 Bing Li
Bing Li Kan Tan
Kan Tan Angelyn R. Lao
Angelyn R. Lao Haiying Wang
Haiying Wang Huiru Zheng
Huiru Zheng Le Zhang
Le Zhang- 1College of Computer Science, Sichuan University, Chengdu, China
- 2Department of Mathematics and Statistics, De La Salle University, Manila, Philippines
- 3School of Computing, Ulster University, Belfast, United Kingdom
With the innovation and advancement of artificial intelligence, more and more artificial intelligence techniques are employed in drug research, biomedical frontier research, and clinical medicine practice, especially, in the field of pharmacology research. Thus, this review focuses on the applications of artificial intelligence in drug discovery, compound pharmacokinetic prediction, and clinical pharmacology. We briefly introduced the basic knowledge and development of artificial intelligence, presented a comprehensive review, and then summarized the latest studies and discussed the strengths and limitations of artificial intelligence models. Additionally, we highlighted several important studies and pointed out possible research directions.
1 Introduction
Artificial intelligence (AI) is defined as the intelligence exhibited by artificial entities to solve complex problems, and is generally considered to be a system of computers or machines (Kumar et al., 2012). With the emergence of big data and the improvement of computing power, machine learning, artificial neural networks, and deep learning (Gao et al., 2022; Song et al., 2022; Gao et al., 2023) have been developing rapidly and continued to integrate other disciplines in recent years, achieving great success in theory and application (Chaturvedula et al., 2019; Brown et al., 2020; Woschank et al., 2020; Alzubaidi et al., 2021; Mohsen et al., 2023). Figure 1 shows the relationship between AI and related concepts such as machine learning, artificial intelligence, and deep learning. Meanwhile, Figure 1 shows the applications of artificial intelligence in pharmacology research.
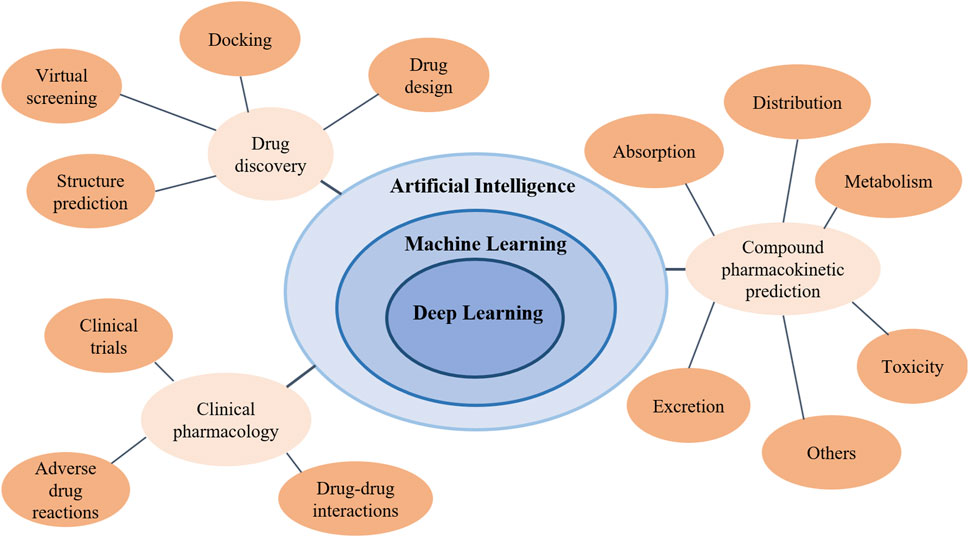
Figure 1. The relationship between artificial intelligence, machine learning, and deep learning and the applications of artificial intelligence in pharmacology research.
The development of AI can be traced back to the 1940s, and its historical process and development have been detailed in many previous reviews (Muggleton, 2014; Haenlein and Kaplan, 2019). In recent decades, the widespread application of neural networks, such as convolutional neural networks (CNNs), recurrent neural networks (RNNs), graph neural networks (GNNs) and deep neural networks (DNNs) (Gao et al., 2021; Lai et al., 2022), as well as the development of deep learning algorithms, such as ResNet (He et al., 2015; Zhang et al., 2024), Attention and Transformer (Vaswani et al., 2017; You et al., 2022b), have driven the development of neural networks and deep learning, and further optimized the application performance of AI algorithms in various fields (Alzubaidi et al., 2021). Figure 2 briefly extracts and exhibits the most important algorithms proposed during the development of AI.
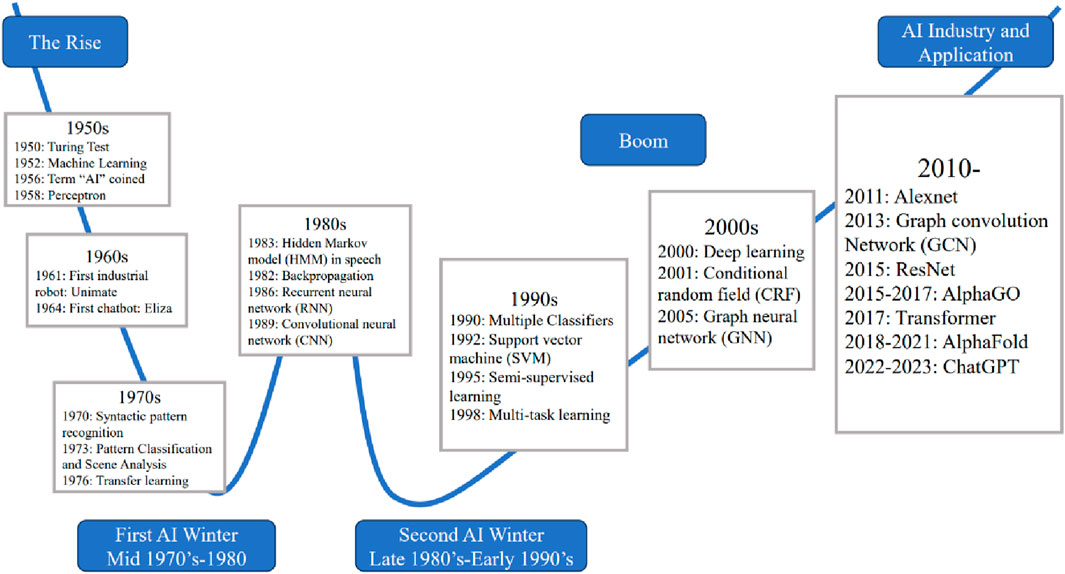
Figure 2. Timeline of the development and application of artificial intelligence.
The study of pharmacology originated in the mid-19th century and it covers a very wide range of fields (Vallance and Smart, 2006). The driving force of pharmacology is to understand and quantify the effects of drugs on physiology, including exploring the action of drugs, the mechanism of action of drugs, and the active ingredients of drugs (Vallance and Smart, 2006). It is generally believed that the scope of pharmacology (Vallance and Smart, 2006) is comprised of drug discovery, design, explanation of mechanisms, drug metabolism and actual clinical research, etc., Therefore, pharmacology is a very complex and comprehensive science.
The contribution of AI in pharmacology research does not appear suddenly, but with the development of AI and pharmacology themselves, mutual promotion and growth. Research on the combination of AI and pharmacology has been proposed for a long time (So and Karplus, 1996a; b). It is worth noting that although methods such as neural networks were proposed for use in QSAR (Quantitative Structure-activity Relationship) models at that time (So and Karplus, 1996a), there are at least some difference or progress for now, namely more abundant and suitable AI models for different situations as mentioned above, more standard data sets and research community building (Su et al., 2019), more various kind of descriptors and wider applications in pharmacology as summarized below.
Pharmacology is a very complex study involving a lot of computing, data statistics and analysis. A number of AI methods (Zhang et al., 2019b; Zhang et al., 2021c; Zhang et al., 2021d; You et al., 2022a; Zhang et al., 2023b) have been used in pharmacology research, where the most widely used fields are AI-assisted drug discovery and design (Paul et al., 2021), prediction of compound pharmacokinetics (Obrezanova, 2023) and clinical pharmacology (Johnson et al., 2023). Thus, this review will focus on the application of AI in these three areas (shown in Figure 1), and introduce latest research methods and models in the following sections.
2 AI-assisted drug discovery and design
Classical molecular drug discovery and design encounters several problems and challenges such as long development time, low clinical success rate and high cost. In general, it takes about 13.5 years for a drug molecule to be developed and approved for marketing, and the total cost to develop a new drug is about $2.6 billion (DiMasi et al., 2016). Moreover, it becomes more difficult to develop a novel clinical drug due to these costs rising every year (DiMasi et al., 2016).
Recently, the development and application of AI has facilitated the research related to drug discovery and drug design, which is reflected in three main aspects: 1. Using AI to predict the structure of proteins and RNA; 2. AI-assisted drug discovery, and 3. Using AI for drug design.
2.1 Using AI to predict the structure of proteins and RNA
The analysis and investigation of the 3D structure of proteins and the related molecules is the precursor for drug discovery and design. It is highly accurate to obtain the 3D structure of proteins and RNA by physical and chemical experimental methods, but it requires a lot of manpower and financial resources. Therefore, recent studies employ computing techniques to predict the 3D structure of molecules (Huang B. et al., 2023).
Classical 3D structure prediction methods consist of de novo modeling, fragment assembly, and homology modeling, the mechanism of which are based on rule-based computing and splicing but not using AI for 3D structure prediction (Li et al., 2020; Huang B. et al., 2023). Thus, before AlphaFold was innovated, the application of AI in structure prediction focused more on the prediction of features related to primary and secondary structures rather than very complicated 3D structures (Kuhlman and Bradley, 2019).
With the release of AlphaFold by DeepMind (Jumper et al., 2021) and RoseTTAFold by David Baker’s team (Baek et al., 2021), scientists proposed many novel ideas for 3D structure prediction of proteins and molecules. A comparison that may be inappropriate but illustrates the significance is: Tunyasuvunakool et al. (Tunyasuvunakool et al., 2021) successfully predicted 98.5% of human proteins by AlphaFold, and 58% of the residues had confident prediction results and 36% of all residues predictions had very high confidence. In contrast, decades of human structural experiments have only determined 17% of all residues.
Moreover, Zhang et al. (2023f) investigated the virtual screening performance for 37 common drug targets, which have AlphaFold2 predicted structures and experimental structures. The AlphaFold2 predicted structures show similar performance with experimental structures in early enrichment in a subset of 27 targets. It demonstrates that AlphaFold2 structures have great potential in virtual screening after proper preparation and refinement.
For more details on the impact and changes AlphaFold has brought to the field of structural biology, Yang et al. (2023) summarized the related studies and applications in structural biology, drug discovery, protein design and so on, and then they considered that AlphaFold has achieved great success and significantly remodeled structural biology (Bertoline et al., 2023; Yang et al., 2023).
Despite the great success, AlphaFold has many limitations. Besides static structures, it is very important for us to study and predict the structural dynamics of unstructured molecules, such as allosteric drugs and their active state, which is the conformational ensemble (Fisher and Stultz, 2011; Nussinov et al., 2023). However, AlphaFold and the related AI methods currently do not provide such solutions.
There is no doubt that the emergence of AlphaFold has brought great changes to the study of protein structure. As for now, an optimized AlphaFold predicted structure can provide a reasonable starting point for physical-based molecular dynamics simulations, making them more effective in drug discovery (Gomes et al., 2022; Schauperl and Denny, 2022; Nussinov et al., 2023). However, there are still many limitations remaining to be solved and optimized. For example, how to further advance and optimize AI methods to predict structure and conformational ensemble for protein complexes and unstructured proteins should be the most important research direction in the future.
2.2 AI-assisted drug discovery
From the random screening and empirical observation of the effects of natural products on disease to discover drugs, to the use of high-throughput screening (HTS) to batch screen drugs against molecular targets (Macarron et al., 2011), and to computer aided drug design (CADD) (Yu and MacKerell, 2017), the approach to discover novel drugs continues to be revolutionized.
With the rapid development of the computational power and algorithms of AI, as well as the rapid expansion of drug-like available chemical space, a new revolution is coming for drug discovery (Maia et al., 2020; Sadybekov and Katritch, 2023). Since computer-aided drug discovery not only can decrease the drug development cycle, but also it can reduce the cost of the clinical trial phase, related studies were carried out to assist and accelerate drug discovery, which include the development of virtual screening (Lavecchia and Di Giovanni, 2013), molecular dynamic simulation (Durrant and McCammon, 2011) and molecular docking (Meng et al., 2011; Fan et al., 2019; Thafar et al., 2019).
In these studies, computer-aided drug development has been categorized into two main approaches according to whether the molecular structure is known or not. One is the structure-based approach and the other is the ligand-based approach. Ligand-based approaches use similarities of known active molecules to carry out modeling and computing, whereas structure-based approaches focus on computing and prediction for binding affinity. Next, we will detail the application of AI methods for these two approaches (Yu and MacKerell, 2017; Yang et al., 2019; Maia et al., 2020; Paul et al., 2021).
For ligand-based approaches, similar to traditional ligand-based QSAR methods, many researchers build up QSAR models (Neves et al., 2018) to realize ligand-based virtual screening by using artificial intelligence methods (Lima et al., 2016; Dai and Guo, 2019). Additionally, compared with traditional machine learning methods, neural networks and other algorithms are used (as shown int Table 1). For example, DNNs were employed to predict QSAR models to screen new dipeptidyl peptidase-4 (DPP-4) inhibitors for the treatment of diabetes mellitus type 2 (Bustamam et al., 2021). Also, DNNs (Xiao et al., 2018) and various AI-driven ligand-based virtual screening tools and platforms have been developed and used (Amendola and Cosconati, 2021; Oliveira et al., 2023).
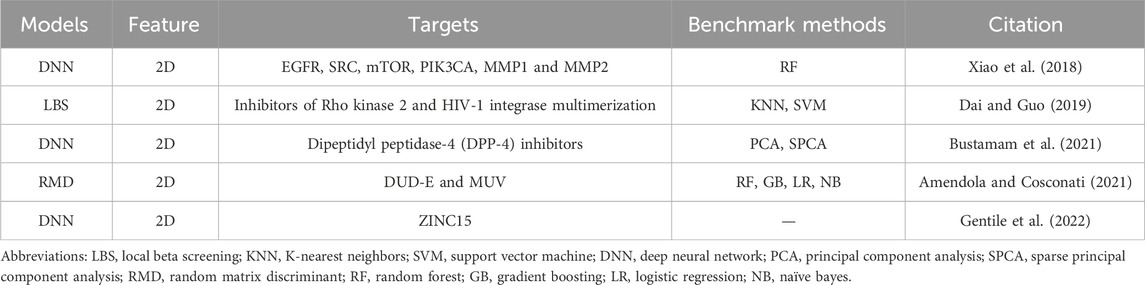
Table 1. Table summaries for some ligand-based models.
In addition to the above approaches, a recent study proposed a deep learning-based deep docking platform (shown in Figure 3), which can train a DNN model by employing a portion of selected data from a huge number of molecular docking libraries. The DNN model is used to predict the docking scores for optional 2-dimensional molecular descriptors and candidate molecules from the molecular docking libraries. According to the predicted score, top-scoring candidate molecules will be selected to carry out further docking (models with higher accuracy), and low-scoring molecules will be filtered out. Since the computing load of virtual screening can be decreased by using a DNN for pre-screening, it provides a novel idea to explore the high-dimensional chemical space efficiently (Gentile et al., 2022).
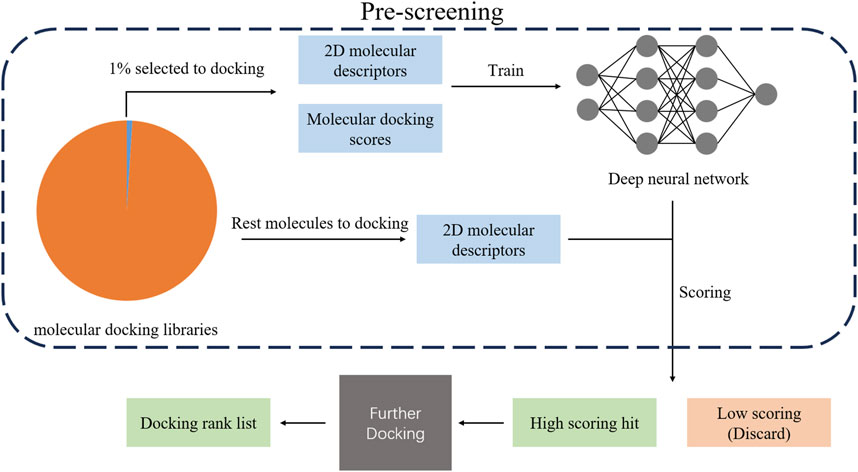
Figure 3. Illustration for pre-screening DNN model.
For structure-based approaches, a hot study direction is to propose the binding affinity models [binding affinity scoring functions (Meli et al., 2022; Sadybekov and Katritch, 2023)] from the known ligand activities and corresponding protein-ligand 3D structural data.
The scoring functions are categorized into the following four types (Meli et al., 2022): Physics-Based (Force-Field Based) scoring function, Empirical (Regression-Based) scoring function, Knowledge-Based (Potential-Based) scoring function, and Descriptor-based or Machine Learning-Based scoring function.
Descriptor-based and traditional machine learning scoring functions have been proposed and used since the 1990s, which are usually based on SVM, random forests, and gradient boosting (Meli et al., 2022). They are often used to explore the nonlinear relationship between descriptors and binding affinities (Meli et al., 2022). With the development of neural networks and deep learning, scientists have proposed many binding affinity models based on feed-forward neural networks (Ashtawy and Mahapatra, 2015; Meli et al., 2021), convolutional neural networks (Jiménez et al., 2018; Stepniewska-Dziubinska et al., 2020), graph neural networks (Gaudelet et al., 2021; Son and Kim, 2021), and other neural networks (Ashtawy and Mahapatra, 2018; Jones et al., 2021).
Both descriptors and models are the key factors for binding affinity prediction, and impact the final prediction ability. Table 2 lists some researches with different descriptors and models, from which it can be found that descriptors with strong expression ability together with appropriate and powerful models, make up good prediction models.
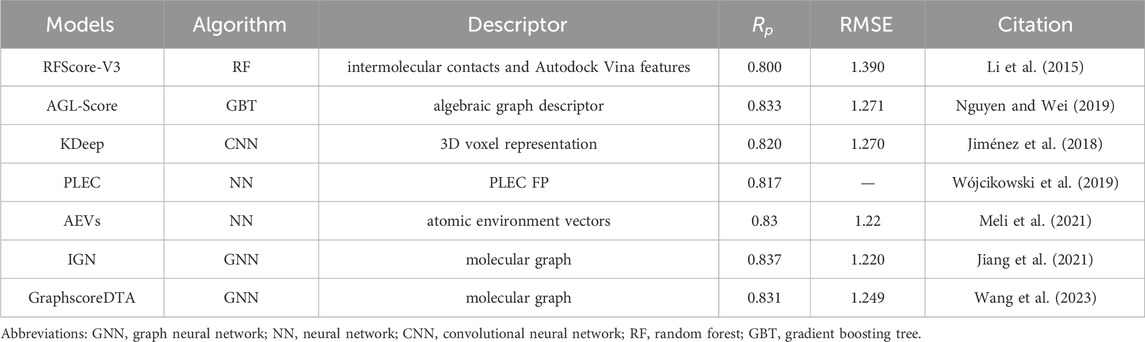
Table 2. Performance comparison for structure-based models.
For descriptors, more detailed and accurate description for protein-ligand interactions could lead to the improvement of prediction ability. For example, Wójcikowski et al. (2019) presented a Protein-Ligand Extended Connectivity (PLEC) Fingerprint to encode protein-ligand interactions and build up different models to predict protein–ligand affinities, including linear regression, random forest and neural network. The Pearson correlation coefficient obtained on the CASF-2016 benchmark is 0.817. Meli et al. (2021) proposed to employ atomic environment vectors (AEVs) and feed-forward neural networks to predict protein-ligand binding affinity, which achieved RMSE of 1.22 and Pearson’s correlation coefficient of 0.83 on the CASF-2016 benchmark. Both researches focus on the enrichment of descriptors, suggesting a research direction, but whether it works remains to be discussed, which will be mentioned later.
Besides the descriptors study, it is very important to build up a suitable and powerful model. Wang et al. (2023) developed GraphscoreDTA, which adopts Vina distance optimization by combining graph neural network, bitransport information mechanism and physics-based distance terms. GraphscoreDTA model obtained RMSE of 1.249 and Pearson’s correlation coefficient of 0.831 on the CASF-2016 benchmark. Jiang et al. (2021) proposed InteractionGraphNet (IGN), stacked by two independent graph convolution modules, which are trained to learn intramolecular and intermolecular interactions. IGN model obtained the RMSE of 1.220 and Pearson’s correlation coefficient of 0.837 on the CASF-2016 benchmark in the best case.
For descriptors, despite numbers of descriptors for protein and ligand presentation are proposed and discussed as mentioned above, Volkov et al. indicated (Volkov et al., 2022) that providing more docking details, such as an explicit description of protein-ligand noncovalent interactions, cannot demonstrate an explicit advantage when training neural network models rather than using only ligand or protein descriptors. Especially, memory largely dominates the learning process of deep neural networks in most cases. Thus, it will become a meaningful research direction to investigate how to represent the structures of ligands and proteins, and how to use optimal descriptors to represent ligands and proteins (Meli et al., 2022; Gu et al., 2023) rather than capturing information about their binding. After that, choosing the suitable models to make better use of the information provided by descriptors will also be an important part of affecting the ability of the model.
Moreover, it is worth noting that the Pearson correlation coefficient is used to evaluate the binding affinity prediction ability of the scoring function, while RMSE is used to evaluate the docking ability of the scoring function, which is, the ability to select the native binding conformation of the ligand from a series of poses (Vittorio et al., 2024). In molecular docking and virtual screening practice, pose prediction and affinity prediction are two complementary tasks. Better molecular docking results can be obtained by constantly adjusting pose and calculating its binding affinity (Meli et al., 2022). Previous study shows that binding affinity prediction ability and docking ability are not closely correlated for many existing scoring functions (Vittorio et al., 2024). Predictions for binding affinity are mostly based on single binding conformation of the ligand found in the experimental complex, which may be partly to blame for the underperformance of these scoring functions in actual virtual screening tasks (Gabel et al., 2014; Shen et al., 2021), and training the scoring functions using the structure of docking pose (Francoeur et al., 2020), or the application of Data Set Augmentation techniques (Scantlebury et al., 2020), may help to improve the robustness of those scoring functions. In addition, most of the current databases provide only well-bound protein and ligand data, i.e., positive data, but lack suboptimal binding affinity, i.e., negative data. Therefore, it will become a hot research direction (David et al., 2020; Xu et al., 2020; Kimber et al., 2021; Sadybekov and Katritch, 2023) to collect and provide these data to improve the performance for AI models.
2.3 Using AI for drug design
Strictly speaking, drug discovery is to discover potential drugs by computational, experimental, and clinical models, whereas drug design is to design and develop new drugs based on known signaling pathways and biological targets, i.e., designing molecules that match their target molecules in shape and charge (Zhou and Zhong, 2017). Here, we will focus on the applications of AI in drug design, namely de novo drug design (Wang et al., 2022).
De novo drug design (Mouchlis et al., 2021) refers to generating a series of new molecules that meet certain constraints by developing generative algorithms. The advantage of this approach is that we can design a drug in such a greater chemical space that could develop more targeted drugs for the treatment of diseases. However, it encounters such a challenge that is how to generate a new molecule, which is stable and easy to produce without a starting template. Traditional de novo drug design is comprised of structure-based, ligand-based, sampling-based, and evolutionary algorithm-based approaches, which are detailed by Mouchlis et al. (2021) due to space limitations.
Generally, there are four basic types of models to do de novo drug design, which are RNN-based model, Autoencoder-based model (AE, also known as encoder-decoder model), GAN-based model (Generative Adversarial Network), and reinforcement learning-based model. In practice, most algorithms are based on one or a combination of these four structures (Wang et al., 2022).
RNN related models (Li et al., 2018; Kotsias et al., 2020; Moret et al., 2020; van Deursen et al., 2020) generate new molecules with the highest probability by taking the output of the previous layer as input, and iterate to continuously optimize its output molecules.
For example, Urbina et al. (2022) recently proposed MegaSyn, which is a tool integrating generative molecular design and automated analog design into synthetic viability prediction. MegaSyn employed SMILES-based RNN generative model and its performance is demonstrated by several case studies (Urbina et al., 2022).
In addition, several studies combine RNN and reinforcement learning (Popova et al., 2018; Liu et al., 2019; Ståhl et al., 2019; Blaschke et al., 2020) to construct de novo drug design models. For example, Hu et al. (2023) proposed a de novo drug design model based on Stack-RNN, multi-objective reward-weighted sum and reinforcement learning. By multi-objective reward-weighted sum, it solved the potential conflicts between different properties of the generated molecules. Moreover, since it is a multi-objective optimization task, it also prevents the generated molecules to be extremely biased towards a certain property. Their model achieved a validity of 97.3%, an internal diversity of 0.8613, and increased desirable molecules from 55.9% to 92%.
Autoencoder is an unsupervised learning model consisting of the encoder and decoder (Gómez-Bombarelli et al., 2018). The encoder converts the input molecules into vectors in the latent space, and the decoder can revert the vectors into molecular representations. Therefore, we can adjust the molecular design by changing the vectors in the latent space. Variational autoencoder (VAE) is the first AE framework for molecule design (Gómez-Bombarelli et al., 2018). Several studies have subsequently made improvements and enhancements based on this framework (Kusner et al., 2017; Skalic et al., 2019; Ye et al., 2021). For example, Lim et al. (2018) introduced several molecular properties into the latent space to carry out conditional control and adjustment for the generated molecules. Liu et al. (2018) introduced graph into variational autoencoder, where both encoder and decoder are graph structured. Moreover, deep generative model was introduced by Samanta et al. (2019), which can effectively discover plausible, diverse and novel molecules and generate molecules that maximize the property of interest.
Furthermore, adversarial autoencoder (AAE) algorithms, which is the combination of VAE and GAN, can generate target-specific molecules (Kadurin et al., 2017; Polykovskiy et al., 2018; Prykhodko et al., 2019). For example, Prykhodko et al. (2019) proposed a deep learning architecture LatentGAN, which is able to generate both drug-like compounds and target-biased compounds. A GAN is trained to generate fake latent vector which is taken as the input for the decoder in VAE and then generates new molecules for their model. Besides the above models, some other studies based on AE include Heteroencoder (Bjerrum and Sattarov, 2018), GTM-RNN (Sattarov et al., 2019) and reinforcement learning based GENTRL (Zhavoronkov et al., 2019).
GAN (Aggarwal et al., 2021) consists of a generator and a discriminator, where the generator generates new molecules and the discriminator distinguish whether the input molecules are real or generated by the generator. Performance of the generator and the discriminator can be improved by continuously training. And in practice, GAN is often used together with other models.
Reinforcement learning (Nian et al., 2020; Pereira et al., 2021; Atance et al., 2022; Korshunova et al., 2022; Fang et al., 2023) consists of a generative model and a drug design agent model. Generative model is generally constructed by a multi-layer neural network, which generates a new state as an output based on the results of the previous generation or the initial state by the neural network. The outputs are evaluated by the drug agent model, enabling iterations to optimize the designed molecule. Reinforcement learning is not only always combined with other generative algorithms like RNN mentioned above, but also works with GAN, such as ORGAN (Guimaraes et al., 2017), ORGANIC (Sanchez-Lengeling et al., 2017) and ATNC (Putin et al., 2018) to construct de novo drug design models. Especially, Abbasi et al. (2022) proposed a framework comprising Encoder–Decoder architecture, Wasserstein GAN with gradient penalty and optimization step based on Feedback GAN (Pereira et al., 2021), which can be regarded as the combination of autoencoder, GAN and reinforcement learning. Their Encoder–Decoder model correctly reconstructed 99% of the datasets, including stereochemical information, and generated compounds with 62.3% validity, 0.88 internal diversity and 0.94 external diversity.
The applications of AI for de novo drug design are still in its beginning stages, and their performance have not significantly surpassed that of traditional and evolutionary algorithm-based models (Wang et al., 2022; Zhang et al., 2023e). We still lack a comprehensive target-specific de novo drug design platform, while a large amount of work is currently on theoretical studies for the development of new algorithms (Bai et al., 2021; Wang et al., 2022).
Additionally, synthetic feasibility is still an important question without enough concerns. A reachable solution is to provide synthesizability scoring, like synthetic accessibility (SA) score (Ertl and Schuffenhauer, 2009) and synthetic complexity (SC) score (Coley et al., 2018), which can easily compute scores of syntheses for a target molecule and exclude unsynthesizable molecules. Besides synthesizability scoring functions, there are also ways to make sure the synthetic feasibility, like synthesis planning, synthesis prediction and Fragment/synthesis-driven molecular construction and generative models, which are detailed in this review (Stanley and Segler, 2023).
In conclusion, AI shows great potential in de novo drug design, but its research is still in its infancy. A great deal of research on algorithmic exploration and practical application is yet to be explored further in depth.
3 Artificial intelligence for compound pharmacokinetics prediction
In scenarios such as drug development, drug design, and drug dosage exploration, the pharmacokinetic studies of candidate compounds, i.e., the studies of properties like drug absorption, distribution, metabolism, excretion, and toxicity (ADMET), are essential, because any drug candidate must be tested for ADMET properties to guarantee the effectiveness and safety of the drug (Tsaioun et al., 2009).
Therefore, we can significantly reduce the chemical searching space, increase the success rate of drug development, and decrease its cost (Tran et al., 2023c) by employing AI technology to build up predictive models for pharmacokinetics, validate ADMET properties for drug candidates in the early stages of drug development, and screen out the undesired drugs, When predicting ADMET and physicochemical properties, each process is corresponding to a number of important features, including but not limited to those shown in Table 3 (Dulsat et al., 2023). Both traditional machine learning and neural network methods received good predictive effect using these features. Due to the limitation of space, the development process and detailed studies can be found in these reviews (Chandrasekaran et al., 2018; Yang et al., 2019; Danishuddin et al., 2022; Dulsat et al., 2023; Tran et al., 2023a; Tran et al., 2023c; Tran et al., 2023b), and the following highlighted several important studies and recent advances of AI in ADMET prediction.

Table 3. Important features for ADMET and Physicochemical properties.
Since previous machine learning studies (Aleksić et al., 2022) indicated that choosing different traditional machine learning models and increasing the amount of training data cannot significantly affect the prediction accuracy, it implied that limited improvement can be achieved by using machine learning methods. Therefore, many studies have started to use various neural network models to predict pharmacokinetic parameters as described below.
For example, DNN (Sakiyama et al., 2021; Kumar et al., 2022; Mazumdar et al., 2023), RNN (Alsenan et al., 2020) and CNN (Alsenan et al., 2021) are used to predict blood-brain barrier permeability. DNN is used to predict CYP450s inhibition (Park et al., 2022). Multi-task CNN is used to predict in vitro clearance from molecular images (Martínez Mora et al., 2022). GCN (Chen et al., 2021) and multi-task DNN (Sharma et al., 2023) are used to predict toxicity. These studies show that modelling with neural networks is commonly used to predict the ADMET and physicochemical properties of compounds.
More importantly, unlike independently constructed models that predict single or partial properties of ADMET, recent studies (Dulsat et al., 2023) can predict multiple important features of ADMET and physicochemical properties by integrating multiple models. Representative works include ADMETlab (Dong et al., 2018), ADMETlab 2.0 (Xiong et al., 2021), admetSAR (Cheng et al., 2012), admetSAR 2.0 (Ye et al., 2019), FAF-Drugs4 (Lagorce et al., 2017), FP-ADMET (Venkatraman, 2021), Interpretable-ADMET (Wei et al., 2022), and HelixADMET (Zhang et al., 2022).
In these works, ADMETlab (Dong et al., 2018) can predict a wide range of coverage with good accuracy and precision (Dulsat et al., 2023), which has 31 ADMET endpoints prediction in ADMETlab Version 1.0, and increases to 88 in ADMETlab Version 2.0. Furthermore, ADMETlab Version 2.0 (Xiong et al., 2021) increases the quality and quantity of data for model construction.
In terms of modeling methods, ADMETlab Version 1.0 uses traditional machine learning algorithms, including random forest (RF) (Cao et al., 2012), support vector machine (SVM) (Cao et al., 2015), recursive partition regression (RP) (Strobl et al., 2009), partial least squares (PLS) (Cao et al., 2010), naïve Bayes (NB) (Jiang et al., 2018), and decision tree (DT) (Xia et al., 2018), to build QSAR regression models and classification models for ADMET properties. ADMETlab Version 2.0 employs attention mechanism and graph convolutional neural network to simultaneously learn the regression and classification tasks in ADMET prediction. And it proposes a multi-task graph attention (MGA) framework, where different attention layers can be generated for various tasks to generate specific feature maps (customized fingerprints).
Compared with Version 1.0, ADMETlab Version 2.0 not only increases its precision and accuracy, but also improves computational efficiency by employing graphs to represent molecules instead of the traditional descriptor-based representation. Table 4 shows the comparisons between ADMETlab Version 1.0 and Version 2.0, which is a portrayal of the comparison between neural network model and traditional machine learning methods. More detailed comparisons and evaluations between different works can be found in this review (Dulsat et al., 2023).
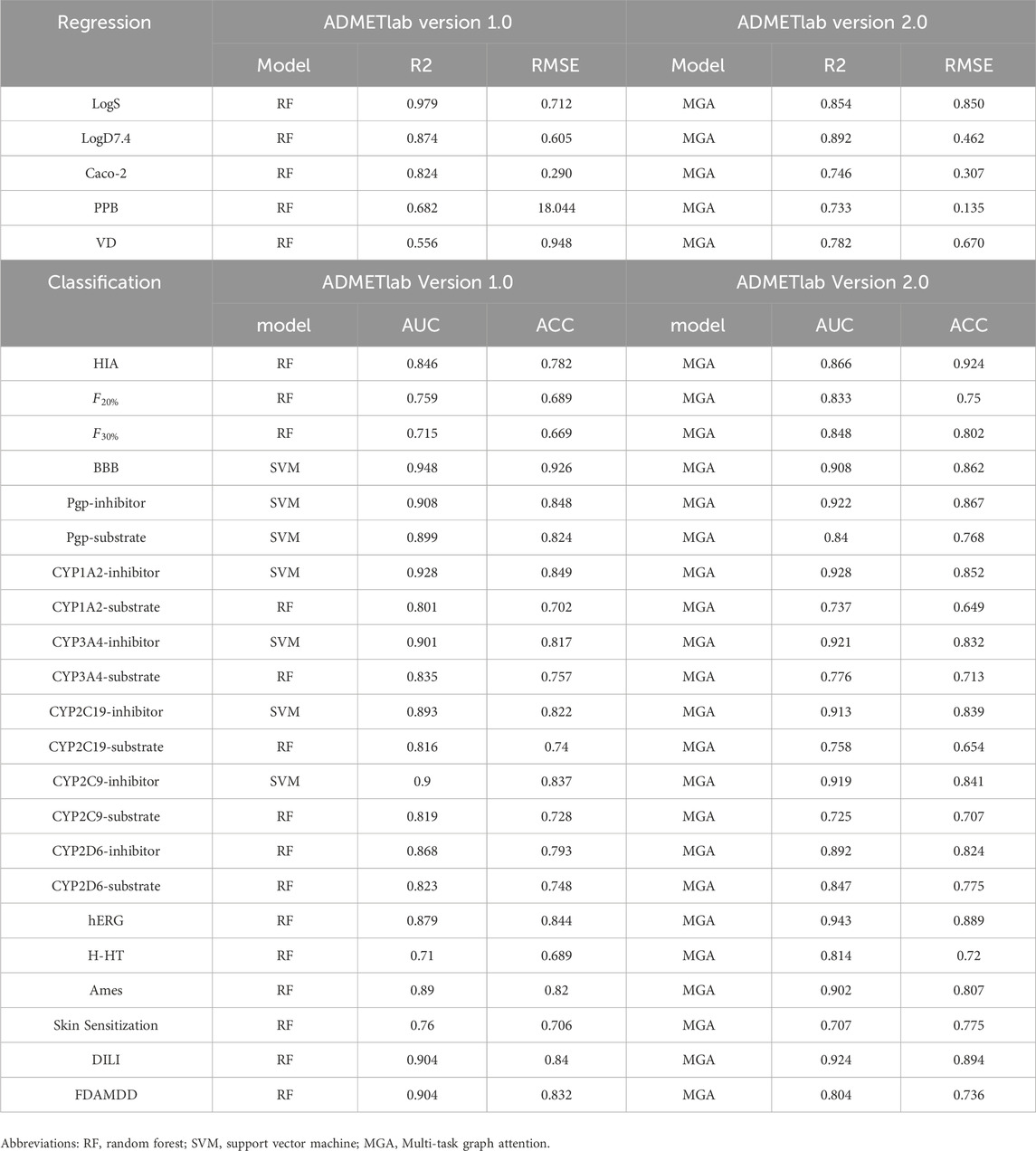
Table 4. Comparisons between ADMETlab Version 1.0 and Version 2.0
ADMETlab Version 1.0 took more than 2 hours while ADMETlab Version 2.0 only took 84 s in the computational test for 1,000 molecules, since ADMETlab Version 2.0 improved the performance of regression and classification for many properties (shown in Table 4). For examples, the
Similar to the modeling for ADMETlab Version 2.0, Interpretable-ADMET uses graph convolutional neural networks and graph attention networks (Wei et al., 2022), and HelixADMET is based on graph neural networks (Zhang et al., 2022). The above methods have made great progress in ADMET prediction, which have also been compared and evaluated by Dulsat et al. (Dulsat et al., 2023) in detail. Since these works employ graph presentation and graph neural networks, it suggests feasible directions for subsequent studies on ADMET prediction for both molecular expression and model selection. The wide use of these ADMET prediction tools also demonstrates the great potential for deep learning and graph neural networks in ADMET prediction.
4 Artificial intelligence for clinical pharmacology
Besides the above work related to drug discovery, drug design and pharmacokinetics prediction, AI has many applications in clinical pharmacology, such as using AI to optimize clinical trial design, simulate clinical trial results, optimize drug treatment process, predict drug interactions and adverse reactions, and so on.
4.1 AI in clinical trials
Clinical trial is an important stage in the development of drugs. The failure during clinical trials will result in a huge loss of time and cost. Thus, using AI to assist clinical trials will effectively improve efficiency and success rate (Askin et al., 2023).
As we know, it is one of the most challenging steps to recruit the relevant patients during the clinical trial design. For this reason, we usually employ machine learning algorithms to screen the patients, match them to the trial’s inclusion criteria through multiple aspects of data, and guarantee that the included patients are suitable for that clinical trial (Harrer et al., 2019; Beck et al., 2020; Beaulieu et al., 2021; Vazquez et al., 2021; Weissler et al., 2021).
Also, AI can be used to predict and select these patients who will progress and reach the endpoint more quickly. And then, the duration of drug trials can be potentially reduced (Lee and Lee, 2020). When the trial is in progress, AI can predictively determine participants who may drop out midway through the electronic medical record, and to improve the completion rate of the trial (Krittanawong et al., 2019) by reminding the experimentalist to pay extra attention to these participants.
More notably, with the development of large language models, AI becomes increasingly capable of simulating human-like responses and behaviors in social science research, to the point where AI can be used to complete certain trials instead of humans (Grossmann et al., 2023). Like oncology drug research, AI algorithms can predict drugs’ performance in clinical trials.
One of the previous studies (Kolla et al., 2021) used Causal AI to build in silico trials, which employed clinical data to construct simulated cohorts to simulate the treatment effects for both control and trial groups. The simulated cohort data not only can provide more information for patient recruitment and determination of the actual trial protocols, but also can increase the success rate and safety of the subsequent trial sessions.
Although we still lack the high-quality datasets and are unable to completely replace clinical trials, it is potential for us to employ drug clinical trials simulation to increase drug development efficiency (Kolla et al., 2021).
4.2 AI in optimizing drug treatment
Besides the applications related to clinical trials, AI can be used to optimize the therapeutic effects of drugs, which is important for clinical pharmacology. These applications include but are not limited to dosage of drug recommendations, individualized medical recommendations and effect prediction, adverse drug reactions, and prediction of drug-drug interactions (Johnson et al., 2023).
For drug dosage recommendations, both traditional machine learning (Vinks et al., 2020; van Gelder and Vinks, 2021; Bououda et al., 2022; Labriffe et al., 2022) and neural network (Yauney and Shah, 2018; Rödle et al., 2020) methods are widely used to estimate the amount of drugs. And then, we can optimize the efficacy of treatment while satisfying various constraints. For example, Rödle et al. (2020) developed an ANN model with backpropagation and genetic learning algorithm to predict the dosages of Ibuprofen, Paracetamol and Cefotaxime. The deviations of predicted dosage from real dosage of each medicine are 13%, 20% and 33%. As discussed by Rödle et al. (2020), it is urgent for this study area to have higher quality datasets, more indicators and outcome parameters to guarantee better development and application for drug dosage recommendation. Additionally, AI models are widely used in individualized treatment both in the static setting and time-dependent setting, including treatment recommendation, treatment outcome prediction, and individualized dose-response estimation. Potential data include patients’ personal information, electronic health records, diagnose data and so on. Detailed algorithms and methods of those studies are listed in the review by Bica et al. (2021).
Adverse drug reactions (ADR) (Martin et al., 2022) are also extremely important in the actual use of drugs, which means unexpected or unwanted effects caused by drugs. Improper use of drugs can lead to adverse reactions, causing additional illnesses or even deaths (Mohsen et al., 2021). Most of adverse drug reactions can be identified by toxicity-related predictions during pharmacokinetic parameter estimation (Basile et al., 2019), but some of them yet need to be predicted by AI models based on patients’ feedback and physiological data (Martin et al., 2022; Liu and Rudd, 2023). For example, Martin et al. (Martin et al., 2022) built up a predictive model for both ADR identification and seriousness assessment from structured and unstructured free-text information filled by patients, which employed TF-IDF + LGBM and Cross-lingual Language Model (XLM) to predict ADR identification. Here, XLM is an attention-based neural network and takes unstructured text data, while TF-IDF + LGBM takes additional structured data, like age, sex and so on. XLM and TF-IDF + LGBM both achieved an AUC of 0.97 on external validation, indicating the possibility to use of AI in the automatic pre-coding of pharmacovigilance reports. Meanwhile, the AI-based prediction and early detection of adverse drug reactions can effectively prevent the occurrence of adverse drug reactions and mitigate their consequences (Syrowatka et al., 2022).
Recently, it has become more and more common to adopt multi-drug combination therapy, but multiple drugs can easily inactivate some of them to affect the efficacy or even produce toxicity and cause additional complications. Therefore, the prediction of drug-drug interactions (DDIs) (Ryu et al., 2018; Zhang et al., 2023d) has become increasingly important.
With the increasing abundance of DDI-related databases, many machine learning and neural network models have been proposed to predict DDI and make great progress, which are detailed reviewed by Zhang et al. (2023d).
It is worth noting that the order of drugs administered may also affect the occurrence of DDIs, leading to asymmetric drug interactions. For example, a recent work by Feng et al. (2022) employs the directed graph attention network model DGAT-DDI to predict asymmetric drug interactions, in which source role encoder, target role encoder and self-role encoder are designed to represent how drugs influence and be influenced by other drugs and their chemical structures. Meanwhile, aggressiveness and impressionability are designed to capture the number of interaction partners and interaction tendencies. DGAT-DDI (Feng et al., 2022) achieved an AUC of 0.951, an AUPRC of 0.943 and an accuracy of 0.886 in the direction-specific task, and achieved an AUC of 0.867, an AUPRC of 0.854 and an accuracy of 0.795 in the direction-blind task. In the case study, seven of the top ten drug candidates in the model are validated by DrugBank, which demonstrates the practical capabilities of the model and the importance of further study on asymmetric drug interaction prediction (Feng et al., 2022; Zhang et al., 2023d).
5 Discussion and conclusion
AI have advanced many researches in biology (Zhang and Zhang, 2017; Zhang et al., 2018; Zhang et al., 2021e), disease (Li et al., 2017; Zhang et al., 2023a), cancer (Zhang et al., 2017a; Zhang et al., 2017b; Zhang et al., 2021b) and so do pharmacology. This review has briefly introduced the basic concepts of AI and the history of its development, and then summarized the applications of AI in pharmacology from three aspects: drug discovery and design, pharmacokinetic parameters estimation, and clinical pharmacology (Xia et al., 2017; Zhang et al., 2019a; Zhang et al., 2021a).
For these three aspects, we have listed relevant applications and major breakthroughs of AI in specific research fields, such as structure prediction, drug discovery, de novo drug design, clinical trial, and clinical drug therapy optimization. Although several research fields have not been mentioned, such as the application of AI in drug repositioning, drug manufacturing, and drug distribution, we listed them in the following articles (Paul et al., 2021; Tanoli et al., 2021; Yang et al., 2022).
It is noteworthy that AlphaFold has made great success in molecular structure prediction. Benefiting from the highly accurate prediction for 3D structures of a large number of molecules, it is easier for us to obtain structural information of targets in downstream drug discovery and design studies, thus providing the necessary prerequisite foundation for the discovery and design of novel drugs (Borkakoti and Thornton, 2023), and in turn, bringing a lot of new opportunities and ideas for drug discovery and design.
Therefore, it has become a widely discussed question: Can AI technologies and models, represented by AlphaFold, completely change the research of drug discovery and design?
Although most of the answers are “No”, we must note that AI research in drug discovery and design is constantly advancing and evolving, and AI models do not need to completely replace the research work of human. It’s a great advancement even if we just use AI models and tools to help accelerate the research for drug discovery and design, which will have great study potential in the distant future (Nussinov et al., 2023).
As discussed above, AI models have many advantages, including but not limited to the following: 1) AI models can perform more efficient calculations and predictions, and it can be demonstrated by ADMET online prediction tools such as AlphaFold and ADMETlab Version; 2) It is easier for us to employ deep learning language models to process unstructured text data than traditional machine learning, which is aforementioned by using XML to predict drug adverse reactions; 3) AI models have the potential to explore novel scientific knowledge and patterns, as evidenced by the graph-based attention network model DGAT-DDI mentioned above, which can be used to compute and explore asymmetric drug interactions.
However, there are still remaining many limitations and problems for AI models to solve. With the progress and success of AI models, how to collect data for AI model training becomes an increasingly important issue. The number of databases containing information on molecular structures, drug parameters, and drug interactions (Danishuddin et al., 2022; Sadybekov and Katritch, 2023; Zhang et al., 2023d) is fast increasing, which not only can provide a greater chemical space to explore new drugs, but also offer more data for better AI model training. However, it is worth noting that the large language model represented by ChatGPT and many other studies have pointed out that the quality of data is one of the most important factors in training an AI model, which suggests while expanding the amount of data, we should pay attention to the screening and quality control of the data (Aldoseri et al., 2023; Huang et al., 2023b; van der Lee and Swen, 2023; Whang et al., 2023). Moreover, with more and more databases available, the problems of overfitting, underfitting (Ying, 2019; Aliferis and Simon, 2024) and data imbalance (Krawczyk, 2016; Werner de Vargas et al., 2023) in AI deserve attention and vigilance. How to use some methods to avoid these problems as much as possible, such as cross-validation (Charilaou and Battat, 2022), regularization (Salehin and Kang, 2023), and data argumentation (Mumuni and Mumuni, 2022; Alomar et al., 2023), is also an important part of AI research.
Meanwhile, the interpretability of AI models deserves attention, though most of the current AI research does not take the model interpretability into consideration, such as face recognition or image processing (Zhang Q. et al., 2023). However, the interpretability of AI models has become a controversial issue for healthcare-related fields (Amann et al., 2020; Kırboğa et al., 2023). Many AI models are complex and lack explanations of the decision-making process causing these models to be termed as “Black-Box,” but explainable AI (XAI) models are trying to enhance transparency (Hassija et al., 2024). Research on XAI not only can alleviate people’s concerns about AI in drug research, but may also help medical and life science researchers discover the mechanisms and theories for the drugs and drug metabolism. Current research on XAI models has made great progress and has been applied in pharmacology related fields, but more exploration is still needed (Jiménez-Luna et al., 2020; Vo et al., 2022; Kırboğa et al., 2023; Hassija et al., 2024).
Also, the representation of molecules and drugs remains an important problem to be further discussed and studied. New algorithmic architecture that uses graph structure to represent molecules and employ graph neural networks to construct models has been wildly investigated with good progress, like ADMETlab, InteractionGraphNet, DGAT-DDI and many other methods mentioned before, but using graph structure to represent molecules still suffers predicament from insufficient expressive ability or too much complexity in some opinions (An et al., 2022). More practices and research are needed to explore the differences and applicable cases for both graph and traditional representation.
Despite the above problems and challenges, the applications of AI in pharmacology and medicine are still very valuable. As AI has made great success and breakthroughs in structure prediction, drug discovery and design, and pharmacokinetic parameter estimation, it is possible for us to build up an automated drug discovery and design platform by integrating these three research directions, a vision for future research. Moreover, it is foreseeable that AI models will gradually replace many previous traditional models, and even part of the work of humans. In this process, how to supervise, control and reasonably develop AI models will be an important issue to address and a future study direction.
Author contributions
BL: Writing–original draft. KT: Writing–original draft. AL: Writing–review and editing. HW: Writing–review and editing. HZ: Writing–review and editing. LZ: Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from National Science and Technology Major Project under Grant (2021YFF1201200), in part by the National Natural Science Foundation of China under Grant (62372316) and in part by Sichuan Science and Technology Program key project under Grant (2024YFHZ0091).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbasi, M., Santos, B. P., Pereira, T. C., Sofia, R., Monteiro, N. R. C., Simões, C. J. V., et al. (2022). Designing optimized drug candidates with generative adversarial network. J. Cheminformatics 14 (1), 40. doi:10.1186/s13321-022-00623-6
Aggarwal, A., Mittal, M., and Battineni, G. (2021). Generative adversarial network: an overview of theory and applications. Int. J. Inf. Manag. Data Insights 1 (1), 100004. doi:10.1016/j.jjimei.2020.100004
Aldoseri, A., Al-Khalifa, K. N., and Hamouda, A. M. (2023). Re-thinking data strategy and integration for artificial intelligence: concepts, opportunities, and challenges. Oppor. Challenges 13 (12), 7082. doi:10.3390/app13127082
Aleksić, S., Seeliger, D., and Brown, J. B. (2022). ADMET predictability at boehringer ingelheim: state-of-the-art, and do bigger datasets or algorithms make a difference? Mol. Inf. 41 (2), 2100113. doi:10.1002/minf.202100113
Aliferis, C., and Simon, G. (2024). “Overfitting, underfitting and general model overconfidence and under-performance pitfalls and best practices in machine learning and AI,” in Artificial intelligence and machine learning in health care and medical sciences: best practices and pitfalls. Editors G. J. Simon, and C. Aliferis (Cham: Springer International Publishing), 477–524.
Alomar, K., Aysel, H. I., and Cai, X. (2023). Data augmentation in classification and segmentation: a survey and new strategies. J. Imaging 9 (2), 46. doi:10.3390/jimaging9020046
Alsenan, S., Al-Turaiki, I., and Hafez, A. (2020). A Recurrent Neural Network model to predict blood–brain barrier permeability. Comput. Biol. Chem. 89, 107377. doi:10.1016/j.compbiolchem.2020.107377
Alsenan, S., Al-Turaiki, I., and Hafez, A. (2021). A deep learning approach to predict blood-brain barrier permeability. PeerJ Comput. Sci. 7, e515. doi:10.7717/peerj-cs.515
Alzubaidi, L., Zhang, J., Humaidi, A. J., Al-Dujaili, A., Duan, Y., Al-Shamma, O., et al. (2021). Review of deep learning: concepts, CNN architectures, challenges, applications, future directions. J. Big Data 8 (1), 53. doi:10.1186/s40537-021-00444-8
Amann, J., Blasimme, A., Vayena, E., Frey, D., Madai, V. I., and the Precise, Q. c. (2020). Explainability for artificial intelligence in healthcare: a multidisciplinary perspective. BMC Med. Inf. Decis. Mak. 20 (1), 310. doi:10.1186/s12911-020-01332-6
Amendola, G., and Cosconati, S. (2021). PyRMD: a new fully automated AI-powered ligand-based virtual screening tool. J. Chem. Inf. Model. 61 (8), 3835–3845. doi:10.1021/acs.jcim.1c00653
An, X., Chen, X., Yi, D., Li, H., and Guan, Y. (2022). Representation of molecules for drug response prediction. Briefings Bioinforma. 23 (1), bbab393. doi:10.1093/bib/bbab393
Ashtawy, H. M., and Mahapatra, N. R. (2015). BgN-Score and BsN-Score: bagging and boosting based ensemble neural networks scoring functions for accurate binding affinity prediction of protein-ligand complexes. BMC Bioinforma. 16 (4), S8. doi:10.1186/1471-2105-16-S4-S8
Ashtawy, H. M., and Mahapatra, N. R. (2018). Task-specific scoring functions for predicting ligand binding poses and affinity and for screening enrichment. J. Chem. Inf. Model 58 (1), 119–133. doi:10.1021/acs.jcim.7b00309
Askin, S., Burkhalter, D., Calado, G., and El Dakrouni, S. (2023). Artificial Intelligence Applied to clinical trials: opportunities and challenges. Health Technol. Berl. 13 (2), 203–213. doi:10.1007/s12553-023-00738-2
Atance, S. R., Diez, J. V., Engkvist, O., Olsson, S., and Mercado, R. (2022). De novo drug design using reinforcement learning with graph-based deep generative models. J. Chem. Inf. Model. 62 (20), 4863–4872. doi:10.1021/acs.jcim.2c00838
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373 (6557), 871–876. doi:10.1126/science.abj8754
Bai, Q., Tan, S., Xu, T., Liu, H., Huang, J., and Yao, X. (2021). MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform 22 (3), bbaa161. doi:10.1093/bib/bbaa161
Basile, A. O., Yahi, A., and Tatonetti, N. P. (2019). Artificial intelligence for drug toxicity and safety. Trends Pharmacol. Sci. 40 (9), 624–635. doi:10.1016/j.tips.2019.07.005
Beaulieu, D., Berry, J. D., Paganoni, S., Glass, J. D., Fournier, C., Cuerdo, J., et al. (2021). Development and validation of a machine-learning ALS survival model lacking vital capacity (VC-Free) for use in clinical trials during the COVID-19 pandemic. Amyotroph. Lateral Scler. Front. Degener. 22 (Suppl. 1), 22–32. doi:10.1080/21678421.2021.1924207
Beck, J. T., Rammage, M., Jackson, G. P., Preininger, A. M., Dankwa-Mullan, I., Roebuck, M. C., et al. (2020). Artificial intelligence tool for optimizing eligibility screening for clinical trials in a large community cancer center. JCO Clin. Cancer Inf. 4, 50–59. doi:10.1200/cci.19.00079
Bertoline, L. M. F., Lima, A. N., Krieger, J. E., and Teixeira, S. K. (2023). Before and after AlphaFold2: an overview of protein structure prediction. Front. Bioinform 3, 1120370. doi:10.3389/fbinf.2023.1120370
Bica, I., Alaa, A. M., Lambert, C., and van der Schaar, M. (2021). From real-world patient data to individualized treatment effects using machine learning: current and future methods to address underlying challenges. Clin. Pharmacol. Ther. 109 (1), 87–100. doi:10.1002/cpt.1907
Bjerrum, E. J., and Sattarov, B. (2018). Improving chemical autoencoder latent space and molecular de novo generation diversity with heteroencoders. Biomolecules 8 (4), 131. doi:10.3390/biom8040131
Blaschke, T., Arús-Pous, J., Chen, H., Margreitter, C., Tyrchan, C., Engkvist, O., et al. (2020). REINVENT 2.0: an AI tool for de novo drug design. J. Chem. Inf. Model. 60 (12), 5918–5922. doi:10.1021/acs.jcim.0c00915
Borkakoti, N., and Thornton, J. M. (2023). AlphaFold2 protein structure prediction: implications for drug discovery. Curr. Opin. Struct. Biol. 78, 102526. doi:10.1016/j.sbi.2022.102526
Bououda, M., Uster, D. W., Sidorov, E., Labriffe, M., Marquet, P., Wicha, S. G., et al. (2022). A machine learning approach to predict interdose vancomycin exposure. Pharm. Res. 39 (4), 721–731. doi:10.1007/s11095-022-03252-8
Brown, N., Ertl, P., Lewis, R., Luksch, T., Reker, D., and Schneider, N. (2020). Artificial intelligence in chemistry and drug design. J. Computer-Aided Mol. Des. 34 (7), 709–715. doi:10.1007/s10822-020-00317-x
Bustamam, A., Hamzah, H., Husna, N. A., Syarofina, S., Dwimantara, N., Yanuar, A., et al. (2021). Artificial intelligence paradigm for ligand-based virtual screening on the drug discovery of type 2 diabetes mellitus. J. Big Data 8 (1), 74. doi:10.1186/s40537-021-00465-3
Cao, D.-S., Dong, J., Wang, N.-N., Wen, M., Deng, B.-C., Zeng, W.-B., et al. (2015). In silico toxicity prediction of chemicals from EPA toxicity database by kernel fusion-based support vector machines. Chemom. Intelligent Laboratory Syst. 146, 494–502. doi:10.1016/j.chemolab.2015.07.009
Cao, D.-S., Xu, Q.-S., Liang, Y.-Z., Chen, X., and Li, H.-D. (2010). Prediction of aqueous solubility of druglike organic compounds using partial least squares, back-propagation network and support vector machine. J. Chemom. 24 (9), 584–595. doi:10.1002/cem.1321
Cao, D.-S., Yang, Y.-N., Zhao, J.-C., Yan, J., Liu, S., Hu, Q.-N., et al. (2012). Computer-aided prediction of toxicity with substructure pattern and random forest. J. Chemom. 26 (1-2), 7–15. doi:10.1002/cem.1416
Chandrasekaran, B., Abed, S. N., Al-Attraqchi, O., Kuche, K., and Tekade, R. K. (2018). “Chapter 21 - computer-aided prediction of pharmacokinetic (ADMET) properties,” in Dosage form design parameters. Editor R. K. Tekade (Academic Press), 731–755.
Charilaou, P., and Battat, R. (2022). Machine learning models and over-fitting considerations. World J. Gastroenterol. 28 (5), 605–607. doi:10.3748/wjg.v28.i5.605
Chaturvedula, A., Calad-Thomson, S., Liu, C., Sale, M., Gattu, N., and Goyal, N. (2019). Artificial intelligence and pharmacometrics: time to embrace. Capitalize, Advance? 8 (7), 440–443. doi:10.1002/psp4.12418
Chen, J., Si, Y.-W., Un, C.-W., and Siu, S. W. I. (2021). Chemical toxicity prediction based on semi-supervised learning and graph convolutional neural network. J. Cheminformatics 13 (1), 93. doi:10.1186/s13321-021-00570-8
Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., et al. (2012). admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 52 (11), 3099–3105. doi:10.1021/ci300367a
Coley, C. W., Rogers, L., Green, W. H., and Jensen, K. F. (2018). SCScore: synthetic complexity learned from a reaction corpus. J. Chem. Inf. Model 58 (2), 252–261. doi:10.1021/acs.jcim.7b00622
Dai, W., and Guo, D. (2019). A ligand-based virtual screening method using direct quantification of generalization ability. Molecules 24 (13), 2414. doi:10.3390/molecules24132414
Danishuddin, , Kumar, V., Faheem, M., and Woo Lee, K. (2022). A decade of machine learning-based predictive models for human pharmacokinetics: advances and challenges. Drug Discov. Today 27 (2), 529–537. doi:10.1016/j.drudis.2021.09.013
David, L., Thakkar, A., Mercado, R., and Engkvist, O. (2020). Molecular representations in AI-driven drug discovery: a review and practical guide. J. Cheminformatics 12 (1), 56. doi:10.1186/s13321-020-00460-5
DiMasi, J. A., Grabowski, H. G., and Hansen, R. W. (2016). Innovation in the pharmaceutical industry: new estimates of R&D costs. J. Health Econ. 47, 20–33. doi:10.1016/j.jhealeco.2016.01.012
Dong, J., Wang, N.-N., Yao, Z.-J., Zhang, L., Cheng, Y., Ouyang, D., et al. (2018). ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminformatics 10 (1), 29. doi:10.1186/s13321-018-0283-x
Dulsat, J., López-Nieto, B., Estrada-Tejedor, R., and Borrell, J. I. (2023). Evaluation of free online ADMET tools for academic or small biotech environments. Biotech. Environ. 28 (2), 776. doi:10.3390/molecules28020776
Durrant, J. D., and McCammon, J. A. (2011). Molecular dynamics simulations and drug discovery. BMC Biol. 9 (1), 71. doi:10.1186/1741-7007-9-71
Ertl, P., and Schuffenhauer, A. (2009). Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform 1 (1), 8. doi:10.1186/1758-2946-1-8
Fan, J., Fu, A., and Zhang, L. (2019). Progress in molecular docking. Quant. Biol. 7 (2), 83–89. doi:10.1007/s40484-019-0172-y
Fang, Y., Pan, X., and Shen, H.-B. (2023). De novo drug design by iterative multiobjective deep reinforcement learning with graph-based molecular quality assessment. Bioinformatics 39 (4), btad157. doi:10.1093/bioinformatics/btad157
Feng, Y.-Y., Yu, H., Feng, Y.-H., and Shi, J.-Y. (2022). Directed graph attention networks for predicting asymmetric drug–drug interactions. Briefings Bioinforma. 23 (3), bbac151. doi:10.1093/bib/bbac151
Fisher, C. K., and Stultz, C. M. (2011). Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 21 (3), 426–431. doi:10.1016/j.sbi.2011.04.001
Francoeur, P. G., Masuda, T., Sunseri, J., Jia, A., Iovanisci, R. B., Snyder, I., et al. (2020). Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model 60 (9), 4200–4215. doi:10.1021/acs.jcim.0c00411
Gabel, J., Desaphy, J., and Rognan, D. (2014). Beware of machine learning-based scoring functions-on the danger of developing black boxes. J. Chem. Inf. Model 54 (10), 2807–2815. doi:10.1021/ci500406k
Gao, J., Lao, Q., Kang, Q., Liu, P., Zhang, L., and Li, K. (2022). “Unsupervised cross-disease domain adaptation by lesion scale matching,” in Miccai 2022 (Springer Nature Switzerland), 660–670.
Gao, J., Lao, Q., Liu, P., Yi, H., Kang, Q., Jiang, Z., et al. (2023). Anatomically guided cross-domain repair and screening for ultrasound fetal biometry. IEEE J. Biomed. Health Inf. 27, 4914–4925. doi:10.1109/JBHI.2023.3298096
Gao, J., Liu, P., Liu, G.-D., and Zhang, L. (2021). Robust needle localization and enhancement algorithm for ultrasound by deep learning and beam steering methods. J. Comput. Sci. Technol. 36 (2), 334–346. doi:10.1007/s11390-021-0861-7
Gaudelet, T., Day, B., Jamasb, A. R., Soman, J., Regep, C., Liu, G., et al. (2021). Utilizing graph machine learning within drug discovery and development. Briefings Bioinforma. 22 (6), bbab159. doi:10.1093/bib/bbab159
Gentile, F., Yaacoub, J. C., Gleave, J., Fernandez, M., Ton, A.-T., Ban, F., et al. (2022). Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 17 (3), 672–697. doi:10.1038/s41596-021-00659-2
Gomes, P. S. F. C., Gomes, D. E. B., and Bernardi, R. C. (2022). Protein structure prediction in the era of AI: challenges and limitations when applying to in silico force spectroscopy. Front. Bioinform. 2, 983306. doi:10.3389/fbinf.2022.983306
Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Sci. 4 (2), 268–276. doi:10.1021/acscentsci.7b00572
Grossmann, I., Feinberg, M., Parker, D. C., Christakis, N. A., Tetlock, P. E., and Cunningham, W. A. (2023). AI and the transformation of social science research. Science 380 (6650), 1108–1109. doi:10.1126/science.adi1778
Gu, S., Shen, C., Yu, J., Zhao, H., Liu, H., Liu, L., et al. (2023). Can molecular dynamics simulations improve predictions of protein-ligand binding affinity with machine learning? Briefings Bioinforma. 24 (2), bbad008. doi:10.1093/bib/bbad008
Guimaraes, G. L., Sanchez-Lengeling, B., Outeiral, C., Farias, P. L. C., and Aspuru-Guzik, A. J. a.p.a. (2017). Objective-reinforced generative adversarial networks (organ) for sequence generation models. arXiv.
Haenlein, M., and Kaplan, A. (2019). A brief history of artificial intelligence: on the past, present, and future of artificial intelligence. Artif. Intell. 61 (4), 5–14. doi:10.1177/0008125619864925
Harrer, S., Shah, P., Antony, B., and Hu, J. (2019). Artificial intelligence for clinical trial design. Trends Pharmacol. Sci. 40 (8), 577–591. doi:10.1016/j.tips.2019.05.005
Hassija, V., Chamola, V., Mahapatra, A., Singal, A., Goel, D., Huang, K., et al. (2024). Interpreting black-box models: a review on explainable artificial intelligence. Cogn. Comput. 16 (1), 45–74. doi:10.1007/s12559-023-10179-8
He, K., Zhang, X., Ren, S., and Sun, J. (2015). “Deep residual learning for image recognition,” in 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 770–778. doi:10.1109/CVPR.2016.90
Hu, P., Zou, J., Yu, J., and Shi, S. (2023). De novo drug design based on Stack-RNN with multi-objective reward-weighted sum and reinforcement learning. J. Mol. Model. 29 (4), 121. doi:10.1007/s00894-023-05523-6
Huang, B., Kong, L., Wang, C., Ju, F., Zhang, Q., Zhu, J., et al. (2023a). Protein structure prediction: challenges, advances, and the shift of research paradigms. Genomics, Proteomics & Bioinforma. 21, 913–925. doi:10.1016/j.gpb.2022.11.014
Huang, H., Zheng, O., Wang, D., Yin, J., Wang, Z., Ding, S., et al. (2023b). ChatGPT for shaping the future of dentistry: the potential of multi-modal large language model. Int. J. Oral Sci. 15 (1), 29. doi:10.1038/s41368-023-00239-y
Jiang, D., Hsieh, C.-Y., Wu, Z., Kang, Y., Wang, J., Wang, E., et al. (2021). InteractionGraphNet: a novel and efficient deep graph representation learning framework for accurate protein–ligand interaction predictions. J. Med. Chem. 64 (24), 18209–18232. doi:10.1021/acs.jmedchem.1c01830
Jiang, W., Shen, Y., Ding, Y., Ye, C., Zheng, Y., Zhao, P., et al. (2018). A naive Bayes algorithm for tissue origin diagnosis (TOD-Bayes) of synchronous multifocal tumors in the hepatobiliary and pancreatic system. Int. J. Cancer 142 (2), 357–368. doi:10.1002/ijc.31054
Jiménez, J., Škalič, M., Martínez-Rosell, G., and De Fabritiis, G. (2018). K(DEEP): protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model 58 (2), 287–296. doi:10.1021/acs.jcim.7b00650
Jiménez-Luna, J., Grisoni, F., and Schneider, G. (2020). Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2 (10), 573–584. doi:10.1038/s42256-020-00236-4
Johnson, M., Patel, M., Phipps, A., van der Schaar, M., Boulton, D., and Gibbs, M. (2023). The potential and pitfalls of artificial intelligence in clinical pharmacology. CPT. Pharmacometrics Syst. Pharmacol. 12 (3), 279–284. doi:10.1002/psp4.12902
Jones, D., Kim, H., Zhang, X., Zemla, A., Stevenson, G., Bennett, W. F. D., et al. (2021). Improved protein–ligand binding affinity prediction with structure-based deep fusion inference. J. Chem. Inf. Model. 61 (4), 1583–1592. doi:10.1021/acs.jcim.0c01306
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kadurin, A., Nikolenko, S., Khrabrov, K., Aliper, A., and Zhavoronkov, A. (2017). druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 14 (9), 3098–3104. doi:10.1021/acs.molpharmaceut.7b00346
Kimber, T. B., Chen, Y., and Volkamer, A. (2021). Deep learning in virtual screening: recent applications and developments. Int. J. Mol. Sci. 22 (9), 4435. doi:10.3390/ijms22094435
Kırboğa, K. K., Abbasi, S., and Küçüksille, E. U. (2023). Explainability and white box in drug discovery. Chem. Biol. & Drug Des. 102 (1), 217–233. doi:10.1111/cbdd.14262
Kolla, L., Gruber, F. K., Khalid, O., Hill, C., and Parikh, R. B. (2021). The case for AI-driven cancer clinical trials - the efficacy arm in silico. Biochim. Biophys. Acta Rev. Cancer 1876 (1), 188572. doi:10.1016/j.bbcan.2021.188572
Korshunova, M., Huang, N., Capuzzi, S., Radchenko, D. S., Savych, O., Moroz, Y. S., et al. (2022). Generative and reinforcement learning approaches for the automated de novo design of bioactive compounds. Commun. Chem. 5 (1), 129. doi:10.1038/s42004-022-00733-0
Kotsias, P.-C., Arús-Pous, J., Chen, H., Engkvist, O., Tyrchan, C., and Bjerrum, E. J. (2020). Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2 (5), 254–265. doi:10.1038/s42256-020-0174-5
Krawczyk, B. (2016). Learning from imbalanced data: open challenges and future directions. Prog. Artif. Intell. 5 (4), 221–232. doi:10.1007/s13748-016-0094-0
Krittanawong, C., Johnson, K. W., and Tang, W. W. (2019). How artificial intelligence could redefine clinical trials in cardiovascular medicine: lessons learned from oncology. Per Med. 16 (2), 83–88. doi:10.2217/pme-2018-0130
Kuhlman, B., and Bradley, P. (2019). Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 20 (11), 681–697. doi:10.1038/s41580-019-0163-x
Kumar, K., Thakur, G. S. M. J. I. J. o.I. T., and Science, C. (2012). Advanced applications of neural networks and artificial intelligence. A Rev. 4, 57–68. doi:10.5815/ijitcs.2012.06.08
Kumar, R., Sharma, A., Alexiou, A., Bilgrami, A. L., Kamal, M. A., and Ashraf, G. M. (2022). DeePred-BBB: a blood brain barrier permeability prediction model with improved accuracy. Front. Neurosci. 16, 858126. doi:10.3389/fnins.2022.858126
Kusner, M. J., Paige, B., and Hern\'ndez-Lobato, J. M. (2017). Grammar variational autoencoder. Icml 17, 1945–1954.
Labriffe, M., Woillard, J. B., Debord, J., and Marquet, P. (2022). Machine learning algorithms to estimate everolimus exposure trained on simulated and patient pharmacokinetic profiles. CPT Pharmacometrics Syst. Pharmacol. 11 (8), 1018–1028. doi:10.1002/psp4.12810
Lagorce, D., Bouslama, L., Becot, J., Miteva, M. A., and Villoutreix, B. O. (2017). FAF-Drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 33 (22), 3658–3660. doi:10.1093/bioinformatics/btx491
Lai, X., Zhou, J., Wessely, A., Heppt, M., Maier, A., Berking, C., et al. (2022). A disease network-based deep learning approach for characterizing melanoma. Int. J. Cancer 150 (6), 1029–1044. doi:10.1002/ijc.33860
Lavecchia, A., and Di Giovanni, C. (2013). Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 20 (23), 2839–2860. doi:10.2174/09298673113209990001
Lee, C. S., and Lee, A. Y. (2020). How artificial intelligence can transform randomized controlled trials. Transl. Vis. Sci. Technol. 9 (2), 9. doi:10.1167/tvst.9.2.9
Li, B., Cao, Y., Westhof, E., and Miao, Z. (2020). Advances in RNA 3D structure modeling using experimental data. Front. Genet. 11, 574485. doi:10.3389/fgene.2020.574485
Li, H., Leung, K. S., Wong, M. H., and Ballester, P. J. (2015). Improving AutoDock Vina using random forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol. Inf. 34 (2-3), 115–126. doi:10.1002/minf.201400132
Li, T., Cheng, Z., and Zhang, L. (2017). Developing a novel parameter estimation method for agent-based model in immune system simulation under the framework of history matching: a case study on influenza A virus infection. Int. J. Mol. Sci. 18 (12), 2592. doi:10.3390/ijms18122592
Li, Y., Zhang, L., and Liu, Z. (2018). Multi-objective de novo drug design with conditional graph generative model. J. Cheminformatics 10 (1), 33. doi:10.1186/s13321-018-0287-6
Lim, J., Ryu, S., Kim, J. W., and Kim, W. Y. (2018). Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminformatics 10 (1), 31. doi:10.1186/s13321-018-0286-7
Lima, A. N., Philot, E. A., Trossini, G. H., Scott, L. P., Maltarollo, V. G., and Honorio, K. M. (2016). Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 11 (3), 225–239. doi:10.1517/17460441.2016.1146250
Liu, J. Y. H., and Rudd, J. A. (2023). Predicting drug adverse effects using a new gastro-intestinal pacemaker activity drug database (GIPADD). Sci. Rep. 13 (1), 6935. doi:10.1038/s41598-023-33655-5
Liu, Q., Allamanis, M., Brockschmidt, M., and Gaunt, A. L. (2018). Constrained graph variational autoencoders for molecule design. Nips 18, 7806–7815.
Liu, X., Ye, K., van Vlijmen, H. W. T., Ijzerman, A. P., and van Westen, G. J. P. (2019). An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: a case for the adenosine A2A receptor. J. Cheminformatics 11 (1), 35. doi:10.1186/s13321-019-0355-6
Macarron, R., Banks, M. N., Bojanic, D., Burns, D. J., Cirovic, D. A., Garyantes, T., et al. (2011). Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 10 (3), 188–195. doi:10.1038/nrd3368
Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A. M., and Taranto, A. G. (2020). Structure-based virtual screening: from classical to artificial intelligence, Front. Chem. 8. 343. doi:10.3389/fchem.2020.00343
Martin, G. L., Jouganous, J., Savidan, R., Bellec, A., Goehrs, C., Benkebil, M., et al. (2022). Validation of artificial intelligence to support the automatic coding of patient adverse drug reaction reports, using nationwide pharmacovigilance data. Drug Saf. 45 (5), 535–548. doi:10.1007/s40264-022-01153-8
Martínez Mora, A., Subramanian, V., and Miljković, F. (2022). Multi-task convolutional neural networks for predicting in vitro clearance endpoints from molecular images. J. Comput. Aided Mol. Des. 36 (6), 443–457. doi:10.1007/s10822-022-00458-1
Mazumdar, B., Deva Sarma, P. K., Mahanta, H. J., and Sastry, G. N. (2023). Machine learning based dynamic consensus model for predicting blood-brain barrier permeability. Comput. Biol. Med. 160, 106984. doi:10.1016/j.compbiomed.2023.106984
Meli, R., Anighoro, A., Bodkin, M. J., Morris, G. M., and Biggin, P. C. (2021). Learning protein-ligand binding affinity with atomic environment vectors. J. Cheminformatics 13 (1), 59. doi:10.1186/s13321-021-00536-w
Meli, R., Morris, G. M., and Biggin, P. C. (2022). Scoring functions for protein-ligand binding affinity prediction using structure-based deep learning: a review. A Rev. 2, 885983. doi:10.3389/fbinf.2022.885983
Meng, X. Y., Zhang, H. X., Mezei, M., and Cui, M. (2011). Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 7 (2), 146–157. doi:10.2174/157340911795677602
Mohsen, A., Tripathi, L. P., and Mizuguchi, K. (2021). Deep learning prediction of adverse drug reactions in drug discovery using open TG–GATEs and FAERS databases. Front. Drug Discov. (Lausanne). 1. doi:10.3389/fddsv.2021.768792
Mohsen, S., Behrooz, A., and Roza, D. (2023). Artificial intelligence, machine learning and deep learning in advanced robotics, a review. Cogn. Robot. 3, 54–70. doi:10.1016/j.cogr.2023.04.001
Moret, M., Friedrich, L., Grisoni, F., Merk, D., and Schneider, G. (2020). Generative molecular design in low data regimes. Nat. Mach. Intell. 2 (3), 171–180. doi:10.1038/s42256-020-0160-y
Mouchlis, V. D., Afantitis, A., Serra, A., Fratello, M., Papadiamantis, A. G., Aidinis, V., et al. (2021). Advances in de novo drug design: from conventional to machine learning methods. Int. J. Mol. Sci. 22 (4), 1676. doi:10.3390/ijms22041676
Muggleton, S. (2014). Alan turing and the development of artificial intelligence. AI Commun. 27, 3–10. doi:10.3233/aic-130579
Mumuni, A., and Mumuni, F. (2022). Data augmentation: a comprehensive survey of modern approaches. Array 16, 100258. doi:10.1016/j.array.2022.100258
Neves, B. J., Braga, R. C., Melo-Filho, C. C., Moreira-Filho, J. T., Muratov, E. N., and Andrade, C. H. (2018). QSAR-based virtual screening: advances and applications in drug discovery. Front. Pharmacol. 9, 1275. doi:10.3389/fphar.2018.01275
Nguyen, D. D., and Wei, G. W. (2019). AGL-score: algebraic graph learning score for protein-ligand binding scoring, ranking, docking, and screening. J. Chem. Inf. Model 59 (7), 3291–3304. doi:10.1021/acs.jcim.9b00334
Nian, R., Liu, J., and Huang, B. (2020). A review on reinforcement learning: introduction and applications in industrial process control. Comput. & Chem. Eng. 139, 106886. doi:10.1016/j.compchemeng.2020.106886
Nussinov, R., Zhang, M., Liu, Y., and Jang, H. (2023). AlphaFold, allosteric, and orthosteric drug discovery: ways forward. Drug Discov. Today 28 (6), 103551. doi:10.1016/j.drudis.2023.103551
Obrezanova, O. (2023). Artificial intelligence for compound pharmacokinetics prediction. Curr. Opin. Struct. Biol. 79, 102546. doi:10.1016/j.sbi.2023.102546
Oliveira, T. A. d., Silva, M. P. d., Maia, E. H. B., Silva, A. M. d., and Taranto, A. G. (2023). Virtual screening algorithms in drug discovery: a review focused on machine and deep learning methods. Methods 2 (2), 311–334. doi:10.3390/ddc2020017
Park, H., Brahma, R., Shin, J.-M., and Cho, K.-H. (2022). Prediction of human cytochrome P450 inhibition using bio-selectivity induced deep neural network. Bull. Korean Chem. Soc. 43 (2), 261–269. doi:10.1002/bkcs.12445
Paul, D., Sanap, G., Shenoy, S., Kalyane, D., Kalia, K., and Tekade, R. K. (2021). Artificial intelligence in drug discovery and development. Drug Discov. Today 26 (1), 80–93. doi:10.1016/j.drudis.2020.10.010
Pereira, T., Abbasi, M., Oliveira, J. L., Ribeiro, B., and Arrais, J. (2021). Optimizing blood–brain barrier permeation through deep reinforcement learning for de novo drug design. Bioinformatics 37 (Suppl. ment_1), i84–i92. doi:10.1093/bioinformatics/btab301
Polykovskiy, D., Zhebrak, A., Vetrov, D., Ivanenkov, Y., Aladinskiy, V., Mamoshina, P., et al. (2018). Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm. 15 (10), 4398–4405. doi:10.1021/acs.molpharmaceut.8b00839
Popova, M., Isayev, O., and Tropsha, A. (2018). Deep reinforcement learning for de novo drug design. Sci. Adv., 4(7), eaap7885. doi:10.1126/sciadv.aap7885
Prykhodko, O., Johansson, S. V., Kotsias, P.-C., Arús-Pous, J., Bjerrum, E. J., Engkvist, O., et al. (2019). A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminformatics 11 (1), 74. doi:10.1186/s13321-019-0397-9
Putin, E., Asadulaev, A., Vanhaelen, Q., Ivanenkov, Y., Aladinskaya, A. V., Aliper, A., et al. (2018). Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 15 (10), 4386–4397. doi:10.1021/acs.molpharmaceut.7b01137
Rödle, W., Caliskan, D., Prokosch, H. U., and Kraus, S. (2020). Evaluation of different learning algorithms of neural networks for drug dosing recommendations in pediatrics. Stud. Health Technol. Inf. 271, 271–276. doi:10.3233/shti200106
Ryu, J. Y., Kim, H. U., and Lee, S. Y. (2018). Deep learning improves prediction of drug–drug and drug–food interactions. Proc. Natl. Acad. Sci. 115 (18), E4304-E4311–E4311. doi:10.1073/pnas.1803294115
Sadybekov, A. V., and Katritch, V. (2023). Computational approaches streamlining drug discovery. Nature 616 (7958), 673–685. doi:10.1038/s41586-023-05905-z
Sakiyama, H., Fukuda, M., and Okuno, T. (2021). Prediction of blood-brain barrier penetration (BBBP) based on molecular descriptors of the free-form and in-blood-form datasets. Molecules 26 (24), 7428. doi:10.3390/molecules26247428
Salehin, I., and Kang, D.-K. (2023). A review on dropout regularization approaches for deep neural networks within the scholarly domain. , 12(14), 3106, doi:10.3390/electronics12143106
Samanta, B., De, A., Jana, G., Chattaraj, P. K., Ganguly, N., and Rodriguez, M. G. (2019). NeVAE: a deep generative model for molecular graphs. Proc. AAAI Conf. Artif. Intell. 33 (01), 1110–1117. doi:10.1609/aaai.v33i01.33011110
Sanchez-Lengeling, B., Outeiral, C., Guimaraes, G. L., and Aspuru-Guzik, A. (2017). Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC). ChemRxiv. doi:10.26434/chemrxiv.5309668.v3
Sattarov, B., Baskin, I. I., Horvath, D., Marcou, G., Bjerrum, E. J., and Varnek, A. (2019). De novo molecular design by combining deep autoencoder recurrent neural networks with generative topographic mapping. J. Chem. Inf. Model 59 (3), 1182–1196. doi:10.1021/acs.jcim.8b00751
Scantlebury, J., Brown, N., Von Delft, F., and Deane, C. M. (2020). Data set augmentation allows deep learning-based virtual screening to better generalize to unseen target classes and highlight important binding interactions. J. Chem. Inf. Model 60 (8), 3722–3730. doi:10.1021/acs.jcim.0c00263
Schauperl, M., and Denny, R. A. (2022). AI-based protein structure prediction in drug discovery: impacts and challenges. J. Chem. Inf. Model. 62 (13), 3142–3156. doi:10.1021/acs.jcim.2c00026
Sharma, B., Chenthamarakshan, V., Dhurandhar, A., Pereira, S., Hendler, J. A., Dordick, J. S., et al. (2023). Accurate clinical toxicity prediction using multi-task deep neural nets and contrastive molecular explanations. Sci. Rep. 13 (1), 4908. doi:10.1038/s41598-023-31169-8
Shen, C., Hu, Y., Wang, Z., Zhang, X., Pang, J., Wang, G., et al. (2021). Beware of the generic machine learning-based scoring functions in structure-based virtual screening. Brief. Bioinform 22 (3), bbaa070. doi:10.1093/bib/bbaa070
Skalic, M., Sabbadin, D., Sattarov, B., Sciabola, S., and De Fabritiis, G. (2019). From target to drug: generative modeling for the multimodal structure-based ligand design. Mol. Pharm. 16 (10), 4282–4291. doi:10.1021/acs.molpharmaceut.9b00634
So, S.-S., and Karplus, M. (1996a). Evolutionary optimization in quantitative Structure−Activity relationship: an application of genetic neural networks. J. Med. Chem. 39 (7), 1521–1530. doi:10.1021/jm9507035
So, S.-S., and Karplus, M. (1996b). Genetic neural networks for quantitative Structure−Activity relationships: improvements and application of benzodiazepine affinity for benzodiazepine/GABAA receptors. J. Med. Chem. 39 (26), 5246–5256. doi:10.1021/jm960536o
Son, J., and Kim, D. (2021). Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PLoS One 16 (4), e0249404. doi:10.1371/journal.pone.0249404
Song, H., Chen, L., Cui, Y., Li, Q., Wang, Q., Fan, J., et al. (2022). Denoising of MR and CT images using cascaded multi-supervision convolutional neural networks with progressive training. Neurocomputing 469, 354–365. doi:10.1016/j.neucom.2020.10.118
Ståhl, N., Falkman, G., Karlsson, A., Mathiason, G., and Boström, J. (2019). Deep reinforcement learning for multiparameter optimization in de novo drug design. J. Chem. Inf. Model. 59 (7), 3166–3176. doi:10.1021/acs.jcim.9b00325
Stanley, M., and Segler, M. (2023). Fake it until you make it? Generative de novo design and virtual screening of synthesizable molecules. Curr. Opin. Struct. Biol. 82, 102658. doi:10.1016/j.sbi.2023.102658
Stepniewska-Dziubinska, M. M., Zielenkiewicz, P., and Siedlecki, P. (2020). Improving detection of protein-ligand binding sites with 3D segmentation. Sci. Rep. 10 (1), 5035. doi:10.1038/s41598-020-61860-z
Strobl, C., Malley, J., and Tutz, G. (2009). An introduction to recursive partitioning: rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol. Methods 14 (4), 323–348. doi:10.1037/a0016973
Su, M., Yang, Q., Du, Y., Feng, G., Liu, Z., Li, Y., et al. (2019). Comparative assessment of scoring functions: the CASF-2016 update. J. Chem. Inf. Model. 59 (2), 895–913. doi:10.1021/acs.jcim.8b00545
Syrowatka, A., Song, W., Amato, M. G., Foer, D., Edrees, H., Co, Z., et al. (2022). Key use cases for artificial intelligence to reduce the frequency of adverse drug events: a scoping review. Lancet Digit. Health 4 (2), e137–e148. doi:10.1016/s2589-7500(21)00229-6
Tanoli, Z., Vähä-Koskela, M., and Aittokallio, T. (2021). Artificial intelligence, machine learning, and drug repurposing in cancer. Expert Opin. Drug Discov. 16 (9), 977–989. doi:10.1080/17460441.2021.1883585
Thafar, M., Raies, A. B., Albaradei, S., Essack, M., and Bajic, V. B. (2019). Comparison study of computational prediction tools for drug-target binding affinities. Front. Chem. 7, 782. doi:10.3389/fchem.2019.00782
Tran, T. T. V., Surya Wibowo, A., Tayara, H., and Chong, K. T. (2023a). Artificial intelligence in drug toxicity prediction: recent advances, challenges, and future perspectives. J. Chem. Inf. Model. 63 (9), 2628–2643. doi:10.1021/acs.jcim.3c00200
Tran, T. T. V., Tayara, H., and Chong, K. T. (2023b). Artificial intelligence in drug metabolism and excretion prediction: recent advances, challenges, and future perspectives. Pharmaceutics 15 (4), 1260. doi:10.3390/pharmaceutics15041260
Tran, T. T. V., Tayara, H., and Chong, K. T. (2023c). Recent studies of artificial intelligence on in silico drug distribution prediction. Int. J. Mol. Sci. 24 (3), 1815. doi:10.3390/ijms24031815
Tsaioun, K., Bottlaender, M., and Mabondzo, A.the Alzheimer's Drug Discovery, F. (2009). ADDME – avoiding Drug Development Mistakes Early: central nervous system drug discovery perspective. BMC Neurol. 9 (1), S1. doi:10.1186/1471-2377-9-S1-S1
Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Žídek, A., et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596 (7873), 590–596. doi:10.1038/s41586-021-03828-1
Urbina, F., Lowden, C. T., Culberson, J. C., and Ekins, S. (2022). MegaSyn: integrating generative molecular design, automated analog designer, and synthetic viability prediction. ACS Omega 7 (22), 18699–18713. doi:10.1021/acsomega.2c01404
Vallance, P., and Smart, T. G. (2006). The future of pharmacology. Br. J. Pharmacol. 147 (Suppl 1), S304–S307. doi:10.1038/sj.bjp.0706454
van der Lee, M., and Swen, J. J. (2023). Artificial intelligence in pharmacology research and practice. Clin. Transl. Sci. 16 (1), 31–36. doi:10.1111/cts.13431
van Deursen, R., Ertl, P., Tetko, I. V., and Godin, G. (2020). GEN: highly efficient SMILES explorer using autodidactic generative examination networks. J. Cheminformatics 12 (1), 22. doi:10.1186/s13321-020-00425-8
van Gelder, T., and Vinks, A. A. (2021). Machine learning as a novel method to support therapeutic drug management and precision dosing. Clin. Pharmacol. Ther. 110 (2), 273–276. doi:10.1002/cpt.2326
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., et al. (2017). “Attention is all you need,” in Proceedings of the 31st international conference on neural information processing systems (Long Beach, California, USA: Curran Associates Inc.).
Vazquez, J., Abdelrahman, S., Byrne, L. M., Russell, M., Harris, P., and Facelli, J. C. (2021). Using supervised machine learning classifiers to estimate likelihood of participating in clinical trials of a de-identified version of ResearchMatch. J. Clin. Transl. Sci. 5 (1), e42. doi:10.1017/cts.2020.535
Venkatraman, V. (2021). FP-ADMET: a compendium of fingerprint-based ADMET prediction models. J. Cheminformatics 13 (1), 75. doi:10.1186/s13321-021-00557-5
Vinks, A. A., Peck, R. W., Neely, M., and Mould, D. R. (2020). Development and implementation of electronic health record-integrated model-informed clinical decision support tools for the precision dosing of drugs. Clin. Pharmacol. Ther. 107 (1), 129–135. doi:10.1002/cpt.1679
Vittorio, S., Lunghini, F., Morerio, P., Gadioli, D., Orlandini, S., Silva, P., et al. (2024). Addressing docking pose selection with structure-based deep learning: recent advances, challenges and opportunities. Comput. Struct. Biotechnol. J. 23, 2141–2151. doi:10.1016/j.csbj.2024.05.024
Vo, T. H., Nguyen, N. T. K., Kha, Q. H., and Le, N. Q. K. (2022). On the road to explainable AI in drug-drug interactions prediction: a systematic review. Comput. Struct. Biotechnol. J. 20, 2112–2123. doi:10.1016/j.csbj.2022.04.021
Volkov, M., Turk, J. A., Drizard, N., Martin, N., Hoffmann, B., Gaston-Mathé, Y., et al. (2022). On the frustration to predict binding affinities from protein-ligand structures with deep neural networks. J. Med. Chem. 65 (11), 7946–7958. doi:10.1021/acs.jmedchem.2c00487
Wang, K., Zhou, R., Tang, J., and Li, M. (2023). GraphscoreDTA: optimized graph neural network for protein–ligand binding affinity prediction. Bioinformatics 39 (6), btad340. doi:10.1093/bioinformatics/btad340
Wang, M., Wang, Z., Sun, H., Wang, J., Shen, C., Weng, G., et al. (2022). Deep learning approaches for de novo drug design: an overview. Curr. Opin. Struct. Biol. 72, 135–144. doi:10.1016/j.sbi.2021.10.001
Wei, Y., Li, S., Li, Z., Wan, Z., and Lin, J. (2022). Interpretable-ADMET: a web service for ADMET prediction and optimization based on deep neural representation. Bioinformatics 38 (10), 2863–2871. doi:10.1093/bioinformatics/btac192
Weissler, E. H., Naumann, T., Andersson, T., Ranganath, R., Elemento, O., Luo, Y., et al. (2021). The role of machine learning in clinical research: transforming the future of evidence generation. Trials 22 (1), 537. doi:10.1186/s13063-021-05489-x
Werner de Vargas, V., Schneider Aranda, J. A., Dos Santos Costa, R., da Silva Pereira, P. R., and Victória Barbosa, J. L. (2023). Imbalanced data preprocessing techniques for machine learning: a systematic mapping study. Knowl. Inf. Syst. 65 (1), 31–57. doi:10.1007/s10115-022-01772-8
Whang, S. E., Roh, Y., Song, H., and Lee, J.-G. (2023). Data collection and quality challenges in deep learning: a data-centric AI perspective. VLDB J. 32 (4), 791–813. doi:10.1007/s00778-022-00775-9
Wójcikowski, M., Kukiełka, M., Stepniewska-Dziubinska, M. M., and Siedlecki, P. (2019). Development of a protein–ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics 35 (8), 1334–1341. doi:10.1093/bioinformatics/bty757
Woschank, M., Rauch, E., and Zsifkovits, H. (2020). A review of further directions for artificial intelligence, machine learning, and deep learning in Smart logistics. Mach. Learn. Deep Learn. Smart Logist. 12 (9), 3760. doi:10.3390/su12093760
Xia, Y., Liu, C., Da, B., and Xie, F. (2018). A novel heterogeneous ensemble credit scoring model based on bstacking approach. Expert Syst. Appl. 93, 182–199. doi:10.1016/j.eswa.2017.10.022
Xia, Y., Yang, C., Hu, N., Yang, Z., He, X., Li, T., et al. (2017). Exploring the key genes and signaling transduction pathways related to the survival time of glioblastoma multiforme patients by a novel survival analysis model. BMC Genomics 18 (Suppl 1), 950. doi:10.1186/s12864-016-3256-3
Xiao, T., Qi, X., Chen, Y., and Jiang, Y. (2018). Development of ligand-based big data deep neural network models for virtual screening of large compound libraries. Mol. Inf. 37 (11), e1800031. doi:10.1002/minf.201800031
Xiong, G., Wu, Z., Yi, J., Fu, L., Yang, Z., Hsieh, C., et al. (2021). ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 49 (W1), W5–W14. doi:10.1093/nar/gkab255
Xu, Y., Verma, D., Sheridan, R. P., Liaw, A., Ma, J., Marshall, N. M., et al. (2020). Deep dive into machine learning models for protein engineering. J. Chem. Inf. Model. 60 (6), 2773–2790. doi:10.1021/acs.jcim.0c00073
Yang, F., Zhang, Q., Ji, X., Zhang, Y., Li, W., Peng, S., et al. (2022). Machine learning applications in drug repurposing. Interdiscip. Sci. 14 (1), 15–21. doi:10.1007/s12539-021-00487-8
Yang, X., Wang, Y., Byrne, R., Schneider, G., and Yang, S. (2019). Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 119 (18), 10520–10594. doi:10.1021/acs.chemrev.8b00728
Yang, Z., Zeng, X., Zhao, Y., and Chen, R. (2023). AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct. Target. Ther. 8 (1), 115. doi:10.1038/s41392-023-01381-z
Yauney, G., and Shah, P. (2018). “Reinforcement learning with action-derived rewards for chemotherapy and clinical trial dosing regimen selection,” in Proceedings of the 3rd Machine Learning for Healthcare Conference, PMLR. Editors D.-V. Finale, F. Jim, J. Ken, K. David, R. Rajesh, W. Byronet al. (Proceedings of Machine Learning Research) 85, 161–226. Available at: https://proceedings.mlr.press/v85/yauney18a.html
Ye, Q., Zhang, X., and Lin, X. (2021). “De novo drug design via multi-label learning and adversarial autoencoder,” in 2021 IEEE international Conference on Bioinformatics and biomedicine (BIBM), 3456–3463.
Ye, Z., Yang, Y., Li, X., Cao, D., and Ouyang, D. (2019). An integrated transfer learning and multitask learning approach for pharmacokinetic parameter prediction. Mol. Pharm. 16 (2), 533–541. doi:10.1021/acs.molpharmaceut.8b00816
Ying, X. (2019). An overview of overfitting and its solutions. J. Phys. Conf. Ser. 1168 (2), 022022. doi:10.1088/1742-6596/1168/2/022022
You, Y., Lai, X., Pan, Y., Zheng, H., Vera, J., Liu, S., et al. (2022a). Artificial intelligence in cancer target identification and drug discovery. Signal Transduct. Target Ther. 7 (1), 156. doi:10.1038/s41392-022-00994-0
You, Y., Zhang, L., Tao, P., Liu, S., and Chen, L. (2022b). Spatiotemporal transformer neural network for time-series forecasting. Entropy (Basel) 24 (11), 1651. doi:10.3390/e24111651
Yu, W., and MacKerell, A. D. (2017). Computer-aided drug design methods. Methods Mol. Biol. 1520, 85–106. doi:10.1007/978-1-4939-6634-9_5
Zhang, L., Badai, J., Wang, G., Ru, X., Song, W., You, Y., et al. (2023a). Discovering hematoma-stimulated circuits for secondary brain injury after intraventricular hemorrhage by spatial transcriptome analysis. Front. Immunol. 14, 1123652. doi:10.3389/fimmu.2023.1123652
Zhang, L., Bai, W., Yuan, N., and Du, Z. (2019a). Comprehensively benchmarking applications for detecting copy number variation. PLoS Comput. Biol. 15 (5), e1007069. doi:10.1371/journal.pcbi.1007069
Zhang, L., Dai, Z., Yu, J., and Xiao, M. (2021a). CpG-island-based annotation and analysis of human housekeeping genes. Brief. Bioinform 22 (1), 515–525. doi:10.1093/bib/bbz134
Zhang, L., Fan, S., Vera, J., and Lai, X. (2023b). A network medicine approach for identifying diagnostic and prognostic biomarkers and exploring drug repurposing in human cancer. Comput. Struct. Biotechnol. J. 21, 34–45. doi:10.1016/j.csbj.2022.11.037
Zhang, L., Fu, C., Li, J., Zhao, Z., Hou, Y., Zhou, W., et al. (2019b). Discovery of a ruthenium complex for the theranosis of glioma through targeting the mitochondrial DNA with bioinformatic methods. Int. J. Mol. Sci. 20 (18), 4643. doi:10.3390/ijms20184643
Zhang, L., Liu, G., Kong, M., Li, T., Wu, D., Zhou, X., et al. (2021b). Revealing dynamic regulations and the related key proteins of myeloma-initiating cells by integrating experimental data into a systems biological model. Bioinformatics 37 (11), 1554–1561. doi:10.1093/bioinformatics/btz542
Zhang, L., Liu, Y., Wang, M., Wu, Z., Li, N., Zhang, J., et al. (2017a). EZH2-CHD4-and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J. Mol. Cell Biol. 9 (6), 477–488. doi:10.1093/jmcb/mjx056
Zhang, L., Lv, J., Xiao, M., Li, Y., and Zhang, L. (2021c). Exploring the underlying mechanism of action of a traditional Chinese medicine formula, Youdujing ointment, for cervical cancer treatment. Quant. Biol. 0 (0), 292–303. doi:10.15302/j-qb-021-0236
Zhang, L., Song, W., Zhu, T., Liu, Y., Chen, W., and Cao, Y. (2024). ConvNeXt-MHC: improving MHC-peptide affinity prediction by structure-derived degenerate coding and the ConvNeXt model. Brief. Bioinform 25 (3), bbae133. doi:10.1093/bib/bbae133
Zhang, L., Xiao, M., Zhou, J., and Yu, J. (2018). Lineage-associated underrepresented permutations (LAUPs) of mammalian genomic sequences based on a Jellyfish-based LAUPs analysis application (JBLA). Bioinformatics 34 (21), 3624–3630. doi:10.1093/bioinformatics/bty392
Zhang, L., Zhang, L., Guo, Y., Xiao, M., Feng, L., Yang, C., et al. (2021d). MCDB: a comprehensive curated mitotic catastrophe database for retrieval, protein sequence alignment, and target prediction. Acta Pharm. Sin. B 11 (10), 3092–3104. doi:10.1016/j.apsb.2021.05.032
Zhang, L., and Zhang, S. (2017). Using game theory to investigate the epigenetic control mechanisms of embryo development: comment on: “epigenetic game theory: how to compute the epigenetic control of maternal-to-zygotic transition” by Qian Wang et al. Phys. Life Rev. 20, 140–142. doi:10.1016/j.plrev.2017.01.007
Zhang, L., Zhao, J., Bi, H., Yang, X., Zhang, Z., Su, Y., et al. (2021e). Bioinformatic analysis of chromatin organization and biased expression of duplicated genes between two poplars with a common whole-genome duplication. Hortic. Res. 8 (1), 62. doi:10.1038/s41438-021-00494-2
Zhang, L., Zheng, C., Li, T., Xing, L., Zeng, H., Li, T., et al. (2017b). Building up a robust risk mathematical platform to predict colorectal cancer. Complexity 2017 (1), 1–14. doi:10.1155/2017/8917258
Zhang, Q., Zhang, H., Zhou, K., and Zhang, L. (2023c). Developing a physiological signal-based, mean threshold and decision-level fusion algorithm (PMD) for emotion recognition. Tsinghua Sci. Technol. 28 (4), 673–685. doi:10.26599/TST.2022.9010038
Zhang, S., Yan, Z., Huang, Y., Liu, L., He, D., Wang, W., et al. (2022). HelixADMET: a robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer. Bioinformatics 38 (13), 3444–3453. doi:10.1093/bioinformatics/btac342
Zhang, Y., Deng, Z., Xu, X., Feng, Y., and Junliang, S. (2023d). Application of artificial intelligence in drug–drug interactions prediction: a review. J. Chem. Inf. Model. 64, 2158–2173. doi:10.1021/acs.jcim.3c00582
Zhang, Y., Li, S., Xing, M., Yuan, Q., He, H., and Sun, S. (2023e). Universal approach to de novo drug design for target proteins using deep reinforcement learning. ACS Omega 8 (6), 5464–5474. doi:10.1021/acsomega.2c06653
Zhang, Y., Vass, M., Shi, D., Abualrous, E., Chambers, J. M., Chopra, N., et al. (2023f). Benchmarking refined and unrefined AlphaFold2 structures for hit discovery. J. Chem. Inf. Model. 63 (6), 1656–1667. doi:10.1021/acs.jcim.2c01219
Zhavoronkov, A., Ivanenkov, Y. A., Aliper, A., Veselov, M. S., Aladinskiy, V. A., Aladinskaya, A. V., et al. (2019). Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37 (9), 1038–1040. doi:10.1038/s41587-019-0224-x
Keywords: artificial intelligence, pharmacology, drug discovery, compound pharmacokinetic prediction, clinical pharmacology
Citation: Li B, Tan K, Lao AR, Wang H, Zheng H and Zhang L (2024) A comprehensive review of artificial intelligence for pharmacology research. Front. Genet. 15:1450529. doi: 10.3389/fgene.2024.1450529
Received: 17 June 2024; Accepted: 26 August 2024;
Published: 03 September 2024.
Edited by:
Nguyen Quoc Khanh Le, Taipei Medical University, TaiwanReviewed by:
Weifan Zheng, North Carolina Central University, United StatesMingsong Shi, University of Electronic Science and Technology of China, China
Copyright © 2024 Li, Tan, Lao, Wang, Zheng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Le Zhang, emhhbmdsZTA2QHNjdS5lZHUuY24=; Huiru Zheng, aC56aGVuZ0B1bHN0ZXIuYWMudWs=