 Yingying Cao1
Yingying Cao1 Yongxing Lai2Zhuxin Li1Shanshan Zhai1Yinghan Dai1Junyu Tao1,3Qing Wang1,4Ziheng Xu1Minjie Jiang1,4Li Yu1,4Jing Leng1,3,5*
Yongxing Lai2Zhuxin Li1Shanshan Zhai1Yinghan Dai1Junyu Tao1,3Qing Wang1,4Ziheng Xu1Minjie Jiang1,4Li Yu1,4Jing Leng1,3,5* Haibo Tang1,3,6*
Haibo Tang1,3,6*- 1Guangxi University of Chinese Medicine, Nanning, China
- 2Guangxi Vocational University of Agriculture, Nanning, China
- 3Guangxi Key Laboratory of Translational Medicine for Treating High-Incidence Infectious Diseases with Integrative Medicine, Nanning, China
- 4Guangxi Zhuang Autonomous Region Engineering Research Center of Graphene Biomedical Application Technology, Nanning, China
- 5Key Laboratory of Characteristic Experimental Animal Models of Guangxi, Nanning, China
- 6Guangxi Health Commission Guangxi Key Laboratory of Molecular Biology of Preventive Medicine of Traditional Chinese Medicine, Nanning, China
Illicium verum (Illiciaceae), an ecologically significant endemic plant, predominantly grows in Guangxi, China, which is the primary region for its cultivation. This area accounts for more than 80% of the total cultivation and yield in China. Despite its importance, comprehensive studies on the chloroplast (cp) genome of I. verum are limited. In our research, we sequenced and analyzed the complete cp genome of I. verum and conducted a comparative analysis with nine related species from the families Magnoliaceae, Schisandraceae, and Illiciaceae. The cp genome of I. verum spans 143,187 base pairs (bp), comprising a large single copy (LSC) region of 100,868 bp, a small single copy (SSC) region of 20,235 bp, and two inverted repeats (IR) regions, each 11,042 bp in length. We identified 81 simple sequence repeats (SSRs) within this genome. The chloroplast genome contains 78 protein-coding genes, 8 ribosomal RNA (rRNA) genes, and 35 transfer RNA (tRNA) genes. Structurally, the IR regions exhibit greater similarity across different genera of Magnoliaceae and Illiciaceae compared to the LSC and SSC regions. Phylogenetic analysis revealed high homology between the cp genome of I. verum and those of Illicium burmanicum, Illicium simonsii, and Illicium anisatum. These findings suggest that the cp genome of I. verum may serve as a valuable genomic resource for elucidating the phylogenetic positions and relationships within the Illiciaceae family. This information will be instrumental for future taxonomic research on Illicium species and for advancing genomic studies of these plants.
1 Introduction
Illicium verum Hook.f., (Illicium verum, Illiciaceae) is an aromatic evergreen tree that produces purple-red flowers and star-shaped fruit with an anise scent. It is classified within the division Magnoliophyta, class Magnoliopsida, subclass Magnoliidae, order Austrobaileyales, and family Illiciaceae (China EBoFo, 2004). Initially, Illicium was placed in the Magnoliaceae family in early taxonomic literature, but later it was reclassified into the Illiciaceae family by Smith based on floral morphology and vegetative anatomy (AC, 1947). Its fruit, also known as star anise, is not only a significant component of traditional Chinese medicine but also a commonly used spice.
Illicium verum, recognized in traditional Chinese medicine as Anisi Stellati Fructus or Bajiaohuixiang, is renowned for its medicinal properties, particularly in China. It has been utilized in various formulations, including crude drugs, powders, and essential oils. The use of I. verum is documented as far back as the Ming Dynasty in the “Compendium of Materia Medica” (Li, 1596). The Chinese Pharmacopoeia describes Anisi Stellati Fructus as having properties that warm yang, dispel cold, and regulate the flow of Qi, thereby alleviating pain or symptoms of the common cold. Clinically, it is applied to treat conditions such as abdominal colic, vomiting, and lower back pain. Furthermore, the crude fruits or their powdered forms have been integrated into traditional teas to mitigate nervousness, insomnia, and to serve as a sedative (Wang et al., 2011).
Illicium verum has been cultivated in the Zuoyou River valley in Guangxi Province since the Ming Dynasty in ancient China. With the development of China’s Maritime Silk Road and the expansion of overseas trade, Fujian Province and Guangdong Province have also become production and sales locations for I. verum. These provinces, located south of 25° north latitude, have a subtropical climate in hilly and mountainous regions with warm winters and cool summers, resulting in good fruit quality. Guangxi Province has maintained its status as a geo-authentic crude drug-producing area of I. verum, particularly in Longzhou, Ningming, Napo, and other counties in the west and south of Guangxi (Qi, 1995). In December 2020, the Administration of Traditional Chinese Medicine of Guangxi Zhuang Autonomous Region and other departments included I. verum in the Guangxi authentic medicinal materials “Ten Herbs of Guangxi,” initiating key development and utilization efforts (Li, 2023).
Because there are too many similarities in the phenotypes (tree shape, branch, stem, leaf, flower, pollen, fruit) of the Illiciaceae plants, the differences are not always obvious (Zhang et al., 2017b). This can make it difficult to identify the species of Illiciaceae, even for professionals. Unfortunately, there is no perfect method for identification. The compounds contained in the plants vary greatly, leading to significant differences in efficacy and toxicity. As a result, there have been cases of ingestion poisoning. Effectively identifying medicinal materials and counterfeit products is a challenging problem. The U.S. Food and Drug Administration (FDA) has developed a GenomeTrakrCP database, a public database of chloroplast genome sequences for plant flavors, plant dietary supplements, etc., including Illiciaceae, to help identify plant species in foods and dietary supplements. Includes plants used as food and dietary supplements, toxin producers, common contaminants and adulterants, and their relatives (Chen et al., 2023; Cui and Fu, 2001; Li and Liu, 2011; Vermaak et al., 2013; Zhang et al., 2017b).
Typically, taxonomic research relies on morphological evaluation, a method prone to environmental influences. Discerning various morphological traits in plants necessitates observations across their entire life cycle, imposing constraints on identifying species lacking comprehensive morphological data. In contrast, molecular identification, which utilizes DNA markers, offers enhanced precision and dependability due to the inherent genetic stability and resilience to external variables. Consequently, this approach is better suited for distinguishing closely related species. The chloroplast genome, generally spanning 100–150 kilobases and rich in evolutionary information, serves as an exemplary model for investigations into molecular biomarkers, phylogenetic analyses, evolutionary studies, and comparative genomics (Wu et al., 2010; Wang et al., 2016; Hong et al., 2017; Zhang et al., 2017a; Zhang et al., 2017b).
Little molecular research has been done on the Illiciaceae. This has hindered further studies of the molecular identification, phylogeny, and evolution of the genus. In this study, we report the sequence, assembly, annotation, and structural analysis of the I. verum chloroplast genome. The publication of more cp genomes from the Illiciaceae will help identify genetic variation through sequence comparison and provide new insights into evolutionary history and interspecific relationships. The study provides a basis for identifying medicinal properties, revising quality standards, and further development and utilization.
2 Materials and methods
2.1 Sampling, DNA extraction, sequencing, and assembly
Illicium verum samples were collected from the nursery at Guangxi University of Chinese Medicine, with the specimen accession number Bankit2753337. These samples were promptly stored at −80°C. Genomic DNA was extracted from the leaves using a modified cetyl trimethyl ammonium bromide (CTAB) protocol (Doyle and Doyle, 1987). Once the DNA quality was confirmed, shotgun sequencing libraries with an insert size of 250 base pairs were prepared according to the standard protocol provided by Illumina Inc. (San Diego, CA, United States). Sequencing was conducted on an Illumina HiSeq platform by Genesky Biotechnologies Inc. (Shanghai, China), employing the PE150 strategy (Feng et al., 2022; Shen et al., 2022).
Quality control of the raw sequencing data was performed using FastQC software, version 0.11.8 (Dierckxsens et al., 2017). High-quality clean reads were generated by eliminating adapters and low-quality sequences from the raw data with Trimmomatic, version 0.35 (Bolger et al., 2014). The chloroplast genome of I. verum was assembled using the metaSPAdes algorithm (Bankevich et al., 2012), with the reference being the previously sequenced I. verum chloroplast genome (NCBI accession number: NC_034689). To confirm the accuracy of the genome, the Illumina reads used for assembly were mapped back to the cp genome using Genious 8.1 (Kearse et al., 2012).
2.2 Annotation of the I. verum cp genome
The chloroplast genome of I. verum was annotated using the CpGAVAS pipeline (Liu et al., 2012). The annotated genome sequence has been deposited in GenBank with the accession number OR668891. Validation of annotated genomes using DOGMA (Dual Organellar Genome Annotator) software (Jansen et al., 2005). Visualization of the circular gene map was achieved using OGDRAW version 1.2 (Greiner et al., 2019), accessed on 12 September 2021, via the website http://ogdraw.mpimp-golm.mpg.de/. The relative synonymous codon usage (RSCU) analysis was conducted using CodonW, version 1.4.4 (http://codonw.sourceforge.net/, accessed on 15 September 2021).
2.3 Chloroplast genome comparison
To verify the possibility of genomic differentiation, we downloaded the chloroplast genomes of eight closely related species from Illiciaceae, Schisandraceae and Magnoliaceae from GenBank, including I. anisatum L., I. henryi Diels, I. lanceolatum A.C.Smith, Kadsura heteroclita (Roxb.) Craib, Liriodendron chinense (Hemsl.) Sarg, Magnolia maudiae (Dunn) Figlar Magnolia ovata (A.St.-Hil.) Spreng, and Schisandra henryi Clarke., and compared them using mVISTA in the shuffle-LAGAN mode (Frazer et al., 2004). The expansion and contraction of the IRs were conducted using IRSCOPE (Amiryousefi et al., 2018). To identify the hypervariable regions within the cp genome of Illiciaceae species, the cp genomes of I. verum (OR668891, OK377288), I. henryi (NC-034699), I. lanceolatum (OL802931) and I. anisatum (KY085919) were aligned using MEGA7. The alignment was manually adjusted using Se-Al 2.04, and the nucleotide diversity (Pi) along the cp genome was calculated using DnaSP version 5 software (Librado and Rozas, 2009) with sliding window analysis.
2.4 Identification of repeats
Tandem Repeats Finder (Benson, 1999) was used to identify repeat sequences. Then, the online microsatellite identification tool (MISA, available online: https://webblast.ipk-gatersleben.de/misa/) (Beier et al., 2017) was applied to predict cpSSRs with default parameters.
2.5 Phylogenetic analysis
To determine the phylogenetic relationships among Illiciaceae species, Maximum-likelihood (ML) phylogeny was built using MEGA 7 with 1000 bootstrap replicates (Kumar et al., 2016). An alignment of 53 cp genomic sequences was created using the MAFFT online version with default parameters.
3 Results
3.1 Genome features of I. verum
After filtering the raw sequencing data, we obtained a total of 646,898 high-quality paired-end reads. These reads comprised 96,506,157 bases, with 88.5% exceeding a quality score of Q30. The complete chloroplast (cp) genome of I. verum measures 143,187 base pairs (bp) in length and exhibits a guanine-cytosine (GC) content of 39.1%. The assembled genome achieved an average read coverage exceeding 700-fold.
The genome of I. verum displays a typical quadripartite structure, containing one large single copy (LSC; 100,868 bp) region, one small single copy (SSC; 20,235 bp) region, and two inverted repeat regions (IRs; 11,042 bp each) (Figure 1). The DNA G + C contents of the LSC, SSC, and IR regions and the whole genome are 38%, 33.9%, 49.1%, and 39.1%, respectively, which is also similar to the chloroplast genomes of other I. verum (Table 1).

Figure 1. Chloroplast genome map of I. verum. Genes inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. Genes of different functions are color-coded. The darker gray in the inner circle shows the GC content, while the lighter gray shows the AT content.

Table 1. Base composition in the cp genome of I. verum.
In the I. verum cp genome, 121 functional genes were predicted, including 8 rRNA genes, 35 tRNA genes, and 78 protein-coding genes (Table 2). Cp genomes have 10 duplicated genes in the IR regions, including approximately 5 tRNA genes (tRNAs), 4 rRNA genes (rRNAs), and 1 protein-coding gene (PCGs) (Figure 1). The LSC region contains 68 protein-coding and 28 tRNA genes, while the SSC region contains 1 tRNA gene and 12 protein-coding genes.
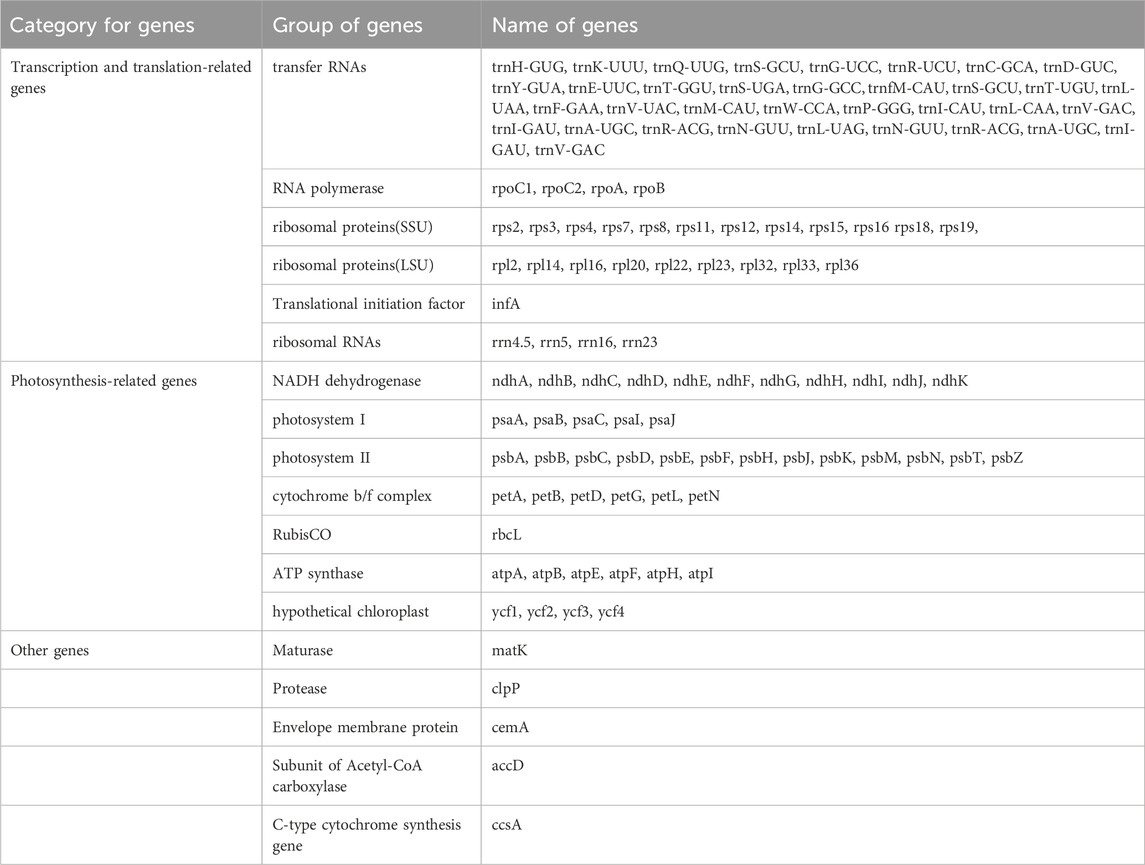
Table 2. Gene contents in the I. verum cp genome.
Within the genome, 20 genes are identified to harbor introns, comprising 12 protein-coding genes and 8 transfer RNA (tRNA) genes as detailed in Table 3. Specifically, 18 genes-encompassing 10 protein-coding genes and 8 tRNA genes-feature at least one intron. Notably, the genes ycf3 and clpP are distinguished by the presence of two introns. Among the intron-containing genes, the trnK-UUU gene is notable for having the largest intron, measuring 2541 base pairs (bp), contrasting with the trnL-UAA gene, which contains the smallest intron at 517 bp. Additionally, the rps12 gene is recognized as a trans-splicing gene, with its 5′ end located in the large single copy (LSC) region and the 3′ end situated in the inverted repeat (IR) region.
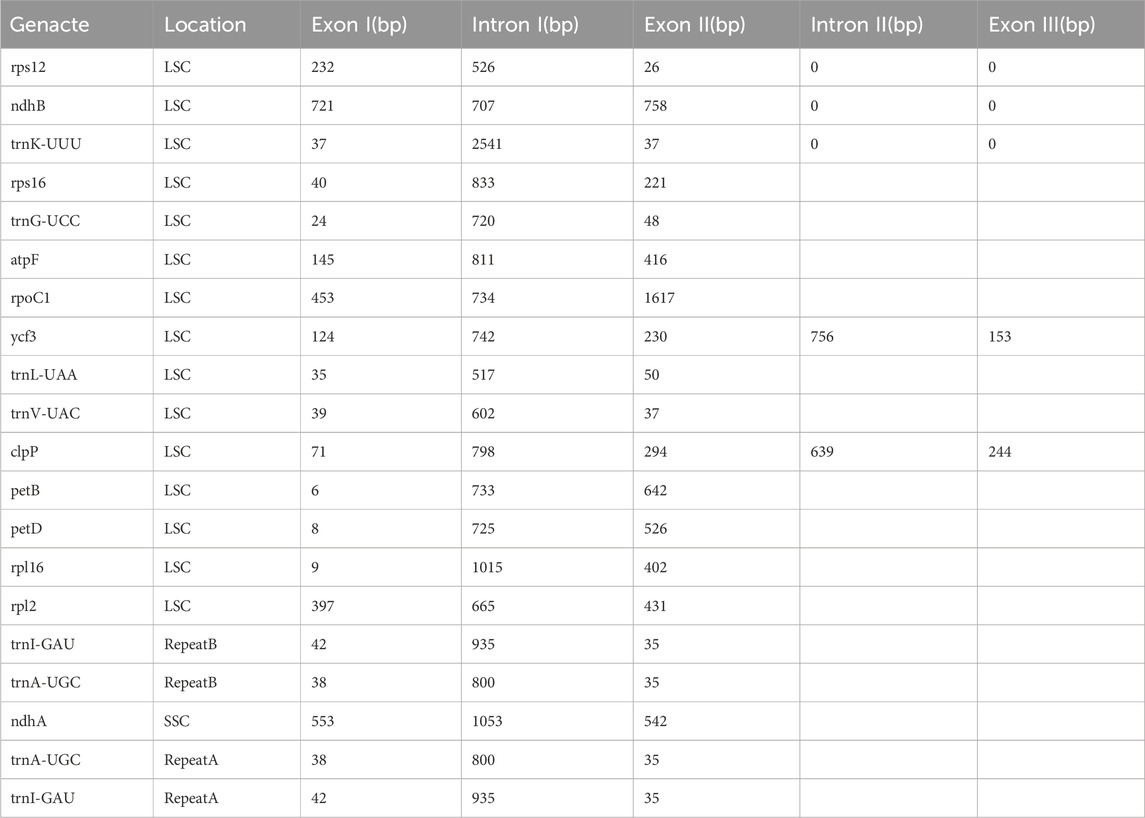
Table 3. Genes with introns in the I. verum cp genome.
3.2 Bias of codon usage
The tRNA and protein-encoding gene sequences of the I. verum cp were analyzed, and the codon usage frequency of the cp genome of I. verum was inferred and summarized. A total of 22,727 codons represent the coding ability of I. verum (Figure 2; Supplementary Table S1), of which 2,319 codons code for leucine (10.2%), and 268 codons code for tryptophan (1.2%), which are the most and least common amino acids in the cp genome of I. verum, respectively. Most of the codons ending with A or T were presented as RSCU >1 (Supplementary Table S1).
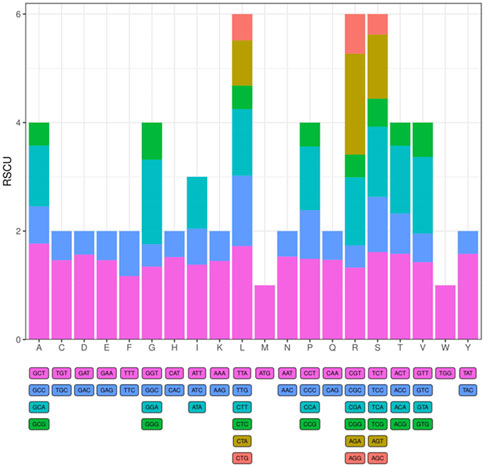
Figure 2. Codon content of 20 amino acids and stop codons in all protein-coding genes of the I. verum chloroplast genome. The codons are represented by different colors in the histogram.
3.3 Comparative analysis of genomic structure
The complete cp genome sequence of I. verum was compared with I. anisatum, I. henryi, I. lanceolatum, K. heteroclita, L. chinense, M. maudiae, M. ovata and S. henryi has the largest cp genome (Figure 3). The results of this comparison revealed that the LSC and SSC regions are more divergent than the IR regions and that higher divergence is found in noncoding than in coding regions. The chloroplast genome sequence of I. verum was not significantly different from that of I. anisatum, I. henryi, and I. lanceolatum, but it was quite different from the five other plants.
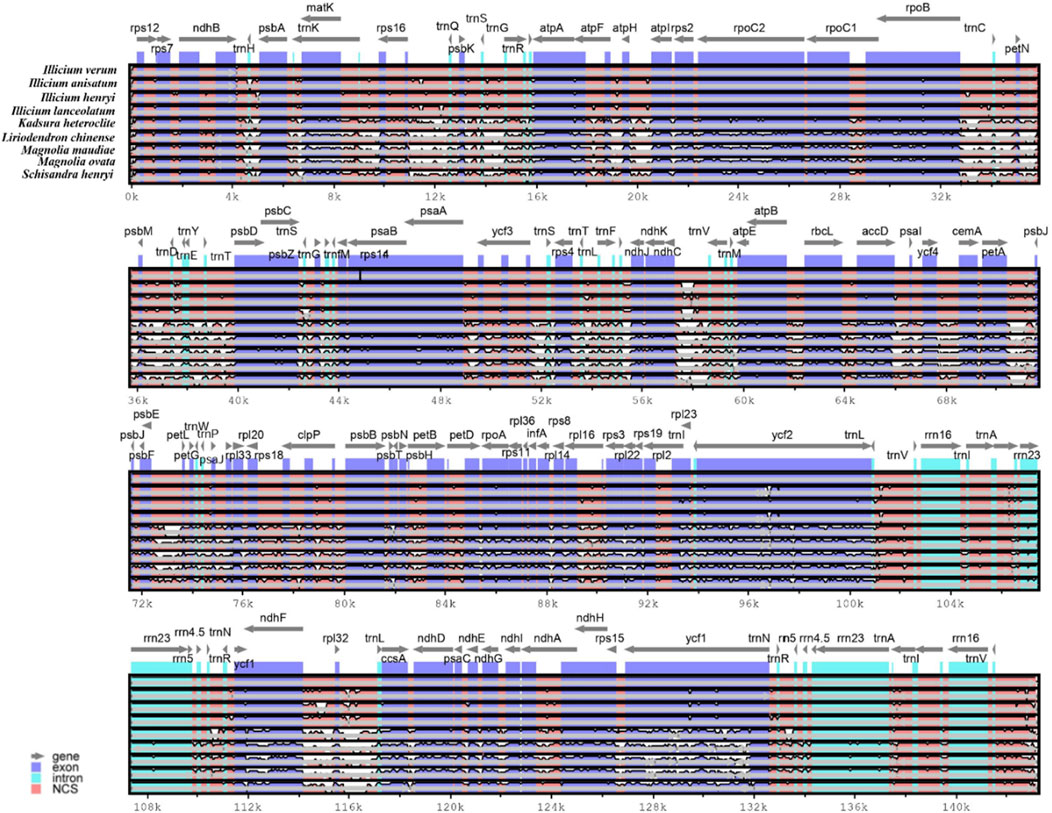
Figure 3. Comparison of the cp genome sequences of nine plants. Comparison of the cp genome sequences of I. verum (NC-034689), Illicium anisatum (KY085919), Illicium henryi (NC-034699), Illicium lanceolatum (OL802931), Kadsura heteroclite (NC-057266), Liriodendron chinense (MK887904), Magnolia maudiae (NC-047409), Magnolia ovata (MT682825) and Schisandra henryi (MH394370) generated with mVISTA. Gray arrows indicate the position and direction of each gene. Red and blue areas indicate the intergenic and genic regions, respectively. The vertical scale indicates the percentage of identity, ranging from 50% to 100%.
To detect highly variable regions, we analyzed variable sites in the Illiciaceae chloroplast genome by sliding window analysis using the software DnaSP (Figure 4). The divergent loci (ndhB-trnH, atpF-atpH, ndhG-ndhI) had a Pi value greater than or equal to 0.02. The ndhB-trnH, atpF-atpH divergent loci were intergenic regions and were present in the LSC region, and ndhG-ndhI occurred in the SSC region, with none detected in the IR region. These results also confirmed that the LSC and SSC regions were less conserved than the IR regions.
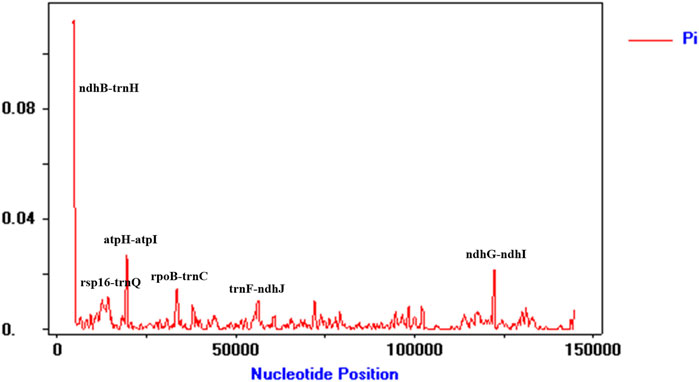
Figure 4. Chloroplast genome comparative analysis between I. verum (OR668891 and OK377288), Illicium henryi (NC-034699), Illicium lanceolatum (OL802931) and Illicium anisatum (KY085919). Sliding window plots of nucleotide diversity (Pi) across the complete cp genomes of Illiciaceae species (window length: 600 bp, step size: 200 bp). Y-axes: nucleotide diversity (Pi) of each window; X-axes: the position of the midpoint of a window.
3.4 Contraction and expansion of the IR regions
To further resolve the structural evolutionary history of the cp genomes of I. verum, we compared the IR/SSC and IR/LSC junctions across eight selected Magnoliaceae, Schisandraceae and Illiciaceae species, including M. maudiae, L. chinense, S. henryi, K. heteroclita, I. henryi, and I. anisatum. The results of the IRscope analysis are presented in Figure 5.
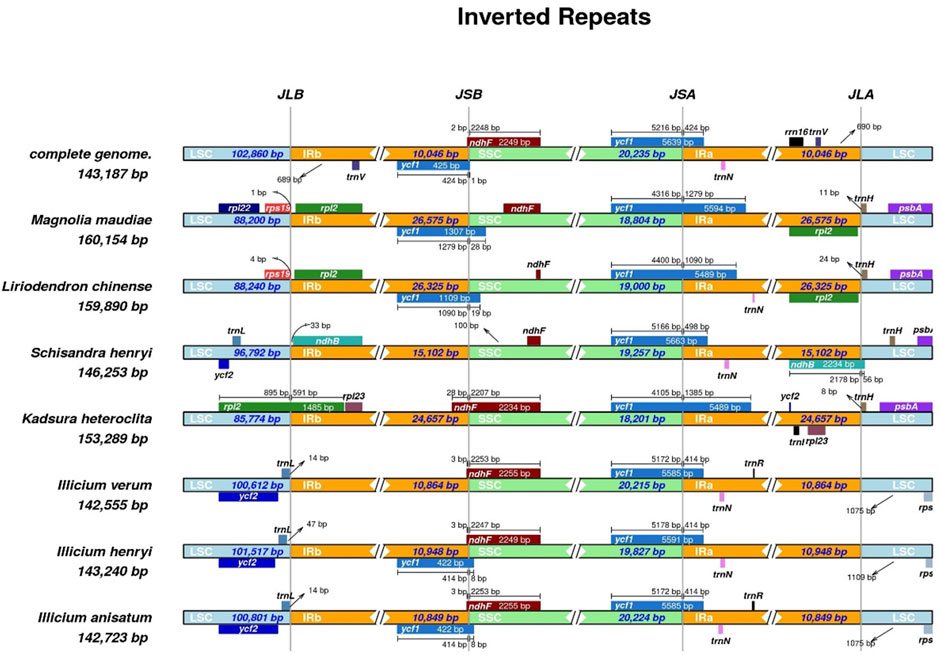
Figure 5. The junctions of IR/SC comparison among 8 plants. Arrows indicated the distance of the gene to the junctions, and the I-shaped line represented the length of the gene on either side of the junctions.
We observed a wide variability in the junction sites of these cp genomes. For example, the ycf1 gene was located at the SSC/IRa junction in all the chloroplast genomes of different species. The length of the ycf1 gene extending into the SSC region varied depending on the genome (I. verum OR668891, 5216 bp; I. verum OK377288, 5172 bp; I. henryi NC-034699, 5178 bp; I. anisatum KY085919, 5172 bp; M. maudiae NC_047409, 4316 bp; L. chinense MK887904, 4400 bp; S. henryi MH394370, 5166 bp; K. heteroclite NC-057266, 4105 bp); the IRa region includes 424, 414, 414, 414, 1279, 1090, 498, and 1385 bp of the ycf1 gene. In the boundary region of JSB (IRb/SSC), except for M. maudiae, L. chinense and S. henryi, the boundaries of JSB (IRb/SSC) in the other five plants were located within the ndhF gene in the SSC region and expanded by 2–28 bp to the IRb boundary.
3.5 Long-repeat and SSR analysis
In the analysis of repetitive structures within the Illiciaceae (I. verum OR668891, I. verum NC_034689, I. verum ON357882, I. verum OK377288, I. anisatum KY085919, I. henryi NC_034699, I. lanceolatum OL802931) chloroplast genomes, 33 inverted repeat sequences were identified in I. verum OR668891 and I. verum NC_034689, 30 inverted repeat sequences in I. verum ON357882 and I. verum OK377288, 28 inverted repeat sequences in I. anisatum KY085919, 37 inverted repeat sequences in I. henryi NC_034699, and 30 inverted repeat sequences in I. lanceolatum OL802931. The majority of these repeats range from 9 to 30 base pairs (bp), as detailed in Figure 6A; Supplementary Table S2. The longest repeat, measuring 30 bp, is situated within the large single copy (LSC) region. The distribution of repeats across the genome is as follows: 25 in the LSC, 6 in the small single copy (SSC), and 2 in the inverted repeat (IR) regions. Predominantly, the repeats in intergenic spacers are concentrated in the LSC region, whereas two repeats are located in the SSC region.
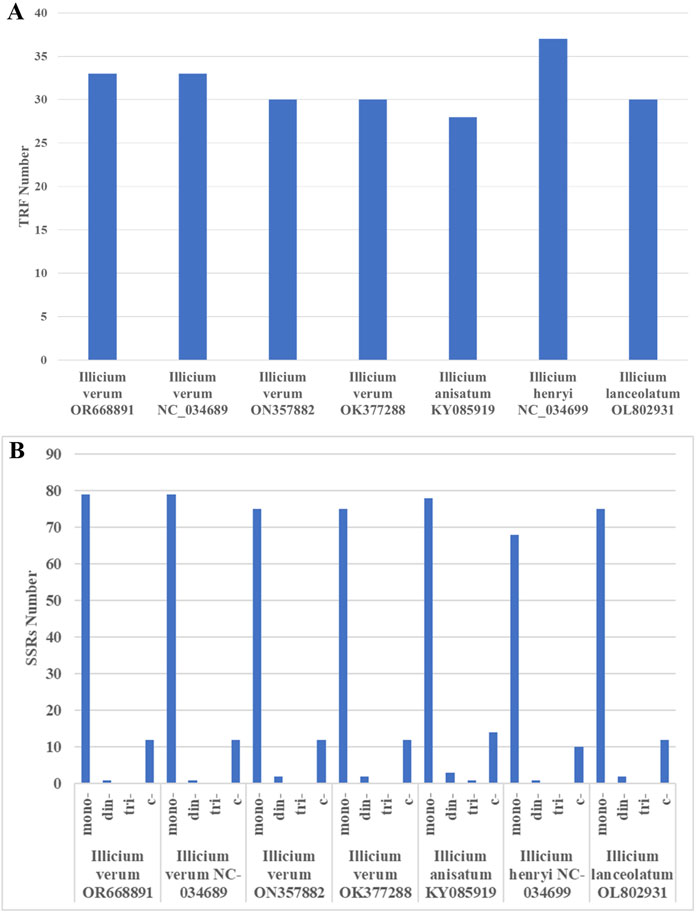
Figure 6. The type of long-repeat and SSRs in the seven Illiciaceae chloroplast genomes. (A) Number of long-repeat. (B) Number of SSR repeat types.
The chloroplast genome is highly conserved, allowing SSR primers to be broadly applicable across different species and genera. Through the analysis of SSRs in the Illiciaceae (I. verum OR668891, I. verum NC_034689, I. verum ON357882, I. verum OK377288, I. anisatum KY085919, I. henryi NC_034699, I. lancolatum OL802931), 92 SSRs were identified in I. verum OR668891 and I. verum NC_034689, 89 SSRs in I. verum OK377288, 96 SSRs in I. anisatum KY085919, 79 SSRs in I. henryi NC_034699, and 89 SSRs in I. lanceolatum OL802931. The majority of these SSRs were distributed in the large single copy (LSC) and small single copy (SSC) regions, with a subset also present in the inverted repeat (IR) regions. The I. verum OR668891 SSRs comprised 79 single nucleotide SSRs (85.8%), and 1 dinucleotide SSRs (1%) (Figure 6B; Table 4). Of these, 12 SSRs were of the C type, while the remainder were A/T types.
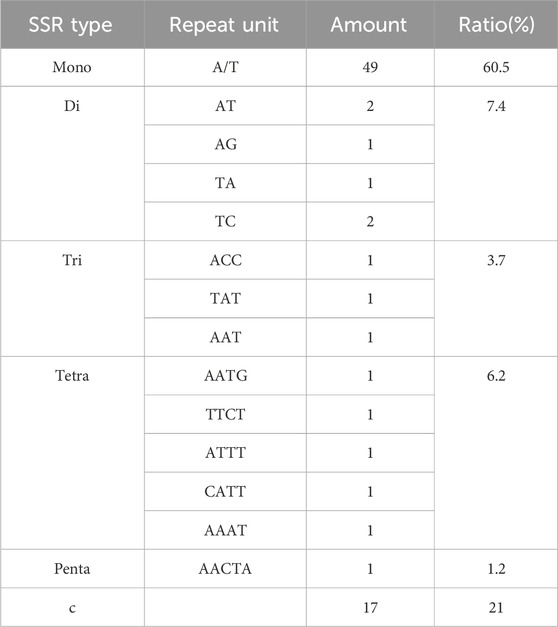
Table 4. Types and amounts of SSRs in the chloroplast genome of I. verum.
3.6 Phylogenetic analysis
We found that the Illiciaceae and the Schisandraceae families are grouped together in the same branch. I. verum was clustered with I. verum (KY085896, NC 034689) into a subclade with 100% supported value and I. verum was grouped into a terminal branch with Illiciaceae plants such as I. anisatum, I. lanceolatum, I. majus, I. burmanicum, I. henryi, I. floridanum and I. tsangii. The family of Schisandriaceae and heteromorphic Schisandriaceae such as K. heteroclita, K. ananosma, S. chinensis, and S. henryi were clustered into another terminal branch. Meanwhile, the Magnoliaceae plants such as M. ovata, M. laevifolia, M. figo, M. dixonii, M. zenii, M. lacei, L. chinense and L. tulipifera formed an independent branch (Figure 7).

Figure 7. Maximum likelihood (ML) tree of 53 species in different genera of Magnoliaceae and Illiciaceae based on the whole chloroplast genomes. The position of I. verum is indicated by “▲.” The cp genomes of other species used in this study were obtained from GenBank. Details for the origin of species were given in Supplementary Table S3.
4 Discussion
The I. verum is predominantly found in provinces near the Tropic of Cancer in China, such as Guangxi province, where it is cultivated extensively. According to the latest 2020 edition of the Chinese Pharmacopoeia, the identification of I. verum is mainly through trait identification, the color reaction of resorcinol hydrochloric acid, and other methods that include a combination of the above methods.
In the present study, we successfully assembled, annotated, and analyzed the complete chloroplast (cp) genome sequence of I. verum. Further analysis focused on its genomic features, guanine-cytosine (GC) content, gene structure, and repetitive sequences. They revealed that the structure and content of I. verum are similar to those of other angiosperms. This phenomenon is also widespread in the chloroplast genomes of other angiosperms. The DNA GC content is an important indicator for evaluating the phylogenetic relationships of I. verum, and its cpDNA GC content is similar to other species within the Illiciaceae. The DNA GC content in the IR regions is higher than in other regions (LSC, SSC), a phenomenon commonly observed in other plants. The higher DNA GC content in the IR regions is primarily attributed to the rRNA and tRNA genes. The lengths of introns and exons are of significant importance in plant chloroplast genomes. The results indicate that in the chloroplast genome of I. verum, only one gene (rps12) contains three exons, while two genes (ycf3 and clpP) contain two introns. The rps12 gene is located at the 5′ end of the LSC region, with the duplicated 3′ end in the IRs region, thus being referred to as a trans-spliced gene. Furthermore, in the codon usage of the I. verum chloroplast genome, 2,319 codons encode leucine (10.2%), the most frequently used amino acid, while 288 codons encode tryptophan, the least common amino acid in this species' cp genome. The high frequency of leucine in the chloroplast genomes can be attributed to its substantial requirement in chloroplasts, as leucine plays a significant role in photosynthesis-related metabolism. Comparative analysis of chloroplast genomes from the Illiciaceae, Magnoliaceae and Schisandraceae using mVISTA revealed the similarity in DNA sequences among closely related species. Although the IR regions is more conserved than the SSC and LSC regions in chloroplast genomes, evolutionary events and size variations in chloroplast genomes of different plants can also be attributed to the expansion and contraction of boundaries between the SC and IR regions (Raubeson et al., 2007; Wang et al., 2008; Wang and Messing, 2011; Meng et al., 2018; Zhao et al., 2018; Androsiuk et al., 2020). In this study, the rps19, ndhF, ycf1, and trnH genes are located at the boundaries of LSC/IRB, IRB/SSC, SSC/IRA, and IRA/LSC, respectively. Similar to most angiosperms, the ndhF and ycf1 genes of I.verum chloroplast genome are located at the IR boundary.
SSRs, known as microsatellites, are important repetitive elements throughout the genome, composed of one or a few tandemly repeated nucleotides. They are identical units located in homologous regions but with different unit numbers, playing a significant role in many applications such as species identification, population genetics, and phylogenetic studies (Yang et al., 2011; Jiao et al., 2012; Zhang et al., 2016; Androsiuk et al., 2020; Zhang et al., 2011). SSRs in chloroplast genomes are typically distributed in intergenic regions (Zhao et al., 2015). Based on transcriptome data, Yu Xinghua et al. (Yu et al., 2022) screened SSRs, where dinucleotide and trinucleotide repeat types dominated in the transcriptome data SSRs, with the highest frequency of AG/CT in dinucleotides and AAG/CTT in trinucleotides. Based on the study of chloroplast genes, it was found that the single nucleotide repeats were the most in the SSRs sequence of the chloroplast genome, and this phenomenon was the same in the octagonal chloroplast genome. A total of 92 SSRs were found in the chloroplast genome of I. verum OR668891 (Figure 6), most of which were located in the LSC region, followed by the SSC region and the IR region. The most abundant are mononucleotide repeat sequences (85.8%), contributing to A/T richness. These results are consistent with most reported angiosperms (Cui et al., 2019; Gao et al., 2019; Li W. et al., 2019; Li Y. et al., 2019). By comparing repetitive sequences with other plants in the Illiciaceae, it was found that I. verum (OR668891) has 33 long-repeat sequences and 92 SSRs, which is consistent with I. verum (NC-034689, KY085896). In contrast, I. verum (OK377288) has 30 long repetitive sequences and 89 SSRs, and its molecular characteristics are more inclined towards I. lanceolatum (OL802931). Comparison of the chloroplast genome SSRs of I. verum with I. anisatum, I. henryi, and I. lanceolatum revealed certain differences in SSRs among the four species. Currently, there are several complete chloroplast genomes of I. verum registered in GenBank, with accession numbers ON357882, OK377288, NC_034689, and KY085896. Among these, the sequences ON357882 and OK377288 were submitted by Nanjing Agricultural University in Jiangsu Province, China. Due to limited information available upon registration, it is unclear whether the I. verum sequences (ON357882 and OK377288) originate from a specific source. Whether a possibility that the I. verum sequences published in Jiangsu (ON357882 and OK377288) might be contamination derived from an I. lanceolatum sample? Further in-depth and extensive sequencing will be required to verify this. Overall, the chloroplast genome SSRs of I. verum exhibit rich variation, which may help detect polymorphisms at the intraspecific level and develop species markers for future evolutionary and genetic diversity studies. The DNA barcoding method, as proposed by Hebert (Hebert et al., 2003), can identify species through DNA sequences, ITS2, matK, psbA-trnH, and rbcL. However, the identification of closely related species (mainly morphologically confused species within the same genus) still presents various challenges. Therefore, finding suitable DNA markers for these species is essential. The chloroplast genome is commonly used for phylogenetic studies and species identification because its evolutionary rate is slower than that of the nuclear genome. In this study, the analysis of chloroplast genome comparisons among five varieties showed that there are many variable sites in the intergenic regions such as ndhB-trnH, atpH-atpI, rpoB-trnC and ndhG-ndhI. The ndhB-trnH region consists of one intergenic space (ndhB-trnH) and a coding gene (trnH); this region is the most variable marker in the Illiciaceae chloroplast genome (Figure 6; Supplementary Figure S1). However, this marker was not extensively used in plant phylogeny and DNA barcoding. The ndhB-trnH can be used in subsequent studies. Thus, these regions could potentially serve as different candidate fragments for the identification of Illiciaceae. Overall, these highly differentiated regions provide abundant molecular marker information for the identification and phylogenetic relationship studies of Illiciaceae.
The phylogenetic positions of 53 cp genomes were successfully analyzed. The Illiciaceae, Schisandraceae, and Magnoliaceae were well separated, and plants of the same species were generally grouped into the same clade. However, when comparing I. verum (OR668891) with I. verum sequences (such as OK377288, NC 357882) deposited in NCBI, some deficiencies were found in the non-coding regions of different sequences, resulting in the OR668891and OK377288, NC 357882 strains being on separate branches in the evolutionary tree. This is consistent with the analysis results of the above long repeat sequences and SSRs. Due to the limited number of chloroplast genome sequences of I. verum published in NCBI, A broader taxon sampling and integration with other molecular makers, e.g., nuclear genes, need to be used to explore the phylogenetic relationship of Illiciaceae. Nevertheless, our phylogenetic studies provide valuable resources for the taxonomic identification, phylogenetics, and evolutionary history research of Illiciaceae.
5 Conclusion
The complete chloroplast (cp) genomes of I. verum exhibit the characteristic quadripartite structure typical of land plants. Comparative analysis of cp genomes across nine species within the tribe Illiciaceae has demonstrated a high degree of conservation in both genome organization and gene order. Notably, the identification of specific repeated sequences, and simple sequence repeat (SSR) loci, and regions of high variability within these genomes suggests their utility as molecular markers for phylogenetic studies within the tribe. Phylogenetic analysis supports will Illiciaceae is classified as an independent branch. Furthermore, the release of additional cp genome sequences from the genus Illicium is anticipated to facilitate the identification of genetic variations, thereby enhancing our understanding of the evolutionary history and interspecific relationships within this group.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OR668891.
Author contributions
YC: Writing–original draft, Methodology. YL: Resources, Writing–original draft. ZL: Methodology, Writing–original draft. SZ: Methodology, Writing–original draft. YD: Methodology, Writing–original draft. JT: Data curation, Writing–original draft. QW: Data curation, Writing–original draft. ZX: Data curation, Writing–review and editing. MJ: Data curation, Writing–original draft. LY: Writing–review and editing. JL: Funding acquisition, Supervision, Writing–review and editing. HT: Funding acquisition, Supervision, Validation, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Scientific Fund from Guangxi Province (2024GXNSFAA010233), The Open Project of Guangxi Health Commission Guangxi Key Laboratory of Molecular Biology of Preventive Medicine of Traditional Chinese Medicine(GXHC-MBTCM-03), High-Level Talent Cultivation and Innovation Team Funding Project of Guangxi University of Chinese Medicine (2022B006), Interdisciplinary innovation research team for key technologies of traditional Chinese medicine emerging biological materials and intelligent equipment development (GZKJ2303) and Training Program for 1,000 Young and Middle aged Backbone Teachers in Colleges and Universities in Guangxi [Phase III] [Gui Jiao normal [2019] No. 81].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1452680/full#supplementary-material
References
Ac, S. (1947). The families Illiciaceae and Schisandraceae. Sargentia 7, 1–224. doi:10.5962/p.265318
Amiryousefi, A., Hyvönen, J., and Poczai, P. I. R. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi:10.1093/bioinformatics/bty220
Androsiuk, P., Jastrze¸bski, J. P., Paukszto, Ł., Makowczenko, K., Okorski, A., Pszczółkowska, A., et al. (2020). Evolutionary dynamics of the chloroplast genome sequences of six Colobanthus species. Sci. Rep. 10, 11522. doi:10.1038/s41598-020-68563-5
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. a J. Comput. Mol. cell Biol. 19, 455–477. doi:10.1089/cmb.2012.0021
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinforma. Oxf. Engl. 33, 2583–2585. doi:10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi:10.1093/nar/27.2.573
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl. 30, 2114–2120. doi:10.1093/bioinformatics/btu170
Chen, S., Xu, M., Wu, Q., Yang, W., Chen, H., and Zheng, L. (2023). Research progress in adulterants identification of star anise. J. Food Saf. Qual. 14, 126–133. doi:10.19812/j.cnki.jfsq11-5956/ts.2023.13.038
Cui, A., and Fu, J. (2001). Identification of star anise and its counterfeit. Lishizhen Med. Mater Med. Res. 12, 513.
Cui, Y., Nie, L., Sun, W., Xu, Z., Wang, Y., Yu, J., et al. (2019). Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 8, 283. doi:10.3390/plants8080283
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novoassembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi:10.1093/nar/gkw955
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Feng, Z., Zheng, Y., Jiang, Y., Miao, Y., Luo, G., and Huang, L. (2022). Complete chloroplast genome of Gentianopsis barbata and comparative analysis with related species from Gentianaceae. Genome 65, 363–375. doi:10.1139/gen-2021-0080
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi:10.1093/nar/gkh458
Gao, C., Deng, Y., and Wang, J. (2019). The complete chloroplast genomes of Echinacanthus species (Acanthaceae): phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front. Plant Sci. 9, 1989. doi:10.3389/fpls.2018.01989
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW(OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59-W64–W64. doi:10.1093/nar/gkz238
Hebert, P. D. N., Cywinska, A., Ball, S. L., and Dewaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 270, 313–321. doi:10.1098/rspb.2002.2218
Hong, S., Cheon, K., Yoo, K. O., Lee, H. O., Cho, K. S., Suh, J. T., et al. (2017). Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front. plant Sci. 8, 1696. doi:10.3389/fpls.2017.01696
Jansen, R. K., Raubeson, L. A., Boore, J. L., Depamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 395, 348–384. doi:10.1016/s0076-6879(05)95020-9
Jiao, Y., Jia, H., Li, X., Chai, M., Jia, H., Chen, Z., et al. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genomics 13, 201. doi:10.1186/1471-2164-13-201
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi:10.1093/bioinformatics/bts199
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi:10.1093/molbev/msw054
Li, F., and Liu, Y. L. (2011). Identification of star anise and its adulterants. Shandong J trad chin med 30, 739–740.
Li, L. (2023). Literature research on Chinese medicine star anise fennel. J. Chin. Med. Mater. 46, 773–779. doi:10.13863/j.issn1001-4454.2023.03.041
Li, S. (1596). Compendium of Materia Medica (bencaogangmu). Compend. Materia Medica (Bencaogangmu), 1636–1639.
Li, W., Zhang, C., Guo, X., Liu, Q., and Wang, K. (2019). Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS One 14, e0216645. doi:10.1371/journal.pone.0216645
Li, Y., Sylvester, S. P., Li, M., Zhang, C., Li, X., Duan, Y., et al. (2019). The complete plastid genome of Magnolia zenii and genetic comparison to Magnoliaceae species. Molecules 24, 261. doi:10.3390/molecules24020261
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi:10.1093/bioinformatics/btp187
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC genomics 13, 715. doi:10.1186/1471-2164-13-715
Meng, X., Xian, Y., Xiang, L., Zhang, D., Shi, Y., Wu, M., et al. (2018). Complete chloroplast genomes from Sanguisorba: identity and variation among four species. Molecules 23, 2137. doi:10.3390/molecules23092137
Qi, L. (1995). The geographical distribution of the family Illiciaceae. J. Trop. subtropical Bot. 3, 1–11.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8, 174. doi:10.1186/1471-2164-8-174
Shen, J., Li, X., Chen, X., Huang, X., and Jin, S. (2022). The complete chloroplast genome of carya cathayensis and phylogenetic analysis. Genes (Basel) 13, 369. doi:10.3390/genes13020369
Vermaak, I., Viljoen, A., and Lindström, S. W. (2013). Hyperspectral imaging in the quality control of herbal medicines - the case of neurotoxic Japanese star anise. J. Pharm. Biomed. analysis 75, 207–213. doi:10.1016/j.jpba.2012.11.039
Wang, G. W., Hu, W. T., Huang, B. K., and Qin, L. P. (2011). Illicium verum: a review on its botany, traditional use, chemistry and pharmacology. J. Ethnopharmacol. 136, 10–20. doi:10.1016/j.jep.2011.04.051
Wang, R., Cheng, C., Chang, C., Wu, C., Su, T., and Chaw, S. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 36. doi:10.1186/1471-2148-8-36
Wang, W., and Messing, J. (2011). High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS One 6, e24670. doi:10.1371/journal.pone.0024670
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016). Complete chloroplast genome sequence of aquilaria sinensis (lour.) gilg and evolution analysis within the malvales order. Front. plant Sci. 7, 280. doi:10.3389/fpls.2016.00280
Wu, F. H., Chan, M. T., Liao, D. C., Hsu, C. T., Lee, Y. W., Daniell, H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC plant Biol. 10, 68. doi:10.1186/1471-2229-10-68
Yang, A., Zhang, J., Yao, X., and Huang, H. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 98, e123–e126. doi:10.3732/ajb.1000532
Yu, X., Yang, Y., Li, Y., Tao, Y., Meng, F., and Teng, J. (2022). Development of SSR molecular markers based on transcriptome sequencing of Illicium verum. Southwest China J. Agric. Sci. 6, 1261–1268. doi:10.16213/j.cnki.scjas.2022.6.004
Zhang, J. M., Liu, J., Sun, H. L., Yu, J., Wang, J. X., and Zhou, S. L. (2011). Nuclear and chloroplast SSR markers in Paeonia delavayi (Paeoniaceae) and cross-species amplification in P. ludlowii. Am. J. Bot. 98, e346–e348. doi:10.3732/ajb.1100240
Zhang, N., Erickson, D. L., Ramachandran, P., Ottesen, A. R., Timme, R. E., Funk, V. A., et al. (2017a). An analysis of Echinacea chloroplast genomes: implications for future botanical identification. Sci. Rep. 7, 216. doi:10.1038/s41598-017-00321-6
Zhang, N., Ramachandran, P., Wen, J., Duke, J. A., Metzman, H., McLaughlin, W., et al. (2017b). Development of a reference standard library of chloroplast genome sequences, GenomeTrakrCP. Planta medica. 83, 1420–1430. doi:10.1055/s-0043-113449
Zhang, Y., Yuan, X., Teng, W., Chen, C., and Wu, J. (2016). Identification and phylogenetic classification of Pennisetum (Poaceae) ornamental grasses based on ssr locus polymorphisms. Plant Mol. Biol. Rep. 34, 1181–1192. doi:10.1007/s11105-016-0990-2
Zhao, J. T., Xu, Y., Xi, L. J., Yang, J. W., Chen, H. W., and Zhang, J. (2018). Characterization of the chloroplast genome sequence of Acer miaotaiense: comparative and phylogenetic analyses. Molecules 23, 1740. doi:10.3390/molecules23071740
Keywords: chloroplast genome sequence, comparative analysis, Illicium verum, phylogenetic relationship, Illiciaceae
Citation: Cao Y, Lai Y, Li Z, Zhai S, Dai Y, Tao J, Wang Q, Xu Z, Jiang M, Yu L, Leng J and Tang H (2024) The complete chloroplast genome of Illicium verum and comparative analysis with related species from Magnoliaceae and Illiciaceae. Front. Genet. 15:1452680. doi: 10.3389/fgene.2024.1452680
Received: 21 June 2024; Accepted: 25 November 2024;
Published: 11 December 2024.
Edited by:
Ahmad M. Alqudah, Qatar University, QatarReviewed by:
Gecele Matos Paggi, Federal University of Mato Grosso do Sul, BrazilXianhan Huang, Chinese Academy of Sciences (CAS), China
Copyright © 2024 Cao, Lai, Li, Zhai, Dai, Tao, Wang, Xu, Jiang, Yu, Leng and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haibo Tang, dGFuZ2hiQGd4dGNtdS5lZHUuY24=; Jing Leng, bGo5ODY3NzE1NThAMTYzLmNvbQ==