuricza, I. G., Cassiano, L. L., Farinha-Arcieri, L. E., Alvim Liberatore, A. M., do Carmo, S. S., et al. (2021). The mgΔlpn mouse model for Marfan syndrome recapitulates the ocular phenotypes of the disease. Exp. Eye Res. 204, 108461. doi:10.1016/j.exer.2021.108461
de Souza, R. B., Lemes, R. B., Foresto-Neto, O., Cassiano, L. L., Reinhardt, D. P., Meek, K. M., et al. (2023). Extracellular matrix and vascular dynamics in the kidney of a murine model for Marfan syndrome. PLoS One 18 (5), e0285418. doi:10.1371/journal.pone.0285418
Dietz, H. C., Loeys, B., Carta, L., and Ramirez, F. (2005). Recent progress towards a molecular understanding of Marfan syndrome. Am. J. Med. Genet. Part C-Seminars Med. Genet. 139C (1), 4–9. doi:10.1002/ajmg.c.30068
Dong, C. X., Malecki, C., Robertson, E., Hambly, B., and Jeremy, R. (2023). Molecular mechanisms in genetic aortopathy-signaling pathways and potential interventions. Int. J. Mol. Sci. 24 (2), 1795. doi:10.3390/ijms24021795
Faivre, L., Collod-Beroud, G., Callewaert, B., Child, A., Binquet, C., Gautier, E., et al. (2009). Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24-32 mutation. Eur. J. Hum. Genet. 17 (4), 491–501. doi:10.1038/ejhg.2008.207
Farag, M., Büsch, C., Rylski, B., Pöling, J., Dohle, D. S., Sarvanakis, K., et al. (2023). Early outcomes of patients with Marfan syndrome and acute aortic type A dissection. J. Thorac. Cardiovasc Surg. 166 (1), 25–34.e8. doi:10.1016/j.jtcvs.2021.07.024
Fici, F., Robles, N. R., Tengiz, I., and Grassi, G. (2023). Beta-blockers and hypertension: some questions and answers. High. Blood Press Cardiovasc Prev. 30 (3), 191–198. doi:10.1007/s40292-023-00576-3
Fontana, B. D., Mezzomo, N. J., Kalueff, A. V., and Rosemberg, D. B. (2018). The developing utility of zebrafish models of neurological and neuropsychiatric disorders: a critical review. Exp. Neurol. 299, 157–171. doi:10.1016/j.expneurol.2017.10.004
Fotopoulos, P., Kim, J., Hyun, M., Qamari, W., Lee, I., and You, Y.-J. (2015a). DPY-17 and MUA-3 interact for connective tissue-like tissue integrity in Caenorhabditis elegans: a model for marfan syndrome. G3-Genes Genomes Genet. 5 (7), 1371–1378. doi:10.1534/g3.115.018465
Fotopoulos, P., Kim, J., Hyun, M., Qamari, W., Lee, I., and You, Y. J. (2015b). DPY-17 and MUA-3 interact for connective tissue-like tissue integrity in Caenorhabditis elegans: a model for marfan syndrome. G3 (Bethesda) 5 (7), 1371–1378. doi:10.1534/g3.115.018465
Fuchs, J. (1997). Marfan syndrome and other systemic disorders with congenital ectopia lentis. A Danish national survey. Acta Paediatr. 86 (9), 947–952. doi:10.1111/j.1651-2227.1997.tb15176.x
Gensicke, N. M., Cavanaugh, N. B., Andersen, N. D., Huang, T., Qian, L., Dyle, M. C., et al. (2020). Accelerated Marfan syndrome model recapitulates established signaling pathways. J. Thorac. Cardiovasc. Surg. 159 (5), 1719–1726. doi:10.1016/j.jtcvs.2019.05.043
Gharraee, N., Sun, Y., Swisher, J. A., and Lessner, S. M. (2022). Age and sex dependency of thoracic aortopathy in a mouse model of Marfan syndrome. Am. J. Physiol. Heart Circ. Physiol. 322 (1), H44–h56. doi:10.1152/ajpheart.00255.2021
Gong, B., Yang, L., Wang, Q., Ye, Z., Guo, X., Yang, C., et al. (2019). Mutation screening in the FBN1 gene responsible for Marfan syndrome and related disorder in Chinese families. Mol. Genet. and Genomic Med. 7 (4), e00594. doi:10.1002/mgg3.594
Goumans, M.-J., and ten Dijke, P. (2018). TGF-Β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 10 (2), a022210. doi:10.1101/cshperspect.a022210
Granata, A., Serrano, F., Bernard, W. G., McNamara, M., Low, L., Sastry, P., et al. (2017). An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 49 (1), 97–109. doi:10.1038/ng.3723
Groth, K. A., Hove, H., Kyhl, K., Folkestad, L., Gaustadnes, M., Vejlstrup, N., et al. (2015). Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J. Rare Dis. 10, 153. doi:10.1186/s13023-015-0369-8
Groth, K. A., Von Kodolitsch, Y., Kutsche, K., Gaustadnes, M., Thorsen, K., Andersen, N. H., et al. (2017). Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet. Med. 19 (7), 772–777. doi:10.1038/gim.2016.181
Gupta, P. A., Putnam, E. A., Carmical, S. G., Kaitila, I., Steinmann, B., Child, A., et al. (2002). Ten novel FBN2 mutations in congenital contractural arachnodactyly:: delineation of the molecular pathogenesis and clinical phenotype. Hum. Mutat. 19 (1), 39–48. doi:10.1002/humu.10017
Gupta, P. A., Wallis, D. D., Chin, T. O., Northrup, H., Tran-Fadulu, V. T., Towbin, J. A., et al. (2004). FBN2 mutation associated with manifestations of Marfan syndrome and congenital contractural arachnodactyly. J. Med. Genet. 41 (5), e56. doi:10.1136/jmg.2003.012880
Habashi, J. P., Doyle, J. J., Holm, T. M., Aziz, H., Schoenhoff, F., Bedja, D., et al. (2011). Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 332 (6027), 361–365. doi:10.1126/science.1192152
Habashi, J. P., Judge, D. P., Holm, T. M., Cohn, R. D., Loeys, B. L., Cooper, T. K., et al. (2006). Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312 (5770), 117–121. doi:10.1126/science.1124287
Halper, J. (2014). Connective tissue disorders in domestic animals. Adv. Exp. Med. Biol. 802, 231–240. doi:10.1007/978-94-007-7893-1_14
Hlavicka, J., Antonov, K., Salem, R., Hecker, F., Marinos, S., Radwan, M., et al. (2022). Long-term outcomes after aortic valve and root replacement in a very high-risk population. J. Cardiovasc. Dev. Dis. 9 (6), 197. doi:10.3390/jcdd9060197
Holm, T. M., Habashi, J. P., Doyle, J. J., Bedja, D., Chen, Y., van Erp, C., et al. (2011). Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332 (6027), 358–361. doi:10.1126/science.1192149
Jack, N., Muto, T., Iemitsu, K., Watanabe, T., Umeyama, K., Ohgane, J., et al. (2022a). Genetically engineered animal models for Marfan syndrome: challenges associated with the generation of pig models for diseases caused by haploinsufficiency. J. Reprod. Dev. 68 (4), 233–237. doi:10.1262/jrd.2022-027
Jack, N., Muto, T., Iemitsu, K., Watanabe, T., Umeyama, K., Ohgane, J., et al. (2022b). Genetically engineered animal models for Marfan syndrome: challenges associated with the generation of pig models for diseases caused by haploinsufficiency. J. Reproduction Dev. 68 (4), 233–237. doi:10.1262/jrd.2022-027
Jespersen, K., Li, C., Batra, R., Stephenson, C. A. A., Harding, P., Sestak, K., et al. (2022). Impact of Notch3 activation on aortic aneurysm development in marfan syndrome. J. Immunol. Res. 2022, 7538649. doi:10.1155/2022/7538649
Johnsen, S. A., Subramaniam, M., Janknecht, R., and Spelsberg, T. C. (2002). TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene 21 (37), 5783–5790. doi:10.1038/sj.onc.1205681
Jondeau, G., Milleron, O., Eliahou, L., Boileau, C., and Ropers, J. (2023). Marfan treatment trialists' collaboration in perspective: sartans and beta-blockers in patients with marfan syndrome. Arch. Cardiovasc Dis. 116 (4), 173–175. doi:10.1016/j.acvd.2023.02.003
Judge, D. P., Biery, N. J., Keene, D. R., Geubtner, J., Myers, L., Huso, D. L., et al. (2004). Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investigation 114 (2), 172–181. doi:10.1172/JCI20641
Koo, H. K., Lawrence, K. A., and Musini, V. M. (2017). Beta-blockers for preventing aortic dissection in Marfan syndrome. Cochrane Database Syst. Rev. 11 (11), Cd011103. doi:10.1002/14651858.CD011103.pub2
Korneva, A., Zilberberg, L., Rifkin, D. B., Humphrey, J. D., and Bellini, C. (2019). Absence of LTBP-3 attenuates the aneurysmal phenotype but not spinal effects on the aorta in Marfan syndrome. Biomechanics Model. Mechanobiol. 18 (1), 261–273. doi:10.1007/s10237-018-1080-1
Kosaki, K., Takahashi, D., Udaka, T., Kosaki, R., Matsumoto, M., Ibe, S., et al. (2006). Molecular pathology of Shprintzen-Goldberg syndrome. Am. J. Med. Genet. Part A 140A (1), 104–108. doi:10.1002/ajmg.a.31006
Kraehenbuehl, E., Maksimovic, S., Sodeck, G., Reineke, D., Schoenhoff, F., Schmidli, J., et al. (2012). What makes the difference between the natural course of a remaining type B dissection after type A repair and a primary type B aortic dissection? Eur. J. Cardio-Thoracic Surg. 41 (5), e110–e116. doi:10.1093/ejcts/ezs121
Kuzmanovic, I. B., Davidovic, L. B., Kostic, D. M., Maksimovic, Z. L., Cinara, I. S., Svetkovic, S. D., et al. (2004). Long-term results after elective and emergency surgery of abdominal aortic aneurysm. Srp. Arh. za Celok. Lek. 132 (9-10), 306–312. doi:10.2298/sarh0410306k
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149 (2), 274–293. doi:10.1016/j.cell.2012.03.017
Lazea, C., Bucerzan, S., Crisan, M., Al-Khzouz, C., Miclea, D., Sufana, C., et al. (2021). Cardiovascular manifestations in Marfan syndrome. Med. Pharm. Rep. 94 (Suppl. No 1), S25–S27. doi:10.15386/mpr-2223
Lima, B. L., Santos, E. J. C., Fernandes, G. R., Merkel, C., Mello, M. R. B., Gomes, J. P. A., et al. (2010). A new mouse model for marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. Plos One 5 (11), e14136. doi:10.1371/journal.pone.0014136
Lin, J., Vora, M., Kane, N. S., Gleason, R. J., and Padgett, R. W. (2019). Human Marfan and Marfan-like Syndrome associated mutations lead to altered trafficking of the Type II TGFβ receptor in Caenorhabditis elegans. Plos One 14 (5), e0216628. doi:10.1371/journal.pone.0216628
Lin, W.-Y., Fordham, S. E., Hungate, E., Sunter, N. J., Elstob, C., Xu, Y., et al. (2021). Genome-wide association study identifies susceptibility loci for acute myeloid leukemia. Nat. Commun. 12 (1), 6233. doi:10.1038/s41467-021-26551-x
Lloyd, R. I. (1934). Arachnodactyly (dystrophia mesodermalis congenita, typus marfans. Marfan's syndrome. Dolichostenomelie). Trans. Am. Ophthalmol. Soc. 32, 361–368.
Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B., et al. (2010). The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 47 (7), 476–485. doi:10.1136/jmg.2009.072785
Loeys, B. L., Schwarze, U., Holm, T., Callewaert, B. L., Thomas, G. H., Pannu, H., et al. (2006). Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 355 (8), 788–798. doi:10.1056/NEJMoa055695
Lumban Tobing, S. D. A., and Akbar, D. L. (2020). Challenges and experiences in correcting scoliosis of a patient with Marfan Syndrome: a case report. Int. J. Surg. case Rep. 76, 85–89. doi:10.1016/j.ijscr.2020.09.166
Manuel Becerra-Munoz, V., Jose Gomez-Doblas, J., Porras-Martin, C., Such-Martinez, M., Generosa Crespo-Leiro, M., Barriales-Villa, R., et al. (2018). The importance of genotype-phenotype correlation in the clinical management of Marfan syndrome. Orphanet J. Rare Dis. 13, 16. doi:10.1186/s13023-017-0754-6
Marzin, P., Rondeau, S., Alessandri, J.-L., Dieterich, K., le Goff, C., Mahaut, C., et al. (2023). Weill-Marchesani syndrome: natural history and genotype-phenotype correlations from 18 news cases and review of literature. J. Med. Genet. 61, 109–116. doi:10.1136/jmg-2023-109288
McKusick, V. A. (1955). The cardiovascular aspects of Marfan's syndrome: a heritable disorder of connective tissue. Circulation 11 (3), 321–342. doi:10.1161/01.Cir.11.3.321
Messerli, F. H., Bangalore, S., and Mandrola, J. M. (2023). β blockers switched to first-line therapy in hypertension. Lancet 402 (10414), 1802–1804. doi:10.1016/s0140-6736(23)01733-6
Milewicz, D. M., and Duvic, M. (1994). Severe neonatal Marfan syndrome resulting from a de novo 3-bp insertion into the fibrillin gene on chromosome 15. Am. J. Hum. Genet. 54 (3), 447–453.
Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T., et al. (2004). Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 36 (8), 855–860. doi:10.1038/ng1392
Morlino, S., Alesi, V., Cali, F., Lepri, F. R., Secinaro, A., Grammatico, P., et al. (2019). LTBP2-related Marfan-like phenotype in two Roma/Gypsy subjects with the LTBP2 homozygous p.R299X variant. Am. J. Med. Genet. Part A 179 (1), 104–112. doi:10.1002/ajmg.a.10
Mu, Y., Gudey, S. K., and Landstrom, M. (2012). Non-Smad signaling pathways. Cell Tissue Res. 347 (1), 11–20. doi:10.1007/s00441-011-1201-y
Nataatmadja, M., West, J., Prabowo, S., and West, M. (2013). Angiotensin II receptor antagonism reduces transforming growth factor beta and Smad signaling in thoracic aortic aneurysm. Ochsner J. 13 (1), 42–48.
Ornelas-Casillas, M. A., and Garcia-Arias, M. R. (2022). Aortic dissection after bentall and Bono surgery. Arch. Cardiol. Mex. 92 (1), 116–117. doi:10.24875/acm.20000514
Paolillo, S., Dell'Aversana, S., Esposito, I., Poccia, A., and Perrone Filardi, P. (2021). The use of β-blockers in patients with heart failure and comorbidities: doubts, certainties and unsolved issues. Eur. J. Intern Med. 88, 9–14. doi:10.1016/j.ejim.2021.03.035
Pereira, L., Andrikopoulos, K., Tian, J., Lee, S. Y., Keene, D. R., Ono, R., et al. (1997). Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet. 17 (2), 218–222. doi:10.1038/ng1097-218
Pereira, L., Lee, S. Y., Gayraud, B., Andrikopoulos, K., Shapiro, S. D., Bunton, T., et al. (1999). Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. U. S. A. 96 (7), 3819–3823. doi:10.1073/pnas.96.7.3819
Pollock, L., Ridout, A., Teh, J., Nnadi, C., Stavroulias, D., Pitcher, A., et al. (2021). The musculoskeletal manifestations of marfan syndrome: diagnosis, impact, and management. Curr. Rheumatol. Rep. 23 (11), 81. doi:10.1007/s11926-021-01045-3
Potter, K. A., and Besser, T. E. (1994). Cardiovascular lesions in bovine Marfan syndrome. Veterinary Pathol. 31 (5), 501–509. doi:10.1177/030098589403100501
Rava, A., Dema, E., Palmisani, M., Palmisani, R., Cervellati, S., and Girardo, M. (2020). Sublaminar fixation versus hooks and pedicle screws in scoliosis surgery for Marfan syndrome. J. craniovertebral junction and spine 11 (1), 26–30. doi:10.4103/jcvjs.JCVJS_12_20
Richards, T. R., and Tobe, S. W. (2014). Combining other antihypertensive drugs with β-blockers in hypertension: a focus on safety and tolerability. Can. J. Cardiol. 30 (5 Suppl. l), S42–S46. doi:10.1016/j.cjca.2013.08.012
Rifkin, D. B., Rifkin, W. J., and Zilberberg, L. (2018). LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 71-72, 90–99. doi:10.1016/j.matbio.2017.11.014
Ritelli, M., and Colombi, M. (2020). Molecular genetics and pathogenesis of ehlers-danlos syndrome and related connective tissue disorders. Genes 11 (5), 547. doi:10.3390/genes11050547
Robinson, P. N., and Godfrey, M. (2000). The molecular genetics of Marfan syndrome and related microfibrillopathies. J. Med. Genet. 37 (1), 9–25. doi:10.1136/jmg.37.1.9
Rouf, R., MacFarlane, E. G., Takimoto, E., Chaudhary, R., Nagpal, V., Rainer, P. P., et al. (2017). Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. Jci Insight 2 (15), e91588. doi:10.1172/jci.insight.91588
Sahay, P., Maharana, P. K., Shaikh, N., Goel, S., Sinha, R., Agarwal, T., et al. (2019). Intra-lenticular lens aspiration in paediatric cases with anterior dislocation of lens. Eye 33 (9), 1411–1417. doi:10.1038/s41433-019-0426-y
Sakai, L. Y., Keene, D. R., and Engvall, E. (1986). Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. cell Biol. 103 (6 Pt 1), 2499–2509. doi:10.1083/jcb.103.6.2499
Salinas, S. D., Farra, Y. M., Amini Khoiy, K., Houston, J., Lee, C. H., Bellini, C., et al. (2022). The role of elastin on the mechanical properties of the anterior leaflet in porcine tricuspid valves. PLoS One 17 (5), e0267131. doi:10.1371/journal.pone.0267131
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307 (5712), 1098–1101. doi:10.1126/science.1106148
Sato, T., Arakawa, M., Tashima, Y., Tsuboi, E., Burdon, G., Trojan, J., et al. (2018). Statins reduce thoracic aortic aneurysm growth in marfan syndrome mice via inhibition of the ras-induced ERK (extracellular signal-regulated kinase) signaling pathway. J. Am. Heart Assoc. 7 (21), e008543. doi:10.1161/jaha.118.008543
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168 (6), 960–976. doi:10.1016/j.cell.2017.02.004
Senser, E. M., Misra, S., and Henkin, S. (2021). Thoracic aortic aneurysm: a clinical review. Cardiol. Clin. 39 (4), 505–515. doi:10.1016/j.ccl.2021.06.003
Sharma, A. K. (2009). Marfan syndrome - diagnosis and management. Curr. Probl. Cardiol. 48, 101834. doi:10.1016/j.cpcardiol.2007.10.001
Shi, Y. G., and Massagué, J. (2003). Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113 (6), 685–700. doi:10.1016/s0092-8674(03)00432-x
Silverman, D. I., Burton, K., Gray, J., Boxer, M., Rosner, M., Kouchoukos, N., et al. (1994). Life expectancy in the marfan-syndrome revisited. J. Am. Coll. Cardiol., A402. doi:10.1016/s0002-9149(00)80066-1
Simpson, C. F., Boucek, R. J., and Noble, N. L. (1980a). Similarity of aortic pathology in Marfan's syndrome, copper deficiency in chicks and B-aminopropionitrile toxicity in turkeys. Exp. Mol. Pathol. 32 (1), 81–90. doi:10.1016/0014-4800(80)90045-3
Simpson, C. F., Boucek, R. J., and Noble, N. L. (1980b). Similarity of aortic pathology in Marfan's syndrome, copper deficiency in chicks and B-aminopropionitrile toxicity in turkeys. Exp. Mol. pathology 32 (1), 81–90. doi:10.1016/0014-4800(80)90045-3
Singh, K. K., Rommel, K., Mishra, A., Karck, M., Haverich, A., Schmidtke, J., et al. (2006). TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum. Mutat. 27 (8), 770–777. doi:10.1002/humu.20354
Somers, A. E., Hinton, R. B., Pilipenko, V., Miller, E., and Ware, S. M. (2016). Analysis of TGFBR1*6A variant in individuals evaluated for marfan syndrome. Am. J. Med. Genet. Part A 170 (7), 1786–1790. doi:10.1002/ajmg.a.37668
Song, H. K., Kindem, M., Bavaria, J. E., Dietz, H. C., Milewicz, D. M., Devereux, R. B., et al. (2012). Long-term implications of emergency versus elective proximal aortic surgery in patients with marfan syndrome in the genetically triggered thoracic aortic aneurysms and cardiovascular conditions consortium registry. J. Thorac. Cardiovasc. Surg. 143 (2), 282–286. doi:10.1016/j.jtcvs.2011.10.024
Sood, S., Eldadah, Z. A., Krause, W. L., McIntosh, I., and Dietz, H. C. (1996). Mutation in fibrillin-1 and the marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat. Genet. 12 (2), 209–211. doi:10.1038/ng0296-209
Spencer, M. (2024). Marfan syndrome. Nursing 54 (4), 19–25. doi:10.1097/01.NURSE.0001007604.09204.9a
Stheneur, C., Collod-Beroud, G., Faivre, L., Gouya, L., Sultan, G., Le Parc, J.-M., et al. (2008). Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with marfan syndrome type I and II, loeys-dietz syndrome and related disorders. Hum. Mutat. 29 (11), E284–E295. doi:10.1002/humu.20871
Sticchi, E., De Cario, R., Magi, A., Giglio, S., Provenzano, A., Nistri, S., et al. (2018). Bicuspid aortic valve: role of multiple gene variants in influencing the clinical phenotype. Biomed Res. Int. 2018, 8386123. doi:10.1155/2018/8386123
Summers, K. M. (2024). Genetic models of fibrillinopathies. Genetics 226 (1), iyad189. doi:10.1093/genetics/iyad189
Tae, H.-J., Petrashevskaya, N., Marshall, S., Krawczyk, M., and Talan, M. (2016). Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am. J. Physiology-Heart Circulatory Physiology 310 (2), H290–H299. doi:10.1152/ajpheart.00354.2015
Tang, P.M.-K., Zhang, Y.-Y., Mak, T.S.-K., Tang, P.C.-T., Huang, X.-R., and Lan, H.-Y. (2018). Transforming growth factor-β signalling in renal fibrosis: from Smads to non-coding RNAs. J. Physiology-London 596 (16), 3493–3503. doi:10.1113/jp274492
Tatu, A. L., Elisei, A. M., Chioncel, V., Miulescu, M., and Nwabudike, L. C. (2019). Immunologic adverse reactions of β-blockers and the skin. Exp. Ther. Med. 18 (2), 955–959. doi:10.3892/etm.2019.7504
Ten Dijke, P. T., Goumans, M. J., Itoh, F., and Itoh, S. (2002). Regulation of cell proliferation by Smad proteins. J. Cell. Physiology 191 (1), 1–16. doi:10.1002/jcp.10066
Todorovic, V., Jurukovski, V., Chen, Y., Fontana, L., Dabovic, B., and Rifkin, D. B. (2005). Latent TGF-beta binding proteins. Int. J. Biochem. and Cell Biol. 37 (1), 38–41. doi:10.1016/j.biocel.2004.03.011
Umeyama, K., Watanabe, K., Watanabe, M., Horiuchi, K., Nakano, K., Kitashiro, M., et al. (2016a). Generation of heterozygous fibrillin-1 mutant cloned pigs from genome-edited foetal fibroblasts. Sci. Rep. 6, 24413. doi:10.1038/srep24413
Umeyama, K., Watanabe, K., Watanabe, M., Horiuchi, K., Nakano, K., Kitashiro, M., et al. (2016b). Generation of heterozygous fibrillin-1 mutant cloned pigs from genome-edited foetal fibroblasts. Sci. Rep. 6, 24413. doi:10.1038/srep24413
van Elsacker, E., Vink, A. S., Menke, L. A., Pals, G., Bokenkamp, R., Backx, A. C. P. M., et al. (2022). Growth of the aortic root in children and young adults with Marfan syndrome. Open heart 9 (2), e002097. doi:10.1136/openhrt-2022-002097
Vargas, F., Ringel, J. B., Yum, B., Levitan, E. B., Mangal, S., Steinman, M. A., et al. (2023). Implications of under-reporting medication side effects: beta-blockers in heart failure as a case example. Drugs Aging 40 (3), 285–291. doi:10.1007/s40266-023-01007-7
Veiga-Fernandez, A., Joigneau Prieto, L., Alvarez, T., Ruiz, Y., Perez, R., Gamez, F., et al. (2020). Perinatal diagnosis and management of early-onset Marfan syndrome: case report and systematic review. J. Maternal-Fetal and Neonatal Med. 33 (14), 2493–2504. doi:10.1080/14767058.2018.1552935
Verstraeten, A., Alaerts, M., Van Laer, L., and Loeys, B. (2016). Marfan syndrome and related disorders: 25 Years of gene discovery. Hum. Mutat. 37 (6), 524–531. doi:10.1002/humu.22977
Wang, L., Zhao, Y., Zhang, W., Shu, X., Wang, E., Guo, D., et al. (2021). Retrograde type A aortic dissection after thoracic endovascular aortic repair: incidence, time trends and risk factors. Seminars Thorac. Cardiovasc. Surg. 33 (3), 639–653. doi:10.1053/j.semtcvs.2020.11.010
Wang, S., Niu, Z., Wang, H., Ma, M., Zhang, W., Wang, S. F., et al. (2017). De novo paternal FBN1 mutation detected in embryos before implantation. Med. Sci. Monit. 23, 3136–3146. doi:10.12659/msm.904546
Wang, Y., Panicker, I. S., Anesi, J., Sargisson, O., Atchison, B., and Habenicht, A. J. R. (2024). Animal models, pathogenesis, and potential treatment of thoracic aortic aneurysm. Int. J. Mol. Sci. 25 (2), 901. doi:10.3390/ijms25020901
Weiss, D., Rego, B. V., Cavinato, C., Li, D. S., Kawamura, Y., Emuna, N., et al. (2023). Effects of age, sex, and extracellular matrix integrity on aortic dilatation and rupture in a mouse model of marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 43 (9), e358–e372. doi:10.1161/atvbaha.123.319122
Williams, J. A., Loeys, B. L., Nwakanma, L. U., Dietz, H. C., Spevak, P. J., Patel, N. D., et al. (2007). Early surgical experience with Loeys-Dietz:: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann. Thorac. Surg. 83 (2), S757–S763. doi:10.1016/j.athoracsur.2006.10.091
Wozniak-Mielczarek, L., Sabiniewicz, R., Drezek-Nojowicz, M., Nowak, R., Gilis-Malinowska, N., Mielczarek, M., et al. (2019). Differences in cardiovascular manifestation of marfan syndrome between children and adults. Pediatr. Cardiol. 40 (2), 393–403. doi:10.1007/s00246-018-2025-2
Yang, H., Ma, Y., Luo, M., Zhao, K., Zhang, Y., Zhu, G., et al. (2018). Identification of gross deletions in FBN1 gene by MLPA. Hum. Genomics 12, 46. doi:10.1186/s40246-018-0178-y
Yildiz, M., Nucera, M., Jungi, S., Heinisch, P. P., Mosbahi, S., Becker, D., et al. (2023). Outcome of Stanford type B dissection in patients with Marfan syndrome. Eur. J. Cardio-Thoracic Surg. 64 (1), ezad178. doi:10.1093/ejcts/ezad178
Yin, X., Hao, J., and Yao, Y. (2021). CRISPR/Cas9 in zebrafish: an attractive model for FBN1 genetic defects in humans. Mol. Genet. and Genomic Med. 9 (10), e1775. doi:10.1002/mgg3.1775
Yu, C., and Jeremy, R. W. (2018). Angiotensin, transforming growth factor β and aortic dilatation in Marfan syndrome: of mice and humans. Int. J. Cardiol. Heart and Vasc. 18, 71–80. doi:10.1016/j.ijcha.2018.02.009
Zhang, C., Qiao, F., Cheng, Q., Luo, C., Zhang, Q., Hu, P., et al. (2023). A novel splice site mutation in the FBN2 gene in a Chinese family with congenital contractural arachnodactyly. Biochem. Genet. 62, 2495–2503. doi:10.1007/s10528-023-10550-2
Zhang, H., Hu, W., and Ramirez, F. (1995). Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J. Cell Biol. 129 (4), 1165–1176. doi:10.1083/jcb.129.4.1165
Keywords: marfan syndrome, fibrillin-1, transforming growth factor-beta signaling pathways, animal models, gene
Citation: Jiang Y, Jia P, Feng X and Zhang D (2025) Marfan syndrome: insights from animal models. Front. Genet. 15:1463318. doi: 10.3389/fgene.2024.1463318
Received: 31 October 2024; Accepted: 13 December 2024;
Published: 06 January 2025.
Edited by:
Darko Bosnakovski, University of Minnesota, United StatesReviewed by:
Elen H. Miyabara, University of São Paulo, BrazilUsuk Jung, University of Minnesota Medical Center, United States
Copyright © 2025 Jiang, Jia, Feng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingding Zhang, emhhbmdkZDI1QDEyNi5jb20=
†These authors have contributed equally to this work